
Semisynthetic Derivatives of Pentacyclic Triterpenes Bearing Heterocyclic Moieties with Therapeutic Potential

 , ,
, , .jpg) , ,
, , 
Abstract
:1. Introduction
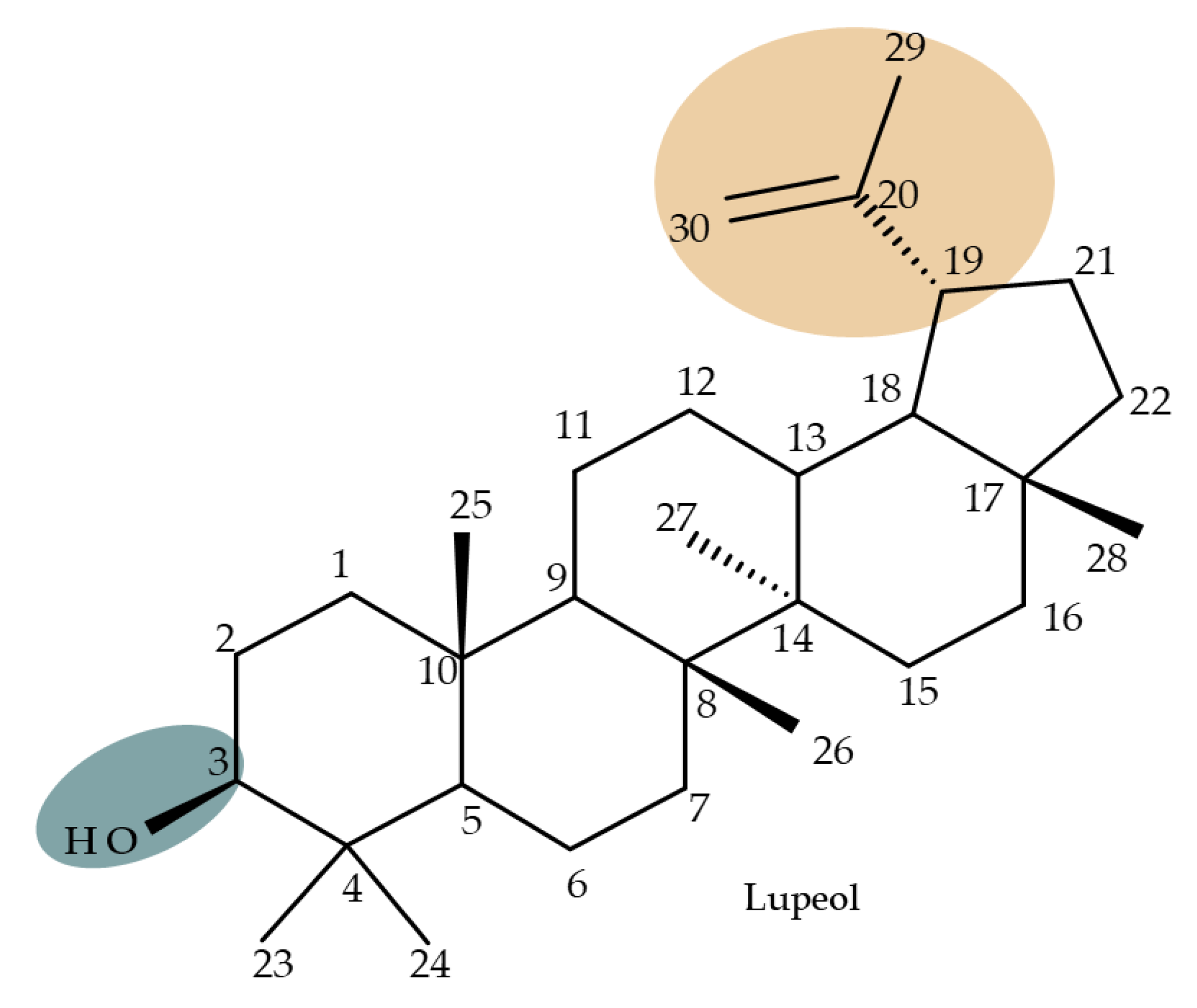
2. Lupeol Derivatives
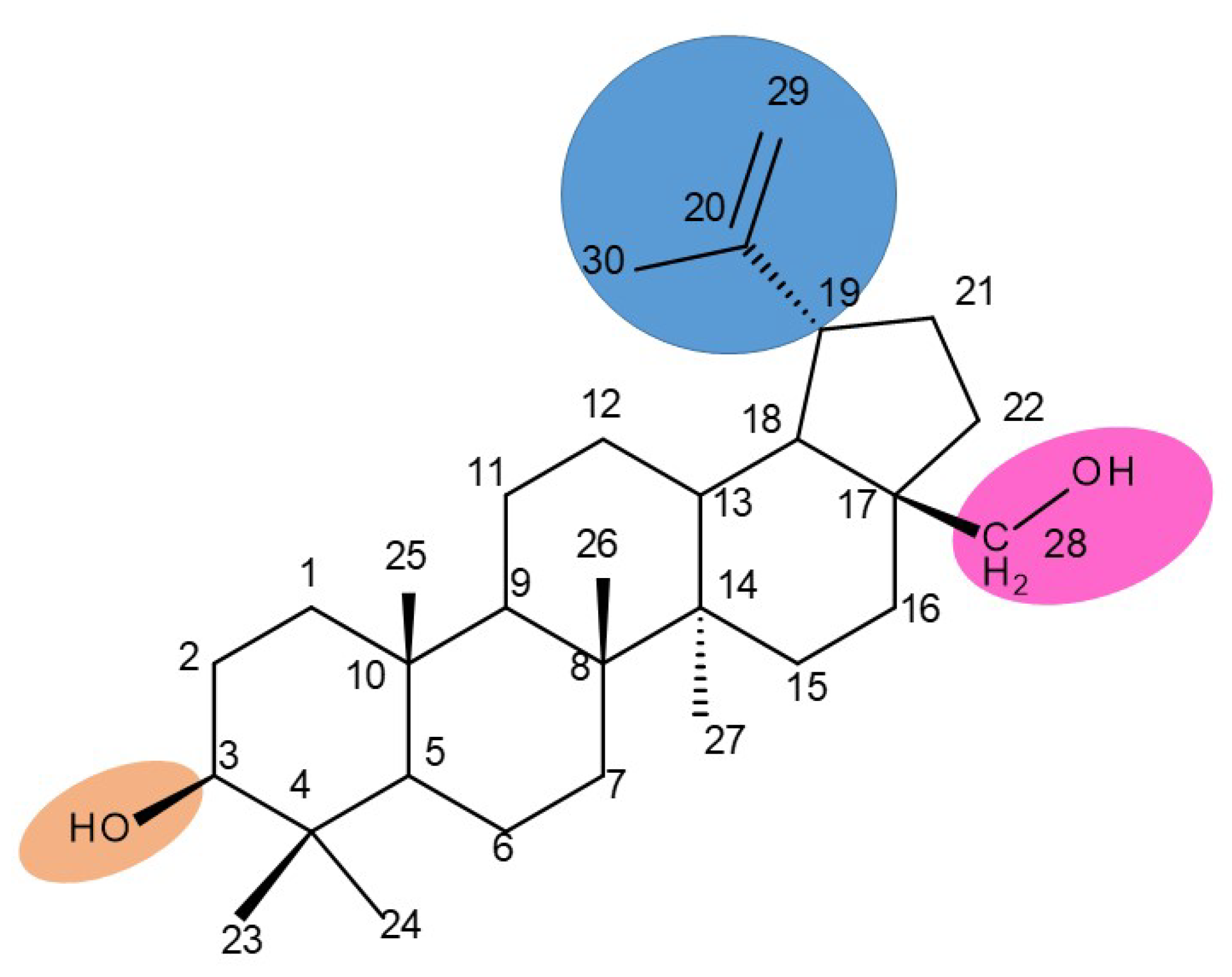
3. Betulin
3.1. C3 Modifications
3.2. C28 Modifications
3.3. C30 (Allylic) Modifications
3.4. C3, C28, C30 Modifications
3.5. C-3 and C28 Modifications
3.6. A-Ring-Fused Heterocyclic Derivatives
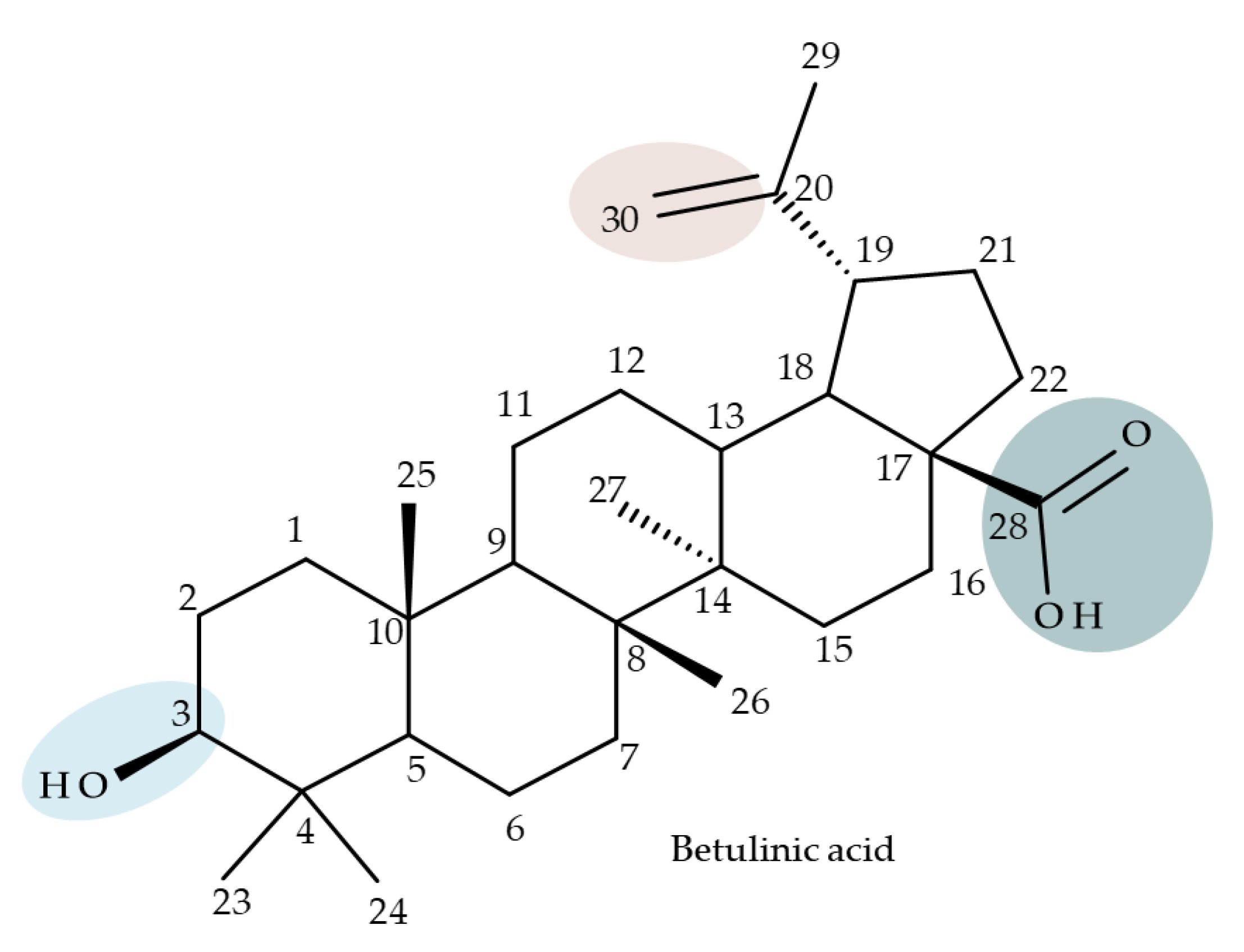
4. Betulinic Acid
4.1. C2 Modifications
4.2. C3 Modifications
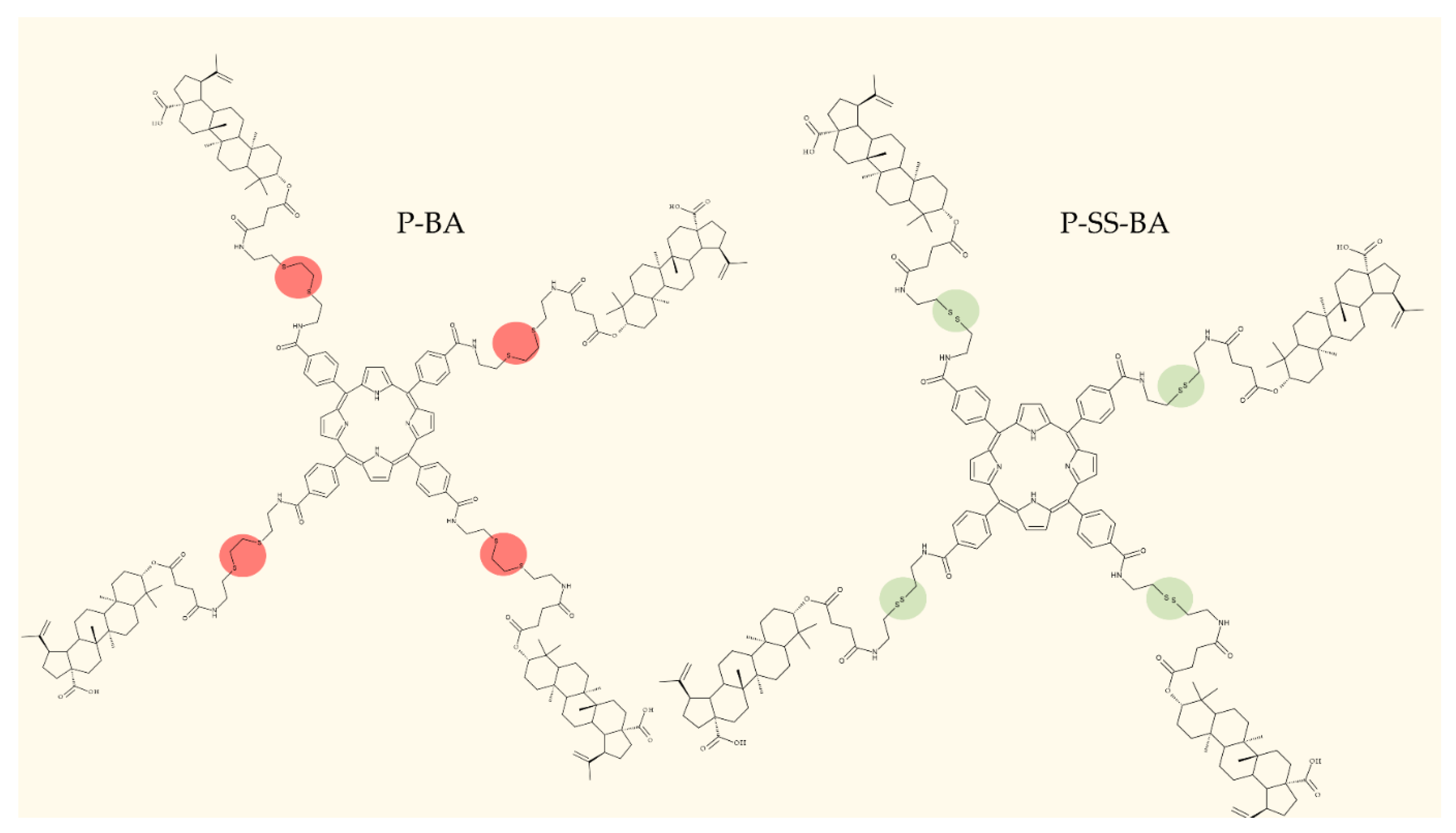
4.3. C28 Modifications
4.4. C30 Modifications
4.5. A-Ring-Fused Heterocyclic Derivatives
