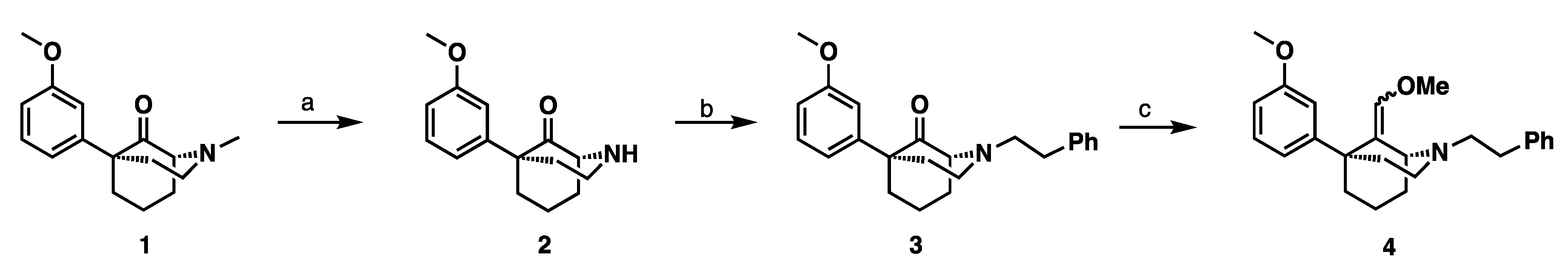
3.2. Syntheses
(1S,5S)-5-(3-Methoxyphenyl)-2-azabicyclo [3.3.1]nonan-9-one (
2): In an oven-dried flask,
1 (2.2 g, 8.3 mmol) and K
2CO
3 (2.3 g, 16.6 mmol) in 15 mL acetonitrile were treated with cyanogen bromide (3.3 mL, 16.6 mmol). The reaction mixture was stirred under a N
2 atmosphere at room temperature for 2 h then heated to reflux. After 2 h, the reaction mixture was extracted with CHCl
3, and the organic phase was washed with brine and concentrated. The residue was taken up in a mixture of 21 mL 3 N HCl and 2.2 mL methanol and stirred at reflux for 17 h. Upon completion, the reaction mixture was cooled and quenched with 7 N NH
4OH in methanol. The mixture was extracted with CHCl
3 and washed with water, brine, dried with Na
2SO
4 and concentrated. Purification by flash column chromatography on silica gel (0–20% CMA in CHCl
3) gave the red oil
2 (1.62 g, 80% yield). The
1H NMR of the product was identical to that of the known compound
2 [
16].
(1S,5S)-5-(3-Methoxyphenyl)-2-phenethyl-2-azabicyclo [3.3.1] nonan-9-one (
3): In an oven-dried flask,
2 (1.62 g, 6.6 mmol) and K
2CO
3 (1.82 g, 13.2 mmol) in 16 mL acetonitrile were treated with phenethyl bromide (1.34 mL, 9.9 mmol). The reaction mixture was stirred at reflux under a N
2 atmosphere. After 16 h, the mixture was cooled, concentrated, and extracted with CHCl
3. Purification by flash column chromatography on silica gel (0–75% EtOAc in Hexanes) gave a brown oil (1.78 g, 77% yield). The
1H NMR of the product
3 was identical to that of the known compound [
16].
(1S,5S)-9-(Methoxymethylene)-5-(3-methoxyphenyl)-2-phenethyl-2-azabicyclo [3.3.1] nonane (
4): An oven-dried flask charged with
3 (1.87 g, 5.1 mmol) and methoxy methylphosphonium chloride (5.25 g, 15.3 mmol) was evacuated and backfilled with N
2 three times. 11 mL of THF was added and the reaction mixture was cooled to 0 °C. LHMDS (13.3 mL, 13.3 mmol) was added dropwise over 15 min. The reaction mixture was stirred at 0 °C for 30 min then allowed to warm to room temperature and stirred for an additional 22 h. Upon completion, the reaction mixture was cooled to 0 °C, quenched with methanol, concentrated, and extracted with CHCl
3, washed with water and brine, and dried with Na
2SO
4. Purification by flash column chromatography on silica gel (0–55% EtOAc in Hexanes) gave an orange oil (1.26 g, 65% yield,
E:
Z 1:4). The
1H NMR spectra of the product were identical to the known compound
4 [
16].
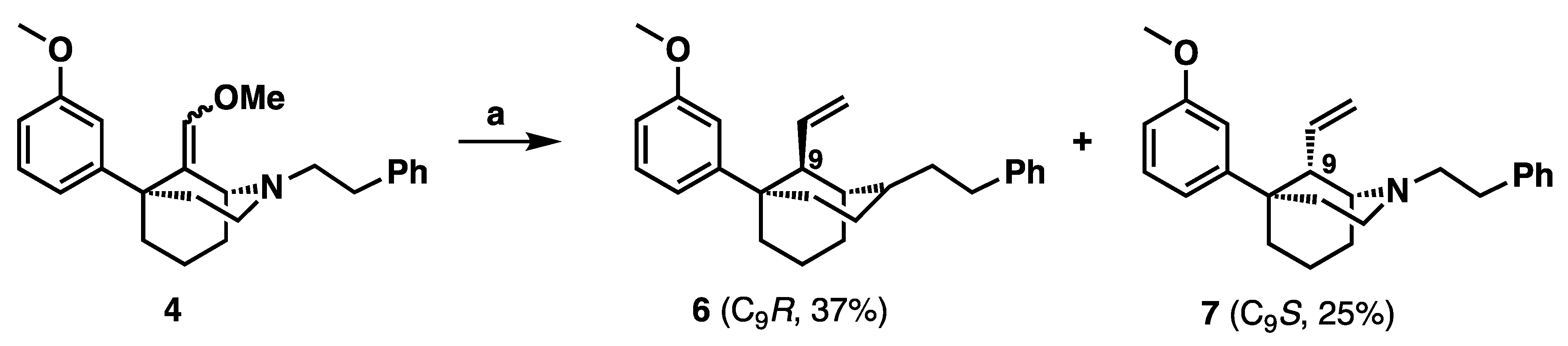
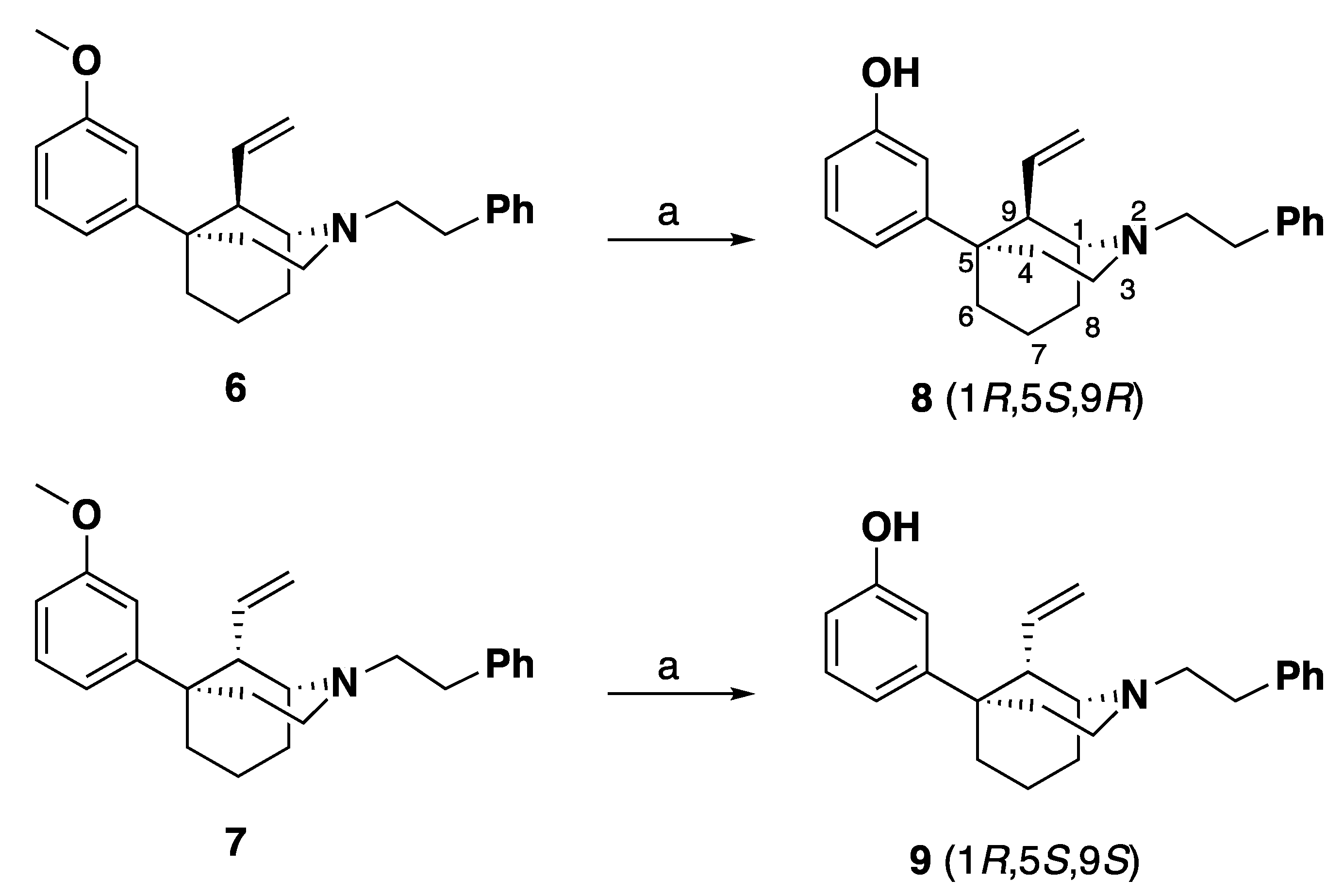
(1R,5S,9R)-5-(3-Methoxyphenyl)-2-phenethyl-9-vinyl-2-azabicyclo [3.3.1] nonane (6): In an oven-dried flask, methoxy methylene 4 (650 mg, 1.56 mmol) was suspended in THF (7 mL) and treated with HCl (6 M, 12 mL) and the reaction mixture was stirred overnight at room temperature under argon. The reaction mixture was quenched with 7 N NH4OH in MeOH, extracted with CHCl3 and washed with water and brine. The organic layer was then dried with sodium sulfate, concentrated, and put under high vacuum for 2 h. An oven-dried round-bottom flask flushed with argon was charged with methyltriphenylphosphonium bromide (1.67 g, 3 equiv, 4.69 mmol) and potassium 2-methylpropan-2-olate (526 mg, 3 equiv, 4.69 mmol) and suspended in THF (15 mL). The reaction mixture was heated to 45 °C for 1 h and turned yellow when the ylide was formed. The dried aldehyde 5 was suspended in THF (10 mL) and was transferred to the ylide mixture. The reaction mixture was stirred at 45 °C for 2 h when