

Involvement of Arsenic Atom of AsF3 in Five Pnicogen Bonds: Differences between X-ray Structure and Theoretical Models




Abstract
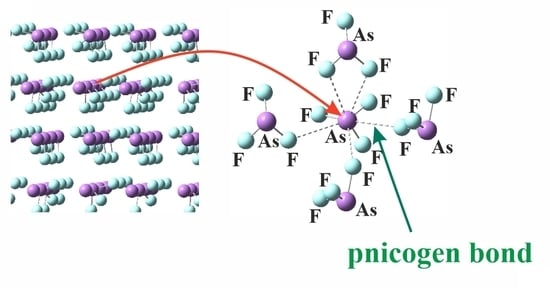
:
1. Introduction
2. Computational Details
3. Results
3.1. Nonamers
3.2. Nature of Bonding
3.3. Building of Clusters
3.4. Maximum Number of Pnicogen Bonds
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Scheiner, S. Characterization of Type I and II Interactions between Halogen Atoms. Cryst. Growth Des. 2022, 22, 2692–2702. [Google Scholar] [CrossRef]
- Scheiner, S. Search for an exothermic halogen bond between anions. Phys. Chem. Chem. Phys. 2022, 24, 6964–6972. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. Biological halogen bonds in protein–ligand complexes: A combined QTAIM and NCIPlot study in four representative cases. Org. Biomol. Chem. 2021, 19, 6858–6864. [Google Scholar] [CrossRef]
- de Azevedo Santos, L.; van der Lubbe, S.C.C.; Hamlin, T.A.; Ramalho, T.C.; Matthias Bickelhaupt, F. A Quantitative Molecular Orbital Perspective of the Chalcogen Bond. ChemistryOpen 2021, 10, 387. [Google Scholar] [CrossRef]
- de Azevedo Santos, L.; Ramalho, T.C.; Hamlin, T.A.; Bickelhaupt, F.M. Chalcogen bonds: Hierarchical ab initio benchmark and density functional theory performance study. J. Comput. Chem. 2021, 42, 688–698. [Google Scholar] [CrossRef]
- Scheiner, S. Origins and properties of the tetrel bond. Phys. Chem. Chem. Phys. 2021, 23, 5702–5717. [Google Scholar] [CrossRef]
- Grabowski, S.J. pi-Hole Tetrel Bonds-Lewis Acid Properties of Metallylenes. Crystals 2022, 12, 112. [Google Scholar] [CrossRef]
- Liu, N.; Li, Q.; Scheiner, S.; Xie, X. Resonance-assisted intramolecular triel bonds. Phys. Chem. Chem. Phys. 2022, 24, 15015–15024. [Google Scholar] [CrossRef]
- Wang, X.; Li, B.; Li, Y.; Wang, H.; Ni, Y.; Wang, H. The influence of monomer deformation on triel and tetrel bonds between TrR3/TR4 (Tr = Al, Ga, In; T = Si, Ge, Sn) and N-base (N-base = HCN, NH3, CN−). Comput Theor Chem 2021, 1201, 113268. [Google Scholar] [CrossRef]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional sigma/-Hole Interactions. Chemphyschem 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The Phosphorus Bond, or the Phosphorus-Centered Pnictogen Bond: The Covalently Bound Phosphorus Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Molecules 2022, 27, 1487. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. On the Importance of σ–Hole Interactions in Crystal Structures. Crystals 2021, 11, 1205. [Google Scholar] [CrossRef]
- Saha, A.; Veluthaparambath, R.V.P.; Saha, B.K. Directionality of P⋯O pnicogen bonding in light of geometry corrected statistical analysis. New J. Chem. 2020, 44, 9607–9610. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. Fluorinated elements of Group 15 as pnictogen bond donor sites. J. Fluor. Chem. 2017, 203, 62–74. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E. Systematic Coupled Cluster Study of Noncovalent Interactions Involving Halogens, Chalcogens, and Pnicogens. J. Phys. Chem. A 2017, 121, 9544–9556. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Halogen bonding versus chalcogen and pnicogen bonding: A combined Cambridge structural database and theoretical study. CrystEngComm 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Chandra, S.; Mahapatra, N.; Ramanathan, N.; Sundararajan, K. Unique O=N…O Pnicogen Interactions in Nitromethane Dimers: Evidence Using Matrix Isolation Infrared Spectroscopy and Computational Methodology. J. Phys. Chem. A 2022, 126, 3511–3520. [Google Scholar] [CrossRef]
