
Pd(II) Binding Strength of a Novel Ambidentate Dipeptide-Hydroxypyridinonate Ligand: A Solution Equilibrium Study


Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Syntheses
2.2.1. Synthesis of Benzyl-(1-((1-((2-(3-(Benzyloxy)-2-methyl-4-oxopyridin-1(4H)-yl)ethyl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)carbamate, (1)
2.2.2. Synthesis of the Trifluoroacetate Salt of 2-Amino-N-(1-((2-(3-Hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)ethyl)amino)-1-oxopropan-2-yl)propanamide, H(L1) CF3COOH
2.3. Solution Studies
3. Results and Discussion
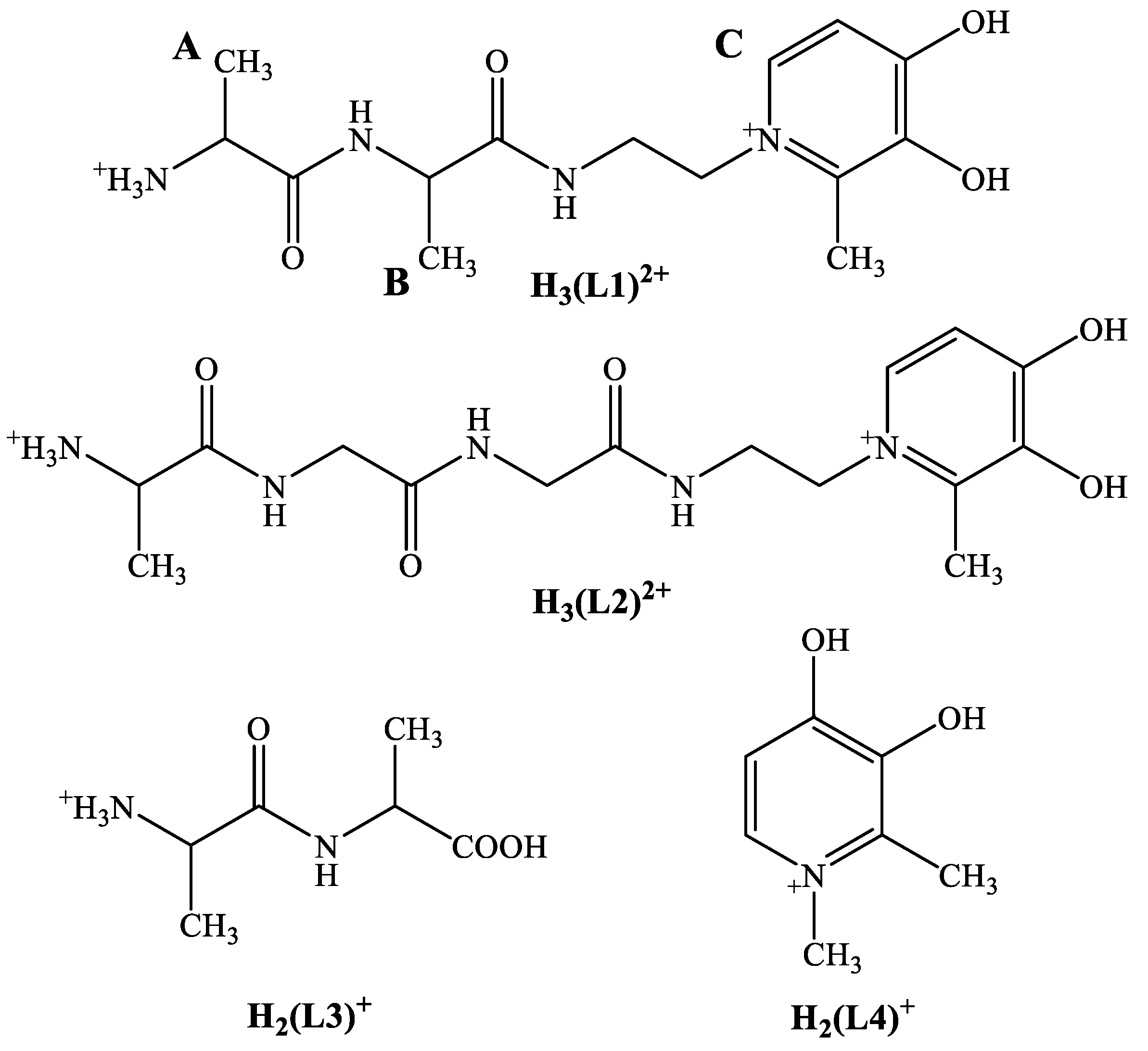
3.1. Synthesis and Characterization of the Novel Peptide Conjugate, H(L1)
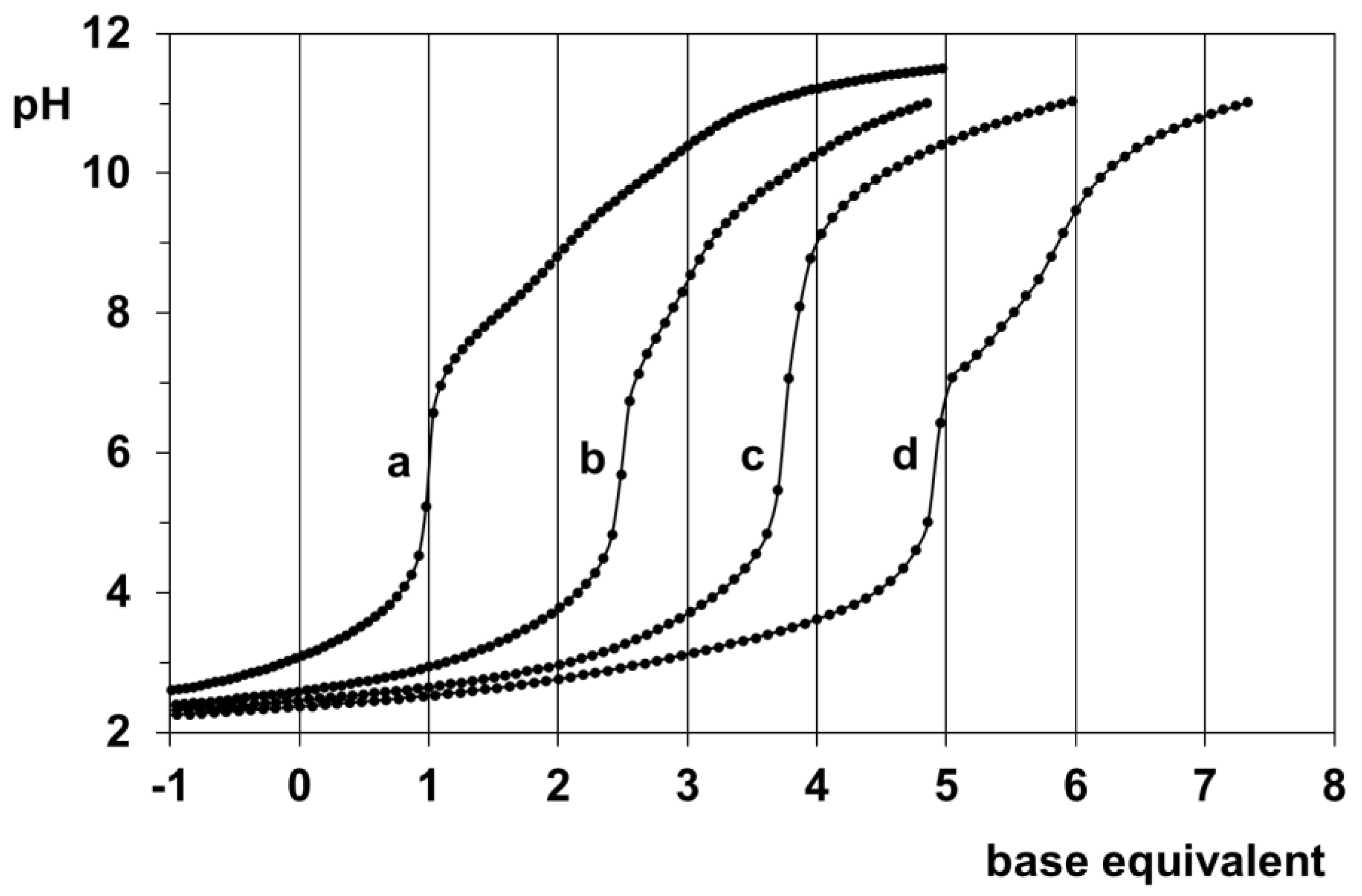
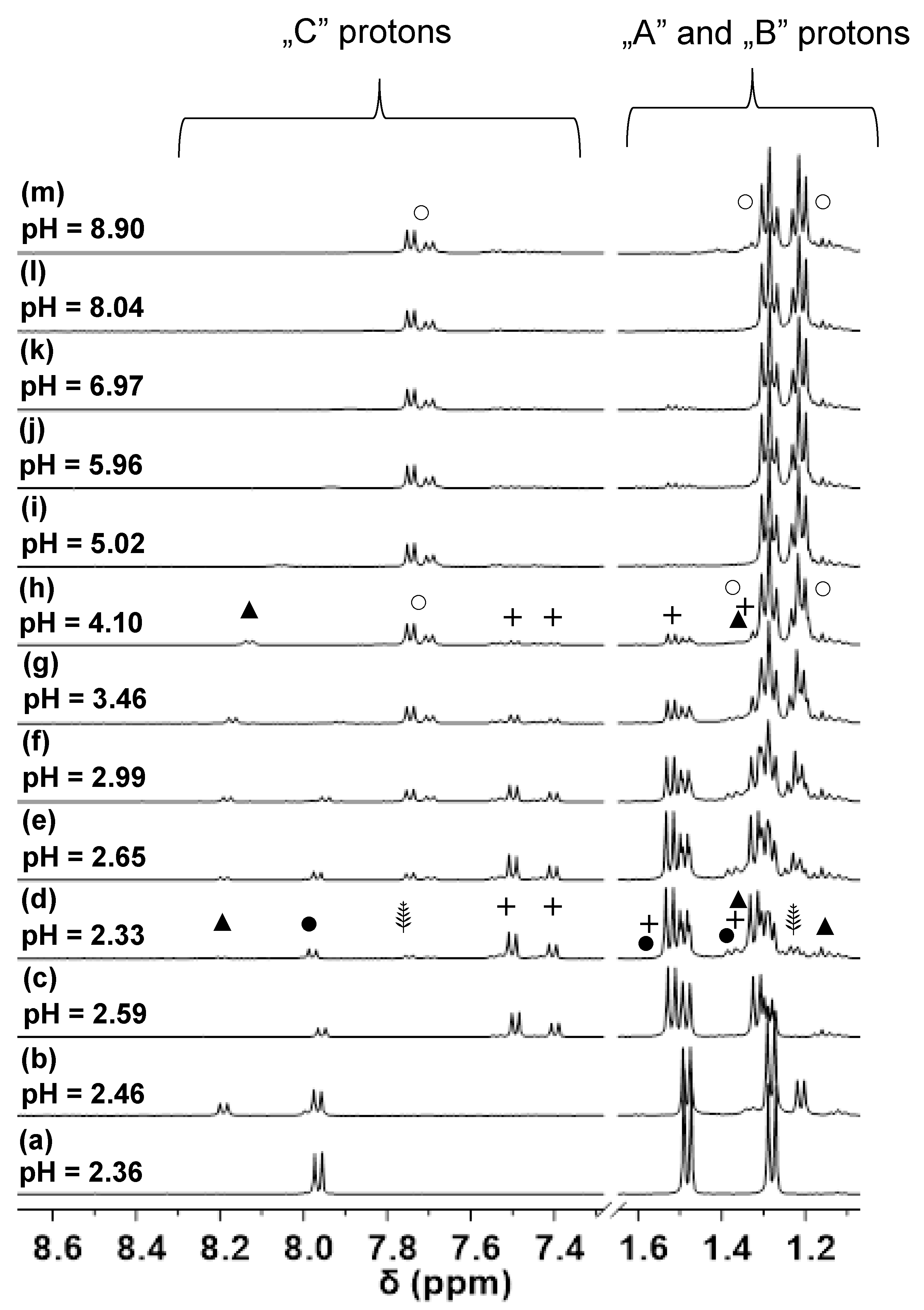
3.2. Proton Dissociation Processes of H(L1)
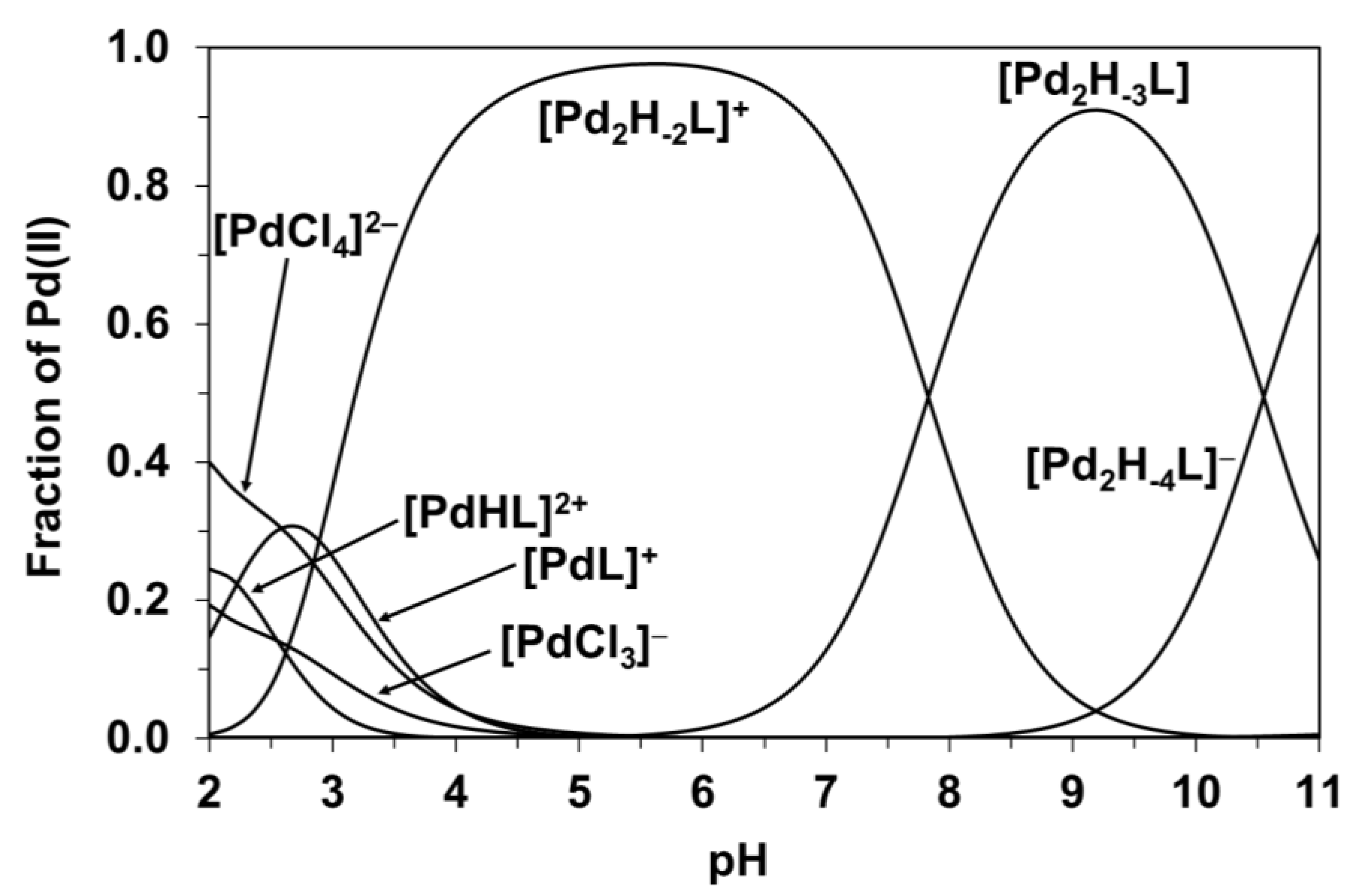
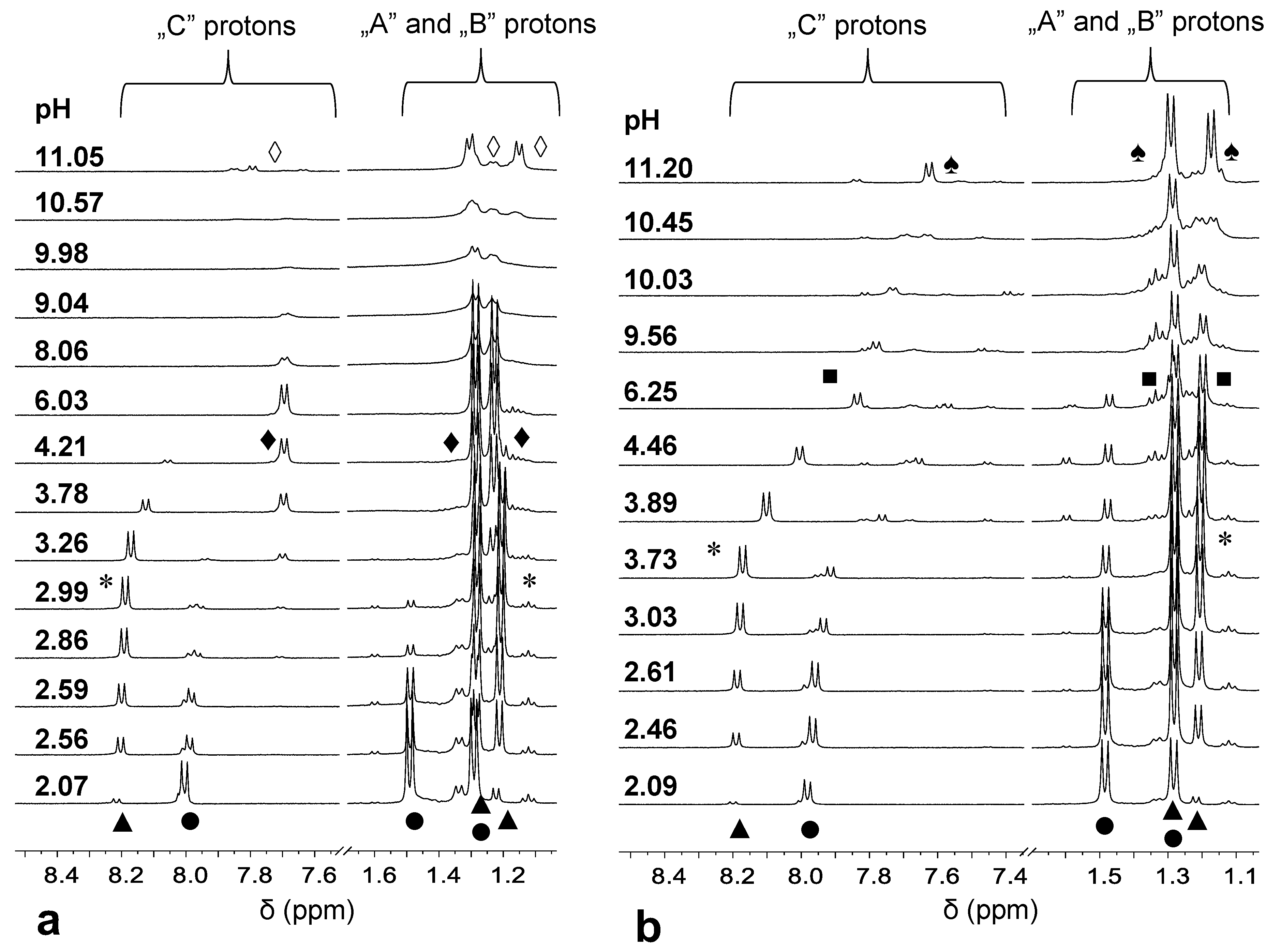
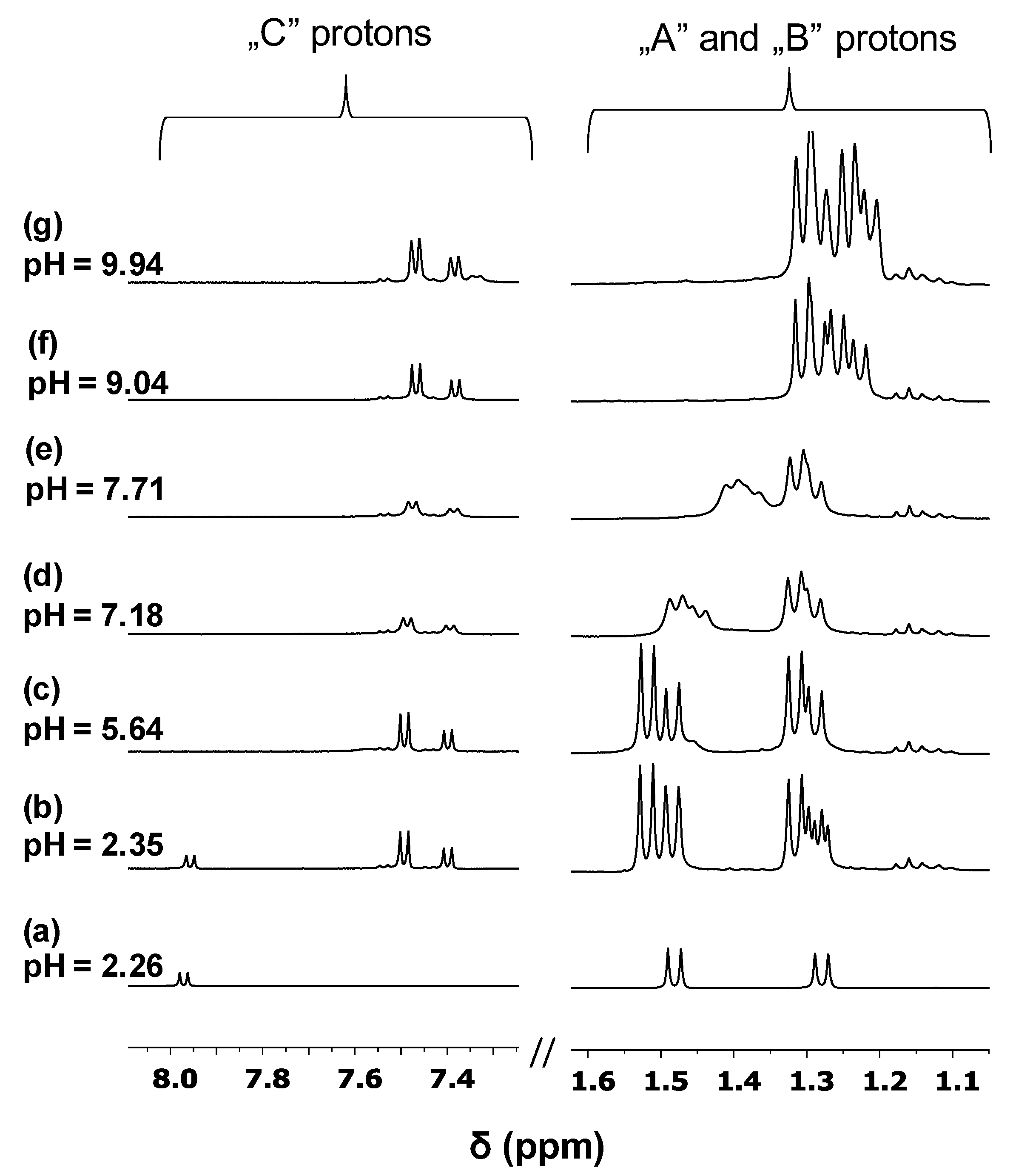
3.3. Pd(II) Complexation of H(L1)
3.4. Formation of Heterobimetallic Complexes with H(L1) in Solution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef]
