
Rapid Quantitation of Adulterants in Premium Marine Oils by Raman and IR Spectroscopy: A Data Fusion Approach



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparations
2.2. Raman Spectroscopy
2.3. ATR-IR Spectroscopy
2.4. Spectral Pre-Processing
2.5. Data Fusion
2.6. Chemometric Analysis
3. Results and Discussion
3.1. Raman and Infrared Spectral Features of Oils
3.2. Principal Component Analysis (PCA)
3.2.1. Raman Analysis
3.2.2. IR Analysis
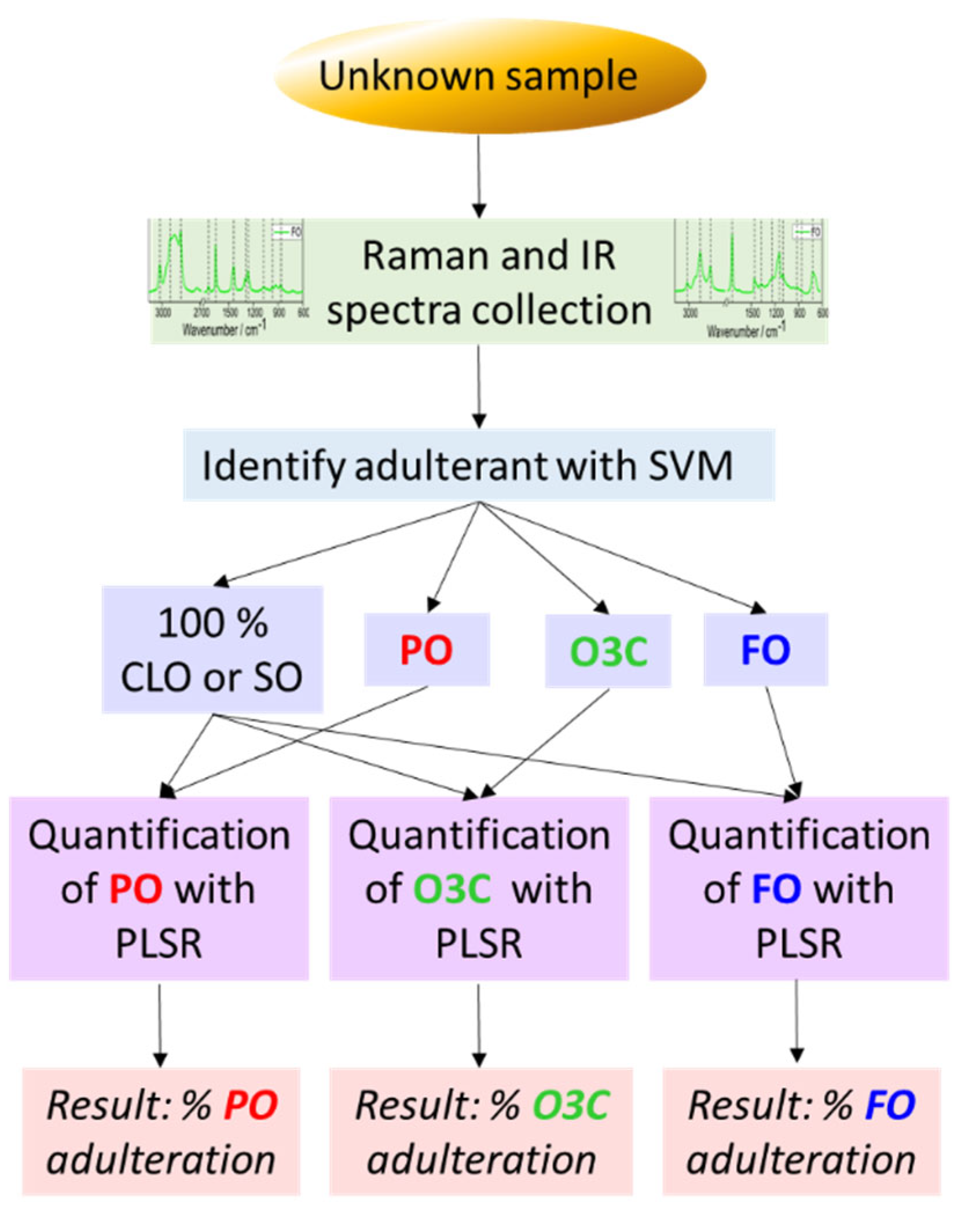
3.3. Identification and Quantification of Adulterants
3.4. Quantitative Measurements of CLO and SO Adulteration
3.4.1. Quantification of PO Adulterant in Cod Liver Oil and Salmon Oil
3.4.2. Spectroscopic Estimation of O3C % in Cod Liver Oil and Salmon Oil
3.4.3. Spectroscopic Estimation of FO % in CLO and SO
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ahmmed, M.K.; Ahmmed, F.; Tian, H.S.; Carne, A.; Bekhit, A.E. Marine omega-3 (n-3) phospholipids: A comprehensive review of their properties, sources, bioavailability, and relation to brain health. Compr. Rev. Food Sci. Food Saf. 2020, 19, 64–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killeen, D.P.; Marshall, S.N.; Burgess, E.J.; Gordon, K.C.; Perry, N.B. Raman spectroscopy of fish oil capsules: Polyunsaturated fatty acid quantitation plus detection of ethyl esters and oxidation. J. Agric. Food Chem. 2017, 65, 3551–3558. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.C.; Johns, C.; Harris, W.S.; Puder, M.; Freedman, S.D.; Thorsteinsson, T.; Daak, A.; Rabinowicz, A.L.; Sancilio, F.D. Bioequivalence Demonstration for Omega-3 Acid Ethyl Ester Formulations: Rationale for Modification of Current Guidance. Clin. Ther. 2017, 39, 652–658. [Google Scholar] [CrossRef]
- Ito, M.K. A comparative overview of prescription omega-3 fatty acid products. Pharmacol. Ther. 2015, 40, 826. [Google Scholar]
- Araujo, P.; Tilahun, E.; Zeng, Y. A novel strategy for discriminating marine oils by using the positional distribution (sn-1, sn-2, sn-3) of omega-3 polyunsaturated fatty acids in triacylglycerols. Talanta 2018, 182, 32–37. [Google Scholar] [CrossRef] [PubMed]
- O’keefe, S.; Ackman, R.G. Vitamins A, D3 and E in Nova Scotian cod liver oils. Proc. Nova Scotian Inst. Sci. 1987, 37, 1–7. [Google Scholar]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications-a review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.C.; Spink, J.; Lipp, M. Development and application of a database of food ingredient fraud and economically motivated adulteration from 1980 to 2010. J. Food Sci. 2012, 77, 118–126. [Google Scholar] [CrossRef]
- Jangaard, P.; Brockerhoff, H.; Burgher, R.; Hoyle, R. Seasonal changes in general condition and lipid content of cod from inshore waters. J. Fish. Res. Board Can. 1967, 24, 607–612. [Google Scholar] [CrossRef]
