1. Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal disorder characterized by a progressive degeneration of the upper and lower motor neurons of the brain, brainstem, and spinal cord, but also in the frontotemporal cortex and hippocampus in a fraction of patients [
1,
2,
3], leading to muscle weakness, wasting and spasticity [
4]. In 2006, the transactive response (TAR) DNA-binding protein (TDP-43) was identified as the main component of intraneuronal cytosolic ubiquitinated protein aggregates found in ALS and tau-negative, ubiquitin-positive frontotemporal lobar degeneration (FTLD) patients [
1,
2]. Numerous post-translational modifications of TDP-43 have been described in the aggregates, such as abnormal polyubiquitination and hyperphosphorylation as well as partial proteolysis to form C-terminal fragments [
1,
2,
5,
6].
The TDP-43 protein contains 414 amino acid residues and the encoding gene
TARDBP is located on chromosome 1. It comprises an N-terminal domain (NTD, aa 1–76), a nuclear localization signal (NLS, aa 82–98), two RNA recognition motifs (RRM1, aa 106–176, and RRM2, aa 191–259), a nuclear export signal (NES, aa 239–250), and a C-terminal domain (CTD, aa 274–414), which encompasses a prion-like glutamine/asparagine-rich (Q/N) region (aa 345–366) and a glycine-rich region (aa 366–414) [
7,
8,
9,
10,
11,
12]. TDP-43 was first proposed to be natively dimeric or at least to exist in a monomer-dimer equilibrium under normal physiological conditions [
13,
14,
15,
16]. The dimerisation of TDP-43 was shown to occur through interactions of the N-terminal domains, and these interactions were proposed to be responsible for the dimerisation of the entire full-length protein [
14,
15,
16]. It was also found that the deletion of the N terminus, or even the first nine residues, is sufficient to abolish NTD folding, dimerisation and the TDP-43–regulated RNA splicing [
15]. With the elucidation of the monomeric structure of NTD with NMR spectroscopy, models of the dimer were proposed [
12,
17,
18]. Later on, the NTD and full-length TDP-43 were found to form oligomers beyond the dimer and that TDP-43 oligomerisation was necessary for its function as mutants destabilising the NTD-NTD interface without affecting the NTD folded structure inhibit the splicing activity of full-length TDP-43 expressed in cells [
19,
20].
Although NTD-mediated TDP-43 dimerisation/oligomerisation appears to be essential for TDP-43 function [
14,
15,
19,
20], extensive NTD-mediated TDP-43 oligomerisation was also found to enhance the liquid-liquid phase separation (LLPS) at physiological concentrations [
20,
21,
22], as well as formation of solid phase cytotoxic inclusions in the cytoplasm [
15,
23].
The NTD monomer consists of seven-eight β-strands and a single α-helix arranged in an axin-1 DIX domain fold [
12,
17,
18,
19,
20], which facilitates dimer formation and further oligomerisation by a head-to-tail disposition of the individual subunits [
19,
20,
24]. The conformational stability of the domain, as measured by the hydrogen/deuterium exchange at pH 3.8 and 25 °C, was found to be 15.9 ± 0.5 kJ mol
−1 [
12]. Using equilibrium urea denaturation curves monitored with intrinsic fluorescence and far-UV circular dichroism at pH 7.4 and 25 °C, higher conformational stability values of 20.1 ± 1.5 kJ mol
−1 and 21.8 ± 1.5 kJ mol
−1 were found, respectively [
24], most probably due to the neutral pH of the measurements. The β-strands, α-helix, and three of four turns are rigid, although the loop formed by residues 47–53 appeared mobile [
17]. All X-Pro peptide bonds adopt a trans configuration and the two cysteine residues at positions 39 and 50 are reduced and distantly separated on the surface of the protein [
12,
17,
19,
20].
The NTD of TDP-43 follows a folding process characterized by a series of steps [
24]. The NTD unfolded state (U) consists of a pre-equilibrium between molecules having all the X-Pro peptide bonds in a native trans configuration and others with one or more X-Pro peptide bonds in a non-native cis configuration. The unfolded state U converts into a collapsed state (CS), which then converts into the fully folded monomer (F) following two parallel pathways: in the first CS converts directly to F, whereas in the second, CS converts into an intermediate state (I) that is on-pathway to F. All these U, CS and I states maintain the partition equilibrium between cis and trans-X-Pro peptide bonds, but folding can occur only from molecules having all-trans X-Pro bonds. Once the fully folded state F is attained, the NTD is able to dimerise into a head-to-tail homodimer that presents the further ability to form higher molecular weight assemblies [
24]. Following urea denaturation at equilibrium, a native-like dimeric state was also detected at low urea concentrations (F*). Moreover, the various NMR studies of TDP-43 NTD show spectral differences in the various experimental conditions in which the protein domain is folded [
10,
12,
14,
17,
18,
20]. Overall, these structural and folding studies have revealed that TDP-43 NTD is a highly plastic protein able to adopt different conformational states.
In this work, we purified the NTD of TDP-43 and identified conditions to obtain two conformational states of the protein—one that is fully folded, native and dimeric and another, obtained through the use of a zwitterionic detergent Sulfobetaine 3-10 (SB3-10), that presents a monomeric, alternative, non-native, partially folded structure. We will show that the SB3-10-stabilised conformational state of TDP-43 NTD retains a cooperative fold and high conformational stability in spite of a remarkably different three-dimensional structure but does not retain the ability to dimerise and has a remarkably lower propensity to oligomerise and aggregate.
3. Discussion
The NTD of TDP-43 is able to dimerise with a head-to-tail arrangement into a dimeric structure that has been solved by both NMR and X-ray crystallography [
19,
20]. Following the head-to-tail interaction, this process does not terminate at the dimeric level but proceeds to form larger oligomers [
19,
20]. The role of NTD dimerisation and oligomerisation in the aggregation process of full-length TDP-43 is still discussed. It has been proposed to be responsible for the oligomerisation of the entire full-length protein [
12,
14,
15,
16,
17,
18,
19,
20]. Oligomerisation of full-length TDP-43 mediated by the NTD is essential for TDP-43 function [
14,
15,
19,
20], but also favours liquid–liquid phase separation and solid-phase inclusion formation of the full-length protein [
20,
21,
22]. The structural plasticity of the TDP-43 NTD resulting in its in dimerisation/oligomerisation is also observed in its folding process from a fully unfolded state, in which the protein domain forms a number of partially folded states before achieving the fully folded dimeric structure, and even populates, at low denaturant concentrations, a native-like dimeric state distinct from the fully native dimeric conformation [
24]. Such conformational heterogeneity is also witnessed by NMR spectral differences in the various experimental conditions in which the protein domain is folded [
10,
12,
14,
17,
18,
20].
