
Functional Bacterial Amyloids: Understanding Fibrillation, Regulating Biofilm Fibril Formation and Organizing Surface Assemblies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Dedication to Sir Chris Dobson by Daniel Otzen
2. The Conundrum of Protein Misfolding and Aggregation
3. Amyloids in Sickness and in Health
4. Functional Bacterial Amyloids
5. The Curli System
6. The Fap System
7. The Major Curli Component, CsgA
8. Predicting FuBA Structure Is Easier Than Predicting Pathological Amyloid Structure
9. The Major Fap Component FapC: A Flexible Number of Imperfect Repeats and Variable Linker Lengths
10. Probing FuBA Fibrillation as Folding Steps with Denaturants: Similar Folding during Nucleation and Elongation Activation Steps
11. Structure-Based Design of Anti-FuBA Peptides
12. Using Small Molecules and Polyphenols to Target FuBA and Biofilm
- (i)
- Biofilm regulation: Impacts on biochemical processes inside or outside the bacterial cell that regulate biofilm formation. For example, EGCG disrupts quorum-sensing (QS) signaling by increasing the binding of pyocyanin (a central QS molecule) to FapC fibrils in P. aeruginosa [40]. EGCG also activates expression of the small non-coding RNA molecule RybB that binds to initiation codon of the csgD mRNA and inhibits expression of the curli transcription factor CsgD, thereby down-regulating production of the two main components of E. coli biofilm, namely, curli and pEtN-cellulose (bacterial cellulose where every second glucose group is modified with phosphoethanolamine) [109,113].
- (ii)
- (iii)
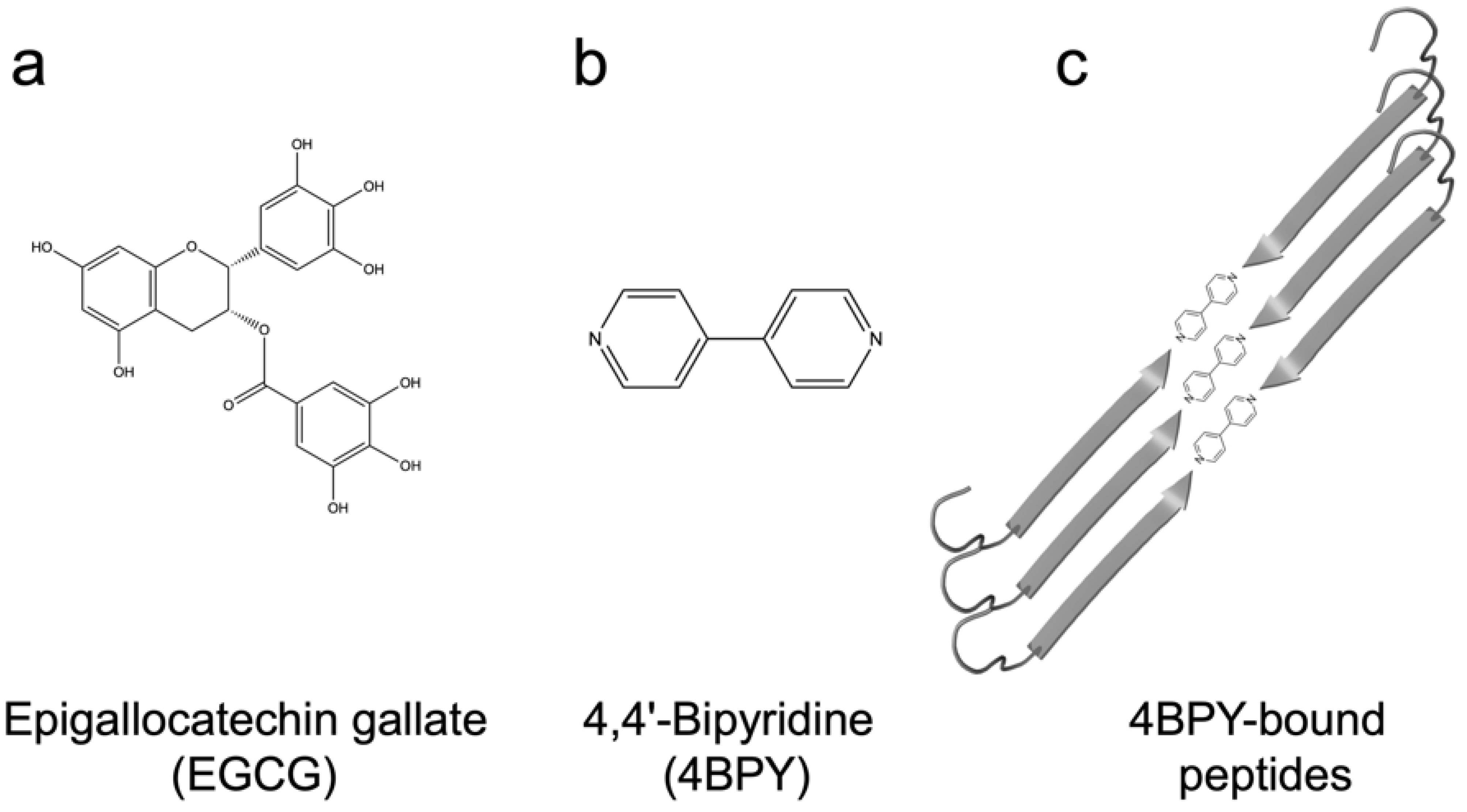
- Direct inhibition: EGCG is known to inhibit fibrillation of amyloidogenic monomers secreted from bacteria and remodeling preformed fibrils to amorphous aggregates [40]. EGCG interacts with FapC monomers and redirects it to relatively stable off-pathway oligomers [117], just as is seen for proteins and peptides involved in neurodegenerative diseases such as α-synuclein and Aβ [118]. As with chaperones [85], the main target of inhibition is the nucleation phase, and FapC/CsgA monomers are redirected to off-pathway oligomers [119]. These off-pathway oligomers are SDS-stable and contain a mixture of β-sheets and random coils [120]. This inhibition of FapC fibrillation can take place even in the presence of amyloid inducers such as SDS, rhamnolipids and LPS [121]. By combining a peptide microarray and the EGCG-binding compound Nitro blue tetrazolium, we have shown that EGCG binds to amyloidogenic hot spots containing the sequence “GVNVAA” in repeats R2 and R3 and even linker regions of FapC sequence [117,122]. Small-angle X-ray scattering measurements revealed a core-shell structure for FapC off-pathway oligomers that consist of ~7 monomers with a 25–26 nm short-axis diameter, which is much bigger than would be expected for on-pathway fibril precursors (~ 10 nm) [117].
