

Molecular and Structural Analysis of Specific Mutations from Saudi Isolates of SARS-CoV-2 RNA-Dependent RNA Polymerase and their Implications on Protein Structure and Drug–Protein Binding

 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval from NCBI Virus Database
2.2. Multiple Sequence Alignment by Clustal Omega Program
2.3. Secondary Structure Predictions
2.4. Structural Homology Modeling
2.5. Molecular Docking Studies
2.6. Molecular Dynamics (MD) Simulation
2.7. Binding Free-Energy Calculations Using MMGBSA
3. Results and Discussion
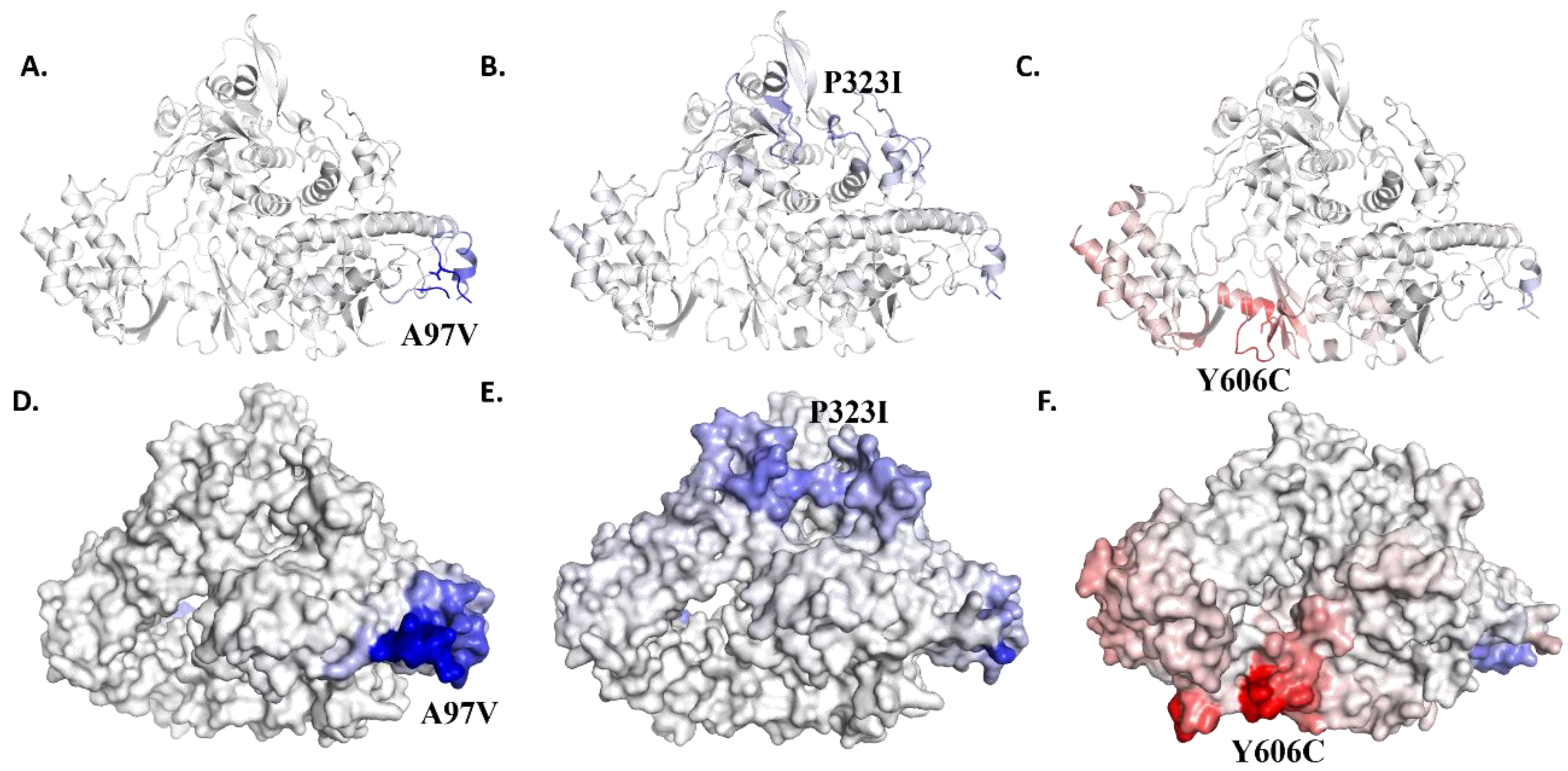
3.1. Identification of Mutations in Structural Proteins Present in Saudi Isolates
3.2. Δ Vibrational Entropy Energy between Wild-Type and Mutant
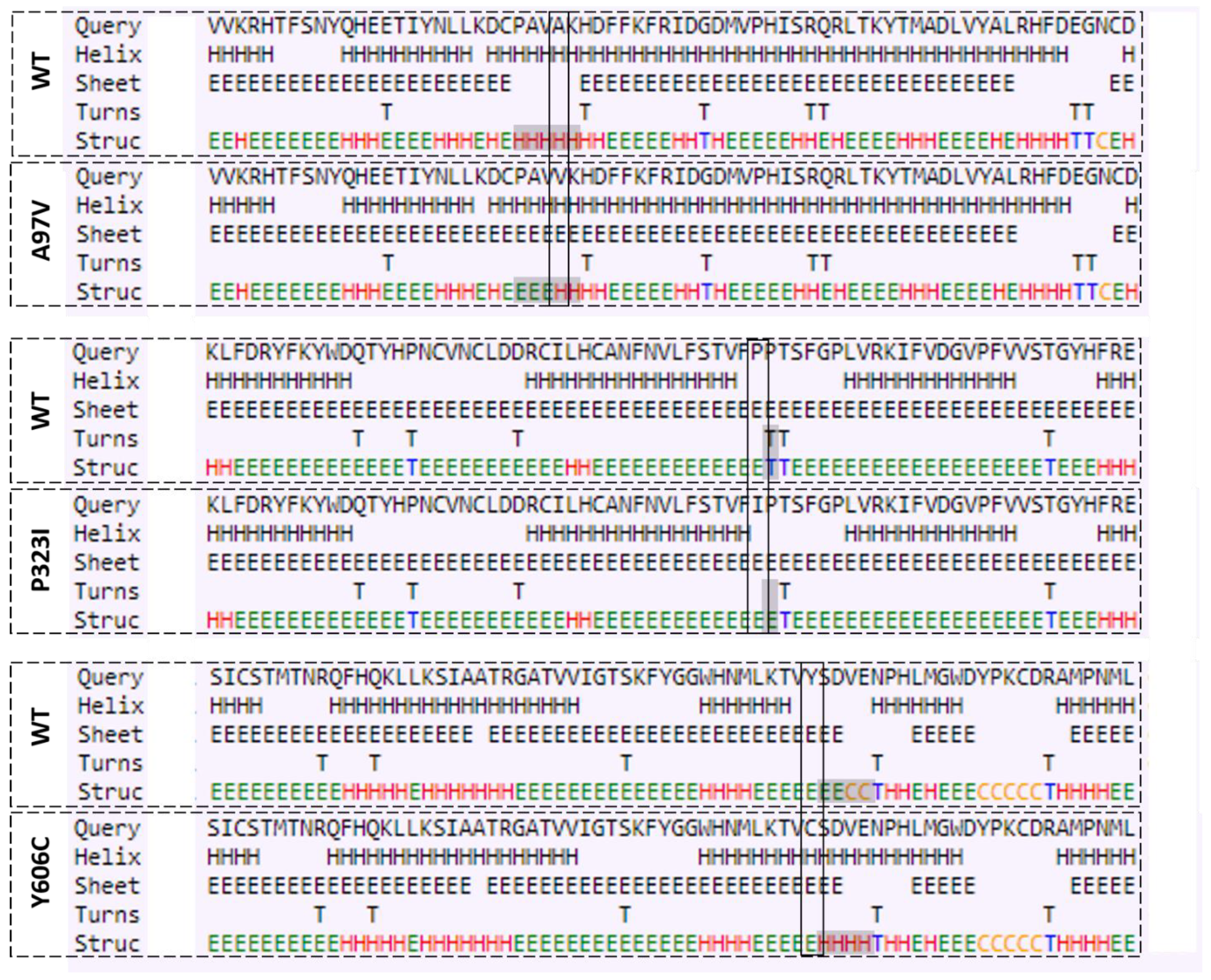
3.3. Mutations Cause Alteration in Secondary Structure of Proteins
