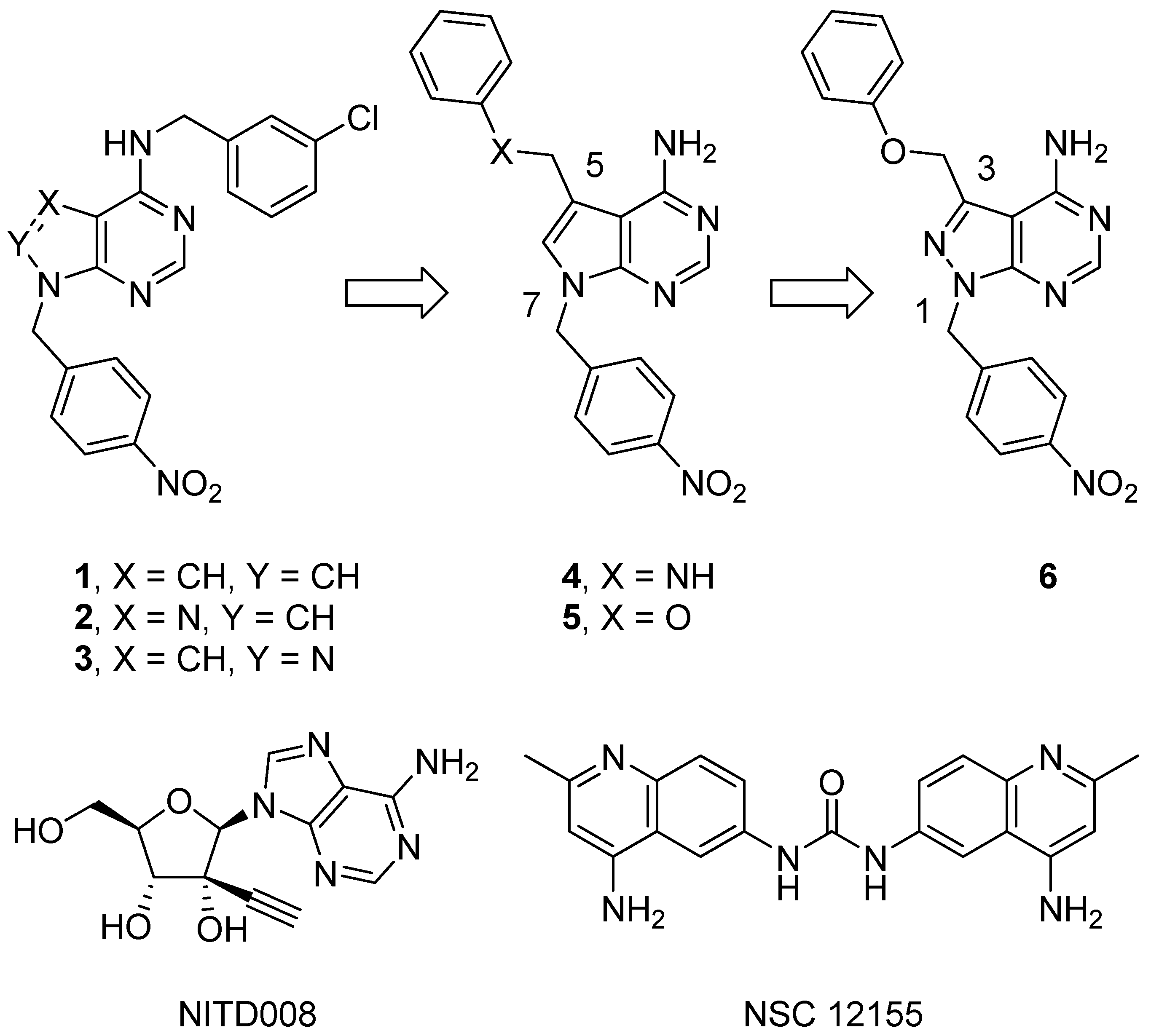
4.2. Chemistry
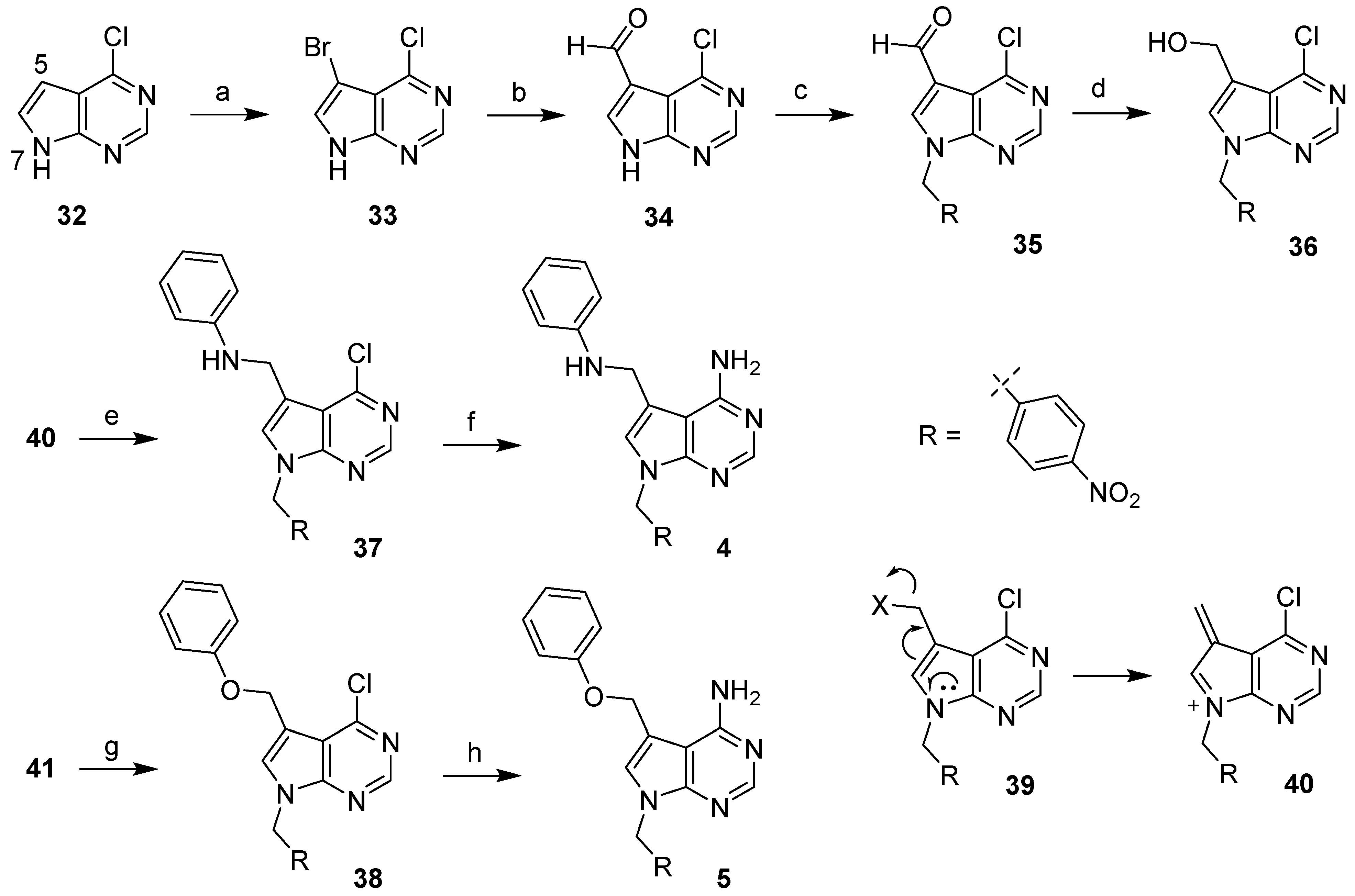
4.2.1. 5-Bromo-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (33) [20]
To a solution of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (32, 2.51 g, 16.3 mmol) in anhydrous DMF (50 mL) was added NBS (2.91 g, 16.4 mmol) in anhydrous DMF (22 mL) in 25 min. The resulting mixture was stirred at rt for 1 h and poured into vigorously stirring water (350 mL). The precipitate was filtered, washed with water, suction-dried, and dried in vacuo to give compound 33 as a pale solid (3.55 g, 94%). 1H NMR (600 MHz, DMSO-d6) δ 13.00 (s, 1H), 8.63 (s, 1H), 7.96 (s, 1H). HRMS (ESI−) m/z calcd for C6H2BrClN3 [M-H]− 229.9126, found 229.9124.