it was complete by TLC, whereupon the reaction mixture was quenched with NH4OH in MeOH and extracted with EtOAc. The mixture was washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (0–100% EtOAc in hexanes) yielded a green oil (233 mg, 37% yield). Crystallization as the oxalate salt from acetone afforded 6 as a white solid, mp 170–175 ˚C. 1H-NMR (400 MHz; CD3OD): δ 7.33 (s, 4H), 7.33–7.20 (m, 3H), 6.97–6.91 (m, 2H), 6.76 (dd, J = 8.1, 1.2 Hz, 1H), 5.76–5.67 (m, 1H), 5.14 (m, 2H), 3.80–3.74 (m, 4H), 3.68–3.61 (m, 2H), 3.56–3.44 (m, 2H), 3.38 (dd, J = 5.5, 0.6 Hz, 1H), 3.14–3.11 (m, 2H), 2.41–1.93 (m, 8H). 13C NMR (101 MHz; CD3OD): δ 164.9, 159.8, 148.2, 136.5, 134.4, 129.0, 128.5, 126.8, 118.6, 117.8, 112.0, 110.9, 60.7, 55.7, 54.2, 49.4, 46.5, 37.8, 36.7, 30.4, 28.0, 19.6, 16.8; HRMS-ESI (m/z): [M + H+] calcd. for C25H32NO 362.2484; found 362; [a]20D 18.2° (c 0.74, CHCl3).
(1R,5S,9S)-5-(3-Methoxyphenyl)-2-phenethyl-9-vinyl-2-azabicyclo [3.3.1] nonane (7): Methoxy methylene 4 was subjected to the same reaction conditions as with 6 to give 7, which was isolated as a yellow oil (160 mg, 26% yield). Crystallization as the oxalate salt from acetone afforded 7 as a white solid, mp 180–184 ˚C. 1H NMR (400 MHz; CD3OD): δ 7.35–7.20 (m, 6H), 6.89–6.84 (m, 2H), 6.76 (dd, J = 8.2, 2.2 Hz, 1H), 5.73–5.64 (m, 1H), 5.35–5.24 (m, 2H), 3.94 (d, J = 8.6 Hz, 1H), 3.78–3.74 (m, 3H), 3.74–3.67 (m, 1H), 3.59–3.50 (m, 2H), 3.41–3.31 (m, 2H), 3.17–3.09 (m, 1H), 2.96–2.88 (m, 1H), 2.52–2.37 (m, 3H), 2.18–1.81 (m, 5H); 13C NMR (101 MHz; CD3OD): δ 166.2, 161.3, 149.7, 137.4, 136.3, 130.5, 130.05, 129.88, 128.4, 120.9, 119.0, 113.4, 112.2, 60.8, 56.6, 55.7, 51.7, 47.4, 41.8, 38.1, 31.5, 29.3, 24.1, 22.0; HRMS-ESI (m/z): [M + H+] calcd. for C25H32NO 362.2484; found 362.2485; [a]20D –49.1° (c 0.2, CHCl3)
3-((1R,5S,9R)-2-Phenethyl-9-vinyl-2-azabicyclo [3.3.1]nonan-5-yl)phenol (8): In an oven-dried round-bottom flask, 6 (280 mg, 1 equiv, 360 µmol) was suspended in dichloromethane (6 mL) and the mixture was cooled to –78 °C. tribromoborane (147 µL, 3 equiv, 1.44 mmol) was added dropwise and the reaction was stirred at –78 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight (16 h). Upon completion, the reaction mixture was cooled to 0 °C and quenched with MeOH and stirred for 30 min. 1N HCl (2 mL) was added, and the reaction mixture was distilled at 100 °C for 1 h. The reaction mixture was then cooled to 0 °C and made basic (>10.5) with NH4OH and extracted with 9:1 CHCl3: MeOH. The combined organic layers were washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (20–100% EtOAc in hexanes) gave a yellow oil (248 mg, 92% yield). The HCl salt of 8 was formed in iPrOH (1 mL) with 37% HCl (0.1 mL) and recrystallized from hot ethanol (3 mL) to give a white solid, mp 233–237 ˚C. 1H NMR (400 MHz; CD3OD): δ 7.38–7.33 (m, 4H), 7.28 (dq, J = 8.8, 4.3 Hz, 1H), 7.14 (t, J = 8.0 Hz, 1H), 6.88–6.83 (m, 2H), 6.63 (dd, J = 8.0, 1.9 Hz, 1H), 5.73 (tt, J = 10.4, 7.0 Hz, 1H), 5.22–5.14 (m, 2H), 3.78 (d, J = 0.2 Hz, 1H), 3.72–3.60 (m, 2H), 3.51 (dd, J = 10.0, 6.8 Hz, 2H), 3.39 (dd, J = 5.5, 0.4 Hz, 1H), 3.16 (dd, J = 10.4, 6.6 Hz, 2H), 2.38–1.93 (m, 8H); 13C NMR (101 MHz; CD3OD): δ 158.6, 149.4, 137.7, 135.8, 130.4, 129.99, 129.89, 128.3, 120.0, 118.0, 114.3, 114.1, 62.0, 57.0, 51.1, 48.0, 39.4, 38.0, 31.8, 29.4, 21.1, 18.1; HRMS-ESI (m/z): [M + H+] calcd. for C24H30NO 348.2327; found 348.2328; Anal. Calcd. for C24H30ClNO: C, 75.08%; H, 7.88%; N, 3.65%. Found C, 75.12%; H 7.84%; N, 3.64%; [a]20D 22.5° (c 0.64, CHCl3).