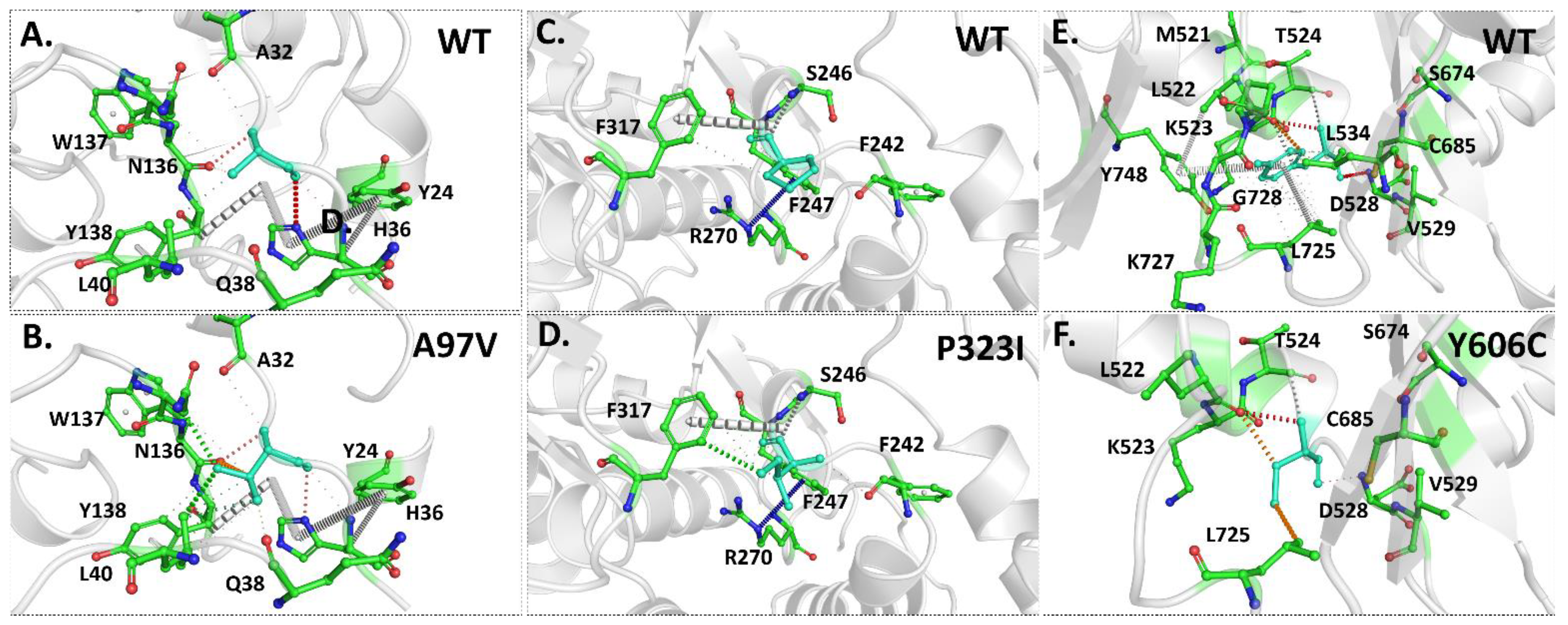
3.4. Intramolecular Interactions Are Altered Due to Mutations in Proteins
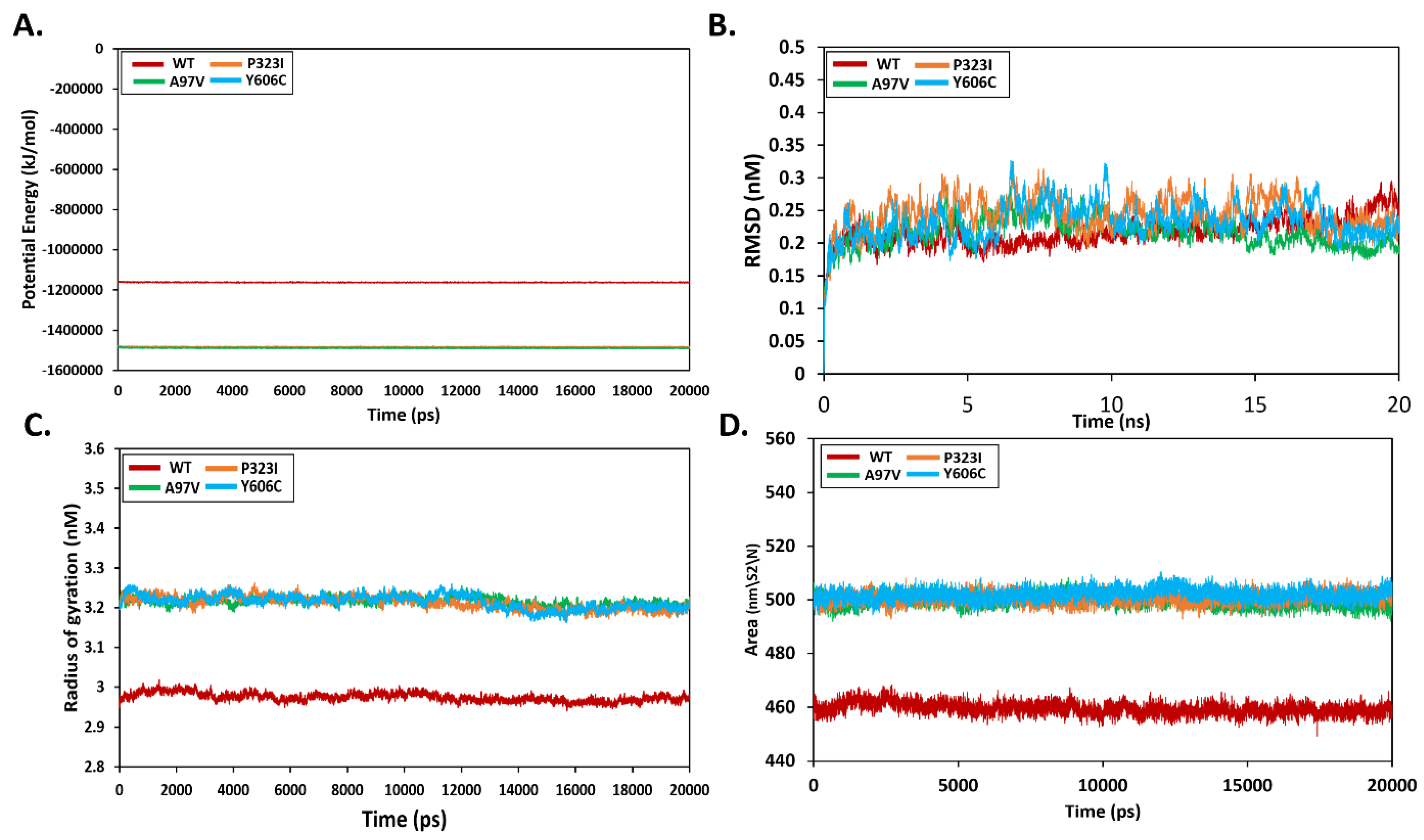
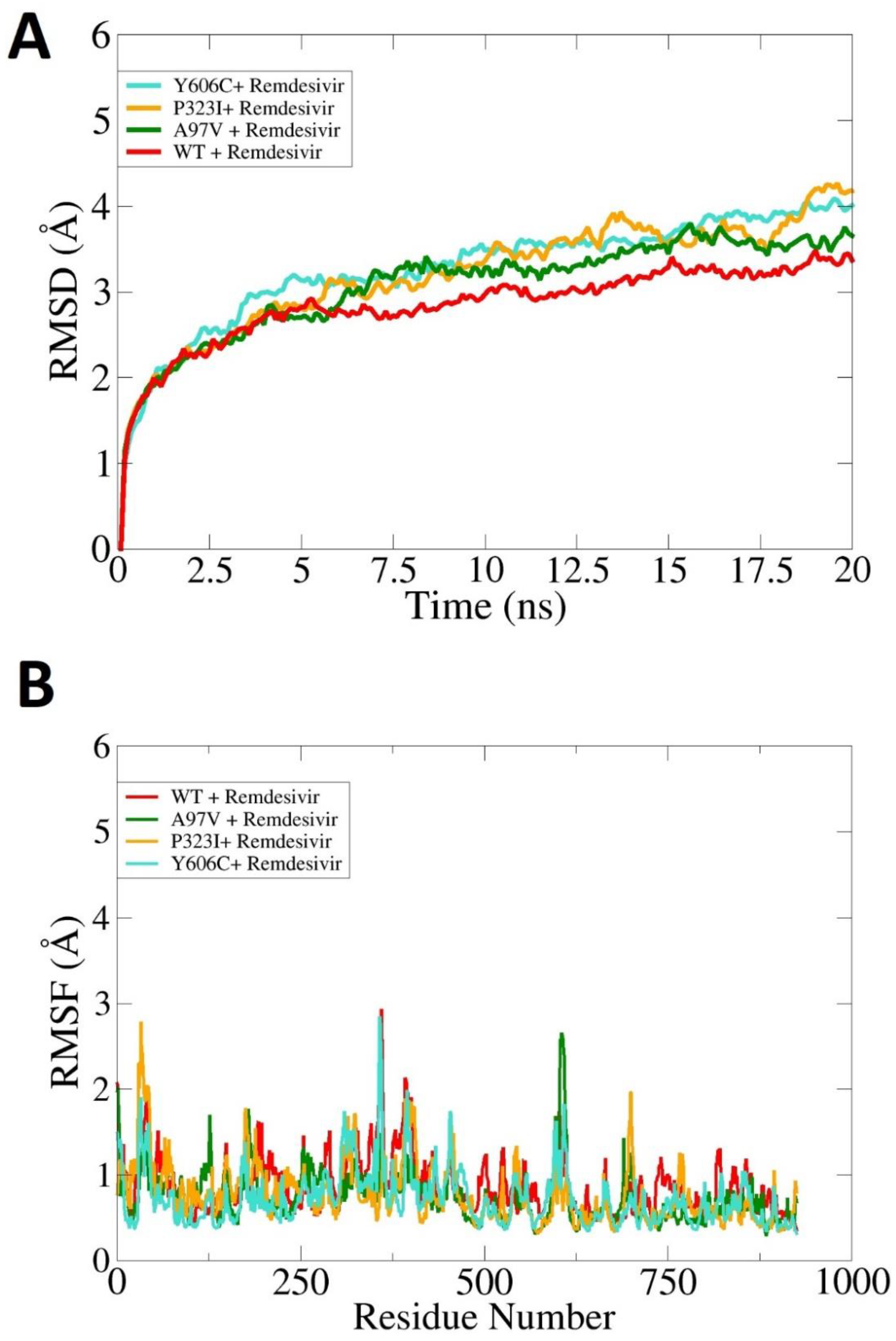
3.5. Effect of Mutations on Protein Structural Conformation and Dynamic Stability
3.6. Effect of Mutations on Binding and Stability of Remdesivir
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amanat, F.; Krammer, F. SARS-CoV-2 vaccines: Status report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L.L.; ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L.L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Mehmood, I.; Ijaz, M.; Ahmad, S.; Ahmed, T.; Bari, A.; Abro, A.; Allemailem, K.S.; Almatroudi, A.; Tahir ul Qamar, M. SARS-CoV-2: An update on genomics, risk assessment, potential therapeutics and vaccine development. Int. J. Environ. Res. Public Health 2021, 18, 1626. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020, 21, 224. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B. 1.1. 7 in England. Science 2021, 372, 3055. [Google Scholar] [CrossRef]
- Chand, M.; Hopkins, S.; Dabrera, G.; Achison, C.; Barclay, W.; Ferguson, N. Investigation of Novel SARS-COV-2 Variant: Variant of Concern 202012/01 (PDF); Public Health England PHE: London, UK, 2020; Volume 21.
