


Electrocatalytic Depolymerization of Self-Immolative Poly(Dithiothreitol) Derivatives

Abstract
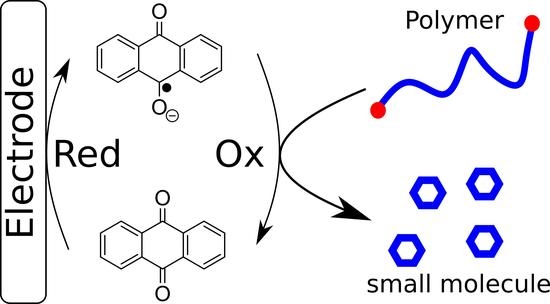
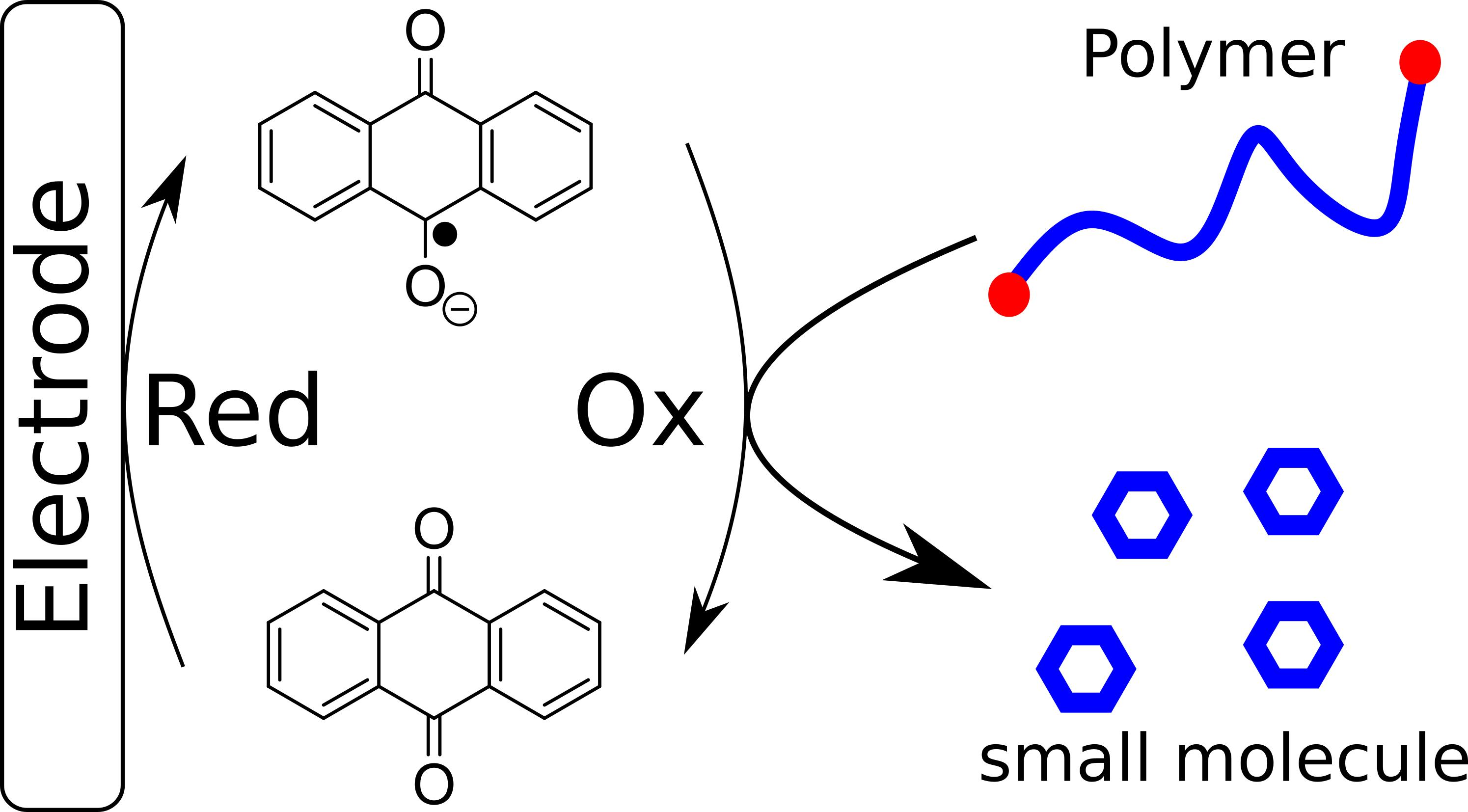
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
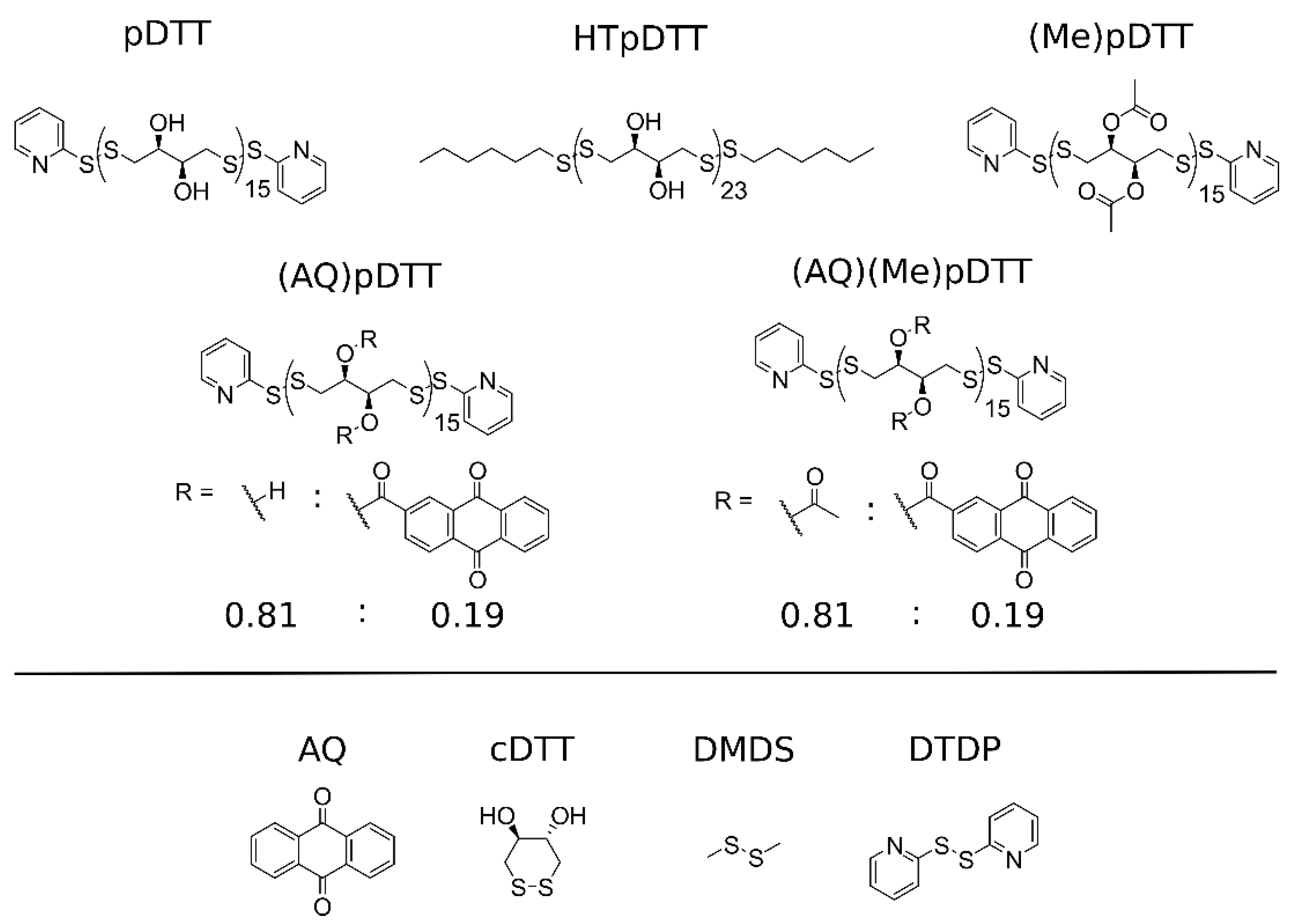
2.1. Polymers
2.2. Electrolyzes of Poly(dithiothreitol) Derivatives
2.3. Time-Resolved Titration of AQ•− with Polymer Solution
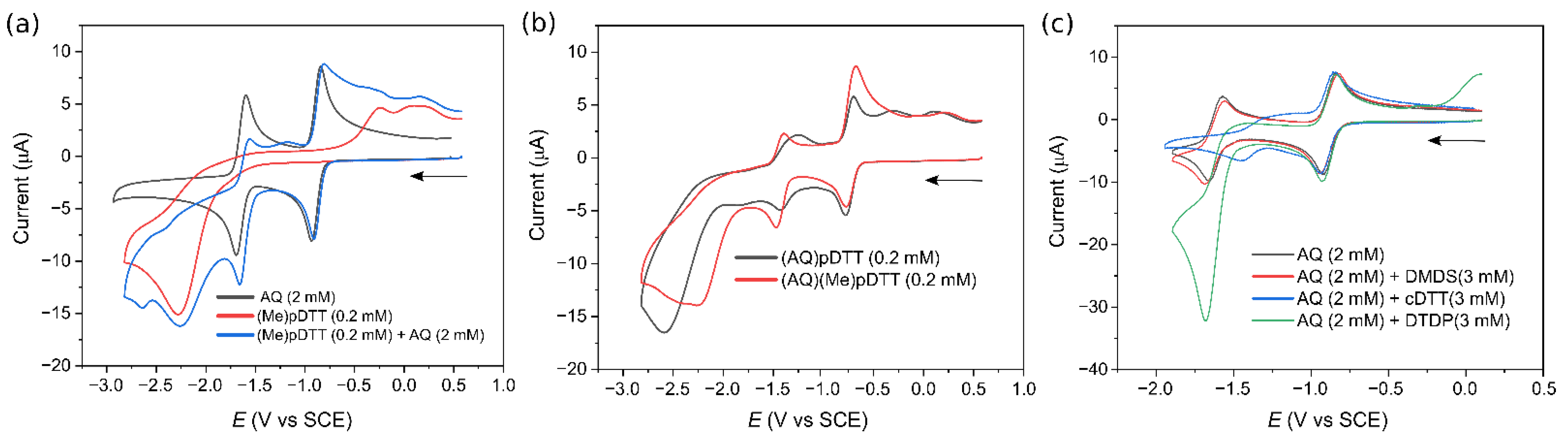
2.4. Cyclic Voltammetry of pDTT Derivatives, Model Compounds, and AQ
3. Discussion
3.1. Depolymerization of pDTT Derivatives Using Redox Catalysis
3.2. Catalytic Depolymerization
3.3. Protonation Pathway
3.4. Effect of End-Cap
3.5. Electrocatalytic Depolymerization Mechanism
4. Materials and Methods
4.1. General Materials
4.2. General Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sagi, A.; Weinstain, R.; Karton, N.; Shabat, D. Self-immolative polymers. J. Am. Chem. Soc. 2008, 130, 5434–5435. [Google Scholar] [CrossRef] [PubMed]
- Yardley, R.E.; Kenaree, A.R.; Gillies, E.R. Triggering Depolymerization: Progress and Opportunities for Self-Immolative Polymers. Macromolecules 2019, 52, 6342–6360. [Google Scholar] [CrossRef]
- Lloyd, E.M.; Lopez Hernandez, H.; Feinberg, E.C.; Yourdkhani, M.; Zen, E.K.; Mejia, E.B.; Sottos, N.R.; Moore, J.S.; White, S.R. Fully Recyclable Metastable Polymers and Composites. Chem. Mater. 2018, 31, 398–406. [Google Scholar] [CrossRef]
- Fan, B.; Trant, J.F.; Wong, A.D.; Gillies, E.R. Polyglyoxylates: A versatile class of triggerable self-immolative polymers from readily accessible monomers. J. Am. Chem. Soc. 2014, 136, 10116–10123. [Google Scholar] [CrossRef] [PubMed]
- Ergene, C.; Palermo, E.F. Self-immolative polymers with potent and selective antibacterial activity by hydrophilic side chain grafting. J. Mater. Chem. B 2018, 6, 7217–7229. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, H.; Zhang, B.; Cheng, Z.; Li, Y.; Tan, X.; Zhang, K. Modulating the Depolymerization of Self-Immolative Brush Polymers with Poly(benzyl ether) Backbones. Macromolecules 2018, 51, 2899–2905. [Google Scholar] [CrossRef]