4.2.2. 4-Chloro-7H-pyrrolo[2,3-d]pyrimidine-5-carbaldehyde (34)
nBuLi (1.6 M in hexanes, 10.5 mL, 16.8 mmol) was added dropwise to a solution of 33 (1.78 g, 7.66 mmol) in anhydrous THF (80 L) at −78 °C. The resulting mixture was stirred at −78 °C for 1 h, and anhydrous DMF (0.66 mL, 8.5 mmol) was added dropwise. After the reaction mixture was stirred at −78 °C for 30 min and then at room temperature overnight, it was cooled at 0 °C and carefully quenched with water (40 mL). The mixture was concentrated to produce a thick syrup, which was treated with saturated NH4Cl (60 mL) and vigorously stirred for 20 min. The solid that formed was filtered, washed with water (10 mL × 3), EtOAc (10 mL × 3), suction-dried, and dried in vacuo to give compound 34 as a pale solid (1.31 g, 94%). 1H NMR (600 MHz, DMSO-d6) δ 13.55 (s, 1H), 10.23 (s, 1H), 8.74 (s, 1H), 8.59 (s, 1H). HRMS (APCI+) m/z calcd for C7H5ClN3O [M + H]+ 182.0116, found 182.0109.
4.2.3. 4-Chloro-7-(4-nitrobenzyl)-7H-nyrrolo[2,3-d]pyrimidine-5-carbaldehyde (35)
To a suspension of 34 (363 mg, 2.00 mmol) in anhydrous CH3CN (20 mL), K2CO3 (829 mg, 6.00 mmol) and 4-nitrobenzyl bromide (561 mg, 2.60 mmol) were added. The resulting mixture was stirred at room temperature for 23 h and then poured into stirring water (100 mL). The precipitate was filtered, washed with water, and suction-dried. The solid was suspended in ether (10 mL) and the mixture was vigorously stirred for 30 min. After filtration, the solid was washed with ether and dried in vacuo to give compound 35 as a light-yellow solid (576 mg, 91%). 1H NMR (600 MHz, CDCl3) δ 10.50 (s, 1H), 8.78 (s, 1H), 8.22 (d, J = 8.7 Hz, 2H), 8.06 (s, 1H), 7.45 (d, J = 8.7 Hz, 2H), 5.62 (s, 2H). HRMS (APCI+) m/z calcd for C14H10ClN4O3 [M + H]+ 317.0436, found 317.0440.
4.2.4. 4-Chloro-7-(4-nitrobenzyl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)Methanol (36)
To a suspension of 35 (561 mg, 1.77 mmol) in THF (20 mL) and MeOH (20 mL) at 0 °C, NaBH4 (420 mg, 11.1 mmol) was added in small portions. After the resulting mixture was stirred at 0 °C for 30 min, the reaction was carefully quenched with an NH4Cl solution (30 mL of saturated NH4Cl and 10 mL of water), and the mixture was extracted with CHCl3 (40 × 2 mL). The combined organic layer was concentrated, and the residue was purified by flash column chromatography (0–10% MeOH/CH2Cl2) to give compound 36 as a yellow solid (473 mg, 84%). 1H NMR (600 MHz, CDCl3) δ 8.65 (s, 1H), 8.14 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 8.7 Hz, 2H), 7.26 (s, 1H), 5.54 (s, 2H), 4.98 (s, 2H), 2.05 (s, 1H). HRMS (APCI+) m/z calcd for C14H12ClN4O3 [M + H]+ 319.0592, found 319.0587.
4.2.5. N-((4-Chloro-7-(4-nitrobenzyl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)pethyl)pniline (37)
To a suspension of 35 (120 mg, 0.379 mmol) in anhydrous dichloroethane (6 mL), aniline (42 µL, 0.46 mmol), Na(OAc)3BH (241 mg, 1.14 mmol), and acetic acid (22 µL, 0.38 mmol) were added. The resulting mixture was stirred at room temperature for 40 h and diluted with CH2Cl2 (20 mL). The organic layer was washed with saturated NaHCO3 (10 mL), water (10 mL), and brine (20 mL). After concentration, the residue was purified by flash column chromatography (30–90% EtOAc/hexanes) to give compound 37 as a yellow solid (68 mg, 46%). 1H NMR (600 MHz, CDCl3) δ 8.64 (s, 1H), 8.15 (d, J = 8.7 Hz, 2H), 7.28 (d, J = 8.7 Hz, 2H), 7.19–7.16 (m, 3H), 6.74 (t, J = 7.2 Hz, 1H), 6.67–6.65 (m, 2H), 5.49 (s, 2H), 4.67 (s, 2H), 4.18 (s, 1H). HRMS (APCI+) m/z calcd for C20H17ClN5O2 [M + H]+ 394.1065, found 394.1060.
4.2.6. 7-(4-Nitrobenzyl)-5-((phenylamino)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (4)
A solution of 37 (61 mg, 0.16 mmol) in dioxane (3 mL) and strong ammonia solution (3 mL) was heated at 120 °C for 6 h in a sealed tube and allowed to cool to room temperature. The mixture was concentrated, and the residue was purified by flash column chromatography (0–10% MeOH/CH2Cl2) to give compound 4 as a yellow solid (11 mg, 18%). 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 8.18 (d, J = 8.7 Hz, 2H), 7.34 (d, J = 8.7 Hz, 2H), 7.28–7.26 (m, 2H), 6.91–6.89 (m, 2H), 6.84–6.82 (m, 2H), 6.08 (s, 2H), 5.46 (s, 2H), 4.35 (s, 2H), 3.85 (s, 1H). HRMS (ESI+) m/z calcd for C20H19N6O2 [M + H]+ 375.1564, found 375.1561.