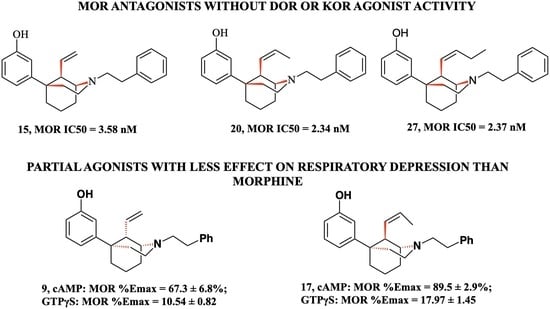
3-((1R,5S,9S)-2-Phenethyl-9-vinyl-2-azabicyclo [3.3.1]nonan-5-yl)phenol (9): In an oven-dried round-bottom flask, 7 (280 mg, 1 equiv, 360 µmol) was suspended in dichloromethane (6 mL) and the mixture was cooled to –78 °C. tribromoborane (147 µL, 3 equiv, 1.44 mmol) was added dropwise and the reaction was stirred at –78 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight (16 h). Upon completion, the reaction mixture was cooled to 0 °C and quenched with MeOH and stirred for 30 min. 1 N HCl (2 mL) was added, and the reaction mixture was distilled at 100 °C for 1 h. The reaction mixture was then cooled to 0 °C and made basic (pH > 10.5) with NH4OH and extracted with 9:1 CHCl3: MeOH. The combined organic layers were washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (20–00% EtOAc in hexanes) gave 9 as a yellow oil (248 mg, 92% yield). The HCl salt of 9 was formed in iPrOH (0.5 mL) with 37% HCl (0.05 mL) and recrystallized from hot ethanol (3 mL) to give a white solid (173 mg, 64% yield), mp 264–267 ˚C. 1H NMR (400 MHz; CD3OD): δ 7.38–7.26 (m, 5H), 7.13 (t, J = 8.0 Hz, 1H), 6.79 (d, J = 7.9 Hz, 1H), 6.75 (s, 1H), 6.63 (dd, J = 8.0, 1.7 Hz, 1H), 5.74 (ddd, J = 17.4, 10.7, 6.7 Hz, 1H), 5.33 (dd, J = 24.4, 14.0 Hz, 2H), 3.97 (d, J = 0.5 Hz, 1H), 3.77–3.69 (m, 1H), 3.57 (td, J = 11.9, 5.9 Hz, 2H), 3.38 (td, J = 12.1, 5.2 Hz, 1H), 3.31 (t, J = 1.5 Hz, 2H), 3.16 (td, J = 12.2, 5.6 Hz, 1H), 2.98–2.90 (m, 1H), 2.51–2.36 (m, 3H), 2.20–2.03 (m, 2H), 2.01–1.83 (m, 3H); 13C NMR (101 MHz; CD3OD): δ 158.6, 149.7, 137.4, 136.4, 130.5, 130.04, 129.9, 128.4, 120.9, 117.8, 114.2, 113.8, 60.5, 56.4, 51.9, 47.3, 41.8, 38.0, 31.5, 29.3, 24.0, 22.0. HRMS-ESI (m/z): [M + H+] calcd. for C24H30NO 348.2327; found 348.2328; Anal. Calcd. for C24H30ClNO·0.5 H2O: C, 73.36%; H, 7.95%; N, 3.56%. Found C, 73.11%; H, 7.68%; N, 3.55%; [a]20D –35.8° (c 0.45, CHCl3).
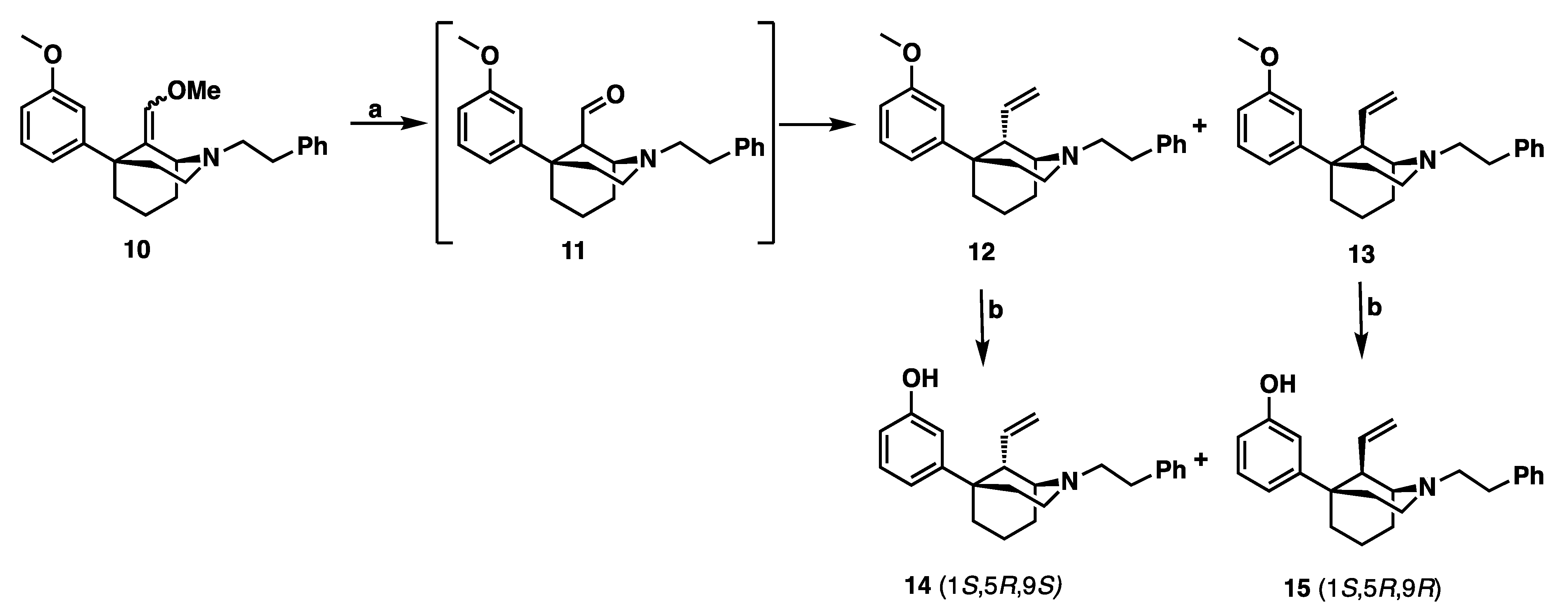
(1S,5R,9S)-5-(3-Methoxyphenyl)-2-phenethyl-9-vinyl-2-azabicyclo [3.3.1]nonane (
12). In an oven-dried flask, methoxy methylene
10 (590 mg, 1.56 mmol) [
12] was suspended in THF (6 mL) and treated with HCl (6 M, 10 mL) and the reaction mixture was stirred overnight at room temperature under argon. The reaction mixture was quenched with 7 N NH
4OH in MeOH, extracted with CHCl
3 and washed with water and brine. The organic layer was then dried with sodium sulfate, concentrated, and put under high vacuum for 2 h. An oven-dried round-bottom flask flushed with argon was charged with methyltriphenylphosphonium bromide (1.67 g, 3 equiv, 4.69 mmol) and potassium 2-methylpropan-2-olate (526 mg, 3 equiv, 4.69 mmol) and suspended in THF (13 mL). The reaction mixture was heated to 45 °C for 1 h and turned yellow when the ylide was formed. The dried aldehyde (
11) was suspended in THF (10 mL) and was transferred to the ylide mixture. The reaction mixture was stirred at 45 °C for 2 h when it was complete by TLC, whereupon the reaction mixture was quenched with NH
4OH in MeOH and extracted with EtOAc. The mixture was washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (0–100% EtOAc in hexanes) gave
12 as a green oil (183 mg, 32% yield). δ 7.32–7.17 (m, 6H), 6.99–6.94 (m, 2H), 6.72–6.69 (m, 1H), 5.76 (ddd,
J = 17.4, 10.4, 7.8 Hz, 1H), 5.09–4.98 (m, 2H), 3.81–3.76 (m, 3H), 3.08 (td,
J = 13.8, 4.3 Hz, 4H), 2.91–2.78 (m, 4H), 2.21–1.77 (m, 7H), 1.60 (ddt,
J = 17.1, 9.6, 4.4 Hz, 1H).
13C NMR (100 MHz; CDCl
3): δ 159.3, 151.4, 140.5, 138.3, 128.9, 128.7, 128.4, 126.0, 118.4, 116.9, 112.4, 110.4, 58.4, 57.9, 55.1, 49.8, 49.4, 41.22, 38.1, 34.6, 29.7, 22.1, 19.3.