- Olah, M.G.; Robbins, J.S.; Baker, M.S.; Phillips, S.T. End-Capped Poly(benzyl ethers): Acid and Base Stable Polymers That Depolymerize Rapidly from Head-to-Tail in Response to Specific Applied Signals. Macromolecules 2013, 46, 5924–5928. [Google Scholar] [CrossRef]
- Seo, W.; Phillips, S.T. Patterned plastics that change physical structure in response to applied chemical signals. J. Am. Chem. Soc. 2010, 132, 9234–9235. [Google Scholar] [CrossRef]
- Taimoory, S.M.; Sadraei, S.I.; Fayoumi, R.A.; Nasri, S.; Revington, M.; Trant, J.F. Preparation and Characterization of a Small Library of Thermally-Labile End-Caps for Variable-Temperature Triggering of Self-Immolative Polymers. J. Org. Chem. 2018, 83, 4427–4440. [Google Scholar] [CrossRef]
- Weinstain, R.; Sagi, A.; Karton, N.; Shabat, D. Self-immolative comb-polymers: Multiple-release of side-reporters by a single stimulus event. Chem. Eur. J. 2008, 14, 6857–6861. [Google Scholar] [CrossRef]
- Hernandez, H.L.; Kang, S.K.; Lee, O.P.; Hwang, S.W.; Kaitz, J.A.; Inci, B.; Park, C.W.; Chung, S.; Sottos, N.R.; Moore, J.S.; et al. Triggered transience of metastable poly(phthalaldehyde) for transient electronics. Adv. Mater. 2014, 26, 7637–7642. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Trant, J.F.; Gillies, E.R. End-Capping Strategies for Triggering End-to-End Depolymerization of Polyglyoxylates. Macromolecules 2016, 49, 9309–9319. [Google Scholar] [CrossRef]
- Robbins, J.S.; Schmid, K.M.; Phillips, S.T. Effects of electronics, aromaticity, and solvent polarity on the rate of azaquinone-methide-mediated depolymerization of aromatic carbamate oligomers. J. Org. Chem. 2013, 78, 3159–3169. [Google Scholar] [CrossRef]
- Lewis, G.G.; Robbins, J.S.; Phillips, S.T. A prototype point-of-use assay for measuring heavy metal contamination in water using time as a quantitative readout. Chem. Commun. 2014, 50, 5352–5354. [Google Scholar] [CrossRef]