4.2.7. 7-(4-Nitrobenzyl)-5-(phenoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (5)
Phenol (58 mg, 0.62 mmol), Ph3P (162 mg, 0.618 mmol), and then DEAD (97 µL, 0.62 mmol, dropwise) were added to a solution of 36 (131 mg, 0.411 mmol) in anhydrous THF (7 mL) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h and then at room temperature for 20 h. After concentration, the residue was purified by flash column chromatography (10–90% EtOAc/hexanes) to give a white solid (121 mg), which contained a small amount of compound 38. The solid was dissolved in dioxane (5 mL) and strong ammonia solution (5 mL) in a sealed tube. The resulting mixture was heated at 120 °C for 6 h and allowed to cool to room temperature. After the mixture was concentrated, the residue was purified by preparative thin layer chromatography (5% MeOH/CH2Cl2) to give compound 5 as a yellowish solid (5 mg). 1H NMR (600 MHz, CDCl3/CD3OD) δ 8.17 (d, J = 8.4 Hz, 2H), 8.11 (s, 1H), 7.38–7.36 (m, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.10–7.04 (m, 2H), 6.90–6.84 (m, 2H), 6.80 (t, J = 8.4 Hz, 1H), 5.45 (s, 2H), 4.10 (s, 2H). HRMS (ESI+) m/z calcd for C20H18N5O3 [M + H]+ 376.1404, found 376.1400.
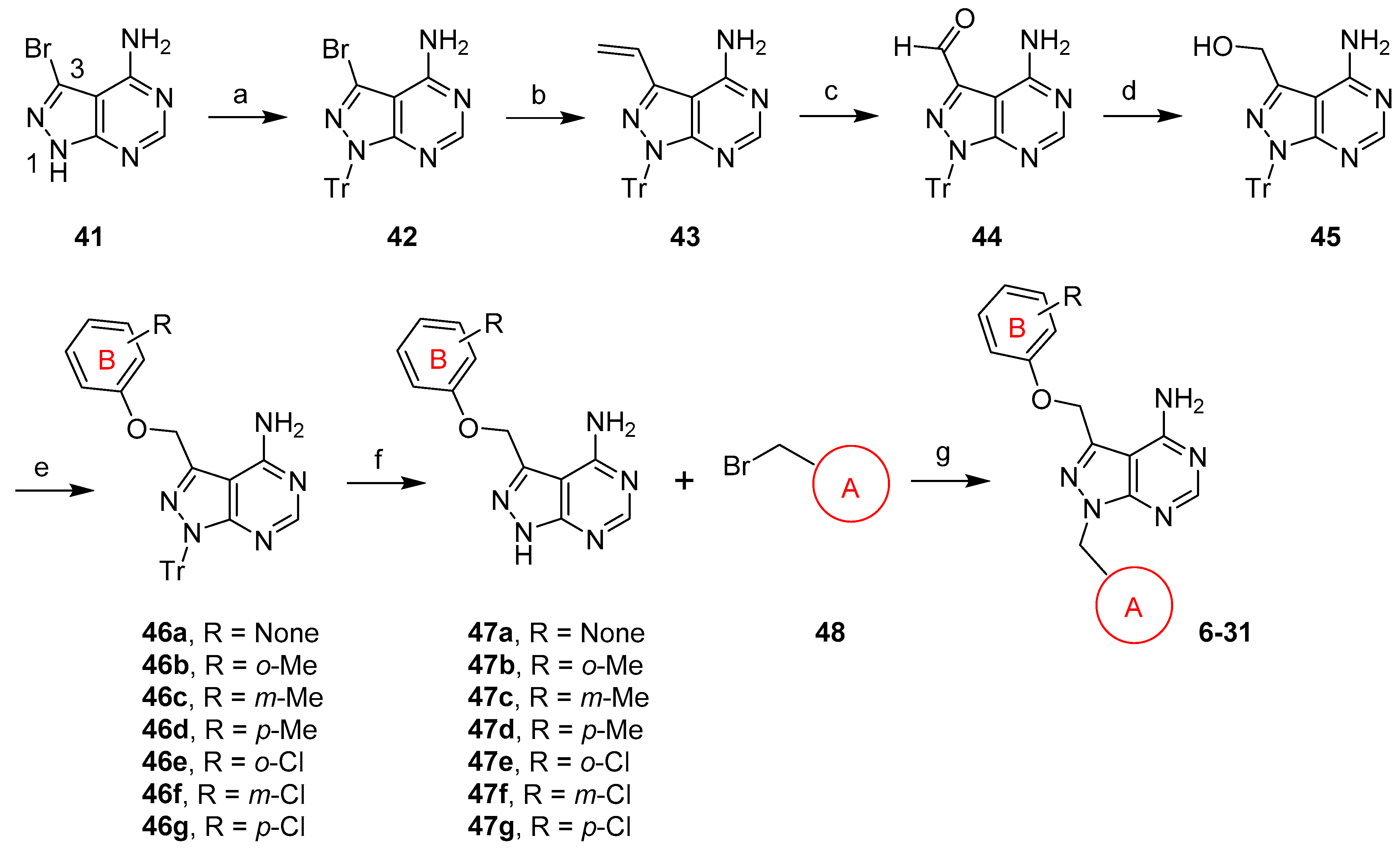
4.2.8. 3-Bromo-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (42)
Cs2CO3 (6.55 g, 20.1 mmol) and trityl chloride (2.94 g, 10.5 mmol) were added to a solution of 3-bromo-1H-pyrazolo[3,4-d]pyrimidin-4-amine (41, 2.14 g, 10.0 mmol) in anhydrous DMF (50 mL). The resulting mixture was heated at 60 °C for 21 h, allowed to cool to room temperature, and poured into ice-water (300 mL). After the mixture was vigorously stirred for 30 min, the precipitate was filtered, washed with water, suction-dried, and dried in vacuo to give compound 42 as a white solid. (4.58 g, quantitative yield). 1H NMR (600 MHz, CDCl3) δ 8.05 (s, 1H), 7.32–7.21 (m, 15H), 5.91 (s, 2H). HRMS (ESI+) m/z calcd for C24H19BrN5 [M + H]+ 456.0818, found 456.0811.
4.2.9. 1-Trityl-3-vinyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (43)
Pd(PPh3)4 (588 mg, 0.509 mmol) was added to a suspension of 42 (4.57 g, 10.0 mmol) in anhydrous toluene (100 mL). After the mixture was evacuated and backfilled with argon for three times, tributyl(vinyl)tin (3.51 mL, 12.0 mml) was added. The resulting mixture was heated at 110 °C for 6 h, allowed to cool to room temperature, diluted with EtOAc (200 mL), and treated with KF (7 g) for 15 min. After Celite was added, the mixture was stirred for 15 min and filtered. The filtrate was concentrated, and the residue was purified by flash column chromatography (20–100% EtOAc/hexanes) to give compound 43 as a yellowish solid (3.89 g, 96%). 1H NMR (600 MHz, CDCl3) δ 8.07 (s, 1H), 7.28–7.23 (m, 15H), 6.94 (dd, J = 17.8, 11.0 Hz, 1H), 5.90 (dd, J = 17.8, 1.3 Hz, 1H), 5.60–5.54 (m, 3H). HRMS (ESI+) m/z calcd for C26H22N5 [M + H]+ 404.1870, found 404.1867.