- Grint, D.J.; Wing, K.; Houlihan, C.; Gibbs, H.P.; Evans, S.J.W.; Williamson, E.; McDonald, H.I.; Bhaskaran, K.; Evans, D.; Walker, A.J.; et al. Severity of SARS-CoV-2 alpha variant (B. 1.1. 7) in England. Clin. Infect. Dis. 2021, 75, ciab754. [Google Scholar]
- Islam, A.; Sayeed, M.A.; Kalam, M.A.; Ferdous, J.; Rahman, M.K.; Abedin, J.; Islam, S.; Shano, S.; Saha, O.; Shirin, T.; et al. Molecular epidemiology of SARS-CoV-2 in diverse environmental samples globally. Microorganisms 2021, 9, 1696. [Google Scholar] [CrossRef]
- Mistry, P.; Barmania, F.; Mellet, J.; Peta, K.; Strydom, A.; Viljoen, I.M.; James, W.; Gordon, S.; Pepper, M.S. SARS-CoV-2 variants, vaccines, and host immunity. Front. Immunol. 2021, 12, 809244. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Khateeb, J.; Li, Y.; Zhang, H. Emerging SARS-CoV-2 variants of concern and potential intervention approaches. Crit. Care 2021, 25, 244. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.M.; Vostok, J.; Johnson, H.; Burns, M.; Gharpure, R.; Sami, S.; Sabo, R.T.; Hall, N.; Foreman, A.; Schubert, P.L. Outbreak of SARS-CoV-2 infections, including COVID-19 vaccine breakthrough infections, associated with large public gatherings—Barnstable County, Massachusetts, July 2021. Morb. Mortal. Wkly. Rep. 2021, 70, 1059. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sharma, K.; Singh, P.; Bhargava, A.; Negi, S.S.; Sharma, P.; Bhise, M.; Tripathi, M.K.; Jindal, A.; Nagarkar, N.M. Genomic characterization unravelling the causative role of SARS-CoV-2 Delta variant of lineage B. 1.617. 2 in 2nd wave of COVID-19 pandemic in Chhattisgarh, India. Microb. Pathog. 2022, 164, 105404. [Google Scholar] [CrossRef] [PubMed]
- Meo, S.A.; Meo, A.S.; Al-Jassir, F.F.; Klonoff, D.C. Omicron SARS-CoV-2 new variant: Global prevalence and biological and clinical characteristics. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 8012–8018. [Google Scholar] [PubMed]
- Eyre, D.W.; Taylor, D.; Purver, M.; Chapman, D.; Fowler, T.; Pouwels, K.; Walker, A.S.; Peto, T.E.A. The impact of SARS-CoV-2 vaccination on Alpha and Delta variant transmission. medRxiv 2021. [Google Scholar] [CrossRef]
- Callaway, E.; Ledford, H. How to redesign COVID vaccines so they protect against variants. Nature 2021, 590, 15–16. [Google Scholar] [CrossRef]
- Elshabrawy, H.A. SARS-CoV-2: An Update on Potential Antivirals in Light of SARS-CoV Antiviral Drug Discoveries. Vaccines 2020, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; Tahir Ul Qamar, M.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M.H.H. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2020, 39, 4936–4948. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Peng, F.; Wang, R.; Guan, K.; Jiang, T.; Xu, G.; Sun, J.; Chang, C. The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun. 2020, 109, 102434. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. J. Biomol. Struct. Dyn. 2020, 39, 3204–3212. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Vanmeert, M.; Froeyen, M.; Ali, A.; Rafique, S.; Idrees, M. In silico structural elucidation of RNA-dependent RNA polymerase towards the identification of potential Crimean-Congo Hemorrhagic Fever Virus inhibitors. Sci. Rep. 2019, 9, 6809. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Marotta, C.; Stefanelli, P.; Tramuto, F.; Angeloni, U.; Maida, C.M.; Cernigliaro, A.; Barone, T.; Vitale, F.; Rezza, G.; Mazzucco, W. The dual/global value of SARS-COV-2 genome surveillance on migrants arriving to Europe via the mediterranean routes. Ann. Glob. Health 2021, 87, 71. [Google Scholar] [CrossRef]
- Robishaw, J.D.; Alter, S.M.; Solano, J.J.; Shih, R.D.; DeMets, D.L.; Maki, D.G.; Hennekens, C.H. Genomic surveillance to combat COVID-19: Challenges and opportunities. Lancet Microbe 2021, 2, e481–e484. [Google Scholar] [CrossRef]
- Yashvardhini, N.; Jha, D.K.; Bhattacharya, S. Identification and characterization of mutations in the SARS-CoV-2 RNA-dependent RNA polymerase as a promising antiviral therapeutic target. Arch. Microbiol. 2021, 203, 5463–5473. [Google Scholar] [CrossRef]
- Bawono, P.; Dijkstra, M.; Pirovano, W.; Feenstra, A.; Abeln, S.; Heringa, J. Multiple sequence alignment. Methods Mol. Biol. 2017, 1525, 167–189. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]
- Kumar, T.A. CFSSP: Chou and Fasman secondary structure prediction server. Wide Spectr. 2013, 1, 15–19. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasi, M.; Tiberti, M.; Arrigoni, A.; Papaleo, E. XPyder: A PyMOL plugin to analyze coupled residues and their networks in protein structures. J. Chem. Inf. Model. 2012, 52, 1865–1874. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Giambasu, G.; et al. Amber 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An accessory software package for molecular mechanical calculations. J. Am. Chem. Soc 2001, 222, U403. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. The FF14SB force field. Amber 2014, 14, 29–31. [Google Scholar]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef] [Green Version]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Studer, R.A.; Dessailly, B.H.; Orengo, C.A. Residue mutations and their impact on protein structure and function: Detecting beneficial and pathogenic changes. Biochem. J. 2013, 449, 581–594. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Accession Number |
|---|---|
| 1 | YP_009724389 |
| 2 | QMS50985 |
| 3 | QMS50997 |
| 4 | QMS51009 |
| 5 | QMS51021 |
| 6 | QMS51033 |
| 7 | QMS51045 |
| 8 | QMS51057 |
| 9 | QMS51069 |
| 10 | QMS51081 |
| 11 | QMS51093 |
| 12 | QMS51105 |
| 13 | QMS51117 |
| 14 | QMS51129 |
| 15 | QMS51141 |
| 16 | QMS51153 |
| 17 | QMS51165 |
| 18 | QMS51177 |
| 19 | QMS51189 |
| 20 | QMS51201 |
| 21 | QMS51213 |
| 22 | QMS51225 |
| 23 | QMS51237 |
| 24 | QMS51249 |
| 25 | QMS51261 |
| 26 | QMS51273 |
| 27 | QMS51285 |
| 28 | QMS51297 |
| 29 | QMS51309 |
| 30 | QMS51321 |
| 31 | QLH56060 |
| 32 | QLH56072 |
| 33 | QLH56084 |
| 34 | QLH56096 |
| 35 | QLH56108 |
| 36 | QLH56120 |
| 37 | QLH56132 |
| 38 | QLH56144 |
| 39 | QLH56156 |