- Sruthi, P.K.; Chandra, S.; Ramanathan, N.; Sundararajan, K. Unusual blue to red shifting of C–H stretching frequency of CHCl3 in co-operatively P⋯Cl phosphorus bonded POCl3–CHCl3 heterodimers at low temperature inert matrixes. J. Chem. Phys. 2020, 153, 174305. [Google Scholar] [CrossRef]
- Chandra, S.; Suryaprasad, B.; Ramanathan, N.; Sundararajan, K. Dominance of unique P⋯π phosphorus bonding with π donors: Evidence using matrix isolation infrared spectroscopy and computational methodology. Phys. Chem. Chem. Phys. 2020, 22, 20771–20791. [Google Scholar] [CrossRef]
- Sruthi, P.K.; Sarkar, S.; Ramanathan, N.; Sundararajan, K. Elusive hypervalent phosphorus⋯π interactions: Evidence for paradigm transformation from hydrogen to phosphorus bonding at low temperatures. Phys. Chem. Chem. Phys. 2019, 21, 12250–12264. [Google Scholar] [CrossRef]
- Leroy, C.; Johannson, R.; Bryce, D.L. 121/123Sb Nuclear Quadrupole Resonance Spectroscopy: Characterization of Non-Covalent Pnictogen Bonds and NQR Crystallography. J. Phys. Chem. A 2019, 123, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Mokrai, R.; Barrett, J.; Apperley, D.C.; Batsanov, A.S.; Benkő, Z.; Heift, D. Weak Pnictogen Bond with Bismuth: Experimental Evidence Based on Bi−P Through-Space Coupling. Chem. Eur. J. 2019, 25, 4017–4024. [Google Scholar] [CrossRef] [Green Version]
- Moaven, S.; Villanueva, O.H.; Unruh, D.K.; Cozzolino, A.F. The complicating role of pnictogen bond formation in the solution-phase and solid-state structures of the heavier pnictogen atranes. Dalton Trans. 2022, 51, 11335. [Google Scholar] [CrossRef]
- Gonzalez, V.M.; Park, G.; Yang, M.; Gabbaï, F.P. Fluoride anion complexation and transport using a stibonium cation stabilized by an intramolecular P=O → Sb pnictogen bond. Dalton Trans. 2021, 50, 17897–17900. [Google Scholar] [CrossRef]
- Schiavo, E.; Bhattacharyya, K.; Mehring, M.; Auer, A.A. Are Heavy Pnictogen-π Interactions Really “π Interactions”? Chem. Eur. J. 2021, 27, 14520–14526. [Google Scholar] [CrossRef]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with Pnictogen, Chalcogen, and Halogen Bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef] [Green Version]
- Yaghoobi, F.; Sohrabi−Mahboub, M. Theoretical Study on the Aza–Diels–Alder Reaction Catalyzed by PHCl2 Lewis Acid via Pnicogen Bonding. J. Phys. Chem. A 2018, 122, 2781–2791. [Google Scholar] [CrossRef]
- Ullah, Z.; Kim, K.; Venkanna, A.; Kim, H.s.; Kim, M.I.; Kim, M.-h. Plausible Pnicogen Bonding of epi-Cinchonidine as a Chiral Scaffold in Catalysis. Front. Chem. 2021, 9, 669515. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, J.; Ding, W.-Y.; Li, S.; Xiang, S.-H.; Tan, B. Asymmetric Pnictogen-Bonding Catalysis: Transfer Hydrogenation by a Chiral Antimony(V) Cation/Anion Pair. J. Am. Chem. Soc. 2021, 143, 6382–6387. [Google Scholar] [CrossRef]