- Luengo, A.; Marzo, I.; Reback, M.; Daubit, I.M.; Fernández-Moreira, V.; Metzler-Nolte, N.; Gimeno, M.C. Luminescent Bimetallic IrIII/AuI Peptide Bioconjugates as Potential Theranostic Agents. Chem. Eur. J. 2020, 26, 12158–12167. [Google Scholar] [CrossRef]
- Albada, B.; Metzler-Nolte, N. Organometallic−Peptide Bioconjugates: Synthetic Strategies and Medicinal Applications. Chem. Rev. 2016, 116, 11797–11839. [Google Scholar] [CrossRef]
- Nagy, S.; Ozsváth, A.; Bényei, A.C.; Farkas, E.; Buglyó, P. Donor Atom Preference of Organoruthenium and Organorhodium Cations on the Interaction with Novel Ambidentate (N,N) and (O,O) Chelating Ligands in Aqueous Solution. Molecules 2021, 26, 3586. [Google Scholar] [CrossRef]
- Ozsváth, A.; Diószegi, R.; Bényei, A.C.; Buglyó, P. Pd(II)-Complexes of a novel pyridinone based tripeptide conjugate: Solution and solid state studies. Dalton Trans. 2020, 49, 9254–9267. [Google Scholar] [CrossRef]
- Nagy, I.; Farkas, E.; Kasparkova, J.; Kostrhunova, H.; Brabec, V.; Buglyó, P. Synthesis and characterization of (Ru(II), Co(III)) heterobimetallic complexes formed with a 1,10-phenanthroline based hydroxamic acid conjugate. J. Organomet. Chem. 2020, 916, 121265. [Google Scholar] [CrossRef]