- Feinberg, A.M.; Davydovich, O.; Lloyd, E.M.; Ivanoff, D.G.; Shiang, B.; Sottos, N.R.; Moore, J.S. Triggered Transience of Plastic Materials by a Single Electron Transfer Mechanism. ACS Cent. Sci. 2020, 6, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.; Kim, H.; Mohapatra, H.; Phillips, S.T. Surface-accessible detection units in self-immolative polymers enable translation of selective molecular detection events into amplified responses in macroscopic, solid-state plastics. J. Am. Chem. Soc. 2015, 137, 5324–5327. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Li, Y.; Zhang, B.; Li, H.; Cheng, Z.; Shi, J.; Xiong, J.; Bai, Y.; Zhang, K. Functionalizable, Side Chain-Immolative Poly(benzyl ether)s. ACS Macro Lett. 2019, 8, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Gillies, E.R. Self-immolative Amphiphilic Diblock Copolymers with Individually Triggerable Blocks. ACS Polym. Au 2022. [Google Scholar] [CrossRef]
- Di Marino, D.; Stöckmann, D.; Kriescher, S.; Stiefel, S.; Wessling, M. Electrochemical depolymerisation of lignin in a deep eutectic solvent. Green Chem. 2016, 18, 6021–6028. [Google Scholar] [CrossRef]
- Dier, T.K.F.; Rauber, D.; Durneata, D.; Hempelmann, R.; Volmer, D.A. Sustainable Electrochemical Depolymerization of Lignin in Reusable Ionic Liquids. Sci. Rep. 2017, 7, 5041. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.S.; Ramasamy, K.K. Electrochemical Oxidation of Lignin and Waste Plastic. ACS Omega 2020, 5, 27735–27740. [Google Scholar] [CrossRef] [PubMed]
- Bawareth, B.; Di Marino, D.; Nijhuis, T.A.; Wessling, M. Unravelling Electrochemical Lignin Depolymerization. ACS Sustain. Chem. Eng. 2018, 6, 7565–7573. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, L.; Chen, Y.; Li, G.; Li, H.; Tang, Y.; Wan, P. Electrochemical depolymerization of lignin into renewable aromatic compounds in a non-diaphragm electrolytic cell. RSC Adv. 2014, 4, 29917–29924. [Google Scholar] [CrossRef]