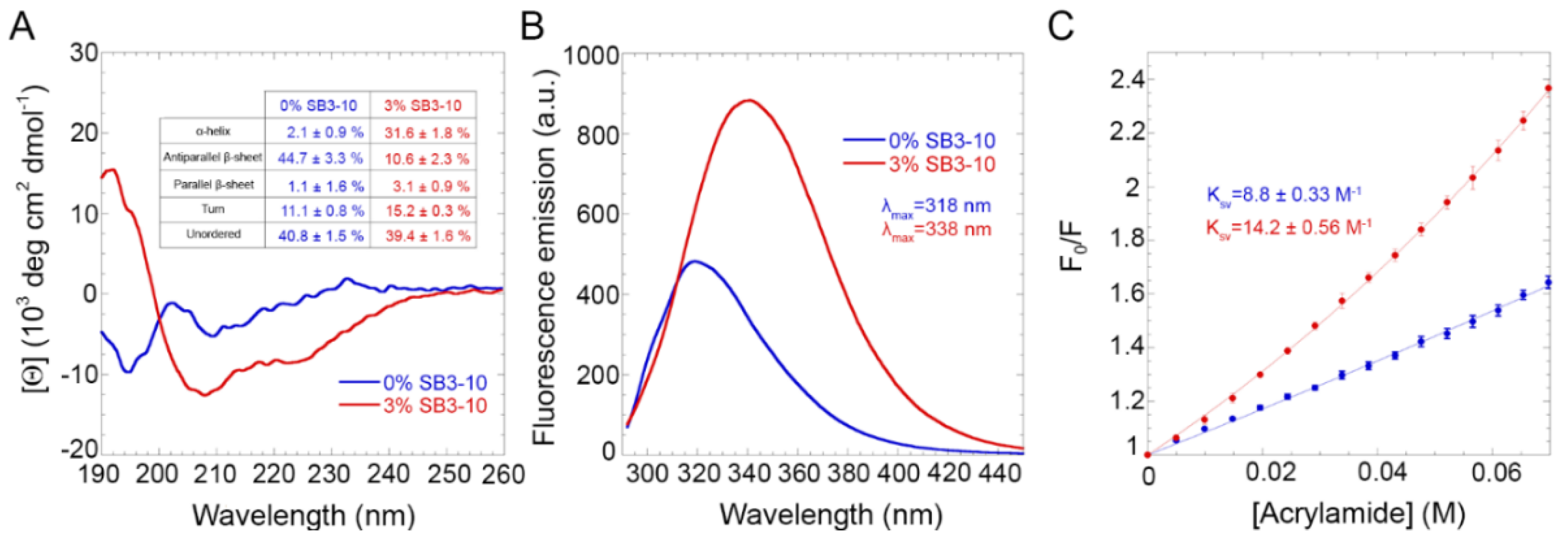
The use of very small SB3-10 percent concentrations, typically in the range of 1.4–3.0% (
w/
v), has allowed us to isolate a stable and well-defined alternative conformation. While the native conformation in the absence of SB3-10 presents a far-UV CD spectrum comparable to those reported in the literature [
14,
18,
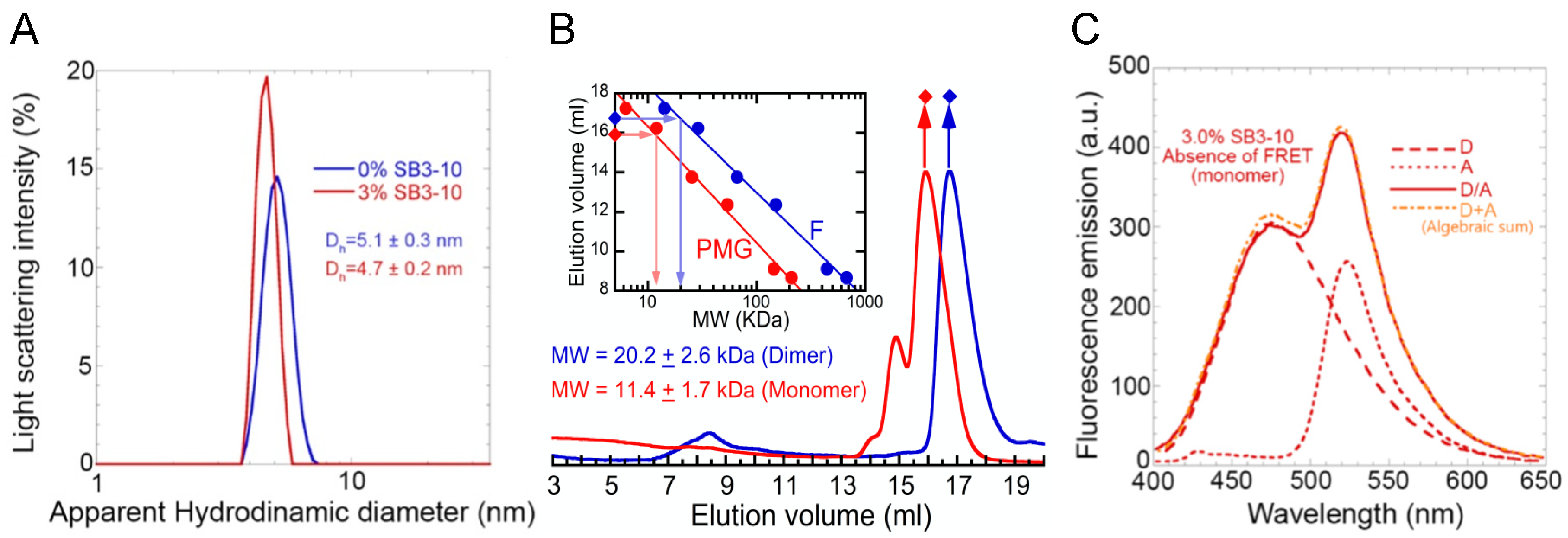
24], the alternative one has a different spectrum characterized by a higher content of α-helical structure, a lower content of β-sheet structure and a slightly higher content of turns. NMR spectroscopy confirmed these observations (discussed below). The intrinsic fluorescence spectrum and Stern–Volmer assay also indicate that the Trp68 residue is more exposed to the solvent in this new conformational state relative to the native structure. The DLS, SEC and FRET analyses all indicate that the NTD in 3.0% SB3-10 is a monomer, unlike the native dimer populated in the absence of this detergent but under identical conditions in terms of protein concentration and solution conditions.