13. Functional Bacterial Amyloid as a Bionanomaterial
14. Steering FuBA Assemblies Using the Structured Surface of Graphene
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.; Dueholm, M.; Christiansen, G.; Nielsen, J.L.; Otzen, D.E.; Nielsen, P.H. Amyloid adhesins are abundant in natural biofilms. Environ. Microbiol. 2007, 9, 3077–3090. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.; Nielsen, J.L.; Otzen, D.E.; Nielsen, P.H. Amyloid-like adhesins in floc-forming and filamentous bacteria in activated sludge. Appl. Environ. Microbiol. 2008, 74, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Greenwald, J.; Riek, R. On the possible amyloid origin of protein folds. J. Mol. Biol. 2012, 421, 417–426. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar] [CrossRef]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarström, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The Biological and Chemical Basis for Tissue-Selective Amyloid Disease. Cell 2005, 121, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Lamb, Y.N.; Deeks, E.D. Tafamidis: A Review in Transthyretin Amyloidosis with Polyneuropathy. Drugs 2019, 79, 863–874. [Google Scholar] [CrossRef]
- Bulawa, C.E.; Connelly, S.; Devit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef] [Green Version]
- Fink, A.L. Factors affecting the fibrillation of α-synuclein, a natively unfolded protein. In Misbehaving Proteins; Springer: New York, NY, USA, 2006; pp. 265–285. [Google Scholar]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermark, P.; Grimelius, L. The pancreatic islet cells in insular amyloidosis in human diabetic and non-diabetic adults. Acta Pathol. Microbiol. Scand. Sect. A Pathol. 1973, 81, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Jahn, T.R.; Makin, O.S.; Morris, K.L.; Marshall, K.E.; Tian, P.; Sikorski, P.; Serpell, L.C. The Common Architecture of Cross-β Amyloid. J. Mol. Biol. 2010, 395, 717–727. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Buehler, M.J. Nanomechanics of functional and pathological amyloid materials. Nat. Nanotechnol. 2011, 6, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Balch, W.E.; Kelly, J.W. Functional amyloid—From bacteria to humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef]
- Coustou, V.; Deleu, C.; Saupe, S.; Begueret, J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. USA 1997, 94, 9773–9778. [Google Scholar] [CrossRef] [Green Version]
- Claessen, D.; Rink, R.; de Jong, W.; Siebring, J.; de Vreugd, P.; Boersma, F.H.; Dijkhuizen, L.; Wösten, H.A. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 2003, 17, 1714–1726. [Google Scholar] [CrossRef] [Green Version]
- Dueholm, M.S.; Petersen, S.V.; Sønderkær, M.; Larsen, P.; Christiansen, G.; Hein, K.L.; Enghild, J.J.; Nielsen, J.L.; Nielsen, K.L.; Nielsen, P.H.; et al. Functional amyloid in Pseudomonas. Mol. Microbiol. 2010, 77, 1009–1020. [Google Scholar] [CrossRef] [Green Version]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, K.; Syed, A.K.; Stephenson, R.E.; Rickard, A.H.; Boles, B.R. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012, 8, e1002744. [Google Scholar] [CrossRef]
- Dueholm, M.S.; Nielsen, P.H.; Chapman, M.; Otzen, D.; Romero, D.; Kolter, R.; Dueholm, M.S.; Nielsen, P.H.; Chapman, M.; Otzen, D. Functional Amyloids in Bacteria; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 411–438. [Google Scholar]
- Hall-Stoodley, L.; Costerton, J.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Hasan, J.; Crawford, R.J.; Ivanova, E.P. Antibacterial surfaces: The quest for a new generation of biomaterials. Trends Biotechnol. 2013, 31, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.F.B.; Schafer, N.; Wolf-Perez, A.; Madsen, D.J.; Otzen, D.E. Bacterial Amyloids: Biogenesis and Biomaterials. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1174, pp. 113–159. [Google Scholar]
- Dueholm, M.S.; Albertsen, M.; Otzen, D.; Nielsen, P.H. Curli Functional Amyloid Systems Are Phylogenetically Widespread and Display Large Diversity in Operon and Protein Structure. PLoS ONE 2012, 7, e51274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawar, D.M.; Rossman, M.L.; Chen, J. Role of curli fimbriae in mediating the cells of enterohaemorrhagic Escherichia coli to attach to abiotic surfaces. J. Appl. Microbiol. 2005, 99, 418–425. [Google Scholar] [CrossRef]
- White, A.P.; Collinson, S.K.; Banser, P.A.; Gibson, D.L.; Paetzel, M.; Strynadka, N.C.J.; Kay, W.W. Structure and characterization of AgfB from Salmonella enteritidis thin aggregative fimbriae. J. Mol. Biol. 2001, 311, 735–749. [Google Scholar] [CrossRef] [Green Version]
- Hammer, N.D.; Schmidt, J.C.; Chapman, M.R. The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc. Natl. Acad. Sci. USA 2007, 104, 12494–12499. [Google Scholar] [CrossRef] [Green Version]
- Barnhart, M.M.; Chapman, M.R. Curli biogenesis and function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef] [Green Version]