- Tatsuma, T.; Yokoyama, Y.; Buttry, D.A.; Oyama, N. Electrochemical Polymerization and Depolymerization of 2,5-Dimercapto-1,3,4-thiadiazole. QCM and Spectroscopic Analysis. J. Phys. Chem. B 1997, 101, 7556–7562. [Google Scholar] [CrossRef]
- Naoi, K.; Oura, Y.; Iwamizu, Y.; Oyama, N. Electrochemistry of Disulfide Compounds: I. Electrochemical Polymerization-Depolymerization Process of 2,5-Dimercapt-1,3,4-thiadiazole. J. Electrochem. Soc. 1995, 142, 354–360. [Google Scholar] [CrossRef]
- Jiang, T.; Zhao, X.; Gu, D.; Yan, C.; Jiang, H.; Wu, H.; Wang, B.; Wang, X. STEP polymer degradation: Solar thermo-coupled electrochemical depolymerization of plastics to generate useful fuel plus abundant hydrogen. Sol. Energy Mater. Sol. Cells 2020, 204, 110208. [Google Scholar] [CrossRef]
- Meng, D.; Li, G.; Liu, Z.; Yang, F. Study of depolymerization of cotton cellulose by Pb/PbO2 anode electrochemical catalysis in sulfuric acid solution. Polym. Degrad. Stab. 2011, 96, 1173–1178. [Google Scholar] [CrossRef]
- Myren, T.H.T.; Stinson, T.A.; Mast, Z.J.; Huntzinger, C.G.; Luca, O.R. Chemical and Electrochemical Recycling of End-Use Poly(ethylene terephthalate) (PET) Plastics in Batch, Microwave and Electrochemical Reactors. Molecules 2020, 25, 2742. [Google Scholar] [CrossRef]
- Hansen-Felby, M.; Henriksen, M.L.; Pedersen, S.U.; Daasbjerg, K. Postfunctionalization of Self-Immolative Poly(dithiothreitol) Using Steglich Esterification. Macromolecules 2022, 55, 5788–5794. [Google Scholar] [CrossRef]
- Pal, S.; Sommerfeldt, A.; Davidsen, M.B.; Hinge, M.; Pedersen, S.U.; Daasbjerg, K. Synthesis and Closed-Loop Recycling of Self-Immolative Poly(dithiothreitol). Macromolecules 2020, 53, 4685–4691. [Google Scholar] [CrossRef]
- Agergaard, A.H.; Sommerfeldt, A.; Pedersen, S.U.; Birkedal, H.; Daasbjerg, K. Dual-Responsive Material Based on Catechol-Modified Self-Immolative Poly(Disulfide) Backbones. Angew. Chem. Int. Ed. 2021, 60, 21543–21549. [Google Scholar] [CrossRef] [PubMed]