4.2.10. 4-Amino-1-trityl-1H-pyrazolo[3,4-d]pyrimidine-3-carbaldehyde (44)
OsO4 solution (2.5 wt% in tBuOH, 0.94 mL, 0.075 mmol) was added to a suspension of 43 (3.00 g, 7.44 mmol) in dioxane (120 mL) and water (40 mL). After the mixture was stirred at room temperature for 30 min, NaIO4 (3.20 g, 15.0 mmol) was added. The resulting mixture was stirred for an additional 4 h, and dioxane was removed in vacuo. Water (60 mL) was added to the resulting slurry and the mixture was stirred for 15 min. The precipitate was filtered, washed with water, suction-dried, and dried in vacuo to give compound 44 as a pale solid (3.00 g, 99%). 1H NMR (600 MHz, CDCl3) δ 9.91 (s, 1H), 8.08 (s, 1H), 7.29–7.27 (m, 12H), 7.19–7.17 (m, 3H). HRMS (ESI−) m/z calcd for C25H18N5O [M-H]− 404.1517, found 404.1515.
4.2.11. 4-Amino-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)methanol (45)
NaBH4 (1.12 g, 29.6 mmol) was added in small portions to a solution of 44 (2.98 g, 7.35 mmol) in THF (50 mL) and MeOH (50 mL) at 0 °C. After the resulting mixture was stirred at 0 °C for 1 h, the reaction was carefully quenched with an NH4Cl solution (100 mL of saturated NH4Cl and 35 mL of water), and the mixture was extracted with CHCl3 (200 mL). The organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated, and the residue was purified by flash column chromatography (0–10% MeOH/CH2Cl2) to give compound 45 as a white fluffy solid (2.42 g, 81%). 1H NMR (600 MHz, DMSO-d6) δ 7.86 (s, 1H), 7.27–7.13 (m, 15H), 6.20 (t, J = 5.3 Hz, 1H), 4.67 (d, J = 5.3 Hz, 2H). HRMS (ESI+) m/z calcd for C25H22N5O [M + H]+ 408.1819, found 408.1818.
4.2.12. 3-(Phenoxymethyl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (46a)
Methanesulfonic anhydride (1.68 g, 9.64 mmol) was added to a suspension of 45 (3.29 g, 8.07 mmol), Et3N (1.35 mL, 9.68 mmol), and DMAP (100 mg, 0.82 mmol) in CH2Cl2 (80 mL) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h and diluted with CHCl3 (240 mL). The organic layer was washed water (80 mL × 2) and brine (300 mL), and then concentrated to give a pale solid. The residue was immediately dissolved in anhydrous DMF (80 mL), then Cs2CO3 (7.90 g, 24.2 mmol) and phenol (1.60 g 17.0 mmol) were added. The resulting mixture was stirred at room temperature for 2 h, and DMF was removed in vacuo. The residue was partitioned between CHCl3 (300 mL) and water (100 mL). The organic layer was washed with water (100 mL) and brine (200 mL) and dried over Na2SO4. After filtration, the filtrate was concentrated, and the residue was purified by flash column chromatography (30–100% EtOAc/hexanes) to give compound 46a as a pale solid (3.09 g, 79%). 1H NMR (600 MHz, CDCl3) δ 8.06 (s, 1H), 7.32–7.23 (m, 17H), 7.04–7.01 (m, 3H), 5.37 (s, 2H). No desired ionization observed in ESI or APCI modes.
4.2.13. 3-((o-Tolyloxy)methyl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (46b)
Compound 46b was prepared from 45 (540 mg, 1.33 mmol) and o-cresol (289 mg, 2.67 mmol) as described for compound 46a. Pale solid, 447 mg, yield 68%. 1H NMR (600 MHz, CDCl3) δ 8.06 (s, 1H), 7.28–7.24 (m, 15H), 7.18–7.16 (m, 1H), 7.14 (td, J = 7.8, 1.4 Hz, 1H), 7.01–6.97 (m, 1H), 6.93 (td, J = 7.8, 1.2 Hz, 1H), 6.11 (s, 2H), 5.36 (s, 2H), 2.29 (s, 3H). No desired ionization was observed in ESI or APCI modes.
4.2.14. 3-((m-Tolyloxy)methyl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (46c)
Compound 46c was prepared from 45 (540 mg, 1.33 mmol) and m-cresol (289 mg, 2.67 mmol) as described for compound 46a. Pale solid, 391 mg, yield 59%. 1H NMR (600 MHz, CDCl3) δ 8.06 (s, 1H), 7.28–7.23 (m, 15H), 7.19 (t, J = 7.8 Hz, 1H), 6.86–6.81 (m, 3H), 6.12 (s, 2H), 5.35 (s, 2H), 2.33 (s, 3H). No desired ionization was observed in ESI or APCI modes.
4.2.15. 3-((p-Tolyloxy)methyl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (46d)
Compound 46d was prepared from 45 (540 mg, 1.33 mmol) and p-cresol (289 mg, 2.67 mmol) as described for compound 46a. White solid, 442 mg, yield 67%. 1H NMR (600 MHz, CDCl3) δ 8.05 (s, 1H), 7.28–7.24 (m, 15H), 7.10 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 6.13 (s, 2H), 5.33 (s, 2H), 2.31 (s, 3H). No desired ionization was observed in ESI or APCI modes.
4.2.16. 3-((2-Chlorophenoxy)methyl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (46e)
Compound 46e was prepared from 45 as described for compound 46a. 1H NMR (600 MHz, CDCl3) δ 8.04 (s, 1H), 7.37 (dd, J = 7.9, 1.6 Hz, 1H), 7.25–7.21 (m, 15H), 7.16 (ddd, J = 8.9, 7.4, 1.6 Hz, 1H), 7.05 (dd, J = 8.4, 1.3 Hz, 1H), 6.94 (td, J = 7.7, 1.3 Hz, 1H), 6.13 (s, 2H), 5.44 (s, 2H). No desired ionization was observed in ESI or APCI modes.
4.2.17. 3-((3-Chlorophenoxy)methyl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (46f)
Compound 46f was prepared from 45 as described for compound 46a. 1H NMR (600 MHz, CDCl3) δ 8.05 (s, 1H), 7.26–7.22 (m, 15H), 7.19 (t, J = 8.1 Hz, 1H), 7.05 (t, J = 2.2 Hz, 1H), 6.99 (dd, J = 8.3, 1.2 Hz, 1H), 6.88 (dd, J = 8.3, 2.5 Hz, 1H), 6.03 (s, 2H), 5.36(s, 2H). No desired ionization was observed in ESI or APCI modes.
4.2.18. 3-((4-Chlorophenoxy)methyl)-1-trityl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (46g)
Compound 46g was prepared from 45 as described for compound 46a. 1H NMR (600 MHz, CDCl3) δ 8.05 (s, 1H), 7.28–7.21 (m, 17H), 6.92 (dd, J = 8.9, 2.4 Hz, 2H), 6.26 (s, 2H), 5.33 (s, 2H). No desired ionization was observed in ESI or APCI modes.