- Ozsváth, A.; Farkas, E.; Diószegi, R.; Buglyó, P. Versatility and trends in the interaction between Pd(II) and peptide hydroxamic acids. New J. Chem. 2019, 43, 8239–8249. [Google Scholar] [CrossRef]
- Kapdi, A.R.; Fairlamb, I.J.S. Anti-cancer palladium complexes: A focus on PdX2L2, palladacycles and related complexes. Chem. Soc. Rev. 2014, 43, 4751–4777. [Google Scholar] [CrossRef]
- Carneiro, T.J.; Martins, A.S.; Marques, M.P.M.; Gil, A.M. Metabolic Aspects of Palladium(II) Potential Anti-Cancer Drugs. Front. Oncol. 2020, 10, 590970. [Google Scholar] [CrossRef]
- Scattolin, T.; Voloshkin, V.A.; Visentin, F.; Nolan, S.P. A critical review of palladium organometallic anticancer agents. Cell Rep. Phys. Sci. 2021, 2, 100446. [Google Scholar] [CrossRef]
- Czarnomysy, R.; Radomska, D.; Szewczyk, O.K.; Roszczenko, P.; Bielawski, K. Platinum and Palladium Complexes as Promising Sources for Antitumor Treatments. Int. J. Mol. Sci. 2021, 22, 8271. [Google Scholar] [CrossRef]
- Failes, T.W.; Hambley, T.W. Models of hypoxia activated prodrugs: Co(III) complexes of hydroxamic acids. Dalton Trans. 2006, 15, 1895–1901. [Google Scholar] [CrossRef]