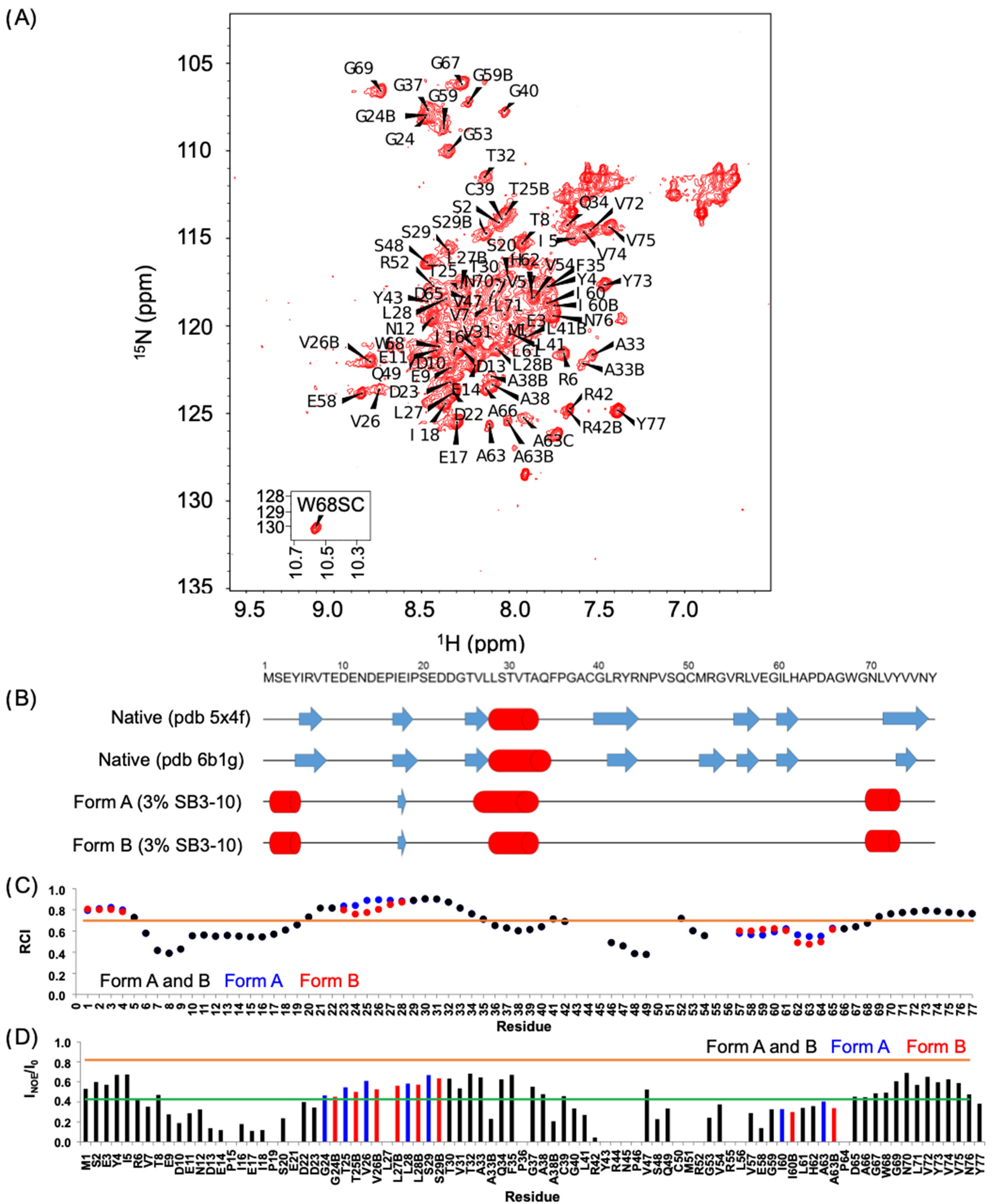
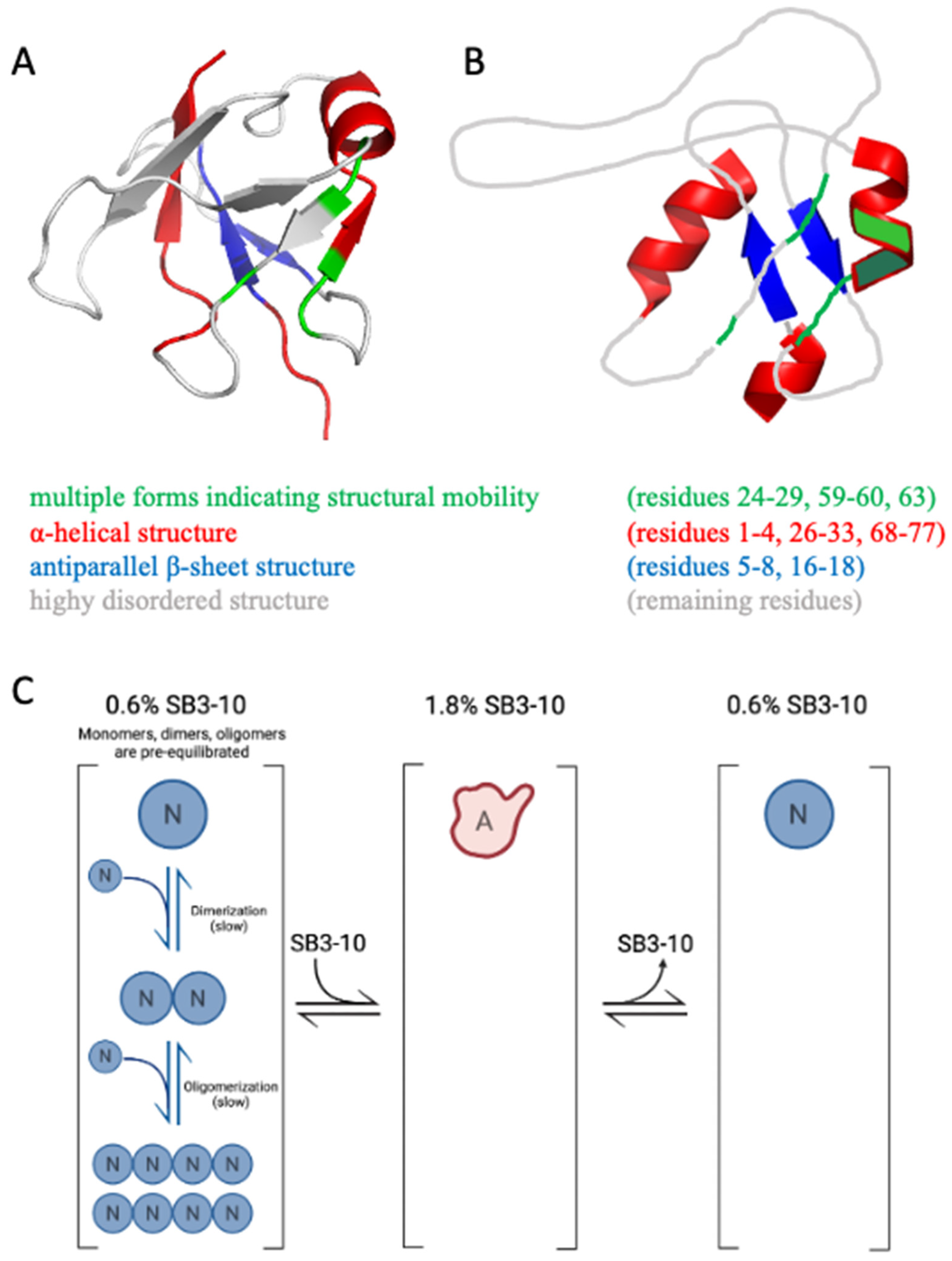
The NMR spectral properties of the SB3-10 dependent conformational state differ from those previously observed for the native NTD, as it clearly appears from the low resonance spreading of its HSQC spectrum. In particular, the NMR characterisation indicates the presence of a partially unfolded state together with the conservation of the native α-helix and of the native antiparallel β-sheet contributed by strands 1-2, although the latter is remarkably more dynamic than the native one (
Figure 8A,B). On the basis of chemical shift values, two other non-native α-helices located in the N- and C-termini are detected encompassing residues 1–4 and 68–77, respectively (
Figure 8A,B). Moreover, the first part of the central native helix along with the preceding β-strand (residues 24–29) and other spatially close residues that in the native structure faces this portion (residues 59,60,63) form a cluster that is present in two different forms in equilibrium, named here A and B (
Figure 8A,B). The intensity of the two forms is temperature dependent, suggesting that the two forms might be related to two conformers with different compactness. For some residues, the two forms show an NH combined chemical shift difference quite marked (between 0.25 and 0.94 ppm) indicating a different chemical environment that reflects distinct conformations and also indicates structure formation under these conditions for at least one form.