4.2.19. 3-(Phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (47a)
Et3SiH (4.4 mL, 28 mmol) and then TFA (5.3 mL, 69 mmol) were added dropwise to a solution of 46a (3.35 g, 6.93 mmol) in anhydrous CH2Cl2 (140 mL) at 0 °C. The resulting mixture was stirred at 0 °C for 30 min and then at room temperature for 2 h. After concentration, the residue was co-evaporated with toluene (100 mL × 2), and the resulting solid was triturated with warm hexanes (100 mL) and saturated NaHCO3 (20 mL). The mixture was filtered and the solid was washed with hexanes (25 mL × 2) and water (10 mL × 2), suction-dried, and dried in vacuo to give compound 47a as a pale solid (1.61 g, 96%). 1H NMR (600 MHz, DMSO-d6) δ 13.56 (s, 1H), 8.21 (s, 1H), 7.35–7.27 (m, 2H), 7.07– 7.05 (m, 2H), 6.96 (t, J = 7.3 Hz, 1H), 5.43 (s, 2H). HRMS (ESI+) m/z calcd for C12H12N5O [M + H]+ 242.1036, found 242.1046.
4.2.20. 3-((o-Tolyloxy)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (47b)
Compound 47b was prepared from 46b (421 mg, 0.846 mmol) as described for compound 47a. Pale solid, 190 mg, yield 88%. 1H NMR (600 MHz, DMSO-d6) δ 13.40 (s, 1H), 8.17 (s, 1H), 7.15–7.10 (m, 3H), 6.85 (t, J = 7.4 Hz, 1H), 5.42 (s, 2H), 2.15 (s, 3H). HRMS (ESI+) m/z calcd for C13H14N5O [M + H]+ 256.1193, found 256.1205.
4.2.21. 3-((m-Tolyloxy)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (47c)
Compound 47c was prepared from 46c (371 mg, 0.746 mmol) as described for compound 47a. Pale solid, 179 mg, yield 94%. 1H NMR (600 MHz, DMSO-d6) δ 13.66 (s, 1H), 8.25 (s, 1H), 7.17 (t, J = 7.9 Hz, 1H), 6.89–6.78 (m, 3H), 5.41 (s, 2H), 2.27 (s, 3H). HRMS (ESI+) m/z calcd for C13H14N5O [M + H]+ 256.1193, found 256.1192.
4.2.22. 3-((p-tolyloxy)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (47d)
Compound 47d was prepared from 46d (417 mg, 0.838 mmol) as described for compound 47a. White solid, 196 mg, yield 92%. 1H NMR (600 MHz, DMSO-d6) δ 13.65 (s, 1H), 8.24 (s, 1H), 7.09 (d, J = 8.1 Hz, 2H), 6.94 (d, J = 8.1 Hz, 2H), 5.40 (s, 2H), 2.22 (s, 3H). HRMS (ESI+) m/z calcd for C13H14N5O [M + H]+ 256.1193, found 256.1201.
4.2.23. 3-((2-Chlorophenoxy)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (47e)
Compound 47e was prepared from 46e as described for compound 47a. 1H NMR (600 MHz, CDCl3) δ 10.80 (s, 1H), 8.34 (s, 1H), 7.41 (dd, J = 8.0, 1.2 Hz, 1H), 7.22 (td, J = 8.0, 1.2 Hz, 1H), 7.11 (d, J = 8.0 Hz, 1H), 6.99 (t, J = 7.8 Hz, 1H), 5.52 (s, 2H). HRMS (ESI+) m/z calcd for C12H11ClN5O [M + H]+ 276.0647, found 276.0637.
4.2.24. 3-((3-Chlorophenoxy)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (47f)
Compound 47f was prepared from 46f as described for compound 47a. 1H NMR (600 MHz, CDCl3) δ 10.54 (s, 1H), 8.39 (s, 1H), 7.23 (t, J = 8.1 Hz, 1H), 7.06 (t, J = 2.1 Hz, 1H), 7.01 (ddd, J = 8.2, 2.1, 0.8 Hz, 1H), 6.94 (ddd, J = 8.1, 2.1, 0.8 Hz, 1H), 6.05 (s, 2H), 5.43 (s, 2H). HRMS (ESI+) m/z calcd for C12H11ClN5O [M + H]+ 276.0647, found 276.0647.
4.2.25. 3-((4-Chlorophenoxy)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (47g)
Compound 47g was prepared from 46g as described for compound 47a. 1H NMR (600 MHz, CDCl3) δ 10.63 (s, 1H), 8.39 (s, 1H), 7.26 (d, J = 9.0 Hz, 2H), 6.98 (d, J = 9.0 Hz, 2H), 5.42 (s, 2H). HRMS (ESI+) m/z calcd for C12H11ClN5O [M + H]+ 276.0647, found 276.0657.
4.2.26. 1-(4-Nitrobenzyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (6)
Cs2CO3 (216 mg, 0.663 mmol) and 4-nitrobenzyl bromide (88 mg, 0.41 mmol) were added to a suspension of 47a (80 mg, 0.33 mmol) in anhydrous CH3CN (4 mL). The resulting mixture was stirred at room temperature for 6 h and concentrated, then the residue was then dissolved in EtOAc (20 mL). The organic layer was washed with water (10 mL × 3) and brine (20 mL) and dried over Na2SO4. After filtration, the filtrate was concentrated, and the residue was purified by flash column chromatography (0–5% MeOH/CH2Cl2) to give compound 6 as a pale solid (70 mg, 56%). 1H NMR (600 MHz, CDCl3) δ 8.36 (s, 1H), 8.16 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.32– 7.29 (m, 2H), 7.05–7.01 (m, 3H), 6.27 (s, 2H), 5.64 (s, 2H), 5.40 (s, 2H). HRMS (ESI+) m/z calcd for C19H17N6O3 [M + H]+ 377.1357, found 377.1360.
4.2.27. 1-Benzyl-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (7)
Compound 7 was prepared from 47a (49 mg, 0.20 mmol) and benzyl bromide (32 µL, 0.27 mmol) as described for compound 6. White solid, 9 mg, yield 13%. 1H NMR (600 MHz, CDCl3) δ 8.37 (s, 1H), 7.31–7.28 (m, 7H), 7.03–7.01 (m, 3H), 6.22 (s, 2H), 5.55 (s, 2H), 5.40 (s, 2H). HRMS (ESI+) m/z calcd for C19H18N5O [M + H]+ 332.1506, found 332.1504.
4.2.28. 1-(2-Nitrobenzyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (8)
Compound 8 was prepared from 47a (100 mg, 0.414 mmol) and 2-nitrobenzyl bromide (107 mg, 0.495 mmol) as described for compound 6. White solid, 84 mg, yield 54%. 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 8.17–8.16 (m, 1H), 7.46–7.43 (m, 2H), 7.33–7.30 (m, 2H), 7.05–7.02 (m, 3H), 6.62–6.60 (m, 1H), 6.15 (s, 2H), 6.03 (s, 2H), 5.43 (s, 2H). HRMS (ESI+) m/z calcd for C19H17N6O3 [M + H]+ 377.1357, found 377.1361.