(1S,5R,9R)-5-(3-Methoxyphenyl)-2-phenethyl-9-vinyl-2-azabicyclo [3.3.1]nonane (13): Methoxy methylene 10 (590 mg, 1.56 mmol) was suspended in THF (6 mL) and treated with HCl (6 M, 10 mL) and the reaction mixture was stirred overnight at room temperature under argon. The reaction mixture was quenched with 7 N NH4OH in MeOH, extracted with CHCl3 and washed with water and brine. The organic layer was then dried with sodium sulfate and concentrated and dried under reduced pressure for 2 h. An oven-dried round-bottom flask flushed with argon was charged with methyltriphenylphosphonium bromide (1.67 g, 3 equiv, 4.69 mmol) and potassium 2-methylpropan-2-olate (526 mg, 3 equiv, 4.69 mmol). THF (13 mL) was added, and the reaction mixture was heated to 45 °C for 1 h. The solution turned yellow when the ylide was formed and the aldehyde mixture 11 was added and stirred at 45 °C for 2 h. Upon completion by TLC, the reaction mixture was quenched with NH4OH in MeOH and extracted with EtOAc. The mixture was washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (0–100% EtOAc in hexanes) gave 13 as a blue oil (138 mg, 24% yield). 1H NMR (400 MHz; CDCl3): δ 7.36–7.32 (m, 2H), 7.27–7.17 (m, 4H), 6.94–6.89 (m, 2H), 6.71 (dd, J = 8.1, 2.0 Hz, 1H), 6.01 (dt, J = 17.4, 8.9 Hz, 1H), 4.94 (t, J = 15.2 Hz, 2H), 3.84–3.79 (m, 3H), 3.16 (dq, J = 18.1, 5.9 Hz, 3H), 2.86–2.71 (m, 5H), 2.46–2.31 (m, 2H), 2.10–1.67 (m, 5H), 1.60–1.49 (m, 1H).
3-((1S,5R,9S)-2-Phenethyl-9-vinyl-2-azabicyclo [3.3.1]nonan-5-yl)phenol (14): In an oven-dried round-bottom flask, 12 (750 mg, 1 equiv, 360 µmol) was suspended in dichloromethane (3 mL) and the mixture was cooled to –78 °C. tribromoborane (136 µL, 3 equiv, 1.44 mmol) was added dropwise and the reaction was stirred at –78 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight (16 h). Upon completion, the reaction mixture was cooled to 0 °C and quenched with MeOH and stirred for 30 min. 1 N HCl (2 mL) was added, and the reaction mixture was distilled at 100 °C for 1 h. The reaction mixture was then cooled to 0 °C and made basic (>10.5) with NH4OH and extracted with 9:1 CHCl3: MeOH. The combined organic layers were washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (20–100% EtOAc in hexanes) gave 14 as a yellow oil (558 mg, 77% yield). The HCl salt of 14 was formed in iPrOH (2 mL) with 37% HCl (0.2 mL) and recrystallized from hot ethanol (4 mL) and cooled, stirring 16 h to give a white solid, mp 231–235 °C. 1H-NMR (400 MHz; CD3OD): δ 7.36 (d, J = 13.0 Hz, 4H), 7.28 (dq, J = 8.5, 4.3 Hz, 1H), 7.13 (t, J = 8.0 Hz, 1H), 6.88–6.84 (m, 2H), 6.63 (dd, J = 8.0, 2.0 Hz, 1H), 5.77–5.68 (m, 1H), 5.22–5.13 (m, 2H), 3.78 (s, 1H), 3.71–3.58 (m, 2H), 3.51 (quintet, J = 8.9 Hz, 2H), 3.41 (d, J = 5.6 Hz, 1H), 3.17 (dd, J = 10.4, 6.5 Hz, 2H), 2.39–1.92 (m, 8H). 13C NMR (101 MHz; CD3OD): δ 158.6, 149.5, 137.8, 135.8, 130.4, 129.98, 129.91, 128.3, 120.0, 118.0, 114.3, 114.1, 62.0, 57.0, 51.0, 47.9, 39.3, 38.0, 31.8, 29.4, 21.1, 18.1. HRMS-ESI (m/z): [M + H+] calcd for C25H32NO 348.2327; found 348.2328; Anal. calcd. for C24H30ClNO: C, 75.08%; H, 7.88%; N, 3.65%. Found C, 75.00%; H, 7.86%; N, 3.64%; [a]20D −24.3° (c 0.64, CHCl3).
3-((1S,5R,9R)-2-Phenethyl-9-vinyl-2-azabicyclo [3.3.1]nonan-5-yl)phenol (15): In an oven-dried round-bottom flask, 13 (750 mg, 1 equiv, 360 µmol) was suspended in dichloromethane (3 mL) and the mixture was cooled to –78 °C. tribromoborane (136 µL, 3 equiv, 1.44 mmol) was added drop-wise and the reaction was stirred at –78 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight (16 h). Upon completion, the reaction mixture was cooled to 0 °C and quenched with MeOH and stirred for 30 min. 1 N HCl (2 mL) was added, and the reaction mixture was distilled at 100 °C for 1 h. The reaction mixture was then cooled to 0 °C and made basic (>10.5) with NH4OH and extracted with 9:1 CHCl3: MeOH. The combined organic layers were washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (20–100% EtOAc in hexanes) gave 15 as a yellow oil (554 mg, 76% yield). The HCl salt of 15 was formed in iPrOH with 37% HCl (0.1 mL) and recrystallized from ethanol to give a white solid, mp 264–266 °C. 1H-NMR (400 MHz; CD3OD): δ 7.38–7.26 (m, 5H), 7.13 (t, J = 8.0 Hz, 1H), 6.79 (d, J = 7.9 Hz, 1H), 6.75 (t, J = 1.9 Hz, 1H), 6.63 (dd, J = 8.0, 2.3 Hz, 1H), 5.74 (ddd, J = 17.4, 10.7, 6.7 Hz, 1H), 5.33 (dd, J = 25.2, 14.0 Hz, 2H), 3.97 (t, J = 0.5 Hz, 1H), 3.73 (td, J = 13.3, 5.9 Hz, 1H), 3.61–3.54 (m, 2H), 3.38 (td, J = 12.1, 5.2 Hz, 1H), 3.16 (td, J = 12.2, 5.6 Hz, 1H), 2.94 (td, J = 12.1, 5.1 Hz, 1H), 2.51–2.36 (m, 3H), 2.20–1.83 (m, 5H); 13C NMR (101 MHz; CD3OD): δ 158.6, 149.7, 137.4, 136.4, 130.5, 130.04, 129.9, 128.4, 120.9, 117.8, 114.2, 113.8, 60.5, 56.4, 51.9, 47.3, 41.8, 38.0, 31.5, 29.3, 24.0, 22.0; HRMS-ESI (m/z): [M + H+] calcd. for C24H30NO 348.2327; found 348.2328; Anal. calcd. for C24H30ClNO: C, 75.08%; H, 7.88%; N, 3.65%. Found C, 74.78%; H, 7.48%; N, 3.61%; [a]20D –35.6° (c 0.95, CHCl3).