- Robinson, L.S.; Ashman, E.M.; Hultgren, S.J.; Chapman, M.R. Secretion of curli fibre subunits is mediated by the outer membrane-localized CsgG protein. Mol. Microbiol. 2006, 59, 870–881. [Google Scholar] [CrossRef] [Green Version]
- Nenninger, A.A.; Robinson, L.S.; Hammer, N.D.; Epstein, E.A.; Badtke, M.P.; Hultgren, S.J.; Chapman, M.R. CsgE is a curli secretion specificity factor that prevents amyloid fibre aggregation. Mol. Microbiol. 2011, 81, 486–499. [Google Scholar] [CrossRef] [Green Version]
- Nenninger, A.A.; Robinson, L.S.; Hultgren, S.J. Localized and efficient curli nucleation requires the chaperone-like amyloid assembly protein CsgF. Proc. Natl. Acad. Sci. USA 2009, 106, 900–905. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chapman, M.R. Sequence Determinants of Bacterial Amyloid Formation. J. Mol. Biol. 2008, 380, 570–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.L.; Chorell, E.; Taylor, J.D.; Aden, J.; Gotheson, A.; Li, F.; Koch, M.; Sefer, L.; Matthews, S.J.; Wittung-Stafshede, P.; et al. The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol. Cell 2015, 57, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.D.; Hawthorne, W.J.; Lo, J.; Dear, A.; Jain, N.; Meisl, G.; Andreasen, M.; Fletcher, C.; Koch, M.; Darvill, N.; et al. Electrostatically-guided inhibition of Curli amyloid nucleation by the CsgC-like family of chaperones. Nat. Publ. Group 2016, 6, 24656. [Google Scholar] [CrossRef] [Green Version]
- Dueholm, M.S.; Otzen, D.; Nielsen, P.H. Evolutionary insight into the functional amyloids of the pseudomonads. PLoS ONE 2013, 8, e76630. [Google Scholar] [CrossRef]
- Rouse, S.L.; Matthews, S.J.; Dueholm, M.S. Ecology and Biogenesis of Functional Amyloids in Pseudomonas. J. Mol. Biol. 2018, 430, 3685–3695. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Vad, B.S.; Dueholm, M.S.; Christiansen, G.; Nilsson, M.; Tolker-Nielsen, T.; Nielsen, P.H.; Meyer, R.L.; Otzen, D.E. Functional bacterial amyloid increases Pseudomonas biofilm hydrophobicity and stiffness. Front. Microbiol. 2015, 6, 1099. [Google Scholar] [CrossRef] [Green Version]
- Stenvang, M.; Dueholm, M.S.; Vad, B.S.; Seviour, T.; Zeng, G.; Geifman-Shochat, S.; Søndergaard, M.T.; Christiansen, G.; Meyer, R.L.; Kjelleberg, S.; et al. Epigallocatechin Gallate Remodels Pseudomonas aeruginosa Functional Amyloids and Increases Biofilm Susceptibility to Antibiotic Treatment. J. Biol. Chem. 2016, 291, 26540–26553. [Google Scholar] [CrossRef] [Green Version]
- Dueholm, M.S.; Sondergaard, M.T.; Nilsson, M.; Christiansen, G.; Stensballe, A.; Overgaard, M.T.; Givskov, M.; Tolker-Nielsen, T.; Otzen, D.E.; Nielsen, P.H. Expression of Fap amyloids in Pseudomonas aeruginosa, P. fluorescens, and P. putida results in aggregation and increased biofilm formation. MicrobiologyOpen 2013, 2, 365–382. [Google Scholar] [CrossRef]
- Rouse, S.L.; Hawthorne, W.J.; Berry, J.L.; Chorev, D.S.; Ionescu, S.A.; Lambert, S.; Stylianou, F.; Ewert, W.; Mackie, U.; Morgan, R.M.L.; et al. A new class of hybrid secretion system is employed in Pseudomonas amyloid biogenesis. Nat. Commun. 2017, 8, 263. [Google Scholar] [CrossRef] [Green Version]
- Goyal, P.; Krasteva, P.V.; Van Gerven, N.; Gubellini, F.; Van den Broeck, I.; Troupiotis-Tsailaki, A.; Jonckheere, W.; Pehau-Arnaudet, G.; Pinkner, J.S.; Chapman, M.R.; et al. Structural and mechanistic insights into the bacterial amyloid secretion channel CsgG. Nature 2014, 516, 250–253. [Google Scholar] [CrossRef] [Green Version]
- Tian, P.; Boomsma, W.; Wang, Y.; Otzen, D.E.; Jensen, M.H.; Lindorff-Larsen, K.; Lindor, K. Structure of a Functional Amyloid Protein Subunit Computed Using Sequence Variation. J. Am. Chem. Soc. 2015, 137, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Louros, N.N.; Bolas, G.M.P.; Tsiolaki, P.L.; Hamodrakas, S.J.; Iconomidou, V.A. Intrinsic aggregation propensity of the CsgB nucleator protein is crucial for curli fiber formation. J. Struct. Biol. 2016, 195, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Debenedictis, E.P.; Ma, D.; Keten, S. Structural predictions for curli amyloid fibril subunits CsgA and CsgB. RSC Adv. 2017, 7, 48102–48112. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Deng, X.; Gonzalez Llamazares, A.; Wagstaff, J.M.; Hale, V.L.; Cannone, G.; McLaughlin, S.H.; Kureisaite-Ciziene, D.; Löwe, J. The structure of bactofilin filaments reveals their mode of membrane binding and lack of polarity. Nat. Microbiol. 2019, 4, 2357–2368. [Google Scholar] [CrossRef]
- Göbel, U.; Sander, C.; Schneider, R.; Valencia, A. Correlated mutations and residue contacts in proteins. Proteins Struct. Funct. Bioinform. 1994, 18, 309–317. [Google Scholar] [CrossRef]
- Gallardo, R.; Ranson, N.A.; Radford, S.E. Amyloid structures: Much more than just a cross-β fold. Curr. Opin. Struct. Biol. 2020, 60, 7–16. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Pinheiro, F.; Santos, J.; Ventura, S. AlphaFold and the amyloid landscape. J. Mol. Biol. 2021, 433, 167059. [Google Scholar] [CrossRef]