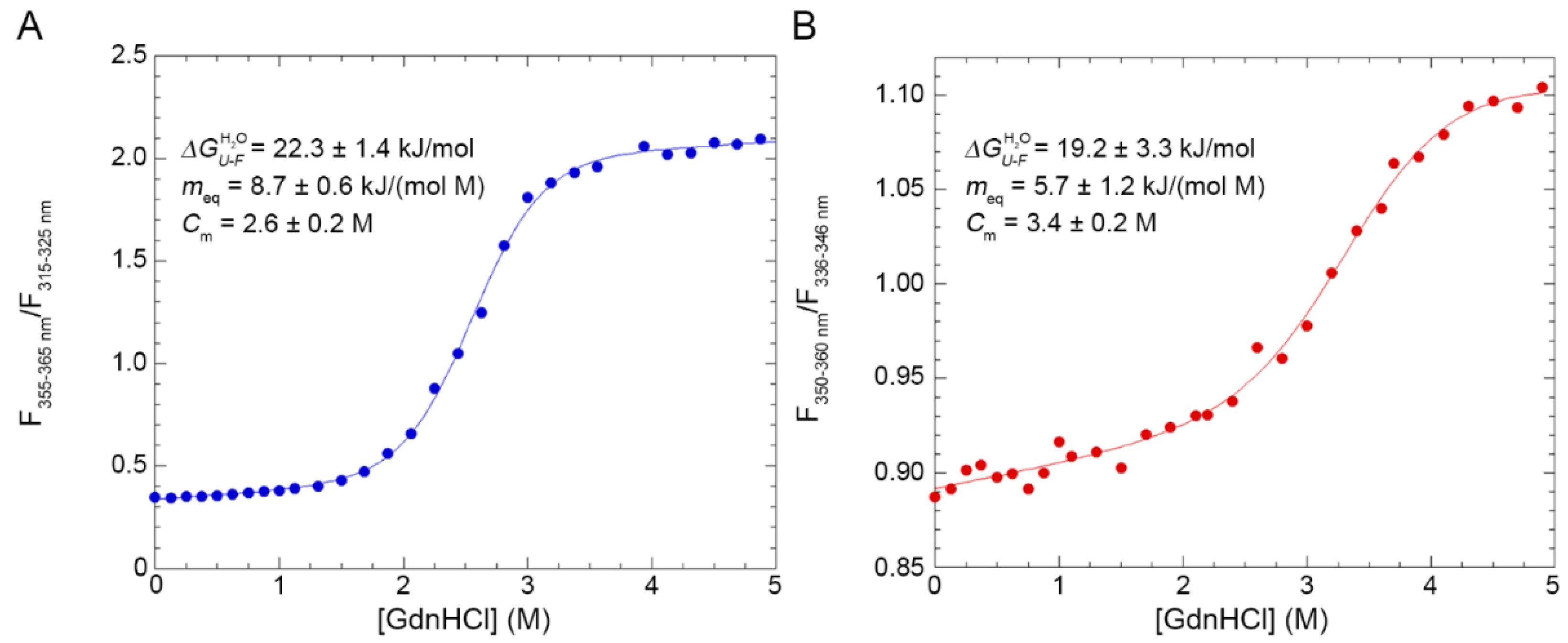
The alternative conformation adopted in 3.0% (w/v) SB3-10 is able to return to its initial native conformation if diluted to a smaller concentration of detergent, conditions in which the native conformation is stable, showing how this process is fully reversible. Its conformational stability () in 3.0% SB3-10 is only approximately 3 kJ/mol lower than that of the native conformation in 0.0% SB3-10, as determined with GdnHCl-induced equilibrium denaturation. The meq value, which reports on the cooperativity of the GdnHCl-induced unfolding transition and on the change of solvent-exposed surface area (∆ASA) upon unfolding, is 35 ± 15% lower than that of the native state but is sufficiently high to indicate a cooperative transition. The lower meq value suggests, however, significantly lower cooperativity, possibly due to the increase of the surface area exposed to the solvent in the folded alternative conformation compared to the folded native conformation.
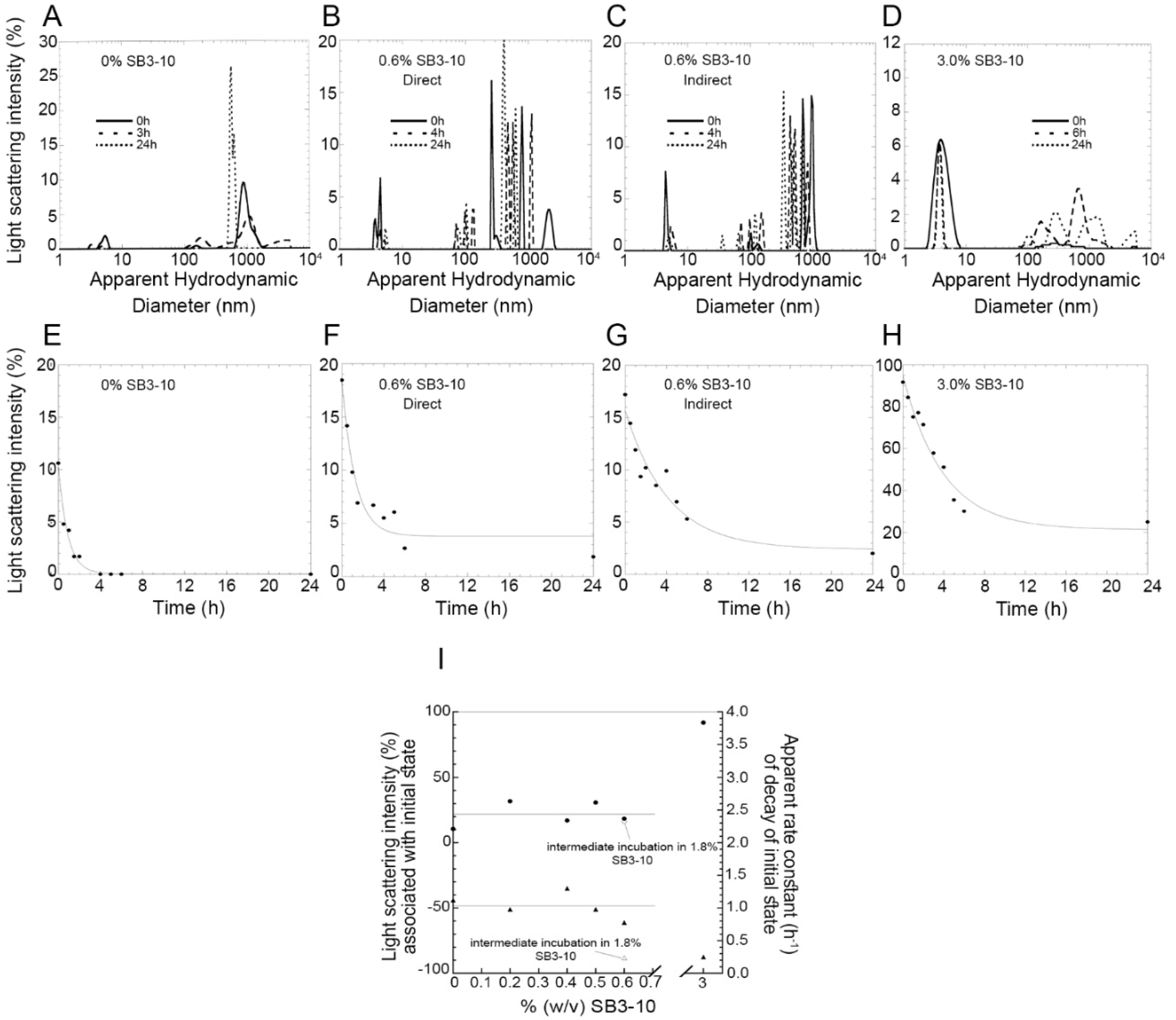
In our time-dependent aggregation assays, we observed a slow, yet detectable, aggregation in the sample left untreated with SB3-10 and in those samples in which the concentration of the detergent was kept low. In both cases, the disappearance of the low molecular weight species (either monomeric, dimeric or oligomeric) was not accompanied by detectable changes in the far-UV CD and intrinsic fluorescence spectra, indicating the maintenance of the initial conformational states in both cases and further supporting the notion that TDP-43 NTD self-assembly consists in a native-like aggregation process similar to that observed for other proteins [
35,
36,
37,
38,
39]. Aggregation is slower in 3.0% (
w/
v) SB3-10, but this effect cannot be attributed necessarily to the SB3-10-mediated conformational change of the TDP-43 NTD, as it may well arise from the effect of the detergent in the formation of intermolecular interactions. However, when the aggregation processes were compared with and without pre-incubation with 1.8% (
w/
v) SB3-10, but with an identical final SB3-10 concentration of 0.6% (
w/
