
Selective Integrin α5β1 Targeting through Spatially Constrained Multivalent DNA-Based Nanoparticles

 , and
, and
Abstract
:
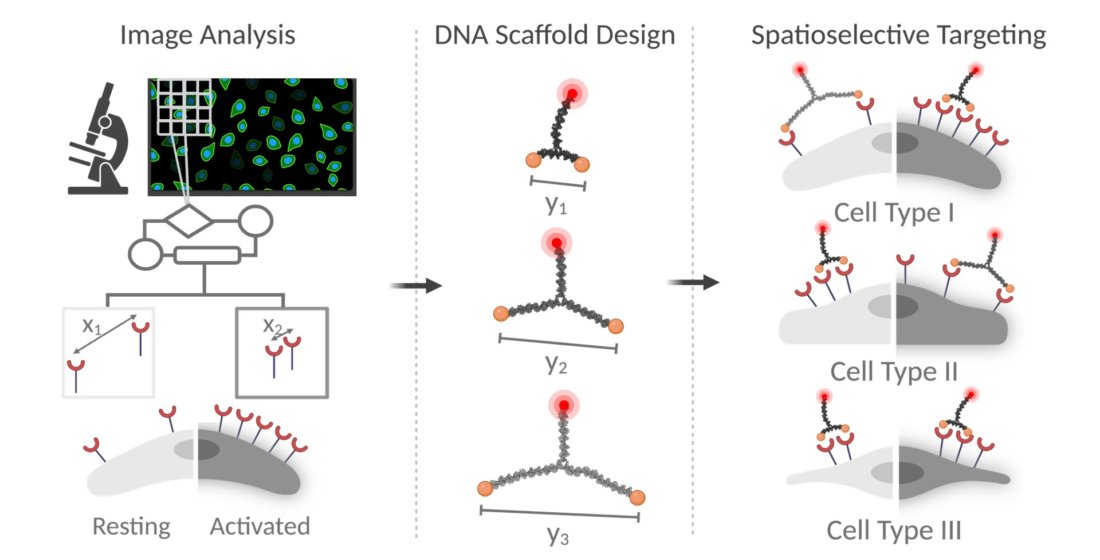{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Analysis of Cell Type Specific Integrin α5β1 Expression Levels
2.1.1. Choice of Cell Types
2.1.2. Activation of α5β1
2.1.3. Model of Integrin α5β1 Distributions
2.2. Design of a Spatially Constrained DNA Nanoparticle Library
2.3. Analysis of α5β1 Targeting Based on Spatial Selectivity
3. Discussion
4. Materials and Methods
4.1. Cell Activation and Antibody Binding Assay
4.2. Ligand Synthesis and DNA Conjugation
4.3. DNA Scaffold Assembly
4.4. Confocal Cell Imaging
4.5. Receptor Distribution Heatmaps
4.6. Selectivity Quantification
4.7. In-Silco Simulation and Analysis of DNA Scaffolds
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An Overview of Active and Passive Targeting Strategies to Improve the Nanocarriers Efficiency to Tumour Sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vhora, I.; Patil, S.; Bhatt, P.; Gandhi, R.; Baradia, D.; Misra, A. Receptor-Targeted Drug Delivery: Current Perspective and Challenges. Ther. Deliv. 2014, 5, 1007–1024. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Sánchez-Madrid, F. Targeting the Integrin Interactome in Human Disease. Curr. Opin. Cell Biol. 2018, 55, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Samaržija, I.; Dekanić, A.; Humphries, J.D.; Paradžik, M.; Stojanović, N.; Humphries, M.J.; Ambriović-Ristov, A. Integrin Crosstalk Contributes to the Complexity of Signalling and Unpredictable Cancer Cell Fates. Cancers 2020, 12, 1910. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Gilcrease, M.Z. Integrin Signaling in Epithelial Cells. Cancer Lett. 2007, 247, 1–25. [Google Scholar] [CrossRef]
- Sales, A.; Ende, K.; Diemer, J.; Kyvik, A.R.; Veciana, J.; Ratera, I.; Kemkemer, R.; Spatz, J.P.; Guasch, J. Cell Type-Dependent Integrin Distribution in Adhesion and Migration Responses on Protein-Coated Microgrooved Substrates. ACS Omega 2019, 4, 1791–1800. [Google Scholar] [CrossRef]
- Schvartzman, M.; Palma, M.; Sable, J.; Abramson, J.; Hu, X.; Sheetz, M.P.; Wind, S.J. Nanolithographic Control of the Spatial Organization of Cellular Adhesion Receptors at the Single-Molecule Level. Nano Lett. 2011, 11, 1306–1312. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-Mediated Mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef]
- Martinez-Veracoechea, F.J.; Frenkel, D. Designing Super Selectivity in Multivalent Nano-Particle Binding. Proc. Natl. Acad. Sci. USA 2011, 108, 10963–10968. [Google Scholar] [CrossRef] [Green Version]
- Mezu-Ndubuisi, O.J.; Maheshwari, A. The Role of Integrins in Inflammation and Angiogenesis. Pediatr. Res. 2021, 89, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Teoh, C.M.; Tan, S.S.L.; Tran, T. Integrins as Therapeutic Targets for Respiratory Diseases. Curr. Mol. Med. 2015, 15, 714–734. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Reddy, D.S. Integrins as Receptor Targets for Neurological Disorders. Pharmacol. Ther. 2012, 134, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, P.; Rinaldi, D.A.; Bondu, V.; Kell, A.M.; Bradfute, S.; Lidke, D.S.; Buranda, T. Integrin Activation Is an Essential Component of SARS-CoV-2 Infection. Sci. Rep. 2021, 11, 20398. [Google Scholar] [CrossRef]