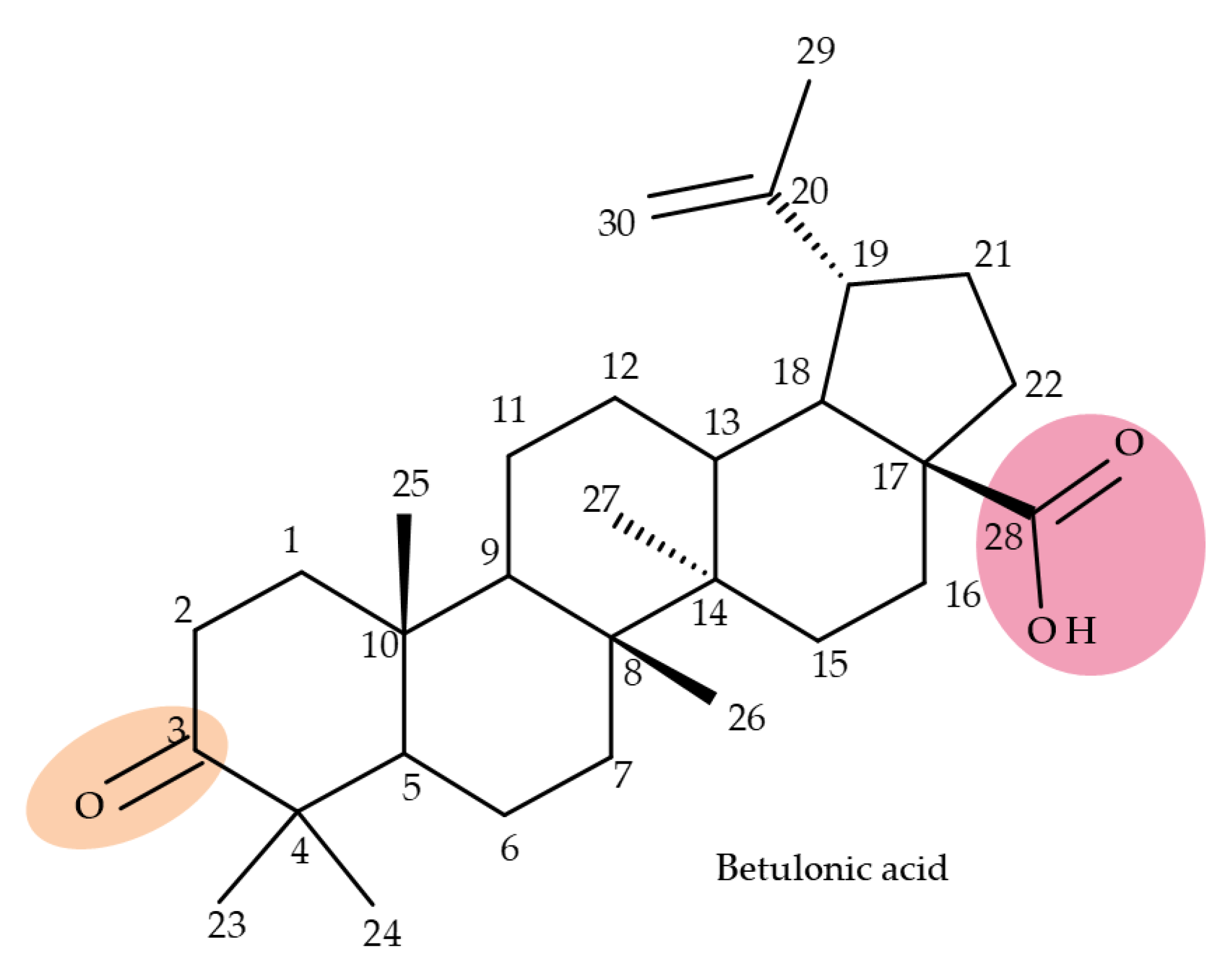
5. Betulonic Acid
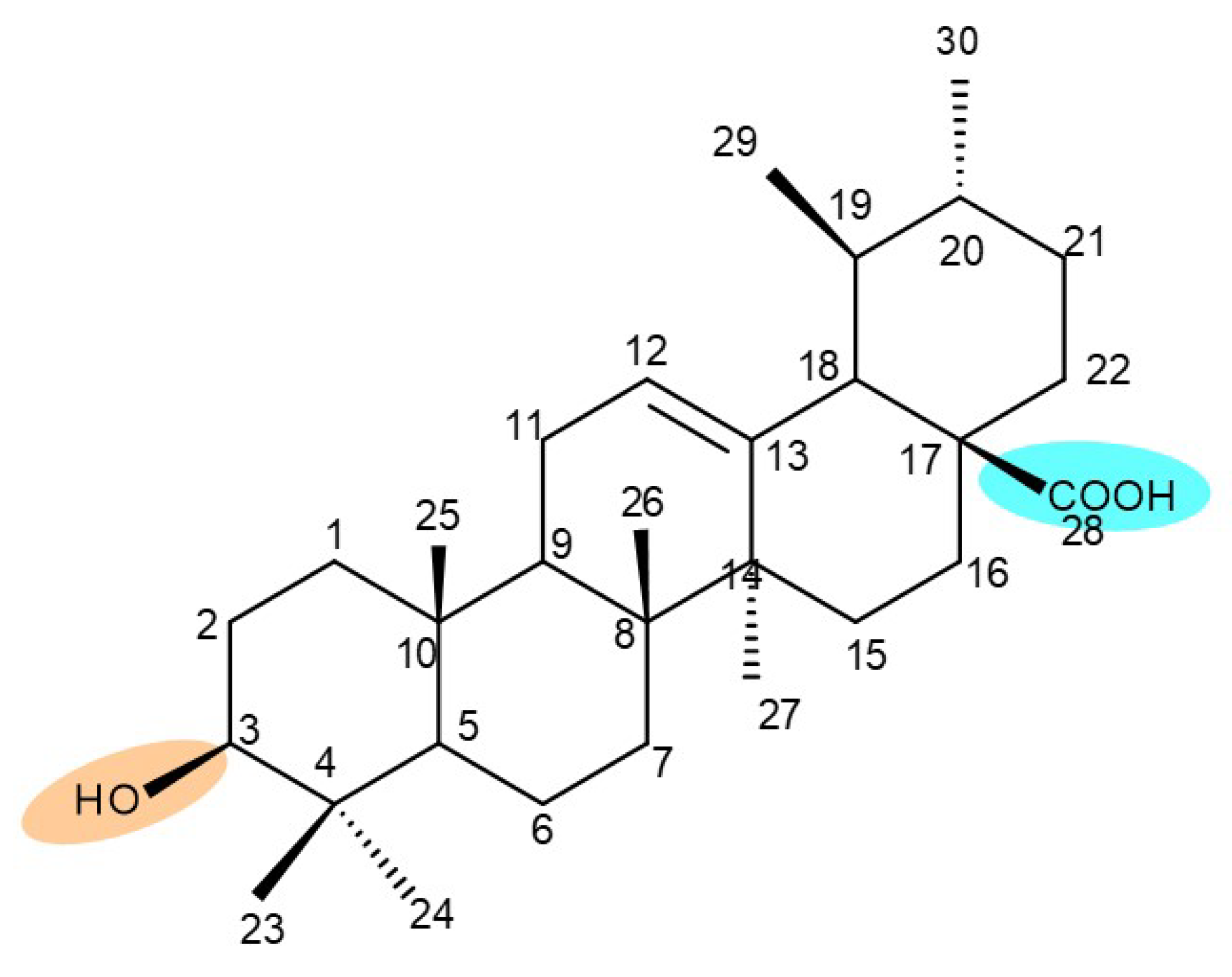
6. Ursolic Acid
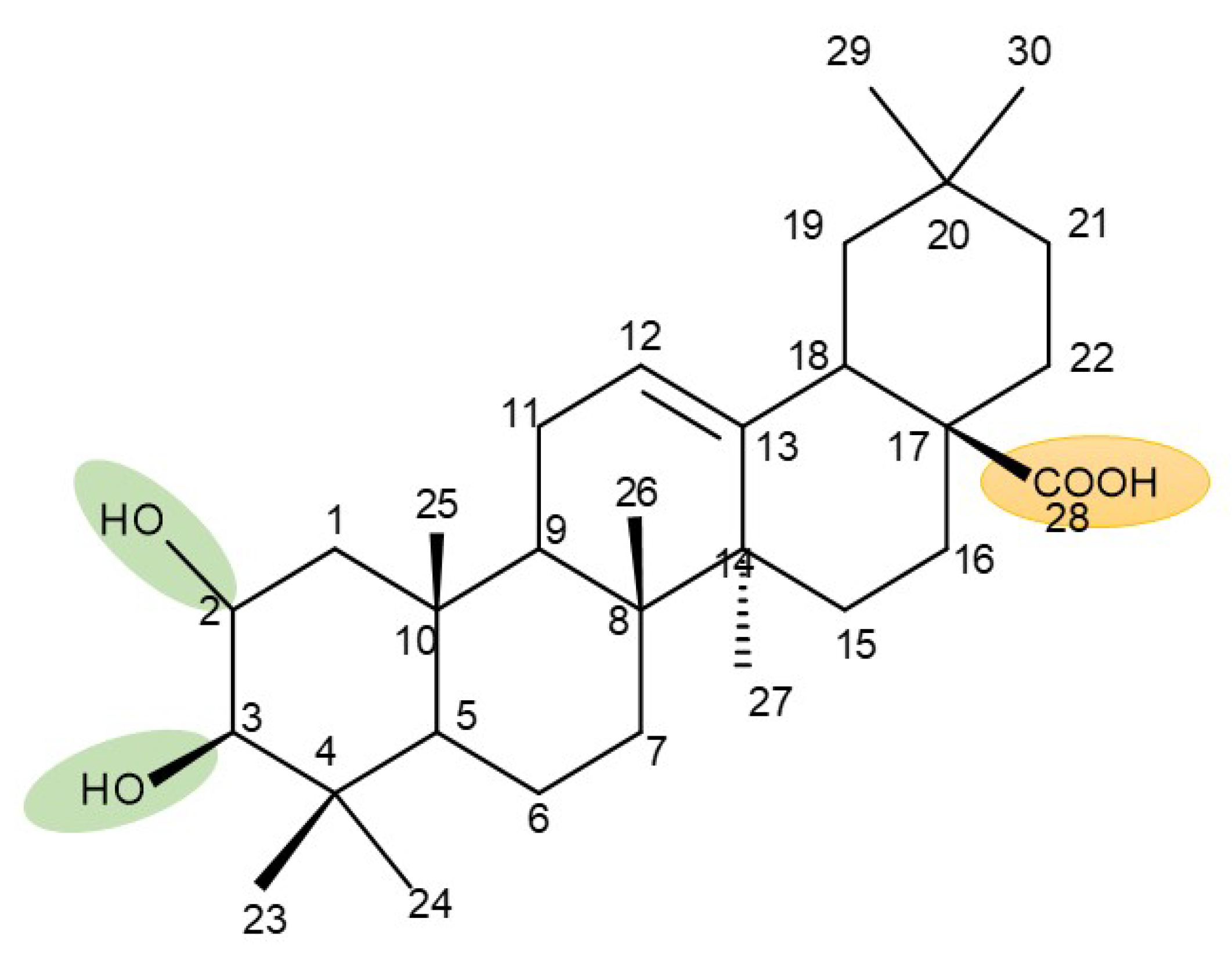
7. Maslinic and Corosolic Acids
8. Oleanolic Acid
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Petrovska, B. Historical review of medicinal plants′ usage. Pharmacogn. Rev. 2012, 6, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; Saxena, J.; Nema, R.; Singh, D.; Gupta, A. Phytochemistry of Medicinal Plants. J. Pharmacogn. Phytochem. 2013, 1, 168–182. [Google Scholar]
- Veeresham, C. Natural products derived from plants as a source of drugs. J. Adv. Pharm. Technol. Res. 2012, 3, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Soica, C.; Voicu, M.; Ghiulai, R.; Dehelean, C.; Racoviceanu, R.; Trandafirescu, C.; Rosca, O.-J.; Nistor, G.; Mioc, M.; Mioc, A. Natural Compounds in Sex Hormone-Dependent Cancers: The Role of Triterpenes as Therapeutic Agents. Front. Endocrinol. 2021, 11, 612396. [Google Scholar] [CrossRef] [PubMed]
- Ghiulai, R.; Roşca, O.J.; Antal, D.S.; Mioc, M.; Mioc, A.; Racoviceanu, R.; Macaşoi, I.; Olariu, T.; Dehelean, C.; Creţu, O.M.; et al. Tetracyclic and pentacyclic triterpenes with high therapeutic efficiency in wound healing approaches. Molecules 2020, 25, 5557. [Google Scholar] [CrossRef]
- Ghante, M.H.; Jamkhande, P.G. Role of Pentacyclic Triterpenoids in Chemoprevention and Anticancer Treatment: An Overview on Targets and Underling Mechanisms. J. Pharmacopunct. 2019, 22, 55–67. [Google Scholar] [CrossRef]
- Furtado, N.A.J.C.; Pirson, L.; Edelberg, H.; Miranda, L.M.; Loira-Pastoriza, C.; Preat, V.; Larondelle, Y.; André, C.M. Pentacyclic Triterpene Bioavailability: An Overview of In Vitro and In Vivo Studies. Molecules 2017, 22, 400. [Google Scholar] [CrossRef] [Green Version]
- Xiao, S.; Tian, Z.; Wang, Y.; Si, L.; Zhang, L.; Zhou, D. Recent progress in the antiviral activity and mechanism study of pentacyclic triterpenoids and their derivatives. Med. Res. Rev. 2018, 38, 951–976. [Google Scholar] [CrossRef] [Green Version]