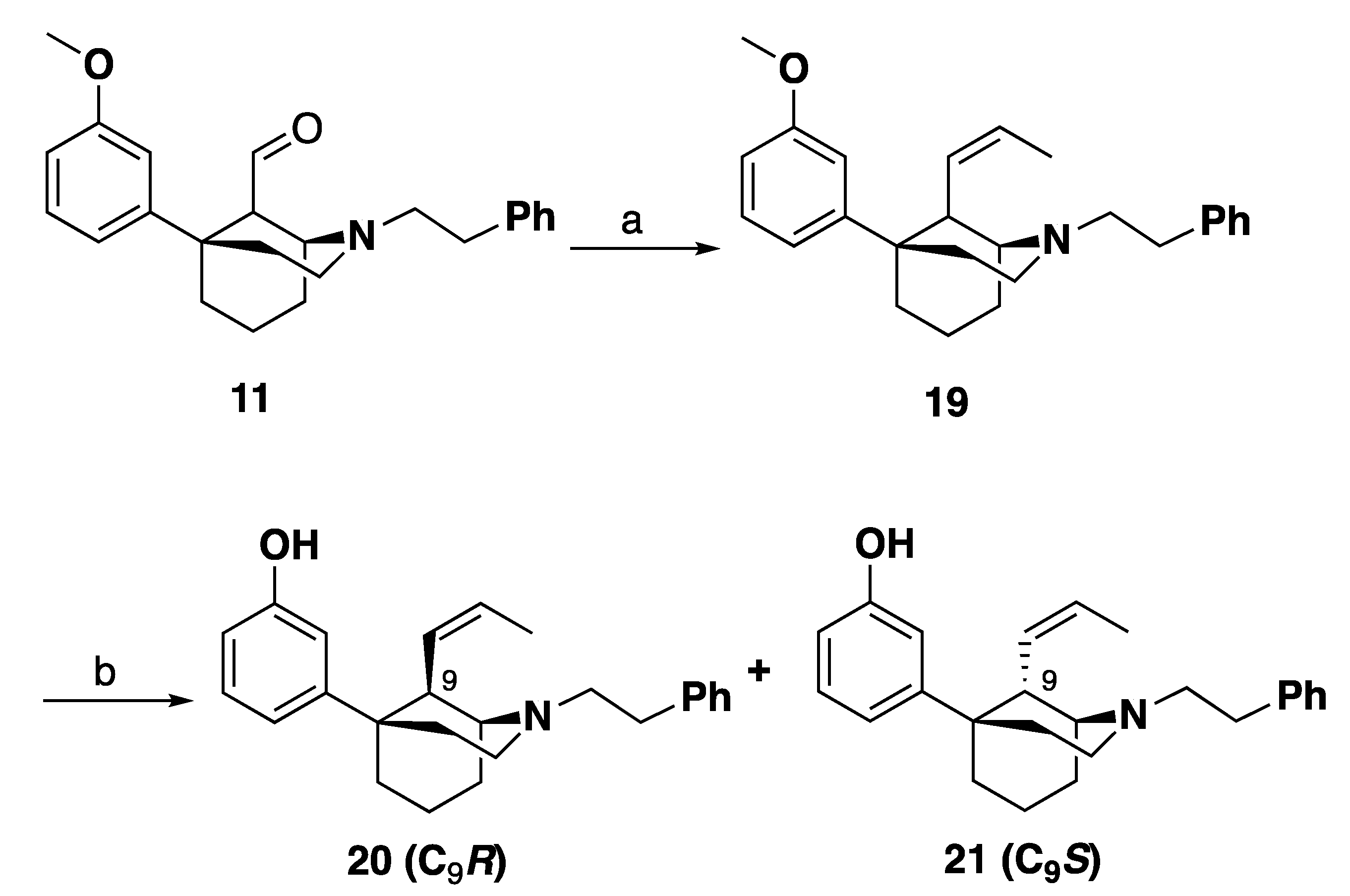
3-((1R,5S,9R)-2-Phenethyl-9-((Z)-prop-1-en-1-yl)-2-azabicyclo [3.3.1]nonan-5-yl)phenol (17): In an oven-dried flask, 4 (1 g, 3 mmol) was suspended in THF (10 mL) and treated with HCl (6 M, 10 mL) and the reaction mixture was stirred overnight at room temperature under nitrogen. The reaction mixture was quenched with 7 N NH4OH in MeOH, extracted with CHCl3 and washed with water and brine. The organic layer was then dried with sodium sulfate, concentrated, to yield the aldehyde intermediate 5. The aldehyde was treated with ethyltriphenylphosphonium iodide (3 g, 3 equiv, 8 mmol) and suspended in THF (10 mL). The reaction mixture was cooled to 0 °C and treated slowly with LiHMDS (1.0 M in THF) (7 mL, 2.6 equiv, 7 mmol). After 30 min, the reaction mixture was warmed to room temperature then the mixture was heated to 45 °C for 16 h. Upon completion by TLC, the reaction mixture was quenched with MeOH and extracted with CHCl3. The mixture was washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (0–50% EtOAc in hexanes) yielded a mixture of C9 epimers 16 which was used without further purification.
In an oven-dried round-bottom flask, 16 (500 mg, 1 equiv, 1.3 mmol) was suspended in dichloromethane (15 mL) and the mixture was cooled to –78 °C. Tribromoborane (667 mg, 253 µL, 2 equiv, 2.66 mmol) was added to drop-wise and the reaction was stirred at –78 °C for 15 min. The reaction mixture was allowed to warm to room temperature and stirred 2 h. Upon completion, the reaction mixture was cooled to 0 °C and quenched with 7 mL MeOH drop wise and stirred for 30 min. subsequently, 10 mL 1 N HCl was added, and the reaction mixture was distilled at 100 °C for 1 h. The reaction mixture was then cooled to 0 °C and made basic (>10.5) with NH4OH and extracted with 9:1 CHCl3: MeOH. The combined organic layers were washed with water and brine, dried with sodium sulfate and concentrated. Purification by silica gel column chromatography 0–60% EtOAc: Hexanes. 17 was isolated as a white foam (153 mg, 32% yield) as the more polar fraction. The HCl salt of 17 was formed in iPrOH with 37% HCl (0.1 mL) and recrystallized from ethanol to give a white solid: mp 271–275 °C; 1H NMR (400 MHz; CD3OD): δ 7.38–7.33 (m, 4H), 7.31–7.26 (m, 1H), 7.11 (t, J = 8.0 Hz, 1H), 6.84–6.79 (m, 2H), 6.61 (dd, J = 8.0, 2.2 Hz, 1H), 5.61–5.53 (m, 1H), 5.39 (ddd, J = 10.6, 8.8, 1.7 Hz, 1H), 3.68 (dt, J = 21.3, 7.7 Hz, 4H), 3.52–3.48 (m, 2H), 3.15 (dt, J = 10.3, 6.0 Hz, 2H), 2.34–2.19 (m, 3H), 2.19–1.91 (m, 5H), 1.76 (dd, J = 6.9, 1.6 Hz, 3H). 13C NMR (100 MHz; CD3OD): δ 158.5, 149.8, 137.7, 130.3, 130.00, 129.91, 129.5, 128.3, 127.4, 117.7, 114.2, 113.8, 61.1, 56.8, 51.1, 41.9, 39.9, 37.8, 31.8, 29.2, 21.0, 18.7, 13.8; HRMS-ESI (m/z): [M + H+] calcd. for C25H32NO 362.2484; found 362.2485; Anal. calcd. for C25H32ClNO·0.1 H2O: C, 75.11%; H, 8.12%; N, 3.5%. Found C, 75.00%; H, 7.87%; N, 3.42%; [a]20D –14.1° (c 0.82, CHCl3).
3-((1R,5S,9S)-2-Phenethyl-9-((Z)-prop-1-en-1-yl)-2-azabicyclo [3.3.1]nonan-5-yl)phenol (18): From the same procedure as in 17, 18 was isolated as a white foam (133 mg, 55% yield) as the less polar fraction. The HCl salt of 18 was formed in iPrOH with 37% HCl (0.1 mL) and recrystallized from ethanol to give a white solid: mp 260–264 °C. 1H NMR (400 MHz; CD3OD): δ 7.35–7.24 (m, 5H), 7.08 (t, J = 8.0 Hz, 1H), 6.73 (d, J = 8.0 Hz, 1H), 6.68 (d, J = 1.8 Hz, 1H), 6.59 (dd, J = 8.0, 1.8 Hz, 1H), 5.70–5.63 (m, 1H), 5.34–5.29 (m, 1H), 3.73–3.67 (m, 2H), 3.59–3.46 (m, 3H), 3.39–3.32 (m, 1H), 3.15–3.07 (m, 1H), 2.93–2.86 (m, 1H), 2.55–2.46 (m, 1H), 2.40–2.35 (m, 2H), 2.17–2.03 (m, 2H), 1.99–1.82 (m, 3H), 1.76 (dd, J = 7.0, 1.3 Hz, 3H). 13C NMR (100 MHz; CD3OD): δ 158.5, 150.0, 137.4, 131.0, 130.3, 130.03, 129.88, 128.4, 127.7, 117.5, 114.1, 113.6, 60.4, 56.5, 51.8, 42.35, 42.19, 37.8, 31.5, 29.1, 23.9, 22.1, 13.7; HRMS-ESI (m/z): [M + H+] calcd. for C25H32NO 362.2484; found 362.2486; C25H32ClNO·1.45 H2O calc. C: 70.8; H: 8.29; N: 3.3; found C: 70.5; H: 7.92; N: 3.3; [a]20D –22.4° (c 1.0, CHCl3).