- Buglyó, P.; Kacsir, I.; Kozsup, M.; Nagy, I.; Nagy, S.; Bényei, A.C.; Kováts, É.; Farkas, E. Tuning the redox potentials of ternary cobalt(III) complexes containing various hydroxamates. Inorg. Chim. Acta 2018, 472, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Nagy, S.; Tóth, E.; Kacsir, I.; Makai, A.; Bényei, A.C.; Buglyó, P. Effect of the replacement of tripodal 4N donors by two 2N chelators on the redox and cytotoxic activity of maltolato and deferipronato containing Co(III) complexes. J. Inorg. Biochem. 2021, 220, 111372. [Google Scholar] [CrossRef]
- Wang, G.; Hazra, T.K.; Mitra, S.; Lee, H.M.; Englander, E.W. Mitochondrial DNA damage and a hypoxic response are induced by CoCl2 in rat neuronal PC12 cells. Nucleic Acids Res. 2000, 28, 2135–2140. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Gan, X.; Zhang, L.; Liu, B.; Zhu, Z.; Li, T.; Zhu, J.; Chen, J.; Yu, H. CoCl2 induces apoptosis via a ROS-dependent pathway and Drp1-mediated mitochondria fission in periodontal ligament stem cells. Cell Physiol. 2018, 315, C389–C397. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. (Eds.) Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann: Oxford, UK, 2009; pp. 88–444. [Google Scholar] [CrossRef]
- Minhua, F.; van der Does, L.; Bantjes, A. Iron(III) chelating resins II. 3-Hydroxy-4(1H)-pyridinones-Sepharose gels. J. Biomater. Sci. Polym. Ed. 1993, 4, 145–154. [Google Scholar] [CrossRef]
- Irto, A.; Cardiano, P.; Chand, K.; Cigala, R.M.; Crea, F.; de Stefano, C.; Gano, L.; Sammartano, S.; Santos, M.A. Bifunctional 3-hydroxy-4-pyridinones as effective aluminium chelators: Synthesis, solution equilibrium studies and in vivo evaluation. J. Inorg. Biochem. 2018, 186, 116–129. [Google Scholar] [CrossRef]
- Ágoston, C.G.; Jankowska, T.K.; Sóvágó, I. Potentiometric and NMR studies on palladium(II) complexes of oligoglycines and related ligands with non-co-ordinating side chains. J. Chem. Soc. Dalton Trans. 1999, 18, 3295–3302. [Google Scholar] [CrossRef]
- Gran, G. Determination of the equivalent point in potentiometric titrations. Acta Chem. Scand. 1950, 4, 559–577. [Google Scholar] [CrossRef] [Green Version]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. SUPERQUAD: An improved general program for computation of formation constants from potentiometric data. J. Chem. Soc. Dalton Trans. 1985, 6, 1195–1200. [Google Scholar] [CrossRef]
- Zékány, L.; Nagypál, I. Computational Methods for the Determination of Formation Constants; Leggett, D.J., Ed.; Springer: Boston, MA, USA, 1985; pp. 291–353. [Google Scholar] [CrossRef]
- Elding, L.I. Palladium(II) halide complexes. I. Stabilities and spectra of palladium(II) chloro and bromo aqua complexes. Inorg. Chim. Acta 1972, 6, 647–651. [Google Scholar] [CrossRef]
- Krężel, A.; Bal, W. A formula for correlating pKa values determined in D2O and H2O. J. Inorg. Biochem. 2004, 98, 161–166. [Google Scholar] [CrossRef]
- Szakács, Z.; Kraszni, M.; Noszál, B. Determination of microscopic acid–base parameters from NMR–pH titrations. Anal. Bioanal. Chem. 2004, 378, 1428–1448. [Google Scholar] [CrossRef]
- Buglyó, P.; Kiss, T.; Kiss, E.; Sanna, D.; Garribba, E.; Micera, G. Interaction between the low molecular mass components of blood serum and the VO(IV)–DHP system (DHP = 1,2-dimethyl-3-hydroxy-4(1H)-pyridinone). J. Chem. Soc. Dalton Trans. 2002, 11, 2275–2282. [Google Scholar] [CrossRef]