- Balcioglu, H.E.; van Hoorn, H.; Donato, D.M.; Schmidt, T.; Danen, E.H.J. The Integrin Expression Profile Modulates Orientation and Dynamics of Force Transmission at Cell–Matrix Adhesions. J. Cell Sci. 2015, 128, 1316–1326. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, D. Functions of Pulmonary Epithelial Integrins: From Development to Disease. Physiol. Rev. 2003, 83, 673–686. [Google Scholar] [CrossRef] [Green Version]
- Taddei, I.; Faraldo, M.M.; Teulière, J.; Deugnier, M.-A.; Thiery, J.P.; Glukhova, M.A. Integrins in Mammary Gland Development and Differentiation of Mammary Epithelium. J. Mammary Gland Biol. Neoplasia 2003, 8, 383–394. [Google Scholar] [CrossRef]
- Watt, F.M. Role of Integrins in Regulating Epidermal Adhesion, Growth and Differentiation. EMBO J. 2002, 21, 3919–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Barraza, J.; Francescone, R.; Luong, T.; Shah, N.; Madhani, R.; Cukierman, G.; Dulaimi, E.; Devarajan, K.; Egleston, B.L.; Nicolas, E.; et al. Matrix-Regulated Integrin Avβ5 Maintains A5β1-Dependent Desmoplastic Traits Prognostic of Neoplastic Recurrence. eLife 2017, 6, e20600. [Google Scholar] [CrossRef]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in Angiogenesis and Lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Qin, X.; Kong, F.; Chen, P.; Pan, G. Improving Cellular Uptake of Therapeutic Entities through Interaction with Components of Cell Membrane. Drug Deliv. 2019, 26, 328–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Zhang, X.-B.; Lv, Y.; Gong, L.; Wang, R.; Zhu, X.; Yang, R.; Tan, W. Functional DNA-Containing Nanomaterials: Cellular Applications in Biosensing, Imaging, and Targeted Therapy. Acc. Chem. Res. 2014, 47, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Sachlos, E.; Risueño, R.M.; Laronde, S.; Shapovalova, Z.; Lee, J.-H.; Russell, J.; Malig, M.; McNicol, J.D.; Fiebig-Comyn, A.; Graham, M.; et al. Identification of Drugs Including a Dopamine Receptor Antagonist That Selectively Target Cancer Stem Cells. Cell 2012, 149, 1284–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Zhao, C.; Spatz, J.P.; Wei, Q. Nanopatterned Adhesive, Stretchable Hydrogel to Control Ligand Spacing and Regulate Cell Spreading and Migration. ACS Nano 2017, 11, 8282–8291. [Google Scholar] [CrossRef]
- Mould, A.P. Getting Integrins into Shape: Recent Insights into How Integrin Activity Is Regulated by Conformational Changes. J. Cell Sci. 1996, 109, 2613–2618. [Google Scholar] [CrossRef]
- Spiess, M.; Hernandez-Varas, P.; Oddone, A.; Olofsson, H.; Blom, H.; Waithe, D.; Lock, J.G.; Lakadamyali, M.; Strömblad, S. Active and Inactive Β1 Integrins Segregate into Distinct Nanoclusters in Focal Adhesions. J. Cell Biol. 2018, 217, 1929–1940. [Google Scholar] [CrossRef] [Green Version]
- Young, J.L.; Hua, X.; Somsel, H.; Reichart, F.; Kessler, H.; Spatz, J.P. Integrin Subtypes and Nanoscale Ligand Presentation Influence Drug Sensitivity in Cancer Cells. Nano Lett. 2020, 20, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, S.; Dedden, D.; Nunez, R.V.; Matoba, K.; Takagi, J.; Biertümpfel, C.; Mizuno, N. Structural Insights into Integrin A5β1 Opening by Fibronectin Ligand. Sci. Adv. 2021, 7, eabe9716. [Google Scholar] [CrossRef]