3-((1S,5R,9R)-2-Phenethyl-9-((Z)-prop-1-en-1-yl)-2-azabicyclo [3.3.1]nonan-5-yl)phenol (20): In an oven-dried flask, 10 (1 g, 3 mmol) was suspended in THF (10 mL) and treated with HCl (6 M, 10 mL) and the reaction mixture was stirred overnight at room temperature under nitrogen. The reaction mixture was quenched with 7 N NH4OH in MeOH, extracted with CHCl3 and washed with water and brine. The organic layer was then dried with sodium sulfate, concentrated, to yield the aldehyde intermediate. The aldehyde was treated with ethyltriphenylphosphonium iodide (3 g, 3 equiv, 8 mmol) and suspended in THF (10 mL). The reaction mixture was cooled to 0 °C and treated slowly with LiHMDS (1.0 M in THF) (7 mL, 2.6 equiv, 7 mmol). After 30 min, the reaction mixture was warmed to room temperature then the mixture was heated to 45 °C for 16 h. Upon completion by TLC, the reaction mixture was quenched with MeOH and extracted with CHCl3. The mixture was washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (0–50% EtOAc in hexanes) yielded a mixture of C9 epimers 19 which was used without further purification.
In an oven-dried round-bottom flask, 19 (700 mg, 1 equiv, 2 mmol) was suspended in dichloromethane (5 mL) and the mixture was cooled to –78 °C. Tribromoborane (900 mg, 400 µL, 2 equiv, 4 mmol) was added to drop-wise and the reaction was stirred at –78 °C for 15 min. The reaction mixture was allowed to warm to room temperature and stirred 2 h. Upon completion, the reaction mixture was cooled to 0 °C and quenched with 7 mL MeOH drop wise and stirred for 30 min. subsequently, 10 mL 1 N HCl was added, and the reaction mixture was distilled at 100 °C for 1 h. The reaction mixture was then cooled to 0 °C and made basic (>10.5) with NH4OH and extracted with 9:1 CHCl3: MeOH. The combined organic layers were washed with water and brine, dried with sodium sulfate and concentrated. Purification by silica gel column chromatography 0–60% EtOAc: Hexanes. 20 was isolated as a white foam (175 mg, 30% yield) as the less polar fraction. The HCl salt of 20 was formed in iPrOH with 37% HCl (0.1 mL) and recrystallized from ethanol to give a white solid: mp 269–274; 1H NMR (400 MHz; CD3OD): δ 7.35–7.24 (m, 5H), 7.10–7.06 (m, 1H), 6.77–6.68 (m, 2H), 6.61–6.57 (m, 1H), 5.71–5.63 (m, 1H), 5.36–5.30 (m, 1H), 3.73–3.66 (m, 2H), 3.59–3.47 (m, 3H), 3.36 (td, J = 12.0, 5.3 Hz, 1H), 3.11 (td, J = 12.0, 5.5 Hz, 1H), 2.94–2.87 (m, 1H), 2.55–2.47 (m, 1H), 2.44–2.35 (m, 2H), 2.18–2.02 (m, 2H), 1.99–1.80 (m, 3H), 1.81–1.74 (m, 3H). 13C NMR (101 MHz; CD3OD): δ 158.5, 150.0, 137.4, 131.0, 130.3, 130.03, 129.9, 128.4, 127.7, 117.5, 114.1, 113.6, 60.4, 56.5, 51.8, 42.4, 42.2, 37.8, 31.5, 29.1, 23.9, 22.1, 13.7. HRMS-ESI (m/z): [M + H+] calcd. for C25H32NO 362.2484; found 362.2487. Anal. calcd. for C25H32ClNO·0.15 H2O: C, 74.94%; H, 8.13%; N, 3.5%. Found C, 74.8%; H, 7.85%; N, 3.36%; [a]20D 22.3° (c 1.0, CHCl3).
3-((1S,5R,9S)-2-Phenethyl-9-((Z)-prop-1-en-1-yl)-2-azabicyclo [3.3.1]nonan-5-yl)phenol (21): From the same reaction as 20. Alkene 21 isolated as a light green foam (157 mg, 20% yield) as the more polar fraction. The HCl salt of 21 was formed in iPrOH with 37% HCl (0.1 mL) and recrystallized from ethanol to give a white solid: mp 277–281; 1H-NMR (400 MHz; CD3OD): δ 7.38–7.33 (m, 4H), 7.29 (tt, J = 9.1, 4.7 Hz, 1H), 7.12–7.09 (m, 1H), 6.84–6.80 (m, 1H), 6.61 (dd, J = 8.0, 2.1 Hz, 1H), 5.61–5.53 (m, 1H), 5.41–5.36 (m, 1H), 3.72–3.63 (m, 4H), 3.50 (dd, J = 10.4, 6.5 Hz, 2H), 3.17–3.13 (m, 2H), 2.34–2.21 (m, 3H), 2.19–1.91 (m, 5H), 1.76 (dd, J = 6.9, 1.1 Hz, 3H); 13C NMR (101 MHz; CD3OD): δ 158.5, 149.8, 137.7, 130.3, 130.00, 129.91, 129.5, 128.3, 127.4, 117.8, 114.2, 113.8, 61.1, 56.8, 51.1, 41.9, 39.8, 37.8, 31.8, 29.2, 21.1, 18.7, 13.8; HRMS-ESI (m/z): [M + H+] calcd. for C25H32NO 362.2484; found 362.2484; Anal. calcd. for C25H32ClNO: C, 75.45%; H, 8.7%; N, 3.52%. Found C, 75.27%; H, 7.73%; N, 3.44%; [a]20D 15.0° (c 0.86, CHCl3).
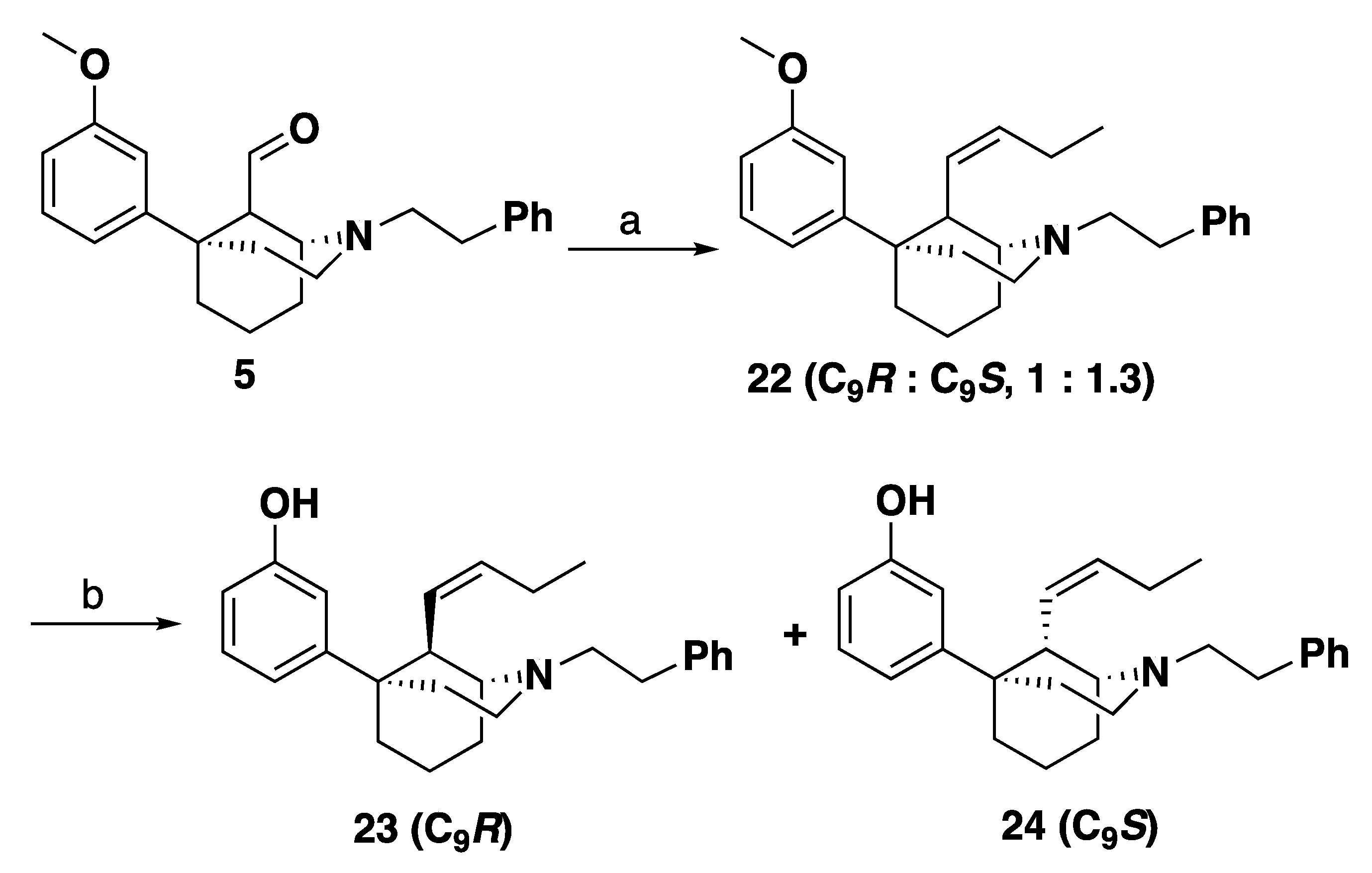
3-((1R,5S,9R)-9-((Z)-But-1-en-1-yl)-2-phenethyl-2-azabicyclo [3.3.1]nonan-5-yl)phenol (23): In an oven-dried flask, 4 (1.2 g, 3.2 mmol) was suspended in THF (12 mL) and treated with HCl (6 M, 12 mL) and the reaction mixture was stirred overnight at room temperature under nitrogen. The reaction mixture was quenched with 7 N NH4OH in MeOH, extracted with CHCl3 and washed with water and brine. The organic layer was then dried with sodium sulfate, concentrated, to yield an aldehyde intermediate. The aldehyde intermediate was treated with propyltriphenylphosphonium bromide (3.7 g, 3 equiv, 9.5 mmol) and suspended in THF (12 mL). The reaction mixture was cooled to 0 °C and treated slowly with LiHMDS (1.0 M in THF) (8.3 mL, 2.6 equiv, 8.3 mmol). After 30 min, the reaction mixture was warmed to room temperature then the mixture was heated to 45 °C for 16 h. Upon completion by TLC, the reaction mixture was quenched with MeOH and extracted with CHCl3. The mixture was washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (0–50% EtOAc in hexanes) yielded a mixture of C9 epimers 22 which were used in the next reaction without further purification or characterization.