- Wang, X.; Smith, D.R.; Jones, J.W.; Chapman, M.R. In vitro polymerization of a functional Escherichia coli amyloid protein. J. Biol. Chem. 2007, 282, 3713–3719. [Google Scholar] [CrossRef] [Green Version]
- Cherny, I.; Rockah, L.; Levy-Nissenbaum, O.; Gophna, U.; Ron, E.Z.; Gazit, E. The formation of Escherichia coli curli amyloid fibrils is mediated by prion-like peptide repeats. J. Mol. Biol. 2005, 352, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Collinson, S.K.; Parker, J.M.R.; Hodges, R.S.; Kay, W.W. Structural predictions of AgfA, the insoluble fimbrial subunit of Salmonella thin aggregative fimbriae. J. Mol. Biol. 1999, 290, 741–756. [Google Scholar] [CrossRef]
- Shewmaker, F.; McGlinchey, R.P.; Thurber, K.R.; McPhie, P.; Dyda, F.; Tycko, R.; Wickner, R.B. The functional curli amyloid is not based on in-register parallel β-sheet structure. J. Biol. Chem. 2009, 284, 25065–25076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhou, Y.; Ren, J.-J.; Hammer, N.D.; Chapman, M.R. Gatekeeper residues in the major curlin subunit modulate bacterial amyloid fi ber biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 163–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otzen, D.E.; Kristensen, P.; Oliveberg, M. Designed protein tetramer zipped together with an Alzheimer sequence: A structural clue to amyloid assembly. Proc. Natl. Acad. Sci. USA 2000, 97, 9907–9912. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hammer, N.D.; Chapman, M.R. The molecular basis of functional bacterial amyloid polymerization and nucleation. J. Biol. Chem. 2008, 283, 21530–21539. [Google Scholar] [CrossRef] [Green Version]
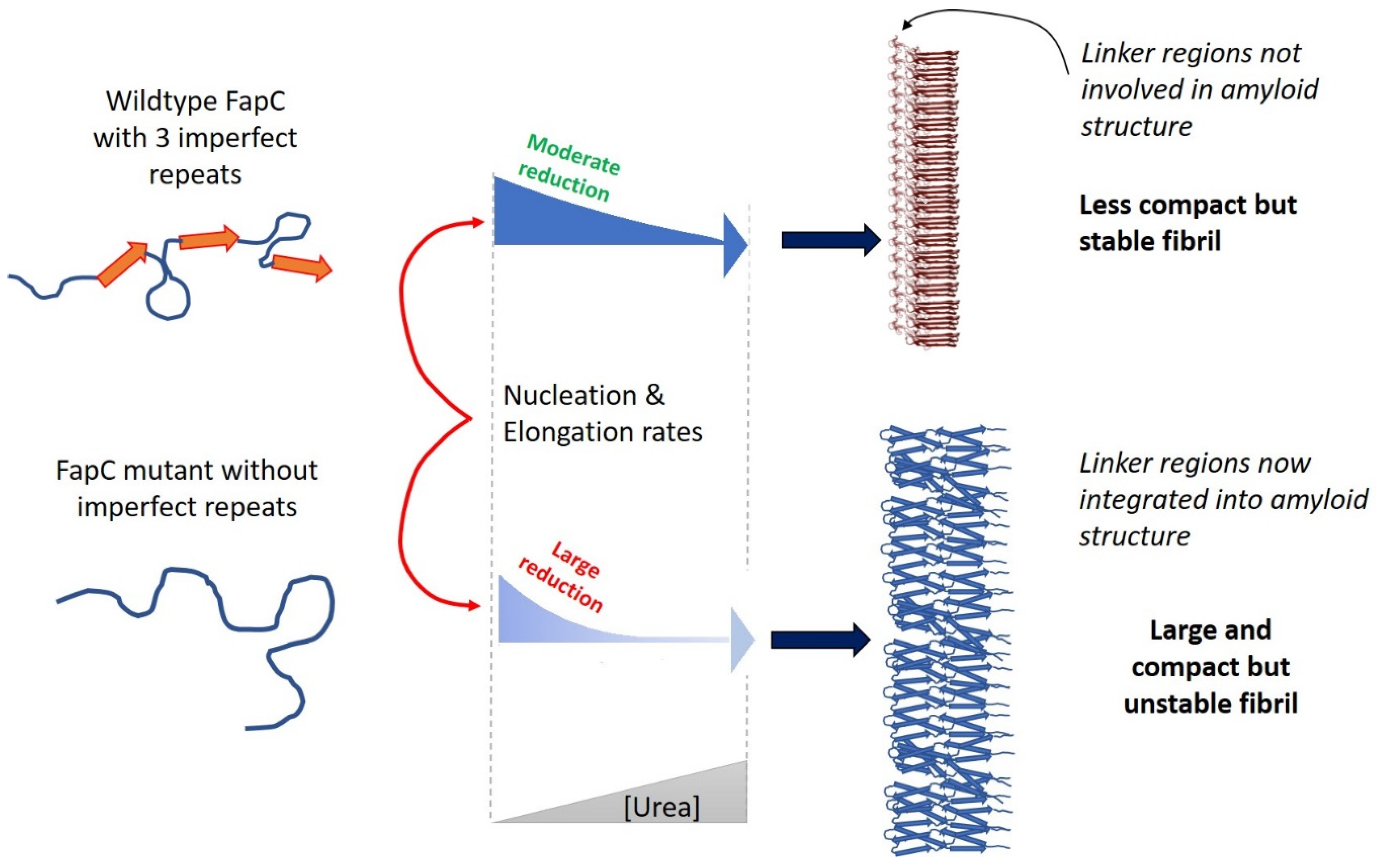
- Christensen, L.F.B.; Nowak, J.S.; Sonderby, T.V.; Frank, S.A.; Otzen, D.E. Quantitating denaturation by formic acid: Imperfect repeats are essential to the stability of the functional amyloid protein FapC. J. Biol. Chem. 2020, 295, 13031–13046. [Google Scholar] [CrossRef]
- Rasmussen, C.B.; Christiansen, G.; Vad, B.S.; Lynggaard, C.; Enghild, J.J.; Andreasen, M.; Otzen, D. Imperfect repeats in the functional amyloid protein FapC reduce the tendency to fragment during fibrillation. Protein Sci. 2019, 28, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Riddick, J.; Bunger, W.; Sakano, T. Techniques of Chemistry, Organic Solvents; John Wiley and Sons: New York, NY, USA, 1985; Volume 2. [Google Scholar]
- Sonderby, T.V.; Rasmussen, H.O.; Frank, S.A.; Skov Pedersen, J.; Otzen, D.E. Folding Steps in the Fibrillation of Functional Amyloid: Denaturant Sensitivity Reveals Common Features in Nucleation and Elongation. J. Mol. Biol. 2021, 434, 167337. [Google Scholar] [CrossRef]
- Christensen, L.F.B.; Jensen, K.F.; Nielsen, J.; Vad, B.S.; Christiansen, G.; Otzen, D.E. Reducing the Amyloidogenicity of Functional Amyloid Protein FapC Increases Its Ability To Inhibit α-Synuclein Fibrillation. ACS Omega 2019, 4, 4029–4039. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Liu, Z. Inherent relationships among different biophysical prediction methods for intrinsically disordered proteins. Biophys. J. 2013, 104, 488–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreasen, M.; Meisl, G.; Taylor, J.D.; Michaels, T.C.T.; Otzen, D.E.; Chapman, M.; Dobson, C.M.; Matthews, S.; Knowles, T.P.J. Physical determinants of amyloid assembly in biofilm formation. mBio 2019, 10, e02279-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caughey, B.; Lansbury Jr, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Vats, A.; Taneja, V. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138. [Google Scholar]
- Wang, H.; Shu, Q.; Rempel, D.L.; Frieden, C.; Gross, M.L. Understanding curli amyloid-protein aggregation by hydrogen–deuterium exchange and mass spectrometry. Int. J. Mass Spectrom. 2017, 420, 16–23. [Google Scholar] [CrossRef]
- Sleutel, M.; Broeck, I.V.D.; Gerven, N.V.; Feuillie, C.; Jonckheere, W.; Valotteau, C.; Dufrêne, Y.F.; Remaut, H. Nucleation and growth of bacterial functional amyloid at single-fiber resolution. Nat. Chem. Biol. 2017, 13, 902–908. [Google Scholar] [CrossRef] [Green Version]
- Dueholm, M.S.; Nielsen, S.B.; Hein, K.L.; Nissen, P.; Chapman, M.; Christiansen, G.; Nielsen, P.H.; Otzen, D.E. Fibrillation of the major curli subunit CsgA under a wide range of conditions implies a robust design of aggregation. Biochemistry 2011, 50, 8281–8290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewell, L.; Stylianou, F.; Xu, Y.; Taylor, J.; Sefer, L.; Matthews, S. NMR insights into the pre-amyloid ensemble and secretion targeting of the curli subunit CsgA. Sci. Rep. 2020, 10, 7896. [Google Scholar] [CrossRef]
- Tian, P.; Lindorff-Larsen, K.; Boomsma, W.; Jensen, M.H.; Otzen, D.E. A Monte Carlo Study of the Early Steps of Functional Amyloid Formation. PLoS ONE 2016, 11, e0146096. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, H.Ø.; Otzen, D.E.; Pedersen, J.S. A multimethod approach for analyzing FapC fibrillation and determining mass per length. Biophys. J. 2021, 120, 2262–2275. [Google Scholar] [CrossRef]