v) promoting the native state, a slower aggregation was observed in the NTD sample pre-incubated in SB3-10, in spite of the rapid reversibility of the conformational change and the native conformation adopted by the protein domain in both cases. This can be attributed to the ability of the condition promoting temporarily the alternative conformation to monomerise the protein and disrupt any intermolecular interaction (
Figure 8C). Under physiological conditions, the native monomers, dimers and oligomers exist initially in a pre-equilibrium where the mutual conversions are slow (
Figure 8, left). This condition is favourable for aggregation as dimers and oligomers are present already. The alternative state has a very low propensity to oligomerise (
Figure 8C, centre), but the lower propensity of the native state to oligomerise and self-assemble is maintained when the alternative state is re-located under native conditions to form the native state (
Figure 8C, right) because the conversion to dimers and oligomers is slow.
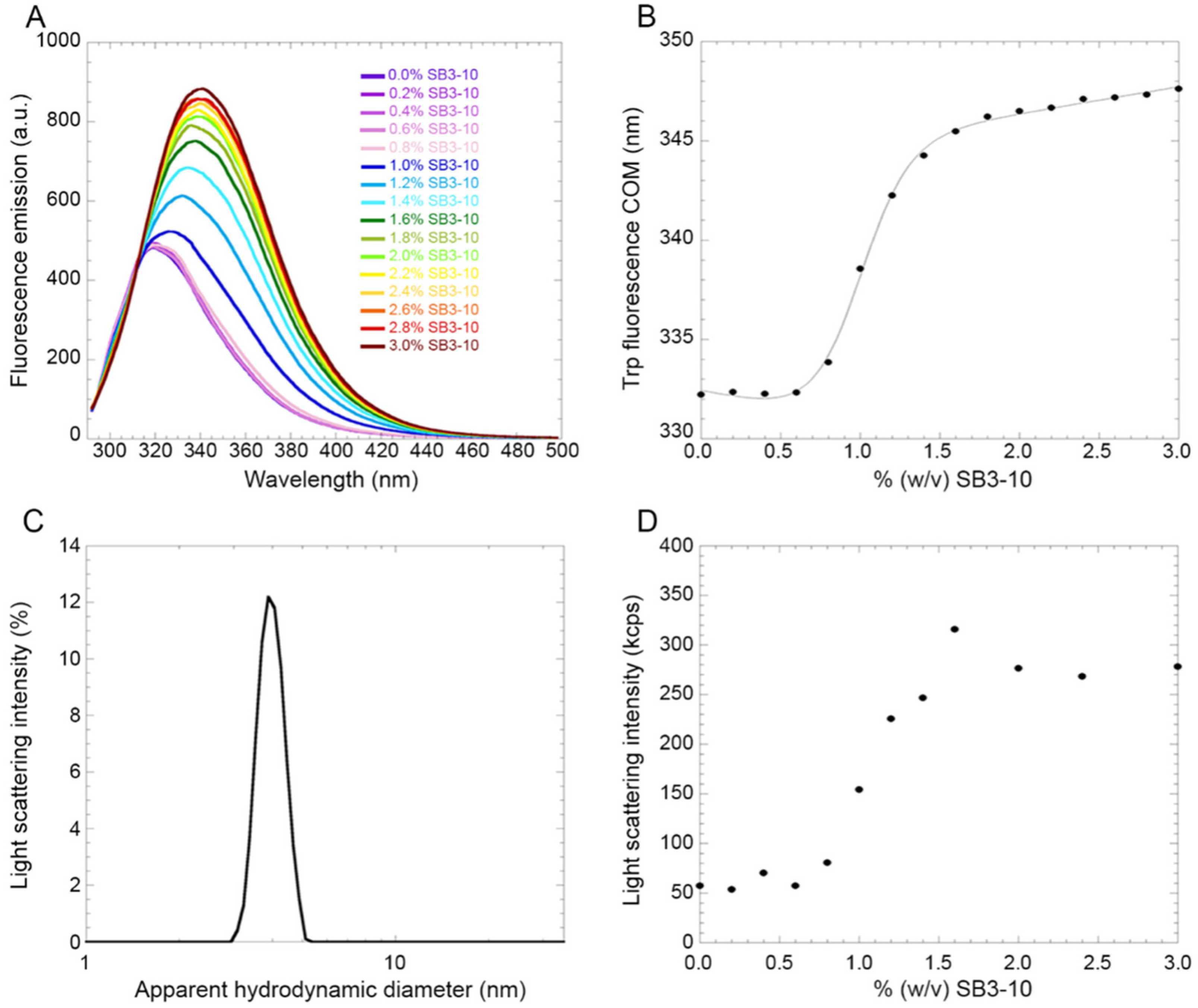
Under conditions in which the alternative conformational state was observed (3.0% SB3-10), the critical micellar concentration (CMC) of the detergent was largely exceeded, leading to hypothesize that the NTD conformational change was induced by the newly formed SB3-10 micelles. The SB3-10 titration of the TDP-43 NTD structural change monitored spectroscopically, on the one hand, and of the SB3-10 micelle formation monitored with light scattering intensity, on the other hand, indicate that the two transitions coincide as they start and end at identical SB3-10 concentrations and also have similar transition midpoints. This indicates that the structural change of TDP-43 NTD is driven by SB3-10 micelle formation. Therefore, the role of biological micelles formed intraneuronally can be decisive in this regard and promote structural conversions into alternative conformations of TDP-43 NTD that may resist oligomerisation, LLPS and inclusion formation, also affecting the full-length protein.
5. Materials and Methods
5.1. Chemicals
SB3-10, acrylamide and ThT were from Sigma-Aldrich (St. Louis, MO, USA). DTT was from Thermo Fisher Scientific (Waltham, MA, USA).