4.2.29. 1-(3-Nitrobenzyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (9)
Compound 9 was prepared from 47a (80 mg, 0.33 mmol) and 3-nitrobenzyl bromide (88 mg, 0.41 mmol) as described for compound 6. White solid, 63 mg, yield 50%. 1H NMR (600 MHz, CDCl3) δ 8.37 (s, 1H), 8.20 (t, J = 2.0 Hz, 1H), 8.15 (ddd, J = 8.2, 2.0, 1.1 Hz, 1H), 7.62 (ddd, J = 7.8, 1.8, 1.1 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.32–7.29 (m, 2H), 7.04–7.01 (m, 3H), 6.21 (s, 2H), 5.64 (s, 2H), 5.41 (s, 2H). HRMS (ESI+) m/z calcd for C19H17N6O3 [M + H]+ 377.1357, found 377.1357.
4.2.30. 1-(2-Aminobenzyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (10)
SnCl2 (198 mg, 1.04 mmol) was added to a suspension of 8 (56 mg, 0.15 mmol) in EtOH (5 mL). The resulting mixture was heated at 70 °C for 18 h, allowed to cool to room temperature, and diluted with EtOAc (12 mL). The reaction was carefully quenched with a NaHCO3 solution (6 mL of saturated NaHCO3 and 1.5 mL of water). The mixture was filtered through a pad of Celite and washed with 5% MeOH/EtOAc. The filtrate was separated, and the organic layer was washed with brine (20 mL × 2) and dried over Na2SO4. After filtration, the filtrate was concentrated, and the residue was purified by flash column chromatography (0–10% MeOH/CH2Cl2) to give compound 10 as a pale solid (41 mg, 79%). 1H NMR (600 MHz, DMSO-d6) δ 8.24 (s, 1H), 7.84 (s, 2H), 7.31–7.28 (m, 2H), 7.06–7.04 (m, 2H), 6.99–6.95 (m, 2H), 6.79 (dd, J = 7.6, 1.6 Hz, 1H), 6.65 (dd, J = 8.0, 1.2 Hz, 1H), 6.47 (td, J = 7.6, 1.2 Hz, 1H), 5.42 (s, 2H), 5.31 (s, 2H), 5.29 (s, 2H). HRMS (ESI+) m/z calcd for C19H19N6O [M + H]+ 347.1615, found 347.1615.
4.2.31. 1-(3-Aminobenzyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (11)
Compound 11 was prepared from 9 (48 mg, 0.13 mmol) as described for compound 10. White solid, 34 mg, yield 77%. 1H NMR (600 MHz, DMSO-d6) δ 8.23 (s, 1H), 7.75 (s, 2H), 7.31–7.28 (m, 2H), 7.06–7.04 (m, 2H), 6.96 (td, J = 7.3, 1.1 Hz, 1H), 6.92 (t, J = 7.7 Hz, 1H), 6.43 (ddd, J = 8.0, 2.2, 1.2 Hz, 1H), 6.40 (s, 1H), 6.36 (dd, J = 7.7, 1.2 Hz, 1H), 5.41 (s, 2H), 5.31 (s, 2H), 5.06 (s, 2H). HRMS (ESI+) m/z calcd for C19H19N6O [M + H]+ 347.1615, found 347.1620.
4.2.32. 1-(4-Aminobenzyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-pmine (12)
Compound 12 was prepared from 6 (58 mg, 0.15 mmol) as described for compound 10. Pale solid, 37 mg, yield 69%. 1H NMR (600 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.83 (s, 2H), 7.30–7.27 (m, 2H), 7.04 (d, J = 8.4 Hz, 2H), 6.97–6.94 (m, 3H), 6.46 (d, J = 8.4 Hz, 2H), 5.40 (s, 2H), 5.27 (s, 2H), 5.04 (2H). HRMS (ESI+) m/z calcd for C19H19N6O [M + H]+ 347.1615, found 347.1616.
4.2.33. 3-(Phenoxymethyl)-1-(4-(trifluoromethyl)benzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (13)
Compound 13 was prepared from 47a (40 mg, 0.17 mmol) and 4-(trifluoromethyl)benzyl bromide (50 mg, 0.21 mmol) as described for compound 6. White solid, 32 mg, yield 48%. 1H NMR (600 MHz, CDCl3) δ 8.37 (s, 1H), 7.56 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 7.31–7.28 (m, 2H), 7.04–7.01 (m, 3H), 6.22 (s, 2H), 5.60 (s, 2H), 5.40 (s, 2H). HRMS (ESI+) m/z calcd for C20H17F3N5O [M + H]+ 400.1380, found 400.1375.
4.2.34. 1-(4-(Methylsulfonyl)Benzyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (14)
Compound 14 was prepared from 47a (40 mg, 0.17 mmol) and 4-(methylsulfonyl)benzyl bromide (52 mg, 0.21 mmol) as described for compound 6. White solid, 23 mg, yield 34%. 1H NMR (600 MHz, CDCl3) δ 8.36 (s, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.4 Hz, 2H), 7.32–7.29 (m, 2H), 7.04–7.01 (m, 3H), 6.25 (s, 2H), 5.63 (s, 2H), 5.40 (s, 2H), 3.01 (s, 3H). HRMS (ESI+) m/z calcd for C20H20N5O3S [M + H]+ 410.1281, found 410.1289.
4.2.35. 4-((4-Amino-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)benzonitrile (15)
Compound 15 was prepared from 47a (40 mg, 0.17 mmol) and 4-cyanobenzyl bromide (39 mg, 0.20 mmol) as described for compound 6. White solid, 34 mg, yield 57%. 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 7.60 (d, J = 8.2 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 7.31–7.29 (m, 2H), 7.04–7.01 (m, 3H), 6.26 (s, 2H), 5.59 (s, 2H), 5.39 (s, 2H). HRMS (ESI+) m/z calcd for C20H17N6O [M + H]+ 357.1458, found 357.1467.
4.2.36. Methyl 2-((4-Amino-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)benzoate (16)
Compound 16 was prepared from 47a (100 mg, 0.41 mmol) and methyl 2-(bromomethyl)benzoate (115 mg, 0.50 mmol) as described for compound 6. White solid, 41 mg, yield 25%. 1H NMR (600 MHz, CDCl3) δ 8.34 (s, 1H), 8.03 (dd, J = 7.2, 2.0 Hz, 1H), 7.35–7.29 (m, 4H), 7.05–7.01 (m, 3H), 6.53 (dd, J = 7.3, 1.7 Hz, 1H), 6.25 (s, 2H), 6.05 (s, 2H), 5.44 (s, 2H), 3.94 (s, 3H). HRMS (ESI+) m/z calcd for C21H20N5O3 [M + H]+ 390.1561, found 390.1567.