- Kauzmann, W. Some factors in the interpretation of protein denaturation. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1959; Volume 14, pp. 1–63. [Google Scholar]
- Canchi, D.R.; García, A.E. Cosolvent effects on protein stability. Annu. Rev. Phys. Chem. 2013, 64, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Fersht, A.R. Folding of chymotrypsin inhibitor 2.1. Evidence for a two-state transition. Biochemistry 1991, 30, 10428–10435. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; Macmillan: New York, NY, USA, 1999. [Google Scholar]
- Kim, J.R.; Muresan, A.; Lee, K.Y.C.; Murphy, R.M. Urea modulation of β-amyloid fibril growth: Experimental studies and kinetic models. Protein Sci. 2004, 13, 2888–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiiman, A.; Krishtal, J.; Palumaa, P.; Tõugu, V. In vitro fibrillization of Alzheimer’s amyloid-β peptide (1-42). AIP Adv. 2015, 5, 092401. [Google Scholar] [CrossRef] [Green Version]
- Knowles, T.P.; Shu, W.; Devlin, G.L.; Meehan, S.; Auer, S.; Dobson, C.M.; Welland, M.E. Kinetics and thermodynamics of amyloid formation from direct measurements of fluctuations in fibril mass. Proc. Natl. Acad. Sci. USA 2007, 104, 10016–10021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Knapp, K.; Le, K.N.; Schafer, N.P.; Safari, M.S.; Davtyan, A.; Wolynes, P.G.; Vekilov, P.G. Frustrated peptide chains at the fibril tip control the kinetics of growth of amyloid-beta fibrils. Proc. Natl. Acad. Sci. USA 2021, 118, e2110995118. [Google Scholar] [CrossRef] [PubMed]
- Otzen, D.E.; Oliveberg, M. Salt-induced detour through compact regions of the protein folding landscape. Proc. Nat. Acad. Sci. USA 1999, 96, 11746–11751. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, R. On-pathway versus off-pathway folding intermediates. Fold. Des. 1996, 1, R1–R8. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, M.; Najarzadeh, Z.; Pansieri, J.; Biverstål, H.; Musteikyte, G.; Smirnovas, V.; Matthews, S.; Emanuelsson, C.; Johansson, J.; Buxbaum, J.N.; et al. Chaperones mainly suppress primary nucleation during formation of functional amyloid required for bacterial biofilm formation. Chem. Sci. 2022, 13, 536–553. [Google Scholar] [CrossRef]
- Chorell, E.; Andersson, E.; Evans, M.L.; Jain, N.; Götheson, A.; Åden, J.; Chapman, M.R.; Almqvist, F.; Wittung-Stafshede, P. Bacterial Chaperones CsgE and CsgC Differentially Modulate Human α-Synuclein Amyloid Formation via Transient Contacts. PLoS ONE 2015, 10, e0140194. [Google Scholar] [CrossRef] [Green Version]
- Armiento, V.; Spanopoulou, A.; Kapurniotu, A. Peptide-Based Molecular Strategies To Interfere with Protein Misfolding, Aggregation, and Cell Degeneration. Angew. Chem. Int. Ed. 2020, 59, 3372–3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Agnaf, O.M.A.; Paleologou, K.E.; Greer, B.; Abogrein, A.M.; King, J.E.; Salem, S.A.; Fullwood, N.J.; Benson, F.E.; Hewitt, R.; Ford, K.J.; et al. A strategy for designing inhibitors of α-synuclein aggregation and toxicity as a novel treatment for Parkinson’s disease and related disorders. FASEB J. 2004, 18, 1315–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjernberg, L.O.; Näslundt, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Tereniust, L.; Nordstedt, C. Arrest of β-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Wang, C.; Song, B.; Zhang, W.; Guo, Y.; Yang, R.; Nie, G.; Yang, Y.; Wang, C. Attenuation of β-Amyloid Toxicity In Vitro and In Vivo by Accelerated Aggregation. Neurosci. Bull. 2017, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
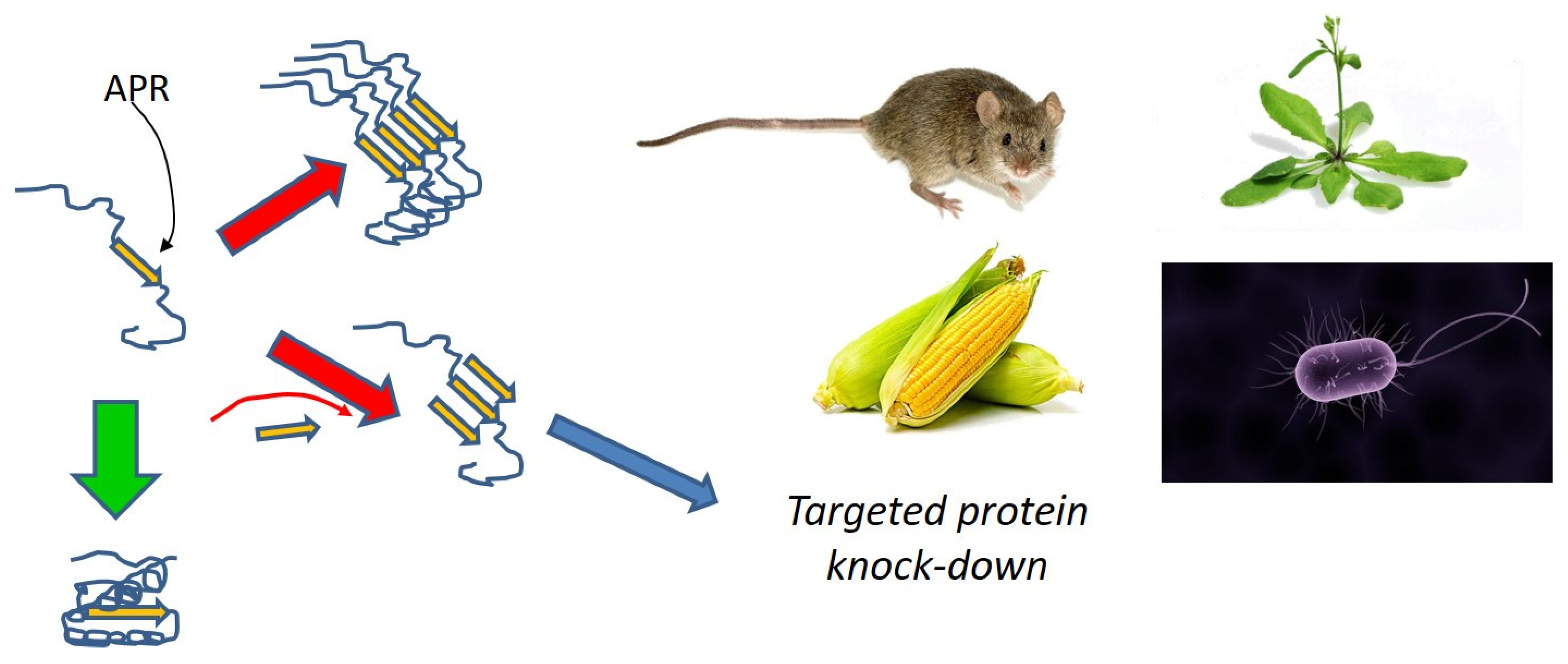
- Beerten, J.; Schymkowitz, J.; Rousseau, F. Aggregation prone regions and gatekeeping residues in protein sequences. Curr. Top. Med. Chem. 2012, 12, 2470–2478. [Google Scholar] [CrossRef]
- Bednarska, N.G.; van Eldere, J.; Gallardo, R.; Ganesan, A.; Ramakers, M.; Vogel, I.; Baatsen, P.; Staes, A.; Goethals, M.; Hammarström, P.; et al. Protein aggregation as an antibiotic design strategy. Mol. Microbiol. 2016, 99, 849–865. [Google Scholar] [CrossRef] [Green Version]
- Khodaparast, L.; Khodaparast, L.; Gallardo, R.; Louros, N.N.; Michiels, E.; Ramakrishnan, R.; Ramakers, M.; Claes, F.; Young, L.; Shahrooei, M.; et al. Aggregating sequences that occur in many proteins constitute weak spots of bacterial proteostasis. Nat. Commun. 2018, 9, 866. [Google Scholar] [CrossRef]
- Fernandez-Escamilla, A.M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. [Google Scholar] [CrossRef]
- Sønderby, T.V.; Louros, N.N.; Khodaparast, L.; Khodaparast, L.; Madsen, D.J.; Nagaraj, M.; Strømgaard, K.; Rousseau, F.; Schymkowitz, J.; Otzen, D.E. Sequence-targeted peptides divert Functional Bacterial Amyloid towards destabilized aggregates and reduce biofilm formation. Biophys. J. in review.