- Paraja, M.; Gini, A.; Sakai, N.; Matile, S. Pnictogen-Bonding Catalysis: An Interactive Tool to Uncover Unorthodox Mechanisms in Polyether Cascade Cyclizations. Chem. Eur. J. 2020, 26, 15471–15476. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Gabbaï, F.P. Redox-controlled chalcogen and pnictogen bonding: The case of a sulfonium/stibonium dication as a preanionophore for chloride anion transport. Chem. Sci. 2020, 11, 10107–10112. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.M.; Tsemperouli, M.; Poblador-Bahamonde, A.I.; Benz, S.; Sakai, N.; Sugihara, K.; Matile, S. Anion Transport with Pnictogen Bonds in Direct Comparison with Chalcogen and Halogen Bonds. J. Am. Chem. Soc. 2019, 141, 810–814. [Google Scholar] [CrossRef]
- Scheiner, S. Comparison of halide receptors based on H, halogen, chalcogen, pnicogen, and tetrel bonds. Faraday Disc. 2017, 203, 213–226. [Google Scholar] [CrossRef]
- Scheiner, S. Highly Selective Halide Receptors Based on Chalcogen, Pnicogen, and Tetrel Bonds. Chem. Eur. J. 2016, 22, 18850–18858. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Pnicogen–p complexes: Theoretical study and biological implications. Phys. Chem. Chem. Phys. 2012, 14, 14061–14066. [Google Scholar] [CrossRef] [PubMed]
- Czarny, R.S.; Ho, A.N.; Shing Ho, P. A Biological Take on Halogen Bonding and Other Non-Classical Non-Covalent Interactions. Chem. Rec. 2021, 21, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Jena, S.; Dutta, J.; Tulsiyan, K.D.; Sahu, A.K.; Choudhury, S.S.; Biswal, H.S. Noncovalent interactions in proteins and nucleic acids: Beyond hydrogen bonding and π-stacking. Chem. Soc. Rev. 2022, 51, 4261–4286. [Google Scholar] [CrossRef]
- Shukla, R.; Chopra, D. Chalcogen and pnictogen bonds: Insights and relevance. Curr. Sci. 2021, 120, 1848–1853. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Aliyeva, V.A.; Resnati, G.; Pombeiro, A.J.L. Pnictogen bonding in coordination chemistry. Coord. Chem. Rev. 2020, 418, 213381. [Google Scholar] [CrossRef]
- Moilanen, J.; Ganesamoorthy, C.; Balakrishna, M.S.; Tuononen, H.M. Weak interactions between trivalent pnictogen centers: Computational analysis of bonding in dimers X3E⋯EX3 (E = Pnictogen, X = Halogen). Inorg. Chem. 2009, 48, 6740–6747. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. A new noncovalent force: Comparison of P⋯N Interaction with hydrogen and halogen bonds. J. Chem. Phys. 2011, 134, 094315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen bonds: A new molecular linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Effects of substituents upon the P⋯N noncovalent interaction: The limits of its strength. J. Phys. Chem. A 2011, 115, 11202–11209. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-Z.; Li, R.; Liu, X.-F.; Li, W.-Z.; Cheng, J.-B. Concerted interaction between pnicogen and halogen bonds in XCl-FH2P-NH3 (X=F, OH, CN, NC, and FCC). ChemPhysChem 2012, 13, 1205–1212. [Google Scholar] [CrossRef]
- Scheiner, S.; Adhikari, U. Abilities of different electron donors (D) to engage in a P⋯D noncovalent interaction. J. Phys. Chem. A 2011, 115, 11101–11110. [Google Scholar] [CrossRef]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Alkorta, I.; Elguero, J. Complexes between dihydrogen and amine, phosphine, and arsine derivatives. Hydrogen bond versus pnictogen interaction. J. Phys. Chem. A 2013, 117, 3243–3251. [Google Scholar] [CrossRef] [Green Version]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Influence of Substituent Effects on the Formation of P···Cl Pnicogen Bonds or Halogen Bonds. J. Phys. Chem. A 2014, 118, 2360–2366. [Google Scholar] [CrossRef] [Green Version]