- Hansen-Felby, M.; Sommerfeldt, A.; Henriksen, M.L.; Pedersen, S.U.; Daasbjerg, K. Synthesis and depolymerization of self-immolative poly(disulfide)s with saturated aliphatic backbones. Polym. Chem. 2022, 13, 85–90. [Google Scholar] [CrossRef]
- Alam, N.; Amatore, C.; Combellas, C.; Thiebault, A.; Verpeaux, J.N. Theory and experimental illustration of preparative electrochemistry using redox catalysis of electron transfer initiated radical chain reactions. Application to the cross-coupling between aryl halides and phenoxide ions. J. Org. Chem. 1990, 55, 6347–6356. [Google Scholar] [CrossRef]
- Amatore, C.; Carré, E.; Jutand, A.; Tanaka, H.; Torii, S.; Chiarotto, I.; Carelli, I. Direct vs indirect route in the activation of aroylpalladium(II) complexes by electron transfer. Electrochim. Acta 1997, 42, 2143–2152. [Google Scholar] [CrossRef]
- Savéant, J.M. Molecular catalysis of electrochemical reactions. Mechanistic aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef]
- Christensen, T.B.; Daasbjerg, K. Investigation of the Direct and Indirect Reduction Processes of Some Disulfides by Electrochemical Means. Acta Chem. Scand. 1997, 51, 307–317. [Google Scholar] [CrossRef]
- Daasbjerg, K.; Jensen, H.; Benassi, R.; Taddei, F.; Antonello, S.; Gennaro, A.; Maran, F. Evidence for Large Inner Reorganization Energies in the Reduction of Diaryl Disulfides: Toward a Mechanistic Link between Concerted and Stepwise Dissociative Electron Transfers? J. Am. Chem. Soc. 1999, 121, 1750–1751. [Google Scholar] [CrossRef]
- Antonello, S.; Daasbjerg, K.; Jensen, H.; Taddei, F.; Maran, F. Formation and cleavage of aromatic disulfide radical anions. J. Am. Chem. Soc. 2003, 125, 14905–14916. [Google Scholar] [CrossRef]
- Meneses, A.B.; Antonello, S.; Arevalo, M.C.; Gonzalez, C.C.; Sharma, J.; Wallette, A.N.; Workentin, M.S.; Maran, F. Electron transfer to sulfides and disulfides: Intrinsic barriers and relationship between heterogeneous and homogeneous electron-transfer kinetics. Chemistry 2007, 13, 7983–7995. [Google Scholar] [CrossRef]