- Louros, N.; Konstantoulea, K.; De Vleeschouwer, M.; Ramakers, M.; Schymkowitz, J.; Rousseau, F. WALTZ-DB 2.0: An updated database containing structural information of experimentally determined amyloid-forming peptides. Nucleic Acids Res. 2020, 48, D389–D393. [Google Scholar] [CrossRef]
- Perov, S.; Lidor, O.; Salinas, N.; Golan, N.; Tayeb-Fligelman, E.; Deshmukh, M.; Willbold, D.; Landau, M. Structural Insights into Curli CsgA Cross-beta Fibril Architecture Inspire Repurposing of Anti-amyloid Compounds as Anti-biofilm Agents. PLoS Pathog. 2019, 15, e1007978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunbar, M.; DeBenedictis, E.; Keten, S. Dimerization energetics of curli fiber subunits CsgA and CsgB. npj Comput. Mater. 2019, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Sleutel, M.; Pradhan, B.; Remaut, H. Structural analysis of the bacterial amyloid curli. bioRxiv 2022. [Google Scholar] [CrossRef]
- Louros, N.; Ramakers, M.; Michiels, E.; Konstantoulea, K.; Morelli, C.; Garcia, T.; Moonen, N.; D’Haeyer, S.; Goossens, V.; Thal, D.R. Mapping the sequence specificity of heterotypic amyloid interactions enables the identification of aggregation modifiers. Nat. Commun. 2022, 13, 1351. [Google Scholar] [CrossRef] [PubMed]
- Louros, N.; Orlando, G.; De Vleeschouwer, M.; Rousseau, F.; Schymkowitz, J. Structure-based machine-guided mapping of amyloid sequence space reveals uncharted sequence clusters with higher solubilities. Nat. Commun. 2020, 11, 3314. [Google Scholar] [CrossRef]
- Hancock, R.E.; Chapple, D.S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar] [CrossRef] [Green Version]
- Mohammad-Beigi, H.; Kjaer, L.; Eskandari, H.; Aliakbari, F.; Christiansen, G.; Ruvo, G.; Ward, J.L.; Otzen, D.E. A possible connection between plant longevity and the absence of protein fibrillation: Basis for identifying aggregation inhibitors in plants. Front. Plant Sci. 2019, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Mohammad-Beigi, H.; Aliakbari, F.; Sahin, C.; Lomax, C.; Tawfike, A.; Schafer, N.P.; Amiri-Nowdijeh, A.; Eskandari, H.; Møller, I.M.; Hosseini-Mazinani, M.; et al. Oleuropein derivatives from olive fruit extracts reduce—Synuclein fibrillation and oligomer toxicity. J. Biol. Chem. 2019, 294, 4215–4232. [Google Scholar] [CrossRef]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [Green Version]
- Youn, K.; Ho, C.-T.; Jun, M. Multifaceted neuroprotective effects of (-)-epigallocatechin-3-gallate (EGCG) in Alzheimer’s disease: An overview of pre-clinical studies focused on β-amyloid peptide. Food Sci. Hum. Wellness 2022, 11, 483–493. [Google Scholar] [CrossRef]
- Sahadevan, R.; Singh, S.; Binoy, A.; Sadhukhan, S. Chemico-biological aspects of (-)-epigallocatechin-3-gallate (EGCG) to improve its stability, bioavailability and membrane permeability: Current status and future prospects. Crit. Rev. Food Sci. Nutr. 2022, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, L.; Cardim-Pires, T.R.; Foguel, D.; Palhano, F.L. Green Tea Polyphenol Epigallocatechin-Gallate in Amyloid Aggregation and Neurodegenerative Diseases. Front. Neurosci. 2021, 15, 1049. [Google Scholar] [CrossRef] [PubMed]
- Serra, D.O.; Mika, F.; Richter, A.M.; Hengge, R. The green tea polyphenol EGCG inhibits E. coli biofilm formation by impairing amyloid curli fibre assembly and downregulating the biofilm regulator CsgD via the σE-dependent sRNA RybB. Mol. Microbiol. 2016, 101, 136–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widyaningrum, N.; Fudholi, A.; Sudarsono, S.E.; Setyowati, E. Stability of epigallocatechin gallate (EGCG) from green tea (Camellia sinensis) and its antibacterial activity against Staphylococcus epidermidis ATCC 35984 and Propionibacterium acnes ATCC 6919. Asian J. Biol. Sci 2015, 8, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Mereles, D.; Hunstein, W. Epigallocatechin-3-gallate (EGCG) for clinical trials: More pitfalls than promises? Int. J. Mol. Sci. 2011, 12, 5592–5603. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Lu, H.; Meng, X.; Ryu, J.-H.; Hara, Y.; Yang, C.S. Stability, cellular uptake, biotransformation, and efflux of tea polyphenol (−)-epigallocatechin-3-gallate in HT-29 human colon adenocarcinoma cells. Cancer Res. 2002, 62, 7241–7246. [Google Scholar]
- Arita-Morioka, K.-i.; Yamanaka, K.; Mizunoe, Y.; Tanaka, Y.; Ogura, T.; Sugimoto, S. Inhibitory effects of Myricetin derivatives on curli-dependent biofilm formation in Escherichia coli. Sci. Rep. 2018, 8, 8452. [Google Scholar] [CrossRef] [Green Version]
- Reygaert, W.C. The antimicrobial possibilities of green tea. Front. Microbiol. 2014, 5, 434. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Rock, C.O. Evaluation of epigallocatechin gallate and related plant polyphenols as inhibitors of the FabG and FabI reductases of bacterial type II fatty-acid synthase. J. Biol. Chem. 2004, 279, 30994–31001. [Google Scholar] [CrossRef] [Green Version]
- Chinnam, N.; Dadi, P.K.; Sabri, S.A.; Ahmad, M.; Kabir, M.A.; Ahmad, Z. Dietary bioflavonoids inhibit Escherichia coli ATP synthase in a differential manner. Int. J. Biol. Macromol. 2010, 46, 478–486. [Google Scholar] [CrossRef] [Green Version]