- Dale, K.; Yadetie, F.; Müller, M.B.; Pampanin, D.M.; Gilabert, A.; Zhang, X.; Tairova, Z.; Haarr, A.; Lille-Langøy, R.; Lyche, J.L. Proteomics and lipidomics analyses reveal modulation of lipid metabolism by perfluoroalkyl substances in liver of Atlantic cod (Gadus morhua). Aquat. Toxicol. 2020, 227, 105590. [Google Scholar] [CrossRef]
- Wu, B.; Wei, F.; Xu, S.; Xie, Y.; Lv, X. Mass spectrometry-based lipidomics as a powerful platform in foodomics research. Trends Food Sci. Technol. 2021, 107, 358–376. [Google Scholar] [CrossRef]
- Giese, E.; Rohn, S.; Fritsche, J. Chemometric tools for the authentication of cod liver oil based on nuclear magnetic resonance and infrared spectroscopy data. Anal. Bioanal. Chem. 2019, 411, 6931–6942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, A.M.; Sousa, C. A review on the application of vibrational spectroscopy to the chemistry of nuts. Food Chem. 2019, 277, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Ahmmed, F.; Fuller, I.D.; Killeen, D.P.; Fraser-Miller, S.J.; Gordon, K.C. Raman and Infrared Spectroscopic Data Fusion Strategies for Rapid, Multicomponent Quantitation of Krill Oil Compositions. ACS Food Sci. Technol. 2021, 1, 570–578. [Google Scholar] [CrossRef]
- Rimai, L.; Heyde, M.; Gill, D. Vibrational spectra of some carotenoids and related linear polyenes. Raman spectroscopic study. J. Am. Chem. Soc. 1973, 95, 4493–4501. [Google Scholar] [CrossRef]
- Rohman, A.; Man, Y.B. Authentication analysis of cod liver oil from beef fat using fatty acid composition and FTIR spectra. Food Addit. Contam. Part. A Chem. Anal. Control. Expo. Risk Assess 2011, 28, 1469–1474. [Google Scholar] [CrossRef]
- Bekhit, M.Y.; Grung, B.; Mjos, S.A. Determination of Omega-3 Fatty Acids in Fish Oil Supplements Using Vibrational Spectroscopy and Chemometric Methods. Appl. Spectrosc. 2014, 68, 1190–1200. [Google Scholar] [CrossRef]
- Gao, B.; Xu, S.; Han, L.; Liu, X. FT-IR-based quantitative analysis strategy for target adulterant in fish oil multiply adulterated with terrestrial animal lipid. Food Chem. 2021, 343, 128420. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Ying, Y. Applications of Raman spectroscopy in agricultural products and food analysis: A review. Appl. Spectrosc. Rev. 2011, 46, 539–560. [Google Scholar] [CrossRef]
- Herrero, A.M. Raman spectroscopy a promising technique for quality assessment of meat and fish: A review. Food Chem. 2008, 107, 1642–1651. [Google Scholar] [CrossRef]
- Vongsvivut, J.; Miller, M.R.; McNaughton, D.; Heraud, P.; Barrow, C.J. Rapid Discrimination and Determination of Polyunsaturated Fatty Acid Composition in Marine Oils by FTIR Spectroscopy and Multivariate Data Analysis. Food Bioproc. Tech. 2014, 7, 2410–2422. [Google Scholar] [CrossRef]
- Killeen, D.P.; Card, A.; Gordon, K.C.; Perry, N.B. First Use of Handheld Raman Spectroscopy to Analyze Omega-3 Fatty Acids in Intact Fish Oil Capsules. Appl. Spectrosc. 2019, 74, 365–371. [Google Scholar] [CrossRef]