- Suvarna, V.; Bore, B.; Bhawar, C.; Mallya, R. Complexation of phytochemicals with cyclodextrins and their derivatives- an update. Biomed. Pharmacother. 2022, 149, 112862. [Google Scholar] [CrossRef]
- Milan, A.; Mioc, A.; Prodea, A.; Mioc, M.; Buzatu, R.; Ghiulai, R.; Racoviceanu, R.; Caruntu, F.; Şoica, C. The Optimized Delivery of Triterpenes by Liposomal Nanoformulations: Overcoming the Challenges. Int. J. Mol. Sci. 2022, 23, 1140. [Google Scholar] [CrossRef]
- Timilsina, U.; Ghimire, D.; Timalsina, B.; Nitz, T.J.; Wild, C.T.; Freed, E.O.; Gaur, R. Identification of potent maturation inhibitors against HIV-1 clade C. Sci. Rep. 2016, 6, 27403. [Google Scholar] [CrossRef] [PubMed]
- Rotella, D.P. Heterocycles in drug discovery: Properties and preparation. In Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 2021; pp. 149–183. [Google Scholar]
- Siddique, H.R.; Saleem, M. Beneficial health effects of lupeol triterpene: A review of preclinical studies. Life Sci. 2011, 88, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Palia, P.; Chaudhary, A.; Verma, K.; Kumar, I. A Review on Pharmacological Activities of Lupeol and its Triterpene Derivatives. J. Drug Deliv. Ther. 2020, 10, 325–332. [Google Scholar] [CrossRef]