- Najarzadeh, Z.; Mohammad-Beigi, H.; Nedergaard Pedersen, J.; Christiansen, G.; Sønderby, T.V.; Shojaosadati, S.A.; Morshedi, D.; Strømgaard, K.; Meisl, G.; Sutherland, D.; et al. Plant Polyphenols Inhibit Functional Amyloid and Biofilm Formation in Pseudomonas Strains by Directing Monomers to Off-Pathway Oligomers. Biomolecules 2019, 9, 659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Pruteanu, M.; Hernández Lobato, J.I.; Stach, T.; Hengge, R. Common plant flavonoids prevent the assembly of amyloid curli fibres and can interfere with bacterial biofilm formation. Environ. Microbiol. 2020, 22, 5280–5299. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, M.; Christiansen, G.; Otzen, D.E. Epigallocatechin Gallate Remodels Fibrils of Lattice Corneal Dystrophy Protein, Facilitating Proteolytic Degradation and Preventing Formation of Membrane-Permeabilizing Species. Biochemistry 2016, 55, 2344–2357. [Google Scholar] [CrossRef]
- Najarzadeh, Z.; Pedersen, J.N.; Christiansen, G.; Shojaosadati, S.A.; Pedersen, J.S.; Otzen, D.E. Bacterial amphiphiles as amyloid inducers: Effect of Rhamnolipid and Lipopolysaccharide on FapC fibrillation. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2019, 1867, 140263. [Google Scholar] [CrossRef]
- Bleem, A.; Christiansen, G.; Madsen, D.J.; Maric, H.; Strømgaard, K.; Bryers, J.D.; Daggett, V.; Meyer, R.L.; Otzen, D.E. Protein engineering reveals mechanisms of functional amyloid formation in Pseudomonas aeruginosa biofilms. J. Mol. Biol. 2018, 430, 3751–3763. [Google Scholar] [CrossRef] [Green Version]
- Hengge, R. Targeting bacterial biofilms by the green tea polyphenol EGCG. Molecules 2019, 24, 2403. [Google Scholar] [CrossRef] [Green Version]
- Eskandari, H.; Ghanadian, M.; Noleto-Dias, C.; Lomax, C.; Tawfike, A.; Christiansen, G.; Sutherland, D.S.; Ward, J.L.; Mohammad-Beigi, H.; Otzen, D.E. Inhibitors of α-synuclein fibrillation and oligomer toxicity in Rosa damascena: The all-pervading powers of flavonoids and phenolic glycosides. ACS Chem. Neurosci. 2020, 11, 3161–3173. [Google Scholar] [CrossRef]
- Young, L.M.; Saunders, J.C.; Mahood, R.A.; Revill, C.H.; Foster, R.J.; Tu, L.-H.; Raleigh, D.P.; Radford, S.E.; Ashcroft, A.E. Screening and classifying small-molecule inhibitors of amyloid formation using ion mobility spectrometry–mass spectrometry. Nat. Chem. 2015, 7, 73–81. [Google Scholar] [CrossRef]
- Doig, A.J.; Derreumaux, P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015, 30, 50–56. [Google Scholar] [CrossRef]
- Mohammad-Beigi, H.; Morshedi, D.; Shojaosadati, S.A.; Pedersen, J.N.; Marvian, A.T.; Aliakbari, F.; Christiansen, G.; Pedersen, J.S.; Otzen, D.E. Gallic acid loaded onto polyethylenimine-coated human serum albumin nanoparticles (PEI-HSA-GA NPs) stabilizes α-synuclein in the unfolded conformation and inhibits aggregation. RSC Adv. 2016, 6, 85312–85323. [Google Scholar] [CrossRef]
- Aliakbari, F.; Mohammad-Beigi, H.; Abbasi, S.; Rezaei-Ghaleh, N.; Lermyte, F.; Parsafar, S.; Becker, S.; Tafreshi, A.P.; O’Connor, P.B.; Collingwood, J.F. Multiple Protective Roles of Nanoliposome-Incorporated Baicalein against Alpha-Synuclein Aggregates. Adv. Funct. Mater. 2021, 31, 2007765. [Google Scholar] [CrossRef]
- Fernandes, L.; Messias, B.; Pereira-Neves, A.; Azevedo, E.P.; Araújo, J.; Foguel, D.; Palhano, F.L. Green tea polyphenol microparticles based on the oxidative coupling of EGCG inhibit amyloid aggregation/cytotoxicity and serve as a platform for drug delivery. ACS Biomater. Sci. Eng. 2020, 6, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Ghosh, K.S. Inhibition of amyloid fibrillation by small molecules and nanomaterials: Strategic development of pharmaceuticals against amyloidosis. Protein Pept. Lett. 2019, 26, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Debnath, K.; Sarkar, A.K.; Jana, N.R.; Jana, N.R. Inhibiting Protein Aggregation by Small Molecule-Based Colloidal Nanoparticles. Acc. Mater. Res. 2021, 3, 54–66. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, L.; Mao, X.; Niu, L.; Yang, Y.; Wang, C. Chaperon-Mediated single molecular approach toward modulating aβ peptide aggregation. Nano Lett. 2009, 9, 4066–4072. [Google Scholar] [CrossRef]
- Dorval Courchesne, N.M.; Duraj-Thatte, A.; Tay, P.K.R.; Nguyen, P.Q.; Joshi, N.S. Scalable Production of Genetically Engineered Nanofibrous Macroscopic Materials via Filtration. ACS Biomater. Sci. Eng. 2017, 3, 733–741. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, M.; Evans, M.L.; Chapman, M.R. Amyloid by Design: Intrinsic Regulation of Microbial Amyloid Assembly; Academic Press: London, UK, 2018; Volume 430, pp. 3631–3641. [Google Scholar]
- Van Gerven, N.; Goyal, P.; Vandenbussche, G.; De Kerpel, M.; Jonckheere, W.; De Greve, H.; Remaut, H. Secretion and functional display of fusion proteins through the curli biogenesis pathway. Mol. Microbiol. 2014, 91, 1022–1035. [Google Scholar] [CrossRef]
- Nguyen, P.Q.; Botyanszki, Z.; Tay, P.K.R.; Joshi, N.S. Programmable biofilm-based materials from engineered curli nanofibres. Nat. Commun. 2014, 5, 4945. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, K.; Wang, X.; Cui, M.; Ge, P.; Zhang, J.; Qiu, F.; Zhong, C. Conformable self-assembling amyloid protein coatings with genetically programmable functionality. Sci. Adv. 2020, 6, eaba1425. [Google Scholar] [CrossRef]
- Yang, X.; Li, Z.; Xiao, H.; Wang, N.; Li, Y.; Xu, X.; Chen, Z.; Tan, H.; Li, J. A Universal and Ultrastable Mineralization Coating Bioinspired from Biofilms. Adv. Funct. Mater. 2018, 28, 1802730. [Google Scholar] [CrossRef]