- Seeman, N.C. Molecular Craftwork with DNA. In Culture of Chemistry: The Best Articles on the Human Side of 20th-Century Chemistry from the Archives of the Chemical Intelligencer; Hargittai, B., Hargittai, I., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 141–152. ISBN 978-1-4899-7565-2. [Google Scholar]
- Seeman, N.C. DNA in a Material World. Nature 2003, 421, 427–431. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Gao, H.; Deng, R.; Li, J. Emerging Applications of Nanotechnology for Controlling Cell-Surface Receptor Clustering. Angew. Chem. Int. Ed. 2019, 58, 4790–4799. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, D.; Wang, H.; Wang, F.; Liu, X. Construction of Smart Stimuli-Responsive DNA Nanostructures for Biomedical Applications. Chem. Eur. J. 2020, 27, 3929–3943. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Groves, B.; Muscat, R.A.; Seelig, G. DNA Nanotechnology from the Test Tube to the Cell. Nat. Nanotechnol. 2015, 10, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Rüegg, C.; Dormond, O.; Mariotti, A. Endothelial Cell Integrins and COX-2: Mediators and Therapeutic Targets of Tumor Angiogenesis. Biochim. Biophys. Acta Rev. Cancer 2004, 1654, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Chen, Z.; Wang, Z.-H.; Wang, H.; Huang, L.-M. Prognostic Significance of A5β1-Integrin Expression in Cervical Cancer. Asian Pac. J. Cancer Prev. 2013, 14, 3891–3895. [Google Scholar] [CrossRef] [Green Version]
- Ali, O.; Guillou, H.; Destaing, O.; Albigès-Rizo, C.; Block, M.R.; Fourcade, B. Cooperativity between Integrin Activation and Mechanical Stress Leads to Integrin Clustering. Biophys. J. 2011, 100, 2595–2604. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Xia, W.; Li, J.; Walz, T.; Humphries, M.J.; Vestweber, D.; Cabañas, C.; Lu, C.; Springer, T.A. Relating Conformation to Function in Integrin A5β1. Proc. Natl. Acad. Sci. USA 2016, 113, E3872–E3881. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; Yamashita, Y.; Kimoto, M. A Calcium- or Manganese-Dependent Epitope on the Integrin Β1 Chain Recognized by a Unique MAb. Int. Immunol. 1994, 6, 1221–1226. [Google Scholar] [CrossRef]
- Clark, K.; Pankov, R.; Travis, M.A.; Askari, J.A.; Mould, A.P.; Craig, S.E.; Newham, P.; Yamada, K.M.; Humphries, M.J. A Specific A5β1-Integrin Conformation Promotes Directional Integrin Translocation and Fibronectin Matrix Formation. J. Cell Sci. 2005, 118, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Smith, J.W. Mechanism of Integrin Activation by Disulfide Bond Reduction. Biochemistry 2001, 40, 8861–8867. [Google Scholar] [CrossRef]
- Ohto, U.; Shibata, T.; Tanji, H.; Ishida, H.; Krayukhina, E.; Uchiyama, S.; Miyake, K.; Shimizu, T. Structural Basis of CpG and Inhibitory DNA Recognition by Toll-like Receptor 9. Nature 2015, 520, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Comberlato, A.; Koga, M.M.; Nüssing, S.; Parish, I.A.; Bastings, M.M.C. Spatially Controlled Activation of Toll-like Receptor 9 with DNA-Based Nanomaterials. Nano Lett. 2022, 22, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Schlichthaerle, T.; Lindner, C.; Jungmann, R. Super-Resolved Visualization of Single DNA-Based Tension Sensors in Cell Adhesion. Nat. Commun. 2021, 12, 2510. [Google Scholar] [CrossRef]