4.2.37. Methyl 3-((4-Amino-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)benzoate (17)
Compound 17 was prepared from 47a (100 mg, 0.41 mmol) and methyl 3-(bromomethyl)benzoate (114 mg, 0.50 mmol) as described for compound 6. White solid, 75 mg, yield 46%. 1H NMR (600 MHz, CDCl3) δ 8.37 (s, 1H), 8.03 (d, J = 1.9 Hz, 1H), 7.95 (dd, J = 7.7, 1.5 Hz, 1H), 7.46 (dd, J = 7.7, 1.6 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 7.31–7.27 (m, 2H), 7.03–7.01 (m, 3H), 6.22 (s, 2H), 5.59 (s, 2H), 5.39 (s, 2H), 3.89 (s, 3H). HRMS (ESI+) m/z calcd for C21H20N5O3 [M + H]+ 390.1561, found 390.1562.
4.2.38. Methyl 4-((4-amino-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)benzoate (18)
Compound 18 was prepared from 47a (100 mg, 0.41 mmol) and methyl 4-(bromomethyl)benzoate (115 mg, 0.50 mmol) as described for compound 6. White solid, 80 mg, yield 50%. 1H NMR (600 MHz, CDCl3) δ 8.36 (s, 1H), 7.97 (d, J = 8.3 Hz, 2H), 7.32–7.28 (m, 4H), 7.03–7.01 (m, 3H), 6.23 (s, 2H), 5.60 (s, 2H), 5.40 (s, 2H), 3.89 (s, 3H). HRMS (ESI+) m/z calcd for C21H20N5O3 [M + H]+ 390.1561, found 390.1566.
4.2.39. 4-((4-Amino-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)benzamide (19)
A mixture of 18 (50 mg, 0.13 mmol), CaCl2 (55 mg, 0.50 mmol), and 7 N NH3/MeOH (8 mL) was heated at 90 °C for 24 h in a sealed tube and then allowed to cool to room temperature. The mixture was concentrated, and the residue was treated with 10% MeOH/CH2Cl2 (20 mL). The organic phase was concentrated, and the residue was purified by flash column chromatography (0–10% MeOH/CH2Cl2) to give compound 19 as a white solid (37 mg, 77%). 1H NMR (600 MHz, DMSO-d6) δ 8.23 (s, 1H), 7.92 (s, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.34 (s, 1H), 7.32–7.28 (m, 2H), 7.24 (d, J = 8.4 Hz, 2H), 7.04 (d, J = 8.4 Hz, 2H), 6.96 (t, J = 7.5 Hz, 1H), 5.55 (s, 2H), 5.43 (s, 2H). HRMS (ESI+) m/z calcd for C20H19N6O2 [M + H]+ 375.1564, found 375.1558.
4.2.40. 1-((2-Chloropyridin-4-yl)methyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (20)
Compound
20 was prepared from
47a (80 mg, 0.33 mmol) and 4-(bromomethyl)-2-chloropyridine [
21] (85 mg, 0.41 mmol) as described for compound
6. Pale solid, 52 mg, yield 43%.
1H NMR (600 MHz, CDCl
3) δ 8.36 (s, 1H), 8.32 (dd,
J = 5.1, 0.7 Hz, 1H), 7.33–7.30 (m, 2H), 7.16 (d,
J = 0.8 Hz, 1H), 7.06 (dd,
J = 5.1, 0.7 Hz, 1H), 7.04–7.01 (m, 3H), 6.24 (s, 2H), 5.53 (s, 2H), 5.41 (s, 2H). HRMS (ESI
+) m/z calcd for C
18H
16ClN
6O [M + H]
+ 367.1069, found 367.1078.
4.2.41. 1-((6-Chloropyridin-3-yl)methyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (21)
Compound
21 was prepared from
47a (80 mg, 0.33 mmol) and 5-(bromomethyl)-2-chloropyridine [
21] (85 mg, 0.41 mmol) as described for compound
6. White solid, 47 mg, yield 38%.
1H NMR (600 MHz, CDCl
3) δ 8.45 (d,
J = 2.5 Hz, 1H), 8.36 (s, 1H), 7.61 (dd,
J = 8.3, 2.5 Hz, 1H), 7.31–7.29 (m, 2H), 7.26 (d,
J = 8.3 Hz, 1H), 7.04–7.01 (m, 3H), 6.20 (s, 2H), 5.52 (s, 2H), 5.37 (s, 2H). HRMS (ESI
+) m/z calcd for C
18H
16ClN
6O [M + H]
+ 367.1069, found 367.1070.
4.2.42. 1-((2-Aminopyridin-4-yl)methyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (22)
TFA (2 mL) was added dropwise to a solution of 24 (29 mg, 0.065 mmol) in anhydrous CH2Cl2 (4 mL) at room temperature. The resulting mixture was stirred at room temperature for 1 h and then concentrated. The residue was treated with strong ammonia (1 mL) and then purified by flash column chromatography (0–15% MeOH/CH2Cl2) to give compound 22 as a white solid (22 mg, 98%). 1H NMR (600 MHz, DMSO-d6) δ 8.25 (s, 1H), 7.87 (d, J = 6.4 Hz, 1H), 7.68 (s, 2H), 7.33–7.30 (m, 2H), 7.07–7.05 (m, 2H), 6.98 (t, J = 7.2 Hz, 1H), 6.57 (d, J = 6.4 Hz, 1H), 6.45 (s, 1H), 5.53 (s, 2H), 5.44 (s, 2H). HRMS (ESI+) m/z calcd for C18H18N7O [M + H]+ 348.1567, found 348.1563.
4.2.43. 1-((6-Aminopyridin-3-yl)methyl)-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (23)
Compound 23 was prepared from 25 (53 mg, 0.12 mmol) as described for compound 22. White solid, 30 mg, yield 73%. 1H NMR (600 MHz, DMSO-d6) δ 8.23 (s, 1H), 7.91 (s, 1H), 7.29–7.27 (m, 4H), 7.04–7.03 (m, 2H), 6.95 (t, J = 7.4 Hz, 1H), 6.36 (d, J = 8.5 Hz, 1H), 5.94 (s, 1H), 5.40 (s, 2H), 5.28 (s, 2H). HRMS (ESI+) m/z calcd for C18H18N7O [M + H]+ 348.1567, found 348.1572.
4.2.44. Tert-Butyl (4-((4-Amino-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyridin-2-yl)carbamate (24)
Compound
24 was prepared from
47a (100 mg, 0.41 mmol) and
tert-butyl (4-(bromomethyl)pyridin-2-yl)carbamate [
22] (143 mg, 0.50 mmol) as described for compound
6 except that DMF was used a reaction solvent. White solid, 42 mg, yield 23%.
1H NMR (600 MHz, CDCl
3) δ 8.36 (s, 1H), 8.12 (d,
J = 6.5 Hz, 1H), 8.00 (s, 1H), 7.93 (s,1H), 7.32–7.27 (m, 2H), 7.04–7.02 (m, 3H), 6.59 (s, 1H), 6.30 (s, 2H), 5.54 (s, 2H), 5.41 (s, 2H), 1.52 (s, 9H). HRMS (ESI
+) m/z calcd for C
23H
26N
7O
3 [M + H]
+ 448.2092, found 448.2097.