- Giese, E.; Winkelmann, O.; Rohn, S.; Fritsche, J. Determining quality parameters of fish oils by means of 1H nuclear magnetic resonance, mid-infrared, and near-infrared spectroscopy in combination with multivariate statistics. Food Res. Int. 2018, 106, 116–128. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Bai, X.; Xiao, Q.; Liu, F.; Zhou, L.; Zhang, C. Detection of adulteration in food based on nondestructive analysis techniques: A review. Crit. Rev. Food Sci. Nutr. 2021, 61, 2351–2371. [Google Scholar] [CrossRef] [PubMed]
- Rifna, E.; Pandiselvam, R.; Kothakota, A.; Rao, K.S.; Dwivedi, M.; Kumar, M.; Thirumdas, R.; Ramesh, S. Advanced process analytical tools for identification of adulterants in edible oils–A review. Food Chem. 2022, 369, 130898. [Google Scholar] [CrossRef]
- Ahmmed, F.; Fraser-Miller, S.J.; Garagoda Arachchige, P.S.; Schallenberg, M.; Novis, P.; Gordon, K.C. Lake snow caused by the invasive diatom Lindavia intermedia can be discriminated from different sites and from other algae using vibrational spectroscopy. J. Raman Spectrosc. 2021, 52, 2597–2608. [Google Scholar] [CrossRef]
- Fraser-Miller, S.J.; Rooney, J.S.; Lau, M.; Gordon, K.C.; Schultz, M. Can Coupling Multiple Complementary Methods Improve the Spectroscopic Based Diagnosis of Gastrointestinal Illnesses? A Proof of Principle Ex Vivo Study Using Celiac Disease as the Model Illness. Anal. Chem. 2021, 93, 6363–6374. [Google Scholar] [CrossRef]
- Fraser, S.J.; Oughton, J.; Batten, W.A.; Clark, A.S.; Schmierer, D.M.; Gordon, K.C.; Strachan, C.J. Simultaneous qualitative and quantitative analysis of counterfeit and unregistered medicines using Raman spectroscopy. J. Raman Spectrosc. 2013, 44, 1172–1180. [Google Scholar] [CrossRef]
- McIntyre, S.M.; Ma, Q.; Burritt, D.J.; Oey, I.; Gordon, K.C.; Fraser-Miller, S.J. Vibrational spectroscopy and chemometrics for quantifying key bioactive components of various plum cultivars grown in New Zealand. J. Raman Spectrosc. 2020, 51, 1138–1152. [Google Scholar] [CrossRef]
- Bērziņš, K.; Harrison, S.D.L.; Leong, C.; Fraser-Miller, S.J.; Harper, M.J.; Diana, A.; Gibson, R.S.; Houghton, L.A.; Gordon, K.C. Qualitative and Quantitative Vibrational Spectroscopic Analysis of Macronutrients in Breast Milk. Spectrochim. Acta A Mol. Biomol. 2020, 246, 118982. [Google Scholar] [CrossRef] [PubMed]
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, Č.; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A. Orange: Data mining toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]
- Robert, C.; Jessep, W.; Sutton, J.J.; Hicks, T.M.; Loeffen, M.; Farouk, M.; Ward, J.F.; Bain, W.E.; Craigie, C.R.; Fraser-Miller, S.J.; et al. Evaluating low- mid- and high-level fusion strategies for combining Raman and infrared spectroscopy for quality assessment of red meat. Food Chem. 2021, 361, 130154. [Google Scholar] [CrossRef]