- Pedersen, S.U.; Bo Christensen, T.; Thomasen, T.; Daasbjerg, K. New methods for the accurate determination of extinction and diffusion coefficients of aromatic and heteroaromatic radical anions in N,N-dimethylformamide. J. Electroanal. Chem. 1998, 454, 123–143. [Google Scholar] [CrossRef]
- Wipf, D.O.; Wehmeyer, K.R.; Wightman, R.M. Disproportionation of quinone radical anions in protic solvents at high pH. J. Org. Chem. 1986, 51, 4760–4764. [Google Scholar] [CrossRef]
- Guin, P.S.; Das, S.; Mandal, P. Electrochemical reduction of sodium 1, 4-dihydroxy-9, 10-anthraquinone-2-sulphonate in aqueous and aqueous dimethyl formamide mixed solvent: A cyclic voltammetric study. Int. J. Electrochem. Sci. 2008, 3, 1016–1028. [Google Scholar]
- Rudolph, M.; Feldberg, S.W. DigiSim, Version 3.05. Bioanalytical Systems, Inc.: West Lafayette, IN, USA, 2004. [Google Scholar]
- Amatore, C.; Capobianco, G.; Farnia, G.; Sandona, G.; Savéant, J.M.; Severin, M.G.; Vianello, E. Kinetics and Mechanism of Self-Protonation Reactions in Organic Electrochemical Processes. J. Am. Chem. Soc. 1985, 107, 1815–1824. [Google Scholar] [CrossRef]
- Davis, F.O.; Fettes, E.M. The Preparation and Polymerization of Monomeric Cyclic Disulfides. J. Am. Chem. Soc. 1948, 70, 2611–2613. [Google Scholar] [CrossRef]
- Fava, A.; Reichenbach, G.; Peron, U. Kinetics of the thiol-disulfide exchange. II. Oxygen-promoted free-radical exchange between aromatic thiols and disulfides. J. Am. Chem. Soc. 1967, 89, 6696–6700. [Google Scholar] [CrossRef]
- Zhao, L.; Almaraz, R.T.; Xiang, F.; Hedrick, J.L.; Franz, A.H. Gas-phase scrambling of disulfide bonds during matrix-assisted laser desorption/ionization mass spectrometry analysis. J. Am. Soc. Mass Spectrom. 2009, 20, 1603–1616. [Google Scholar] [CrossRef]
- Gammelgaard, S.K.; Petersen, S.B.; Haselmann, K.F.; Nielsen, P.K. Direct Ultraviolet Laser-Induced Reduction of Disulfide Bonds in Insulin and Vasopressin. ACS Omega 2020, 5, 7962–7968. [Google Scholar] [CrossRef]