- Esrafili, M.D.; Vakili, M.; Solimannejad, M. Cooperative effects in pnicogen bonding: (PH2F)2–7 and (PH2Cl)2–7 clusters. Chem. Phys. Lett. 2014, 609, 37–41. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Description of pnicogen bonding with the help of vibrational spectroscopy—The missing link between theory and experiment. Chem. Phys. Lett. 2014, 614, 136–142. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The pnicogen bond in review: Structures, energies, bonding properties, and spin-spin coupling constants of complexes stabilized by pnicogen bonds. In Noncovalent Forces; Scheiner, S., Ed.; Springer: Dordrecht, The Netherlands, 2015; Volume 19, pp. 191–263. [Google Scholar]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. σ-Hole Opposite to a Lone Pair: Unconventional Pnicogen Bonding Interactions between ZF3 (Z=N, P, As, and Sb) Compounds and Several Donors. ChemPhysChem 2016, 17, 1608–1614. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Zierkiewicz, W.; Švec, P.; Růžičková, Z.; Řezáč, J.; Michalczyk, M.; Růžička, A.; Michalska, D.; Hobza, P. Pnictogen bonding in pyrazine•PnX5 (Pn = P, As, Sb and X = F, Cl, Br) complexes. J. Mol. Model. 2017, 23, 328. [Google Scholar] [CrossRef] [PubMed]
- McDowell, S.A.C.; Buckingham, A.D. A Computational Study of Chalcogen-containing H2X…YF and (CH3)2X…YF (X=O, S, Se; Y=F, Cl, H) and Pnicogen-containing H3X′…YF and (CH3)3X′…YF (X′=N, P, As) Complexes. ChemPhysChem 2018, 19, 1756–1765. [Google Scholar] [CrossRef]
- Scheiner, S.; Lu, J. Halogen, Chalcogen, and Pnicogen Bonding Involving Hypervalent Atoms. Chem. Eur. J. 2018, 24, 8167–8177. [Google Scholar] [CrossRef]
- Gomila, R.M.; Frontera, A. Charge assisted halogen and pnictogen bonds: Insights from the Cambridge Structural Database and DFT calculations. CrystEngComm 2020, 22, 7162–7169. [Google Scholar] [CrossRef]
- de Azevedo Santos, L.; Hamlin, T.A.; Ramalho, T.C.; Bickelhaupt, F.M. The pnictogen bond: A quantitative molecular orbital picture. Phys. Chem. Chem. Phys. 2021, 23, 13842–13852. [Google Scholar] [CrossRef]
- Chandra, S.; Ramanathan, N.; Sundararajan, K. Is nitrogen in ammonia an elusive electron accepting pnicogen in a predominantly phosphorus bonded PCl3:NH3 dimer? Chem. Phys. Lett. 2022, 786, 139192. [Google Scholar] [CrossRef]
- Chandra, S.; Suryaprasad, B.; Ramanathan, N.; Sundararajan, K. Nitrogen as a pnicogen?: Evidence for π-hole driven novel pnicogen bonding interactions in nitromethane–ammonia aggregates using matrix isolation infrared spectroscopy and ab initio computations. Phys. Chem. Chem. Phys. 2021, 23, 6286–6297. [Google Scholar] [CrossRef]
- Bursch, M.; Kunze, L.; Vibhute, A.M.; Hansen, A.; Sureshan, K.M.; Jones, P.G.; Grimme, S.; Werz, D.B. Quantification of Noncovalent Interactions in Azide–Pnictogen, –Chalcogen, and –Halogen Contacts. Chem. Eur. J. 2021, 27, 4627–4639. [Google Scholar] [CrossRef]
- Li, W.; Spada, L.; Tasinato, N.; Rampino, S.; Evangelisti, L.; Gualandi, A.; Cozzi, P.G.; Melandri, S.; Barone, V.; Puzzarini, C. Theory Meets Experiment for Noncovalent Complexes: The Puzzling Case of Pnicogen Interactions. Angew. Chem. Int. Ed. 2018, 57, 13853–13857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, G.; Badi-uz-zama, K.; Ramanathan, G. N…N pnicogen bonds in Boc-DOPA-OMe. Chem. Phys. Lett. 2016, 653, 117–121. [Google Scholar] [CrossRef]