- Gergely, A.; Nagypál, I. Studies on transition-metal–peptide complexes. Part 1. Equilibrium and thermochemical study of the copper(II) complexes of glycylglycine, glycyl-DL-α-alanine, DL-α-alanylglycine, and DL-α-alanyl-DL-α-alanine. J. Chem. Soc. Dalton Trans. 1977, 11, 1104–1108. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H(L1) | H(L2) [5] | H(L3) [29] | H(L4) [28] | ||
|---|---|---|---|---|---|
| pH-Potentiometry | 1H NMR | pH-Potentiometry | |||
| pKCOOH | - | - | - | 3.08 | - |
| pKOH(4) | 3.22(3) | 3.46(5) | 3.32 | - | 3.70 |
| pKNH3+ | 7.88(2) | 7.70(2) | 7.74 | 8.26 | - |
| pKOH(3) | 9.64(1) | 9.56(4) | 9.46 | - | 9.76 |
| H(L1) | H(L2) [5] | |
|---|---|---|
| [PdHL]2+ | 26.84(1) | 28.18 |
| [PdL]+ | 24.62(1) | 24.54 |
| [PdH–1L] | 20.35(3) | 21.01 |
| [PdH–2L]− | 10.39(5) | 12.7 |
| [PdH–3L]2− | −0.71(5) | 2.4 |
| [Pd2H–1L]2+ | - | 32.71 |
| [Pd2H–2L]+ | 28.66(2) | 28.99 |
| [Pd2H–3L] | 20.82(4) | 21.8 |
| [Pd2H–4L]− | 10.27(5) | 11.8 |
| pK PdLH | 2.22 | 3.64 |
| pK PdL | 4.27 | 3.53 |
| pK PdH-1L | 9.96 | 8.31 |