In an oven-dried round-bottom flask, 22 (800 mg, 1 equiv, 2.05 mmol) was suspended in dichloromethane (20 mL) and the mixture was cooled to –78 °C. Tribromoborane (1.03 g, 390 µL, 2 equiv, 4.11 mmol) was added to drop-wise and the reaction was stirred at –78 °C for 15 min. The reaction mixture was allowed to warm to room temperature and stirred 2 h. Upon completion, the reaction mixture was cooled to 0 °C and quenched with 10 mL MeOH drop wise and stirred for 30 min. subsequently, 15 mL 1 N HCl was added, and the reaction mixture was distilled at 100 °C for 1 h. The reaction mixture was then cooled to 0 °C and made basic (>10.5) with NH4OH and extracted with 9:1 CHCl3: MeOH. The combined organic layers were washed with water and brine, dried with sodium sulfate and concentrated. Purification by silica gel column chromatography 0–60% EtOAc: Hexanes. 23 was isolated as a white foam (305 mg, 40% yield) as the more polar fraction. The HCl salt of 23 was formed in iPrOH (1.5 mL) with 37% HCl (0.15 mL) and recrystallized from hot ethanol (5 mL) to give a white solid: mp 265–268 °C. 1H-NMR (400 MHz; CD3OD): δ 7.33 (d, J = 4.3 Hz, 4H), 7.29–7.23 (m, 1H), 7.08 (t, J = 7.9 Hz, 1H), 6.82–6.78 (m, 2H), 6.59 (dd, J = 7.9, 1.7 Hz, 1H), 5.45–5.38 (m, 1H), 5.32–5.27 (m, 1H), 3.65–3.56 (m, 4H), 3.49–3.45 (m, 2H), 3.12 (dd, J = 10.6, 5.9 Hz, 2H), 2.35–1.90 (m, 11H), 0.95 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz; CD3OD): δ 158.5, 149.8, 137.7, 137.1, 130.2, 129.99, 129.91, 128.3, 125.6, 117.8, 114.2, 113.9, 61.6, 56.8, 51.1, 42.3, 39.7, 37.8, 31.8, 29.1, 22.2, 21.1, 18.6, 14.3. HRMS-ESI (m/z): [M + H+] calcd. for C26H34NO 376.2640; found 376.2642; Anal. calcd. for C26H34ClNO·0.05 C2H6O: calc. C: 75.66%; H: 8.34%; N: 3.38%; found C: 75.65%; H: 8.31%; N: 3.34%; [a]20D° −4.1 (c 0.96, CHCl3).
3-((1R,5S,9S)-9-((Z)-But-1-en-1-yl)-2-phenethyl-2-azabicyclo [3.3.1]nonan-5-yl)phenol (24): From the same reaction as 23, alkene 24 was isolated as a white foam (250 mg, 32% yield) as the less polar fraction. The HCl salt of 24 was formed in iPrOH (1.5 mL) with 37% HCl (0.15 mL) and recrystallized from hot ethanol (5 mL) to give a white solid: mp 262–265 °C. 1H NMR (400 MHz; CDCl3): δ 7.38–7.25 (m, 5H), 7.14–7.07 (m, 1H), 6.79–6.74 (m, 1H), 6.73–6.69 (m, 1H), 6.64–6.60 (m, 1H), 5.60–5.54 (m, 1H), 5.36–5.27 (m, 1H), 3.77–3.67 (m, 2H), 3.62–3.52 (m, 2H), 3.52–3.45 (m, 1H), 3.43–3.33 (m, 1H), 3.17–3.09 (m, 1H), 2.96–2.87 (m, 1H), 2.59–2.49 (m, 1H), 2.46–2.34 (m, 2H), 2.30–1.82 (m, 8H), 1.09–1.00 (m, 3H). 13C NMR (101 MHz; CD3OD): δ 158.5, 150.0, 138.5, 137.4, 130.3, 130.0, 129.9, 128.4, 125.9, 117.6, 114.1, 113.7, 60.9, 56.5, 51.8, 42.7, 42.1, 37.8, 31.5, 29.0, 23.9, 22.2, 22.1, 14.2. HRMS-ESI (m/z): [M + H+] calcd. for C26H34NO 376.2640; found 376.2641; Anal. calcd. for C26H34ClNO·0.1 H2O·0.1 C2H6O: C, 75.21%; H, 8.38%; N, 3.35%. Found C, 75.23%; H, 8.4%; N, 3.38%; [a]20D° −14.2 (c 0.8, CHCl3).