- Nelyubina, Y.V.; Korlyukov, A.A.; Lyssenko, K.A. Experimental Charge Density Evidence for Pnicogen Bonding in a Crystal of Ammonium Chloride. ChemPhysChem 2015, 16, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Can two trivalent N atoms engage in a direct N⋯N noncovalent interaction? Chem. Phys. Lett. 2011, 514, 32–35. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Sensitivity of pnicogen, chalcogen, halogen and H-bonds to angular distortions. Chem. Phys. Lett. 2012, 532, 31–35. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Wysokiński, R.; Scheiner, S. On the ability of pnicogen atoms to engage in both σ and π-hole complexes. Heterodimers of ZF2C6H5 (Z = P, As, Sb, Bi) and NH3. J. Mol. Model. 2019, 25, 152. [Google Scholar] [CrossRef] [Green Version]
- Scheiner, S. Detailed comparison of the pnicogen bond with chalcogen, halogen and hydrogen bonds. Int. J. Quantum Chem. 2013, 113, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- Esrafili, M.D.; Mohammadirad, N. Characterization of σ-hole interactions in 1:1 and 1:2 complexes of YOF2X (X = F, Cl, Br, I; Y = P, As) with ammonia: Competition between halogen and pnicogen bonds. Struct. Chem. 2016, 27, 939–946. [Google Scholar] [CrossRef]
- Davydova, E.I.; Virovets, A.; Peresypkina, E.; Pomogaeva, A.V.; Lisovenko, A.S.; Timoshkin, A.Y. Unusual molecular complexes of antimony fluoride dimers with acetonitrile and pyridine: Structures and bonding. Dalton Trans. 2021, 50, 13357–13367. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. The Pnictogen Bond: The Covalently Bound Arsenic Atom in Molecular Entities in Crystals as a Pnictogen Bond Donor. Molecules 2022, 27, 3421. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0-new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Galy, J.; Enjalbert, R. Crystal-Chemistry of the Va Element Trihalides-Lone Pair, Stereochemistry, and Structural Relationships. J. Solid State Chem. 1982, 44, 1–23. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. Calculation of Small Molecular Interactions by Differences of Separate Total Energies-Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bader, R. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Keith, A.T. AIMAll, version 14.11.23; TK Gristmill Software: Overland Park, KS, USA, 2014.
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Bulat, F.A.; Toro-Labbe, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R. Identifying intermolecular atom…atom interactions that are not just bonding but also competitive. CrystEngComm 2020, 22, 7145–7151. [Google Scholar] [CrossRef]
- Jabłoński, M. Counterintuitive bond paths: An intriguing case of the C(NO2)3− ion. Chem. Phys. Lett. 2020, 759, 137946. [Google Scholar] [CrossRef]
- Wick, C.R.; Clark, T. On bond-critical points in QTAIM and weak interactions. J. Mol. Model. 2018, 24, 142. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.; Murray, J.S.; Politzer, P. A perspective on quantum mechanics and chemical concepts in describing noncovalent interactions. Phys. Chem. Chem. Phys. 2018, 20, 30076–30082. [Google Scholar] [CrossRef]