5.2. Gene Cloning, Expression and Purification
Gene cloning, expression and purification of TDP-43 NTD and the Cys50Ser (C50S) single-point mutant were performed as previously described [
24]. The purified protein contained 77 residues and the MHHHHHHSSGVDLGTENLYFQS sequence fused to the N-terminus for a total of 99 residues. It was maintained at 1.6–3.0 mg/mL (150–270 µM) in 5 mM sodium phosphate buffer, 50 mM NaCl, 1 mM DTT, pH 7.4, −20 °C. Protein purity was checked with SDS page. Protein concentration was measured using a SHIMADZU UV-1900 UV-Vis spectrophotometer at a wavelength of 280 nm with an extinction coefficient (ε
280) of 12,950 M
−1 cm
−1.
5.3. Far-UV Circular Dichroism Spectroscopy
The TDP-43 NTD sample was centrifuged at 18,000×
g for 15 min, 4 °C, and diluted to prepare two samples containing 0.5 mg/mL (45 µM) NTD, in 5 mM sodium phosphate buffer, 50 mM NaCl, 1 mM DTT, pH 7.4, one in the absence and the other in the presence of 3.0% (
w/
v) SB3-10. Spectra were acquired at 25 °C in the far-UV between 190 and 260 nm using a 0.1 mm path length cell on a Jasco J-810 spectropolarimeter (Tokyo, Japan) equipped with a thermostated cell holder attached to a Thermo Haake C25P water bath (Karlsruhe, Germany). Spectra were then blank subtracted, truncated when the high tension (HT) signal was higher than 700 V and normalized to mean residue ellipticity using
where [
θ] is the mean residue ellipticity in deg cm
2 dmol
−1,
θ is the ellipticity in mdeg, the optical path is in cm, concentration is in g/L, and the molecular weight is in g/mol.
5.4. Fluorescence Spectroscopy
The TDP-43 NTD sample was centrifuged at 18,000× g for 15 min, at 4 °C, and two samples containing 0.5 mg/mL (45µM) NTD, in 5 mM sodium phosphate buffer, 50 mM NaCl and 1 mM DTT, pH 7.4, were prepared, one in the absence and the other in the presence of 3.0% SB3-10. Fluorescence spectra were acquired at 25 °C from 290 to 500 nm (excitation at 280 nm) using a 3 × 3 mm black wall quartz cell cuvette on an Agilent Cary Eclipse spectrofluorimeter (Agilent Technologies, Santa Clara, CA, USA) equipped with a thermostated cell holder attached to an Agilent PCB 1500 water Peltier system (Agilent Technologies, Santa Clara, CA, USA). Excitation and emission slits were 5 nm. Spectra were then blank subtracted.
5.5. Acrylamide Quenching Experiment
The TDP-43 NTD sample was centrifuged at 18,000×
g for 15 min, at 4 °C, and two 1 mL samples containing 0.05 mg/mL (4.5 µM) NTD, in 5 mM sodium phosphate buffer, 50 mM NaCl and 1 mM DTT, pH 7.4, were prepared, one in the absence and the other in the presence of 3.0% SB3-10. For each sample, a first fluorescence spectrum was acquired at 25 °C from 300 to 420 nm (excitation at 280 nm) using a 10 × 4 mm quartz cuvette under magnetic stirring on an Agilent Cary Eclipse spectrofluorimeter (Agilent Technologies, Santa Clara, CA, USA) equipped with a thermostated cell holder attached to an Agilent PCB 1500 water Peltier system. Excitation and emission slits were 5 nm. Then, 5 µL of 1 M acrylamide was added directly to the cuvette and another spectrum was recorded. This step was repeated 15 times. For each recorded spectrum, the total fluorescence was corrected to take account of the dilution with the acrylamide solution. The quenching of acrylamide in the 0.0% SB3-10 sample was analyzed with the Stern-Volmer equation
where
F0 and
F are the integrated fluorescence intensity areas at 300–400 nm in the absence and presence of acrylamide, respectively,
KSV is the Stern-Volmer constant and [
Q] is the concentration of the quencher (acrylamide) in the cuvette. The quenching of acrylamide in the 3.0% SB3-10 sample was instead analysed using
where
KST is a constant that considers the static quenching caused by the binding of acrylamide to tryptophan residues.
5.6. Nuclear Magnetic Resonance Spectroscopy
NMR spectra were acquired with a Bruker Avance 500 MHz and a Bruker Avance Neo 700 MHz NMR spectrometers on
13C,
15N uniformly labelled TDP-43 NTD samples (250 µM) in 5 mM sodium phosphate, 50 mM NaCl, 3% (
w/
v) SB3-10, 1 mM DTT, 5% (
v/
v) D
2O. For reference purposes, trimethylsilylpropanoic acid (TSP) was added [
40]. For backbone assignment a series of 2D HSQC and 3D HNCA, HN(CO)CA, HNCACB, HNCO, and HN(CA)CO spectra were acquired at 25 °C and 17 °C with 2048 × 256, 2048 × 50 × 96, 2048 × 50 × 96, 2048 × 50 × 128, 2048 × 50 × 72, 2048 × 50 × 72 complex points, respectively. The spectral widths were 13.8 ppm (
1H), 35 ppm (
15N) and 30 ppm (
13C in HNCA and HN(CO)CA), 16 ppm (
13C in HNCO and HN(CA)CO), 80 ppm (
13C in HNCACB). Secondary structures of individual residues were assessed by chemical shift index (CSI) [
27], Talos+ [
26] and Secondary Structure Propensity (SSP) [
28]. According to Wishart [
27], helix (H) or
®-strand (E) were indicated in
Table S1 only if present a consensus for CSI for Cα, Cβ, C
®, and CO. A series of 2D HSQC spectra were acquired at different temperature values (290–312K every 2 K). 2D
15N{
1H} NOE measurements were also performed using a 700 MHz spectrometer at 25 °C and 17 °C, with a 5 s relaxation or saturation delay, 256 t
1 increments of 2048 complex data points, and 48 scans/t
1. The spectra were processed with Topspin 3.5 (Bruker Biospin) and analysed in Sparky [
41]. NOE values were calculated as the ratio of peak height with and without saturation.
5.7. Dynamic Light Scattering
The TDP-43 NTD sample was centrifuged at 18,000× g for 15 min, at 4 °C, and filtered with Whatman Anotop 0.02 µm cut-off filters. Two samples containing 1.35 mg/mL (121 µM) NTD, in 5 mM sodium phosphate buffer, 50 mM NaCl and 1 mM DTT, pH 7.4, were prepared, one in the absence and the other in the presence of 3.0% SB3-10. Their size distributions (distribution of apparent hydrodynamic diameter by light scattering intensity) were then acquired on a Malvern Panalytical Zetasizer Nano S DLS device (Malvern Panalytical, Malvern, UK), thermostated at 25 °C with a Peltier temperature controller using a 3 × 3 mm black wall quartz cell cuvette. The refractive index and viscosity, acquired using a 2WAJ ABBE bench refractometer from Optika Microscopes (Bergamo, Italy) and a Viscoball viscometer (Fungilab, Barcelona, Spain), were 1.331 and 0.8998 cp for the 0.0% SB3-10 sample and 1.338 and 1.0530 cp for the 3.0% SB3-10 sample, respectively. The measurements were acquired with the cell position 4.20 and attenuator index 10. The light scattering intensity was also measured for a blank sample containing 3.0% SB3-10 without protein and was found to be negligible relative to the corresponding sample with protein, ruling out that the apparent hydrodynamic diameter measured for the protein sample under these conditions is affected by SB3-10 micelles. In another experiment, the same buffer solutions containing SB3-10 concentrations ranging from 0.0 to 3.0%, in the absence of protein were also analysed. Their light scattering intensities were then plotted as a function of SB3-10 concentration to monitor micelle formation.