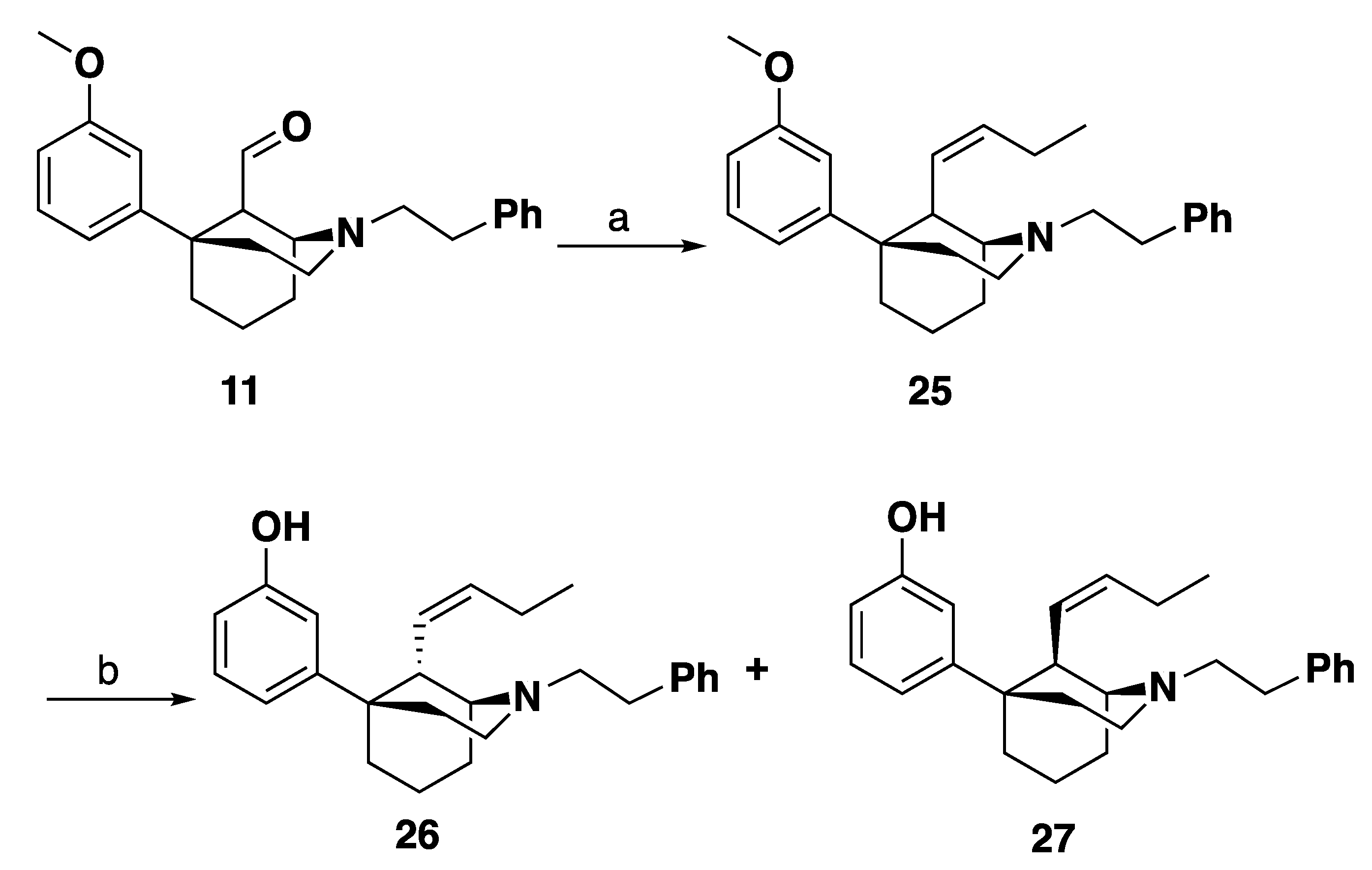
3-((1S,5R,9S)-9-((Z)-But-1-en-1-yl)-2-phenethyl-2-azabicyclo [3.3.1]nonan-5-yl)phenol (26): In an oven-dried flask, 10 (2 g, 5.3 mmol) was suspended in THF (20 mL) and treated with HCl (6 M, 20 mL) and the reaction mixture was stirred overnight at room temperature under nitrogen. The reaction mixture was quenched with 7 N NH4OH in MeOH, extracted with CHCl3 and washed with water and brine. The organic layer was then dried with sodium sulfate, concentrated, to yield the aldehyde intermediate. The aldehyde was treated with propyltriphenylphosphonium bromide (6.1 g, 3 equiv, 16 mmol) and suspended in THF (20 mL). The reaction mixture was cooled to 0 °C and treated slowly with LiHMDS (1.0 M in THF) (14 mL, 2.6 equiv, 14 mmol). After 30 min, the reaction mixture was warmed to room temperature then the mixture was heated to 45 °C for 16 h. Upon completion by TLC, the reaction mixture was quenched with MeOH and extracted with CHCl3. The mixture was washed with water and brine, dried with sodium sulfate and concentrated. Purification by flash column chromatography on silica gel (0–50% EtOAc in hexanes) yielded a mixture of C9 epimers 25 which was used without further purification.
In an oven-dried round-bottom flask, 25 (1.09 g, 1 equiv, 2.8 mmol) was suspended in dichloromethane (15 mL) and the mixture was cooled to –78 °C. Tribromoborane (1.4 g, 531 µL, 2 equiv, 5.6 mmol) was added to drop-wise and the reaction was stirred at –78 °C for 15 min. The reaction mixture was allowed to warm to room temperature and stirred 2 h. Upon completion, the reaction mixture was cooled to 0 °C and quenched with 7 mL MeOH drop wise and stirred for 30 min. subsequently, 10 mL 1 N HCl was added, and the reaction mixture was distilled at 100 °C for 1 h. The reaction mixture was then cooled to 0 °C and made basic (>10.5) with NH4OH and extracted with 9:1 CHCl3: MeOH. The combined organic layers were washed with water and brine, dried with sodium sulfate and concentrated. Purification by silica gel column chromatography 0–60% EtOAc: Hexanes. 26 was isolated as a white foam (347 mg, 33% yield) as the more polar fraction. The HCl salt of 26 was formed in iPrOH (1 mL) with 37% HCl (0.1 mL) and recrystallized from hot ethanol (4 mL) to give a white solid: mp 259–262 °C. 1H-NMR (400 MHz; CD3OD): δ 7.35 (t, J = 6.5 Hz, 4H), 7.28 (dq, J = 8.6, 4.3 Hz, 1H), 7.10 (t, J = 7.9 Hz, 1H), 6.84–6.81 (m, 2H), 6.61 (dd, J = 8.0, 2.3 Hz, 1H), 5.43 (dt, J = 10.9, 7.2 Hz, 1H), 5.32 (dd, J = 10.7, 9.3 Hz, 1H), 3.68–3.62 (m, 3H), 3.59 (d, J = 13.4 Hz, 1H), 3.49 (t, J = 8.4 Hz, 2H), 3.15 (dd, J = 10.3, 5.8 Hz, 2H), 2.38–1.93 (m, 10H), 0.97 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz; CD3OD): δ 158.5, 149.8, 137.7, 137.1, 130.2, 129.9, 129.9, 128.3, 125.6, 117.8, 114.2, 113.9, 61.6, 56.8, 51.1, 42.3, 39.7, 37.8, 31.7, 29.1, 22.2, 21.1, 18.6, 14.3. HRMS-ESI (m/z): [M + H+] calcd. for C26H34NO 376.2640; found 376.2642; Anal. calcd. for C26H34ClNO: C, 75.79%; H, 8.32%, N, 3.4%. Found C26H34ClNO: C, 75.89%; H, 8.18%; N, 3.47%; [a]20D° 4.1 (c 1.08, CHCl3).
3-((1S,5R,9R)-9-((Z)-But-1-en-1-yl)-2-phenethyl-2-azabicyclo [3.3.1]nonan-5-yl)phenol (27). From the same reaction as 26, 27 was isolated as a white foam (294 mg, 28% yield) as the less polar fraction. The HCl salt of 27 was formed in iPrOH (1 mL) with 37% HCl (0.1 mL) and recrystallized from hot ethanol (4 mL) to give a white solid: mp 264–267 °C. 1H-NMR (400 MHz; CD3OD): δ 7.37–7.26 (m, 5H), 7.10 (t, J = 8.0 Hz, 1H), 6.75 (d, J = 7.9 Hz, 1H), 6.71 (s, 1H), 6.61 (dd, J = 8.0, 2.1 Hz, 1H), 5.59–5.53 (m, 1H), 5.30 (dd, J = 10.7, 9.4 Hz, 1H), 3.76–3.68 (m, 2H), 3.56 (dt, J = 13.6, 7.0 Hz, 2H), 3.49–3.47 (m, 1H), 3.38 (td, J = 12.1, 5.2 Hz, 1H), 3.13 (td, J = 12.1, 5.5 Hz, 1H), 2.92 (ddd, J = 12.3, 11.6, 5.3 Hz, 1H), 2.60–2.51 (m, 1H), 2.42–2.35 (m, 2H), 2.28–1.83 (m, 7H), 1.04 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz; CD3OD): δ 158.5, 150.0, 138.5, 137.4, 130.3, 130.0, 129.9, 128.4, 125.9, 117.6, 114.1, 113.7, 60.9, 56.5, 51.8, 42.7, 42.1, 37.8, 31.5, 29.0, 23.9, 22.16, 22.08, 14.2. HRMS-ESI (m/z): [M + H+] calcd. for C26H34NO 376.2640; found 376.2644; Anal. calcd. for C26H34ClNO·0.1 H2O: C, 75.46%; H, 8.33%; N, 3.38%; Found C, 75.47%; H, 8.2%; N, 3.28%; [a]20D° 12.9 (c 0.79, CHCl3).



 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

