- Myburgh, D.; von Berg, S.; Dillen, J. A comparison of energetic criteria to probe the stabilizing interaction resulting from a bond path between congested atoms. J. Comput. Chem. 2018, 39, 2273–2282. [Google Scholar] [CrossRef] [PubMed]
- Escudero-Adán, E.C.; Bauzá, A.; Lecomte, C.; Frontera, A.; Ballester, P. Boron triel bonding: A weak electrostatic interaction lacking electron-density descriptors. Phys. Chem. Chem. Phys. 2018, 20, 24192–24200. [Google Scholar] [CrossRef]
- Shahbazian, S. Why Bond Critical Points Are Not “Bond” Critical Points. Chem. Eur. J. 2018, 24, 5401–5405. [Google Scholar] [CrossRef]
- Keyvani, Z.A.; Shahbazian, S.; Zahedi, M. To What Extent are “Atoms in Molecules” Structures of Hydrocarbons Reproducible from the Promolecule Electron Densities? Chem. Eur. J. 2016, 22, 5003–5009. [Google Scholar] [CrossRef]
- Spackman, M.A. How Reliable Are Intermolecular Interaction Energies Estimated from Topological Analysis of Experimental Electron Densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Solimannejad, M.; Bayati, E.; Esrafili, M.D. Enhancement effect of lithium bonding on the strength of pnicogen bonds: XH2P···NCLi···NCY as a working model (X = F, Cl; Y = H, F, Cl, CN). Mol. Phys. 2014, 112, 2058–2062. [Google Scholar] [CrossRef]
- Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. Pnicogen Bonds between X=PH3 (X = O, S, NH, CH2) and Phosphorus and Nitrogen Bases. J. Phys. Chem. A 2014, 118, 1527–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radha, A.; Kumar, S.; Sharma, D.; Jassal, A.K.; Zaręba, J.K.; Franconetti, A.; Frontera, A.; Sood, P.; Pandey, S.K. Indirect influence of alkyl substituent on sigma-hole interactions: The case study of antimony(III) diphenyldithiophosphates with covalent Sb-S and non-covalent Sb⋯S pnictogen bonds. Polyhedron 2019, 173, 114126. [Google Scholar] [CrossRef]
- Moaven, S.; Andrews, M.C.; Polaske, T.J.; Karl, B.M.; Unruh, D.K.; Bosch, E.; Bowling, N.P.; Cozzolino, A.F. Triple-Pnictogen Bonding as a Tool for Supramolecular Assembly. Inorg. Chem. 2019, 58, 16227–16235. [Google Scholar] [CrossRef]
- Moaven, S.; Watson, B.T.; Polaske, T.J.; Karl, B.M.; Unruh, D.K.; Bowling, N.P.; Cozzolino, A.F. Self-Assembly of Complementary Components Using a Tripodal Bismuth Compound: Pnictogen Bonding or Coordination Chemistry? Inorg. Chem. 2021, 60, 11242–11250. [Google Scholar] [CrossRef]
- Wysokiński, R.; Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. How Many Pnicogen Bonds can be Formed to a Central Atom Simultaneously? J. Phys. Chem. A 2020, 124, 2046–2056. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Coordination of a Central Atom by Multiple Intramolecular Pnicogen Bonds. Inorg. Chem. 2020, 59, 9315–9324. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal | Cluster | ||||
|---|---|---|---|---|---|
| R, Å | ρ, au | R, Å | ρ, au | ||
| As1–F7 | 2.890 | 0.0114 | As5–F16 | 3.095 | 0.0085 |
| As1–F20 | 2.990 | 0.0113 | As5–F35 | 3.150 | 0.0082 |
| As1–F18 | 3.202 | 0.0071 | As5–F10 | 2.862 | 0.0125 |
| As1–F32 | 3.182 | 0.0079 | F6–As29 | 3.204 | 0.0064 |
| As1–F26 | 3.437 | 0.0044 | F6–F18 | 3.235 | 0.0033 |
| F2–As9 | 3.202 | 0.0071 | F7–As29 | 3.462 | 0.0044 |
| F2–As33 | 3.437 | 0.0045 | F7–As25 | 3.139 | 0.0078 |
| F3–As21 | 2.890 | 0.0114 | F8–As17 | 3.027 | 0.0096 |