- Zhong, C.; Gurry, T.; Cheng, A.A.; Downey, J.; Deng, Z.; Stultz, C.M.; Lu, T.K. Strong underwater adhesives made by self-assembling multi-protein nanofibres. Nat. Nanotechnol. 2014, 9, 858–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, B.; Wang, X.; Cui, M.; Gui, X.; Mao, X.; Liu, Y.; Li, K.; Chu, C.; Pu, J.; Ren, S.; et al. Diverse Supramolecular Nanofiber Networks Assembled by Functional Low-Complexity Domains. ACS Nano 2017, 11, 6985–6995. [Google Scholar] [CrossRef] [PubMed]
- Duraj-Thatte, A.M.; Courchesne, N.D.; Praveschotinunt, P.; Rutledge, J.; Lee, Y.; Karp, J.M.; Joshi, N.S. Genetically Programmable Self-Regenerating Bacterial Hydrogels. Adv. Mater. 2019, 31, e1901826. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, K.; Wang, X.; An, B.; Cui, M.; Pu, J.; Wei, S.; Xue, S.; Ye, H.; Zhao, Y.; et al. Patterned Amyloid Materials Integrating Robustness and Genetically Programmable Functionality. Nano Lett. 2019, 19, 8399–8408. [Google Scholar] [CrossRef]
- DeBenedictis, E.P.; Liu, J.; Keten, S. Adhesion mechanisms of curli subunit CsgA to abiotic surfaces. Sci. Adv. 2016, 2, e1600998. [Google Scholar] [CrossRef] [Green Version]
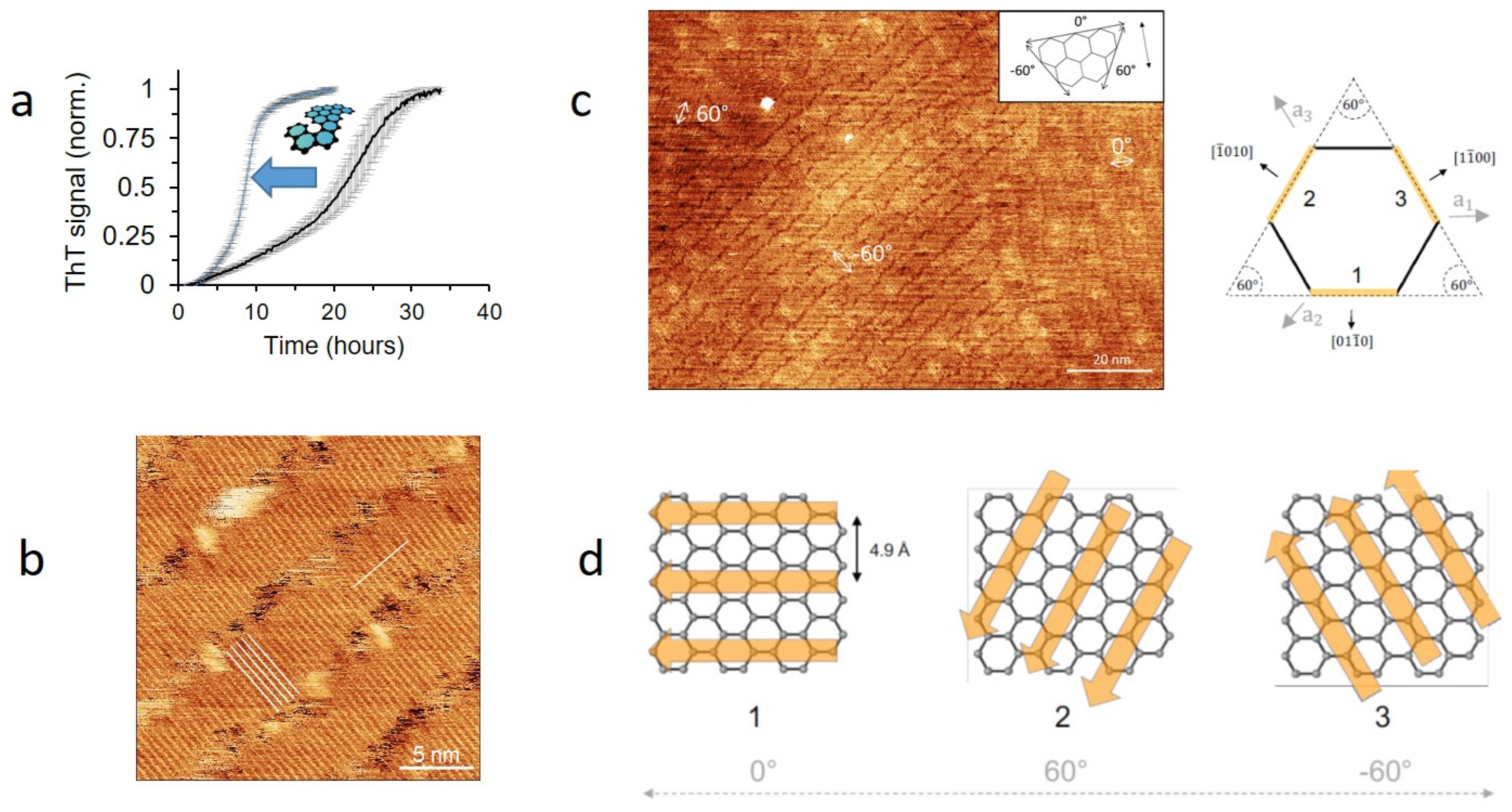
- So, C.R.; Hayamizu, Y.; Yazici, H.; Gresswell, C.; Khatayevich, D.; Tamerler, C.; Sarikaya, M. Controlling Self-Assembly of Engineered Peptides on Graphite by Rational Mutation. ACS Nano 2012, 6, 1648–1656. [Google Scholar] [CrossRef] [Green Version]
- Walsh, T.R.; Knecht, M.R. Biomolecular Material Recognition in Two Dimensions: Peptide Binding to Graphene, h-BN, and MoS2 Nanosheets as Unique Bioconjugates. Bioconjug. Chem. 2019, 30, 2727–2750. [Google Scholar] [CrossRef]
- Lipson, H.S.; Stokes, A. The structure of graphite. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1942, 181, 101–105. [Google Scholar]
- Oliveira, S.F.; Bisker, G.; Bakh, N.A.; Gibbs, S.L.; Landry, M.P.; Strano, M.S. Protein functionalized carbon nanomaterials for biomedical applications. Carbon 2015, 95, 767–779. [Google Scholar] [CrossRef] [Green Version]
- Surovtseva, D.; Crossin, E.; Pell, R.; Stamford, L. Toward a life cycle inventory for graphite production. J. Ind. Ecol. 2022, 26, 964–976. [Google Scholar] [CrossRef]
- Li, C.; Mezzenga, R. The interplay between carbon nanomaterials and amyloid fibrils in bio-nanotechnology. Nanoscale 2013, 5, 6207–6218. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, M. Fullerene inhibits β-amyloid peptide aggregation. Biochem. Biophys. Res. Commun. 2003, 303, 576–579. [Google Scholar] [CrossRef]
- Mao, X.; Wang, Y.; Liu, L.; Niu, L.; Yang, Y.; Wang, C. Molecular-level evidence of the surface-induced transformation of peptide structures revealed by scanning tunneling microscopy. Langmuir 2009, 25, 8849–8853. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mao, X.; Wang, C.; Zeng, W.; Zhang, C.; Li, Z.; Fang, Y.; Yang, Y.; Liang, W.; Wang, C. The effect of graphene oxide on conformation change, aggregation and cytotoxicity of HIV-1 regulatory protein (Vpr). Biomaterials 2013, 34, 1383–1390. [Google Scholar] [CrossRef]
- Mao, X.; Ma, X.; Liu, L.; Niu, L.; Yang, Y.; Wang, C. Structural characteristics of the beta-sheet-like human and rat islet amyloid polypeptides as determined by scanning tunneling microscopy. J. Struct. Biol. 2009, 167, 209–215. [Google Scholar] [CrossRef]
- Brown, C.L.; Aksay, I.A.; Saville, D.A.; Hecht, M.H. Template-Directed Assembly of a de Novo Designed Protein. J. Am. Chem. Soc. 2002, 124, 6846–6848. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhou, C.; Wang, C.; Wan, L.; Fang, X.; Bai, C. AFM and STM study of β-amyloid aggregation on graphite. Ultramicroscopy 2003, 97, 73–79. [Google Scholar] [CrossRef]
- Valle-Delgado, J.J.; Liepina, I.; Lapidus, D.; Sabaté, R.; Ventura, S.; Samitier, J.; Fernàndez-Busquets, X. Self-assembly of human amylin-derived peptides studied by atomic force microscopy and single molecule force spectroscopy. Soft Matter 2012, 8, 1234–1242. [Google Scholar] [CrossRef]
- Sønderby, T.V.; Zou, Y.; Wang, P.; Wang, C.; Otzen, D.E. Molecular-level insights into the surface-induced assembly of Functional Bacterial Amyloid. Biophys. J. in review.
- Liu, L.; Zhang, L.; Niu, L.; Xu, M.; Mao, X.; Yang, Y.; Wang, C. Observation of reduced cytotoxicity of aggregated amyloidogenic peptides with chaperone-like molecules. ACS Nano 2011, 5, 6001–6007. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sønderby, T.V.; Najarzadeh, Z.; Otzen, D.E. Functional Bacterial Amyloids: Understanding Fibrillation, Regulating Biofilm Fibril Formation and Organizing Surface Assemblies. Molecules 2022, 27, 4080. https://doi.org/10.3390/molecules27134080
Sønderby TV, Najarzadeh Z, Otzen DE. Functional Bacterial Amyloids: Understanding Fibrillation, Regulating Biofilm Fibril Formation and Organizing Surface Assemblies. Molecules. 2022; 27(13):4080. https://doi.org/10.3390/molecules27134080
Chicago/Turabian StyleSønderby, Thorbjørn Vincent, Zahra Najarzadeh, and Daniel Erik Otzen. 2022. "Functional Bacterial Amyloids: Understanding Fibrillation, Regulating Biofilm Fibril Formation and Organizing Surface Assemblies" Molecules 27, no. 13: 4080. https://doi.org/10.3390/molecules27134080
APA StyleSønderby, T. V., Najarzadeh, Z., & Otzen, D. E. (2022). Functional Bacterial Amyloids: Understanding Fibrillation, Regulating Biofilm Fibril Formation and Organizing Surface Assemblies. Molecules, 27(13), 4080. https://doi.org/10.3390/molecules27134080