- Kizil, R.; Irudayaraj, J. Spectroscopic Technique: Fourier Transform Raman (FT-Raman) Spectroscopy. In Modern Techniques for Food Authentication; Elservier: Amsterdam, The Netherlands, 2018; pp. 193–217. [Google Scholar]
- Afseth, N.K.; Wold, J.P.; Segtnan, V.H. The potential of Raman spectroscopy for characterisation of the fatty acid unsaturation of salmon. Anal. Chim. Acta 2006, 572, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Nettleton, J.A. Omega-3 Fatty Acids and Health; Springer: New York, NY, USA, 1995; pp. 64–76. [Google Scholar]
- Rohman, A.; Che Man, Y.B. The use of Fourier transform mid infrared (FT-MIR) spectroscopy for detection and quantification of adulteration in virgin coconut oil. Food Chem. 2011, 129, 583–588. [Google Scholar] [CrossRef]
- Guillen, M.D.; Cabo, N. Characterization of edible oils and lard by Fourier transform infrared spectroscopy. Relationships between composition and frequency of concrete bands in the fingerprint region. JAOCS 1997, 74, 1281–1286. [Google Scholar] [CrossRef]
- Rohman, A.; Che Man, Y.B. Application of Fourier transform infrared (FT-IR) spectroscopy combined with chemometrics for authentication of cod-liver oil. Vib. Spectrosc. 2011, 55, 141–145. [Google Scholar] [CrossRef]
- Putri, A.R.; Rohman, A.; Riyanto, S. Authentication of Patin (Pangasius Micronemus) Fish Oil Adulterated with Palm Oil Using Ftir Spectroscopy Combined with Chemometrics. Int. J. Appl. Pharm. 2019, 11, 195–199. [Google Scholar] [CrossRef]
- Failloux, N.; Bonnet, I.; Baron, M.-H.; Perrier, E. Quantitative analysis of vitamin A degradation by Raman spectroscopy. Appl. Spectrosc. 2003, 57, 1117–1122. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Cross Validated Model Set Accuracy (%) | Test Set Accuracy (%) |
|---|---|---|
| Raman | 94 | 76 |
| IR | 99 | 82 |
| Fused data | 97 | 85 |
| Instrument Used | Model Name | No. Factors | Prediction (Test Set) | Prediction (Test Set after SVM Classification) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| r2 | Slope | Offset | RMSEP (%) | r2 | Slope | Offset | RMSEP (%) | |||
| Raman (model range: 0 to 50 %) | CLOSO_PO% | 3 | 0.95 | 0.84 | 2.0 | 4.1 | 0.95 | 0.86 | 1.2 | 4.4 |
| CLO_PO% | 1 | 0.96 | 1 | −1.3 | 3.5 | 0.98 | 0.93 | 1.9 | 2.6 | |
| SO_PO% | 2 | 0.91 | 0.76 | 4.2 | 5.6 | 0.88 | 0.77 | 4 | 5.7 | |
| IR (model range: 0 to 50 %) | CLOSO_PO% | 2 | 0.92 | 1 | −3.5 | 5.3 | 0.94 | 0.99 | −2.4 | 4.6 |
| CLO_PO% | 1 | 0.94 | 1 | −1.6 | 4.6 | 0.96 | 0.9 | 3 | 3.5 | |
| SO_PO% | 2 | 0.97 | 1 | −1.2 | 3.1 | 0.96 | 1 | −1.7 | 3.4 | |
| Low-level fusion (model range: 0 to 50 %) | CLOSO_PO% | 2 | 0.95 | 0.91 | −0.18 | 4.3 | 0.96 | 0.84 | 2.1 | 3.9 |
| CLO_PO% | 1 | 0.96 | 1.01 | −1.4 | 3.7 | 0.98 | 0.94 | 1.4 | 2.5 | |
| SO_PO% | 2 | 0.96 | 0.92 | 1.1 | 3.8 | 0.95 | 0.87 | 3.1 | 4.1 | |
| Instrument Used | Model Name | No. Factors | Prediction (Test Set) | Prediction (Test Set after SVM Classification) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| r2 | Slope | Offset | RMSEP (%) | r2 | Slope | Offset | RMSEP (%) | |||