4.2.45. Tert-Butyl (5-((4-Amino-3-(phenoxymethyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)pyridin-2-yl)carbamate (25)
Compound
25 was prepared from
47a (100 mg, 0.41 mmol) and
tert-butyl (5-(bromomethyl)pyridin-2-yl)carbamate [
22] (143 mg, 0.50 mmol) as described for compound
6. White solid, 72 mg, yield 39%.
1H NMR (600 MHz, CDCl
3) δ 8.37 (s, 1H), 8.30 (d,
J = 2.4 Hz, 1H), 7.97 (s, 1H), 7.89 (d,
J = 8.7 Hz, 1H), 7.67 (dd,
J = 8.7, 2.4 Hz, 1H), 7.31–7.28 (m, 2H), 7.02–7.00 (m, 3H), 6.27 (s, 2H), 5.47 (s, 2H), 5.37 (s, 2H), 1.51 (s, 9H). HRMS (ESI
+) m/z calcd for C
23H
26N
7O
3 [M + H]
+ 448.2092, found 448.2096.
4.2.46. 1-(4-Nitrobenzyl)-3-((o-tolyloxy)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (26)
Compound 26 was prepared from 47b (45 mg, 0.176 mmol) and 4-nitrobenzyl bromide (46 mg, 0.21 mmol) as described for compound 6. Pale solid, 47 mg, yield 68%. 1H NMR (600 MHz, CDCl3) δ 8.37 (s, 1H), 8.17 (d, J = 8.7 Hz, 2H), 7.44 (d, J = 8.7 Hz, 2H), 7.18 (dd, J = 7.5, 1.7 Hz, 1H), 7.14 (td, J = 7.5, 1.7 Hz, 1H), 6.99 (d, J = 8.1 Hz, 1H), 6.94 (td, J = 7.5, 0.9 Hz, 1H), 6.22 (s, 2H), 5.65 (s, 2H), 5.39 (s, 2H), 2.28 (s, 3H). HRMS (ESI+) m/z calcd for C20H19N6O3 [M + H]+ 391.1513, found 391.1519.
4.2.47. 1-(4-Nitrobenzyl)-3-((m-tolyloxy)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (27)
Compound 27 was prepared from 47c (45 mg, 0.176 mmol) and 4-nitrobenzyl bromide (46 mg, 0.21 mmol) as described for compound 6. White solid, 36 mg, yield 52%. 1H NMR (600 MHz, CDCl3) δ 8.36 (s, 1H), 8.16 (d, J = 8.7 Hz, 2H), 7.42 (d, J = 8.7 Hz, 2H), 7.17 (td, J = 7.6, 0.9 Hz, 1H), 6.85–6.81 (m, 3H), 6.27 (s, 2H), 5.64 (s, 2H), 5.38 (s, 2H), 2.31 (s, 3H). HRMS (ESI+) m/z calcd for C20H19N6O3 [M + H]+ 391.1513, found 391.1519.
4.2.48. 1-(4-Nitrobenzyl)-3-((p-tolyloxy)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (28)
Compound 28 was prepared from 47d (45 mg, 0.176 mmol) and 4-nitrobenzyl bromide (46 mg, 0.21 mmol) as described for compound 6. White solid, 43 mg, yield 62%. 1H NMR (600 MHz, CDCl3) δ 8.36 (s, 1H), 8.16 (d, J = 8.7 Hz, 2H), 7.41 (d, J = 8.7 Hz, 2H), 7.09 (d, J = 8.6 Hz, 2H), 6.91 (d, J = 8.6 Hz, 2H), 6.28 (s, 2H), 5.63 (s, 2H), 5.26 (s, 2H), 2.29 (s, 3H). HRMS (ESI+) m/z calcd for C20H19N6O3 [M + H]+ 391.1513, found 391.1522.
4.2.49. 3-((2-Chlorophenoxy)methyl)-1-(4-nitrobenzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (29)
Compound 29 was prepared from 47e (84 mg, 0.30 mmol) and 4-nitrobenzyl bromide (100 mg, 0.46 mmol) as described for compound 6. Pale solid, 103 mg, yield 84%. 1H NMR (400 MHz, CDCl3) δ 8.37 (s, 1H), 8.17 (d, J = 8.6 Hz, 2H), 7.43 (d, J = 8.6 Hz, 2H), 7.39 (dd, J = 8.0, 1.6 Hz, 1H), 7.18 (td, J = 8.0, 1.6 Hz, 1H), 7.08 (dd, J = 8.0, 1.6 Hz, 1H), 6.97 (td, J = 7.7, 1.5 Hz, 1H), 6.24 (s, 2H), 5.64 (s, 2H), 5.48 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 158.2, 157.1, 155.5, 152.7, 147.8, 143.7, 141.4, 130.8, 128.8, 128.2, 124.1, 123.1, 122.7, 114.1, 99.9, 65.7, 49.9. HRMS (ESI−) m/z calcd for C19H14ClN6O3 [M-H]− 409.0821, found 409.0837.
4.2.50. 3-((3-Chlorophenoxy)methyl)-1-(4-nitrobenzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (30)
Compound 30 was prepared from 47f (45 mg, 0.16 mmol) and 4-nitrobenzyl bromide (53 mg, 0.24 mmol) as described for compound 6. Pale solid, 41 mg, yield 63%. 1H NMR (400 MHz, CDCl3) δ 8.37 (s, 1H), 8.17 (d, J = 8.7 Hz, 2H), 7.43 (d, J = 8.7 Hz, 2H), 7.21 (t, J = 8.1 Hz, 1H), 7.25–6.97 (m, 2H), 6.90 (dd, J = 8.3, 0.9 Hz, 1H), 6.14 (s, 2H), 5.64 (s, 2H), 5.40 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 158.0, 157.9, 157.0, 155.4, 147.8, 143.6, 141.5, 135.5, 130.8, 128.7, 124.2, 122.8, 115.6, 113.5, 99.8, 65.3, 49.9. HRMS (ESI−) m/z calcd for C19H14ClN6O3 [M-H]− 409.0821, found 409.0839.
4.2.51. 3-((4-Chlorophenoxy)methyl)-1-(4-nitrobenzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (31)
Compound 31 was prepared from 47g (47 mg, 0.17 mmol) and 4-nitrobenzyl bromide (60 mg, 0.28 mmol) as described for compound 6. Pale solid, 39 mg, yield 57%. 1H NMR (400 MHz, DMSO-d6) δ 8.23 (s, 1H), 8.17 (d, J = 8.8 Hz, 2H), 7.41 (d, J = 8.8 Hz, 2H), 7.33 (d, J = 9.0 Hz, 2H), 7.06 (d, J = 9.0 Hz, 2H), 5.65 (s, 2H), 5.43 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 157.9, 156.5, 156.4, 154.7, 146.9, 144.6, 140.8, 129.2, 128.5, 125.0, 123.7, 116.9, 98.5, 63.8, 49.0. HRMS (ESI−) m/z calcd for C19H14ClN6O3 [M-H]− 409.0821, found 409.0831.








{kind=link}
{kind=link}
{kind=link}




