- Arganda-Carreras, I.; Kaynig, V.; Rueden, C.; Eliceiri, K.W.; Schindelin, J.; Cardona, A.; Sebastian Seung, H. Trainable Weka Segmentation: A Machine Learning Tool for Microscopy Pixel Classification. Bioinformatics 2017, 33, 2424–2426. [Google Scholar] [CrossRef]
- Gajos, K.; Szafraniec, K.; Petrou, P.; Budkowski, A. Surface Density Dependent Orientation and Immunological Recognition of Antibody on Silicon: TOF-SIMS and Surface Analysis of Two Covalent Immobilization Methods. Appl. Surf. Sci. 2020, 518, 146269. [Google Scholar] [CrossRef]
- Adamczyk, Z. Modeling Adsorption of Colloids and Proteins. Curr. Opin. Colloid Interface Sci. 2012, 17, 173–186. [Google Scholar] [CrossRef]
- Pierschbacher, M.D.; Ruoslahti, E. Cell Attachment Activity of Fibronectin Can Be Duplicated by Small Synthetic Fragments of the Molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef]
- Ruoslahti, E. RGD and Other Recognition Sequences for Integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [Google Scholar] [CrossRef] [PubMed]
- Hersel, U.; Dahmen, C.; Kessler, H. RGD Modified Polymers: Biomaterials for Stimulated Cell Adhesion and Beyond. Biomaterials 2003, 24, 4385–4415. [Google Scholar] [CrossRef]
- Mitchell, J.S.; Glowacki, J.; Grandchamp, A.E.; Manning, R.S.; Maddocks, J.H. Sequence-Dependent Persistence Lengths of DNA. J. Chem. Theory Comput. 2017, 13, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Šulc, P.; Romano, F.; Ouldridge, T.E.; Rovigatti, L.; Doye, J.P.K.; Louis, A.A. Sequence-Dependent Thermodynamics of a Coarse-Grained DNA Model. J. Chem. Phys. 2012, 137, 135101. [Google Scholar] [CrossRef] [PubMed]
- Doye, J.P.K.; Ouldridge, T.E.; Louis, A.A.; Romano, F.; Šulc, P.; Matek, C.; Snodin, B.E.K.; Rovigatti, L.; Schreck, J.S.; Harrison, R.M.; et al. Coarse-Graining DNA for Simulations of DNA Nanotechnology. Phys. Chem. Chem. Phys. 2013, 15, 20395–20414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepzelter, D.; Bates, O.; Zaman, M. Integrin Clustering in Two and Three Dimensions. Langmuir 2012, 28, 5379–5386. [Google Scholar] [CrossRef]
- Moreno-Layseca, P.; Icha, J.; Hamidi, H.; Ivaska, J. Integrin Trafficking in Cells and Tissues. Nat. Cell Biol. 2019, 21, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Yan, D.; Liu, Y.; Huang, P.; Cui, H. The Roles of Integrin A5β1 in Human Cancer. Onco. Targets Ther. 2020, 13, 13329–13344. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Möckl, L.; Hirn, S.; Torrano, A.A.; Uhl, B.; Bräuchle, C.; Krombach, F. The Glycocalyx Regulates the Uptake of Nanoparticles by Human Endothelial Cells in Vitro. Nanomedicine 2017, 12, 207–217. [Google Scholar] [CrossRef]
- Peccati, F.; Jiménez-Osés, G. Enthalpy–Entropy Compensation in Biomolecular Recognition: A Computational Perspective. ACS Omega 2021, 6, 11122–11130. [Google Scholar] [CrossRef]
- Morzy, D.; Bastings, M. Significance of Receptor Mobility in Multivalent Binding on Lipid Membranes. Angew. Chem. Int. Ed. 2022, 61, e202114167. [Google Scholar] [CrossRef]