5.8. Analytical SEC
The mother solution containing TDP-43 NTD was centrifuged at 18,000×
g for 15 min, at 4 °C, and two samples containing 0.5 mg/mL (45µM) NTD, in 5 mM sodium phosphate buffer, 50 mM NaCl and 1 mM DTT, pH 7.4, were prepared, one in the absence and the other in the presence of 3.0% SB3-10. 100 µL of NTD sample were loaded in a Superdex
® 200 Increase 10/300 GL column (GE Healthcare, Chicago, IL, USA) pre-equilibrated at 4 °C with the corresponding protein buffer and run using an Akta Pure 25L System (GE Healthcare, Chicago, IL, USA). The experimental
Ve was determined as the volume of the buffer passed through the column between sample injection and the point of highest absorbance at 280 nm. A calibration curve was determined by loading protein standards (100 µL) of known molecular weight (MW) separately, such as thyroglobulin (669 kDa), apoferritin (443 kDa), alcohol dehydrogenase (150 kDa), albumin (66 kDa), carbonic anhydrase (29 kDa) and α-lactalbumin (14.2 kDa), and obtaining their experimental
Ve. A plot of
Ve versus log[MW] was obtained with the 6 standard data points and fitted to a linear regression curve. This calibration curve was then used to interpolate the experimental
Ve (y axis) of NTD samples and to obtain the corresponding MW (x axis) and, thus, their oligomeric states. The MW values determined with this approach are valid only for globular proteins. For the NTD sample in 3.0% SB3-10, the calibration curve was re-determined mathematically to account for the differences between the hydrodynamic radii of folded and pre-molten globule states [
42]. Indeed, the relationships between the hydrodynamic radii of a folded protein (
), or a pre-molten globule (
), and the MW in Da, can be obtained using previously published equations [
41]. The experimental
Ve obtained for the NTD sample in 3.0% SB3-10 was interpolated into the re-determined calibration curve to obtain its MW.
5.9. Förster Resonance Energy Transfer (FRET)
The C50S mutant of TDP-43 NTD (containing only Cys39) was centrifuged at 18,000× g for 15 min, 4 °C, and diluted to prepare two samples containing 0.5 mg/mL (45 µM) C50S NTD, in 5 mM sodium phosphate buffer, 50 mM NaCl, 1 mM DTT, pH 7.4. The C50S NTD samples were labelled using the thiol-reactive probes 1,5-IAEDANS as a donor (D) and 6-IAF as an acceptor (A), respectively. Both fluorescent probes (Thermo Fisher Scientific, Waltham, MA, USA) were dissolved in dimethylformamide (DMF) at 30 mM concentration and added separately to the two C50S samples to a final probe:protein molar ratio of 10:1, for 22 h at 20 °C, under gentle and constant shaking. Then, gravity chromatography was performed to remove the excess of the unreacted probes using 6 mL of G-15 resin (Pharmacia, Uppsala, Sweden), previously equilibrated with 5 mM sodium phosphate buffer, 50 mM NaCl, pH 7.4.Labelledd protein fractions were collected and the concentration of the probe in each fraction was measured on a SHIMADZU UV-1900 UV-Vis spectrophotometer using ε336 = 5700 M−1cm−1 and ε491 = 8200 M−1cm−1 for 1,5-IAEDANS and 6-IAF, respectively. The C50S NTD concentration was determined using ε280 = 12,950 M−1cm−1 after subtraction of the absorbance contribution of the probes at 280 nm. Fractions where the probe:protein ratio was 1:1 were pooled and concentrated using centrifugal filter devices with a 3 kDa molecular weight cut-off (MWCO) cellulose membrane (Millipore, Burlington, MA, USA).
Two samples containing 0.1 mg/mL (9 µM) C50S NTD labelled with D and A, respectively, were prepared in 5 mM sodium phosphate buffer, 50 mM NaCl, 1 mM DTT and 3% SB3-10, pH 7.4, and mixed at 1:1 molar ratio (D:A). Two samples containing only C50S NTD labelled with D and C50S NTD labelled with A at the concentration of 0.05 mg/mL (4.5 µM) were also prepared. Fluorescence spectra of the various samples were acquired at 25 °C from 400 to 650 nm (excitation at 336 nm) using a 10 × 2 mm quartz cuvette on an Agilent Cary Eclipse spectrofluorimeter (Agilent Technologies, Santa Clara, CA, USA) equipped with a thermostated cell holder attached to an Agilent PCB 1500 water Peltier system. Excitation and emission slits were 5 nm. Spectra were then blank-subtracted.
5.10. SB3-10 Titration on NTD
The TDP-43 NTD sample was centrifuged at 18,000×
g for 15 min, at 4 °C. The experiment was performed by preparing 16 samples containing NTD at the concentration of 0.5 mg/mL (45µM), in 5 mM sodium phosphate buffer, 50 mM NaCl and 1 mM DTT, pH 7.4, and SB3-10 concentrations ranging from 0.0 to 3.0%. Fluorescence spectra were acquired at 25 °C from 290 to 500 nm (excitation at 280 nm) using a 3 × 3 mm black wall quartz cell cuvette on an Agilent Cary Eclipse spectrofluorimeter (Agilent Technologies, Santa Clara, CA, USA) equipped with a thermostated cell holder attached to an Agilent PCB 1500 water Peltier system. Excitation and emission slits were 5 nm. Spectra were then subtracted from blanks containing only buffers. For each spectrum, COM was calculated according to
where
Fi is the fluorescence emission at a wavenumber of
νi. The resulting COM values were then plotted as a function of SB3-10 concentration and fitted using the model edited by Santoro and Bolen [
31].
5.11. Guanidine Hydrochloride (GdnHCl)-Induced Denaturation
The TDP-43 NTD sample was centrifuged at 18,000×
g for 15 min, at 4 °C. The experiment was performed by preparing 30 samples containing NTD at the concentration of 0.05 mg/mL (4.5 µL), in 5 mM sodium phosphate buffer, 50 mM NaCl and 1 mM, pH 7.4, and GdnHCl concentrations ranging from 0.0 to 4.9 M. In a second experiment, 30 additional samples were prepared in the presence of 3.0% SB3-10. Fluorescence spectra were acquired at 25 °C from 290 to 500 nm (excitation at 280 nm) using a 10 × 2 mm quartz cuvette on an Agilent Cary Eclipse spectrofluorimeter (Agilent Technologies, Santa Clara, CA, USA), equipped with a thermostated cell holder attached to an Agilent PCB 1500 water Peltier system. Excitation and emission slits were 5 nm. Spectra were then subtracted from blanks containing only buffers. For each spectrum, the ratio between the sum of the fluorescence in bands of 10 nm in the post-transition region and the pre-transition region was calculated. These specific bands were chosen to contain the NTD fluorescence peak observed at 0.0 M GdnHCl and 4.9 M GdnHCl for the pre-transition and the post-transition, respectively. The resulting fluorescence ratios were then plotted as a function of the GdnHCl concentration and the traces obtained were then fitted to the model edited by Santoro and Bolen [
31].