| F4–As9 | 2.990 | 0.0111 | |||
| F4–As13 | 3.182 | 0.0078 | |||
| sum | 0.0839 | sum | 0.0607 | ||
| Crystal | Cluster | |
|---|---|---|
| Σc-p a | 13.92 (15.3) b | 15.78 |
| Σp-p | 6.22 | 35.67 |
| all pairs (sum of c-p and p-p) | 20.14 | 51.45 |
| actual Eint | 19.98 | 54.87 |
| Crystal | Cluster | ||||
|---|---|---|---|---|---|
| R, Å | ρ, au | R, Å | ρ, au | ||
| As21–F35 | 3.624 | 0.0040 | As9–F15 | 3.086 | 0.0082 |
| F11–As13 | 3.624 | 0.0039 | F2–As13 | 2.968 | 0.0106 |
| F12–F35 | 3.227 | 0.0030 | As13–F22 | 3.062 | 0.0088 |
| F14–F19 | 3.282 | 0.0023 | F10–F16 | 3.103 | 0.0041 |
| F14–F20 | 3.424 | 0.0018 | As9–F20 | 3.153 | 0.0070 |
| F15–F24 | 3.227 | 0.0030 | F12–F20 | 2.915 | 0.0073 |
| F20–F27 | 3.227 | 0.0030 | As1–F20 | 3.157 | 0.0077 |
| F22–F27 | 3.282 | 0.0022 | F3–As21 | 3.112 | 0.0072 |
| F22–F28 | 3.424 | 0.0018 | F16–F23 | 2.923 | 0.0059 |
| F26–F31 | 3.282 | 0.0023 | F23–As33 | 3.035 | 0.0092 |
| F26–F32 | 3.424 | 0.0019 | As25–F36 | 3.189 | 0.0075 |
| F31–As33 | 3.624 | 0.0040 | As21–F26 | 2.988 | 0.0094 |
| F7–As9 | 3.624 | 0.0040 | F18–As29 | 3.421 | 0.0043 |
| F7–F32 | 3.227 | 0.0032 | F3–F30 | 3.201 | 0.0034 |
| F8–F18 | 3.424 | 0.0018 | F28–As29 | 3.068 | 0.0084 |
| As21–F30 | 3.098 | 0.0082 | |||
| F26–F30 | 3.019 | 0.0049 | |||
| As1–F32 | 3.141 | 0.0087 | |||
| F24–As33 | 3.419 | 0.0048 | |||
| sum | 0.0423 | sum | 0.1357 | ||
| −Eint (kcal/mol) | −Eint/n (kcal/mol) | |||
|---|---|---|---|---|
| n | Crystal | Cluster | Crystal | Cluster |
| 2 | 3.13 | 4.44 | 1.57 | 2.22 |
| 3 | 4.82 | 10.00 | 1.61 | 3.33 |
| 4 | 7.06 | 16.95 | 1.77 | 4.24 |
| 5 | 9.25 | 24.73 | 1.85 | 4.95 |
| 6 | 11.52 | 31.55 | 1.92 | 5.26 |
| 7 | 16.06 | 40.14 | 2.29 | 5.73 |
| 8 | 17.97 | 46.49 | 2.25 | 5.81 |
| 9 | 19.98 | 54.87 | 2.22 | 6.10 |
| As1 | R, Å | θ(F-As··F), degs | ρ, au | E2, kcal/mol |
|---|---|---|---|---|
| As1–F14 | 2.731 | 165.9 | 0.0167 | 4.47 |
| As1–F47 | 2.921 | 163.5 | 0.0113 | 2.02 |
| As1–F18 | 3.117 | 133.2 | 0.0082 | 1.04 |
| As1–F27 | 3.207 | 139.6 | 0.0069 | 0.94 |
| As1–F50 | 3.328 | 131.5 | 0.0061 | 0.49 |
| F3 | ||||
| F3–As33 | 2.975 | 166.8 | 0.0104 | 1.03 |
| F3–As21 | 3.738 | 139.3 | 0.0030 | 0 |
| F3–F26 | 2.914 | - | 0.0068 | 0.29 |
| F3–F12 | 3.396 | - | 0.0024 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheiner, S.; Michalczyk, M.; Zierkiewicz, W. Involvement of Arsenic Atom of AsF3 in Five Pnicogen Bonds: Differences between X-ray Structure and Theoretical Models. Molecules 2022, 27, 6486. https://doi.org/10.3390/molecules27196486
Scheiner S, Michalczyk M, Zierkiewicz W. Involvement of Arsenic Atom of AsF3 in Five Pnicogen Bonds: Differences between X-ray Structure and Theoretical Models. Molecules. 2022; 27(19):6486. https://doi.org/10.3390/molecules27196486
Chicago/Turabian StyleScheiner, Steve, Mariusz Michalczyk, and Wiktor Zierkiewicz. 2022. "Involvement of Arsenic Atom of AsF3 in Five Pnicogen Bonds: Differences between X-ray Structure and Theoretical Models" Molecules 27, no. 19: 6486. https://doi.org/10.3390/molecules27196486
APA StyleScheiner, S., Michalczyk, M., & Zierkiewicz, W. (2022). Involvement of Arsenic Atom of AsF3 in Five Pnicogen Bonds: Differences between X-ray Structure and Theoretical Models. Molecules, 27(19), 6486. https://doi.org/10.3390/molecules27196486