| pK PdH-2L | 11.10 | 10.30 |
| pK Pd2H-1L | - | 3.72 |
| pK Pd2H-2L | 7.84 | 7.20 |
| pK Pd2H-3L | 10.55 | 10.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bíró, L.; Ozsváth, A.; Kapitány, R.; Buglyó, P. Pd(II) Binding Strength of a Novel Ambidentate Dipeptide-Hydroxypyridinonate Ligand: A Solution Equilibrium Study. Molecules 2022, 27, 4667. https://doi.org/10.3390/molecules27144667
Bíró L, Ozsváth A, Kapitány R, Buglyó P. Pd(II) Binding Strength of a Novel Ambidentate Dipeptide-Hydroxypyridinonate Ligand: A Solution Equilibrium Study. Molecules. 2022; 27(14):4667. https://doi.org/10.3390/molecules27144667
Chicago/Turabian StyleBíró, Linda, András Ozsváth, Réka Kapitány, and Péter Buglyó. 2022. "Pd(II) Binding Strength of a Novel Ambidentate Dipeptide-Hydroxypyridinonate Ligand: A Solution Equilibrium Study" Molecules 27, no. 14: 4667. https://doi.org/10.3390/molecules27144667
APA StyleBíró, L., Ozsváth, A., Kapitány, R., & Buglyó, P. (2022). Pd(II) Binding Strength of a Novel Ambidentate Dipeptide-Hydroxypyridinonate Ligand: A Solution Equilibrium Study. Molecules, 27(14), 4667. https://doi.org/10.3390/molecules27144667