5.12. Aggregation Kinetics Using DLS, Far-UV CD and Intrinsic Fluorescence Spectroscopies
The TDP-43 NTD sample was centrifuged at 18,000× g for 15 min, at 4 °C. Samples containing NTD at the concentration of 0.5 mg/mL (45 µM), in 5 mM sodium phosphate buffer, 50 mM NaCl, 1 mM DTT, pH 7.4, 25 °C were prepared using different SB3-10 concentrations (0.0, 0.2, 0.4, 0.6, 1.8 and 3.0%). One sample was obtained by treating the protein, initially in 0.0% SB3-10, with 1.8% SB3-10 for 1 h and then diluting it down to 0.6% SB3-10. Samples were analyzed every 30 min for the first 2 h, then hourly up to 6 h and the next day at 24 h. Their size distributions were acquired at 25 °C using a 3 × 3 mm black wall quartz cell cuvette on a Malvern Panalytical Zetasizer Nano S DLS device (Malvern, Worcestershire, UK), thermostated at 25 °C with a Peltier temperature controller. The refractive index and viscosity set on the instrument were changed according to the SB3-10 content of the sample. The measurements were acquired with the cell position 4.20 and attenuator index 10. The light scattering intensity percentage of the soluble protein peak was then calculated for each sample and plotted as a function of time. Their far-UV CD spectra were acquired at 25 °C between 190 and 260 nm using a 0.1 mm path length cell on the same Jasco J-810 spectropolarimeter described above. Spectra were then blank subtracted, masked when the high tension (HT) signal was higher than 700 V and normalized to mean residue ellipticity ([θ]). Their fluorescence spectra were acquired at 25 °C from 290 to 500 nm (excitation at 280 nm) using a 10 × 2 mm quartz cuvette on an Agilent Cary Eclipse spectrofluorimeter (Agilent Technologies, Santa Clara, CA, USA), equipped with a thermostated cell holder attached to an Agilent PCB 1500 water Peltier system. Excitation and emission slits were 5 nm. Spectra were then blank subtracted.
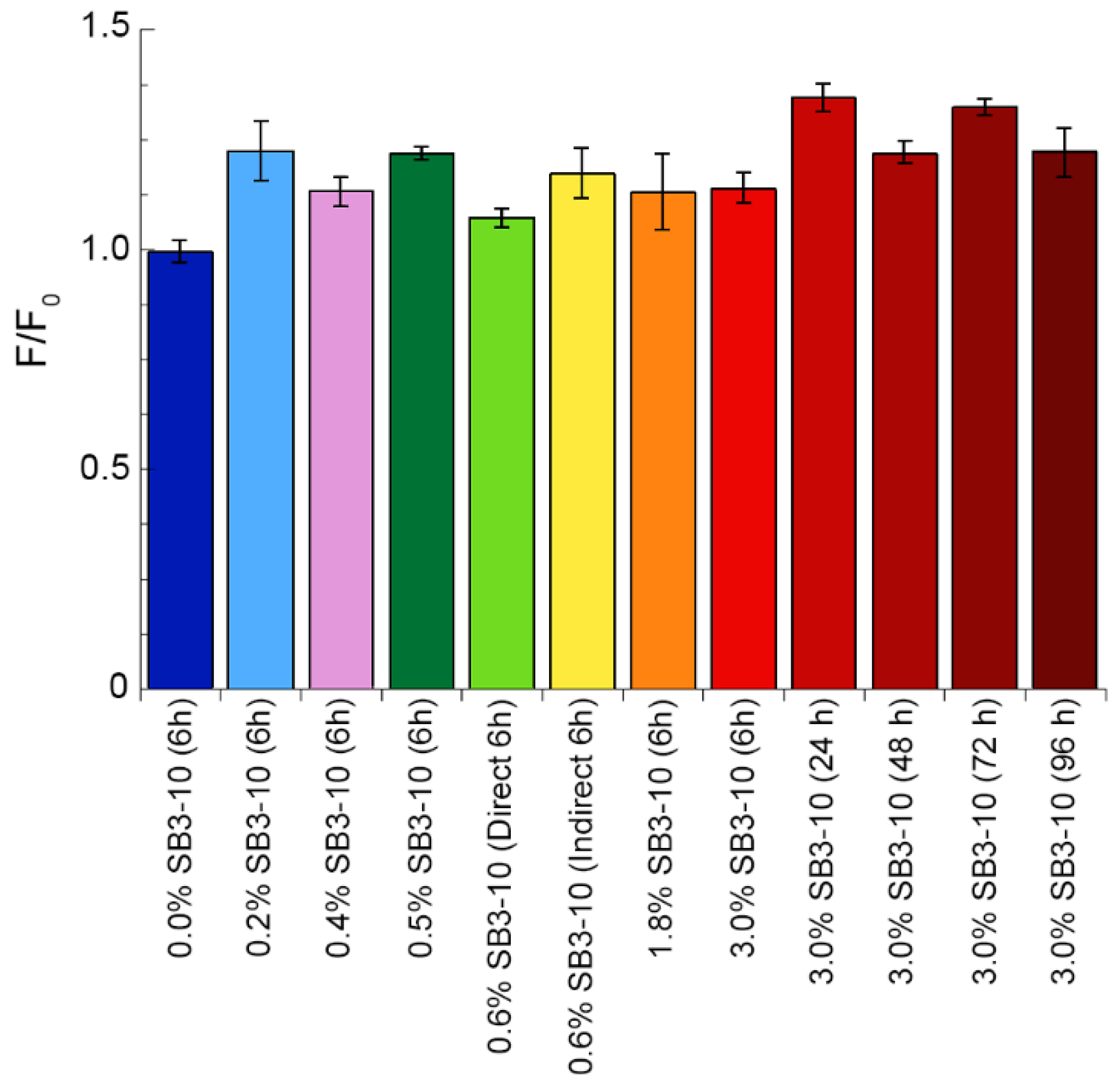
5.13. Thioflavin T (ThT) Assay
TDP-43 NTD samples were prepared at various SB3-10 concentrations, as described in the previous subsection and then incubated for 6 h at 25 °C, while the sample in 3.0% SB3-10 was incubated also for 24, 48, 72 and 96 h. 100 µL of buffer or protein sample were then mixed with 400 µL of 25 µM ThT. Fluorescence spectra were acquired at 25 °C from 450 to 600 nm (excitation at 440 nm) using a 10 × 2 mm quartz cuvette on an Agilent Cary Eclipse spectrofluorimeter (Agilent Technologies, Santa Clara, CA, USA) equipped with a thermostated cell holder attached to an Agilent PCB 1500 water Peltier system. Excitation and emissions slits were 5 and 10 nm, respectively. All spectra were blank subtracted (using only PBS as a blank). The ratio
F0/
F was then calculated for each sample, where
F and
F0 are the blank-subtracted fluorescence values at 485 nm of ThT+protein and free ThT, respectively. An over 5-fold ThT fluorescence increase was considered to be diagnostic for amyloid [
33,
34,
43,
44].
5.14. Statistical Analysis
Data were expressed as means ± SEM. Data pairs were compared using the Student’s t-test and the resulting p values were indicated in the text for each comparison. A p value < 0.05 was considered to be statistically significant.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