| 40 | QLH56168 |
| 41 | QLH56180 |
| 42 | QLH56192 |
| 43 | QLH56204 |
| 44 | QLH56216 |
| 45 | QLH56228 |
| 46 | QLH56240 |
| 47 | QLH56252 |
| 48 | QKU37019 |
| 49 | QKU37031 |
| 50 | QKU37043 |
| 51 | QKU37055 |
| 52 | QKU37067 |
| 53 | QKU37079 |
| 54 | QKU37091 |
| 55 | QKU37103 |
| 56 | QKU37115 |
| 57 | QKU37127 |
| 58 | QKU37139 |
| 59 | QKU37151 |
| Mutation ID | Wild-Type Residue | Position of Mutation | Mutated Residue | Frequency |
|---|---|---|---|---|
| A97V | A | 97 | V | 1 |
| P323I | P | 323 | I | 56 |
| Y606C | Y | 606 | C | 1 |
| S. No | Wuhan Isolate | Saudi Isolate | AA Position | ΔΔS ENCoM | ΔΔG DynaMut | ΔΔG mCSM | ΔΔG SDM | ΔΔG DUET | Effect |
|---|---|---|---|---|---|---|---|---|---|
| 1 | A | V | 97 | 4.117 | 1.397 | −0.271 | −1.270 | −0.242 | Stabilizing |
| 2 | P | I | 323 | 0.406 | 1.017 | −0.251 | 1.500 | 0.454 | Stabilizing |
| 3 | Y | C | 606 | −0.984 | −0.675 | −1.675 | −1.200 | −1.721 | Destabilizing |
| Energy Parameter | WT-RdRp Complex with Remdesivir | A97V-RdRp Complex with Remdesivir | P232I -RdRp Complex with Remdesivir | Y606C-RdRp Complex with Remdesivir |
|---|---|---|---|---|
| VDWAALS | −40.68 | −39.33 | −41.49 | −37.51 |
| EEL | −25.28 | −22.00 | −23.22 | −19.67 |
| Delta G gas | −65.96 | −61.33 | −64.71 | −57.18 |
| Delta G solv | 15.20 | 16.37 | 17.51 | 15.84 |
| Delta Total | −50.76 | −44.96 | −47.2 | −41.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alamri, M.A.; Tahir ul Qamar, M.; Alabbas, A.B.; Alqahtani, S.M.; Alossaimi, M.A.; Azam, S.; Hashmi, M.H.; Rajoka, M.S.R. Molecular and Structural Analysis of Specific Mutations from Saudi Isolates of SARS-CoV-2 RNA-Dependent RNA Polymerase and their Implications on Protein Structure and Drug–Protein Binding. Molecules 2022, 27, 6475. https://doi.org/10.3390/molecules27196475
Alamri MA, Tahir ul Qamar M, Alabbas AB, Alqahtani SM, Alossaimi MA, Azam S, Hashmi MH, Rajoka MSR. Molecular and Structural Analysis of Specific Mutations from Saudi Isolates of SARS-CoV-2 RNA-Dependent RNA Polymerase and their Implications on Protein Structure and Drug–Protein Binding. Molecules. 2022; 27(19):6475. https://doi.org/10.3390/molecules27196475
Chicago/Turabian StyleAlamri, Mubarak A., Muhammad Tahir ul Qamar, Alhumaidi B. Alabbas, Safar M. Alqahtani, Manal A. Alossaimi, Sikandar Azam, Muhammad Harris Hashmi, and Muhammad Shahid Riaz Rajoka. 2022. "Molecular and Structural Analysis of Specific Mutations from Saudi Isolates of SARS-CoV-2 RNA-Dependent RNA Polymerase and their Implications on Protein Structure and Drug–Protein Binding" Molecules 27, no. 19: 6475. https://doi.org/10.3390/molecules27196475
APA StyleAlamri, M. A., Tahir ul Qamar, M., Alabbas, A. B., Alqahtani, S. M., Alossaimi, M. A., Azam, S., Hashmi, M. H., & Rajoka, M. S. R. (2022). Molecular and Structural Analysis of Specific Mutations from Saudi Isolates of SARS-CoV-2 RNA-Dependent RNA Polymerase and their Implications on Protein Structure and Drug–Protein Binding. Molecules, 27(19), 6475. https://doi.org/10.3390/molecules27196475