| Raman (model range: 0 to 50 %) | CLOSO_O3C% | 2 | 0.97 | 0.91 | 0.22 | 3.2 | 0.97 | 0.89 | 0.77 | 3.4 |
| CLO_O3C % | 1 | 0.98 | 0.97 | 1.2 | 2.3 | 0.99 | 1 | −0.8 | 1.5 | |
| SO_O3C % | 2 | 0.98 | 0.92 | −0.1 | 2.5 | 0.97 | 0.89 | 0.94 | 3.4 | |
| IR (model range: 0 to 50 %) | CLOSO_O3C% | 1 | 0.97 | 0.92 | 0.2 | 3.2 | 0.96 | 0.89 | 1.7 | 3.8 |
| CLO_O3C % | 1 | 0.99 | 0.93 | 0.02 | 3.3 | 0.99 | 0.95 | −0.7 | 2.1 | |
| SO_O3C % | 2 | 0.99 | 0.95 | −0.4 | 1.7 | 0.99 | 0.95 | −0.6 | 1.6 | |
| Low-level fusion (model range: 0 to 50 %) | CLOSO_O3C% | 2 | 0.99 | 0.93 | −0.32 | 2.8 | 0.99 | 0.92 | 0.1 | 2.4 |
| CLO_O3C % | 1 | 0.99 | 0.95 | 0.54 | 1.6 | 0.99 | 0.98 | −0.5 | 1.5 | |
| SO_O3C % | 2 | 0.99 | 0.94 | −0.3 | 1.8 | 0.99 | 0.93 | 0.22 | 1.9 | |
| Instrument Used | Model Name | No. Factors | Prediction (Test Set) | Prediction (Test Set after SVM Classification) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| r2 | Slope | Offset | RMSEP (%) | r2 | Slope | Offset | RMSEP (%) | |||
| Raman (model range: 0 to 50 %) | CLOSO_FO% | 3 | 0.75 | 0.61 | 3.9 | 9.3 | 0.76 | 0.59 | 5.3 | 8.6 |
| CLO _FO% | 1 | 0.79 | 0.88 | 5.9 | 8.4 | 0.64 | 0.83 | 7.8 | 10.6 | |
| SO _FO% | 3 | NA | 0.69 | −15.6 | 23 | NA | 0.64 | −11.9 | 21.8 | |
| IR (model range: 0 to 50 %) | CLOSO_FO% | 2 | 0.75 | 0.65 | 9.3 | 9.4 | 0.72 | 0.66 | 8.7 | 9.5 |
| CLO _FO% | 2 | 0.88 | 0.62 | 2.3 | 6.3 | 0.85 | 0.96 | −9.9 | 6.9 | |
| SO _FO% | 2 | 0.88 | 0.66 | 5.8 | 6.5 | 0.88 | 0.69 | 4.9 | 6.2 | |
| Low-level fusion (model range: 0 to 50 %) | CLOSO_FO% | 3 | 0.79 | 0.76 | 5.2 | 8.5 | 0.77 | 0.75 | 5.5 | 8.6 |
| CLO _FO% | 2 | 0.82 | 0.89 | 5.2 | 7.9 | 0.77 | 0.89 | 5.3 | 8.5 | |
| SO _FO% | 2 | 0.87 | 0.71 | 1.3 | 6.9 | 0.79 | 0.78 | −1.4 | 8.7 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmmed, F.; Killeen, D.P.; Gordon, K.C.; Fraser-Miller, S.J. Rapid Quantitation of Adulterants in Premium Marine Oils by Raman and IR Spectroscopy: A Data Fusion Approach. Molecules 2022, 27, 4534. https://doi.org/10.3390/molecules27144534
Ahmmed F, Killeen DP, Gordon KC, Fraser-Miller SJ. Rapid Quantitation of Adulterants in Premium Marine Oils by Raman and IR Spectroscopy: A Data Fusion Approach. Molecules. 2022; 27(14):4534. https://doi.org/10.3390/molecules27144534
Chicago/Turabian StyleAhmmed, Fatema, Daniel P. Killeen, Keith C. Gordon, and Sara J. Fraser-Miller. 2022. "Rapid Quantitation of Adulterants in Premium Marine Oils by Raman and IR Spectroscopy: A Data Fusion Approach" Molecules 27, no. 14: 4534. https://doi.org/10.3390/molecules27144534
APA StyleAhmmed, F., Killeen, D. P., Gordon, K. C., & Fraser-Miller, S. J. (2022). Rapid Quantitation of Adulterants in Premium Marine Oils by Raman and IR Spectroscopy: A Data Fusion Approach. Molecules, 27(14), 4534. https://doi.org/10.3390/molecules27144534