- Paszek, M.J.; DuFort, C.C.; Rossier, O.; Bainer, R.; Mouw, J.K.; Godula, K.; Hudak, J.E.; Lakins, J.N.; Wijekoon, A.C.; Cassereau, L.; et al. The Cancer Glycocalyx Mechanically Primes Integrin-Mediated Growth and Survival. Nature 2014, 511, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.M.J.; oude Egbrink, M.G.A. The Endothelial Glycocalyx: Composition, Functions, and Visualization. Pflügers Arch. Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möckl, L. The Emerging Role of the Mammalian Glycocalyx in Functional Membrane Organization and Immune System Regulation. Bastings 2020, 8, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, N.J.; Radhakrishnan, R. The Role of Glycocalyx in Nanocarrier-Cell Adhesion Investigated Using a Thermodynamic Model and Monte Carlo Simulations. J. Phys. Chem. 2007, 111, 15848–15856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, V.M.; Semetey, V.; Bracher, P.J.; Shen, N.; Whitesides, G.M. Dependence of Effective Molarity on Linker Length for an Intramolecular Protein−Ligand System. J. Am. Chem. Soc. 2007, 129, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Bastings, M.M.; Helms, B.A.; Van Baal, I.; Hackeng, T.M.; Merkx, M.; Meijer, E.W. From Phage Display to Dendrimer Display: Insights into Multivalent Binding. J. Am. Chem. Soc. 2011, 133, 6636–6641. [Google Scholar] [CrossRef] [PubMed]
- Mohri, K.; Nishikawa, M.; Takahashi, N.; Shiomi, T.; Matsuoka, N.; Ogawa, K.; Endo, M.; Hidaka, K.; Sugiyama, H.; Takahashi, Y.; et al. Design and Development of Nanosized DNA Assemblies in Polypod-like Structures as Efficient Vehicles for Immunostimulatory CpG Motifs to Immune Cells. ACS Nano 2012, 6, 5931–5940. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Autin, L.; Olson, A.J. Illustrate: Software for Biomolecular Illustration. Structure 2019, 27, 1716–1720. [Google Scholar] [CrossRef]
- Benson, E.; Mohammed, A.; Gardell, J.; Masich, S.; Czeizler, E.; Orponen, P.; Högberg, B. DNA Rendering of Polyhedral Meshes at the Nanoscale. Nature 2015, 523, 441–444. [Google Scholar] [CrossRef] [Green Version]
- Suma, A.; Poppleton, E.; Matthies, M.; Šulc, P.; Romano, F.; Louis, A.A.; Doye, J.P.K.; Micheletti, C.; Rovigatti, L. TacoxDNA: A User-Friendly Web Server for Simulations of Complex DNA Structures, from Single Strands to Origami. J. Comput. Chem. 2019, 40, 2586–2595. [Google Scholar] [CrossRef]
- Snodin, B.E.K.; Randisi, F.; Mosayebi, M.; Šulc, P.; Schreck, J.S.; Romano, F.; Ouldridge, T.E.; Tsukanov, R.; Nir, E.; Louis, A.A.; et al. Introducing Improved Structural Properties and Salt Dependence into a Coarse-Grained Model of DNA. J. Chem. Phys. 2015, 142, 234901. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurisinkal, E.E.; Caroprese, V.; Koga, M.M.; Morzy, D.; Bastings, M.M.C. Selective Integrin α5β1 Targeting through Spatially Constrained Multivalent DNA-Based Nanoparticles. Molecules 2022, 27, 4968. https://doi.org/10.3390/molecules27154968
Kurisinkal EE, Caroprese V, Koga MM, Morzy D, Bastings MMC. Selective Integrin α5β1 Targeting through Spatially Constrained Multivalent DNA-Based Nanoparticles. Molecules. 2022; 27(15):4968. https://doi.org/10.3390/molecules27154968
Chicago/Turabian StyleKurisinkal, Eva E., Vincenzo Caroprese, Marianna M. Koga, Diana Morzy, and Maartje M. C. Bastings. 2022. "Selective Integrin α5β1 Targeting through Spatially Constrained Multivalent DNA-Based Nanoparticles" Molecules 27, no. 15: 4968. https://doi.org/10.3390/molecules27154968
APA StyleKurisinkal, E. E., Caroprese, V., Koga, M. M., Morzy, D., & Bastings, M. M. C. (2022). Selective Integrin α5β1 Targeting through Spatially Constrained Multivalent DNA-Based Nanoparticles. Molecules, 27(15), 4968. https://doi.org/10.3390/molecules27154968