- Sohn, C.H.; Gao, J.; Thomas, D.A.; Kim, T.Y.; Goddard, W.A., III; Beauchamp, J.L. Mechanisms and energetics of free radical initiated disulfide bond cleavage in model peptides and insulin by mass spectrometry. Chem. Sci. 2015, 6, 4550–4560. [Google Scholar] [CrossRef]
- Nevejans, S.; Ballard, N.; Miranda, J.I.; Reck, B.; Asua, J.M. The underlying mechanisms for self-healing of poly(disulfide)s. Phys. Chem. Chem. Phys. 2016, 18, 27577–27583. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, Y.; Wu, Q.; Moore, J.S. Architecture-Controlled Ring-Opening Polymerization for Dynamic Covalent Poly(disulfide)s. J. Am. Chem. Soc. 2019, 141, 17075–17080. [Google Scholar] [CrossRef]
- Zhang, Q.; Deng, Y.; Shi, C.-Y.; Feringa, B.L.; Tian, H.; Qu, D.-H. Dual closed-loop chemical recycling of synthetic polymers by intrinsically reconfigurable poly(disulfides). Matter 2021, 4, 1352–1364. [Google Scholar] [CrossRef]
- Montasell, M.C.; Monge, P.; Carmali, S.; Dias Loiola, L.M.; Andersen, D.G.; Lovschall, K.B.; Sogaard, A.B.; Kristensen, M.M.; Putz, J.M.; Zelikin, A.N. Chemical zymogens for the protein cysteinome. Nat. Commun. 2022, 13, 4861. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen-Felby, M.; Pedersen, S.U.; Daasbjerg, K. Electrocatalytic Depolymerization of Self-Immolative Poly(Dithiothreitol) Derivatives. Molecules 2022, 27, 6292. https://doi.org/10.3390/molecules27196292
Hansen-Felby M, Pedersen SU, Daasbjerg K. Electrocatalytic Depolymerization of Self-Immolative Poly(Dithiothreitol) Derivatives. Molecules. 2022; 27(19):6292. https://doi.org/10.3390/molecules27196292
Chicago/Turabian StyleHansen-Felby, Magnus, Steen U. Pedersen, and Kim Daasbjerg. 2022. "Electrocatalytic Depolymerization of Self-Immolative Poly(Dithiothreitol) Derivatives" Molecules 27, no. 19: 6292. https://doi.org/10.3390/molecules27196292
APA StyleHansen-Felby, M., Pedersen, S. U., & Daasbjerg, K. (2022). Electrocatalytic Depolymerization of Self-Immolative Poly(Dithiothreitol) Derivatives. Molecules, 27(19), 6292. https://doi.org/10.3390/molecules27196292