- Khan, M.F.; Maurya, C.K.; Dev, K.; Arha, D.; Rai, A.K.; Tamrakar, A.K.; Maurya, R. Design and synthesis of lupeol analogues and their glucose uptake stimulatory effect in L6 skeletal muscle cells. Bioorg. Med. Chem. Lett. 2014, 24, 2674–2679. [Google Scholar] [CrossRef]
- Khan, M.F.; Mishra, D.P.; Ramakrishna, E.; Rawat, A.K.; Mishra, A.; Srivastava, A.K.; Maurya, R. Design and synthesis of lupeol analogues and their in vitro PTP-1B inhibitory activity. Med. Chem. Res. 2014, 23, 4156–4166. [Google Scholar] [CrossRef]
- Tamrakar, A.K.; Maurya, C.K.; Rai, A.K. PTP1B inhibitors for type 2 diabetes treatment: A patent review (2011–2014). Expert Opin. Ther. Pat. 2014, 24, 1101–1115. [Google Scholar] [CrossRef]
- Castro, M.J.; Careaga, V.P.; Sacca, P.A.; Faraoni, M.B.; Murray, A.P.; Calvo, J.C. Lupane Triterpenoids and New Derivatives as Antiproliferative Agents Against Prostate Cancer Cells. Anticancer Res. 2019, 39, 3835–3845. [Google Scholar] [CrossRef]
- Cui, J.; Wang, H.; Huang, Y.; Xin, Y.; Zhou, A. Synthesis and cytotoxic analysis of some disodium 3β,6β-dihydroxysterol disulfates. Steroids 2009, 74, 1057–1060. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Cui, J.; Li, Y.; Fan, L.; Jiao, Y.; Su, S. Syntheses and antiproliferative activity of some sulfated hydroximinosterols. Med. Chem. Res. 2013, 22, 409–414. [Google Scholar] [CrossRef]
- Şoica, C.; Dehelean, C.; Danciu, C.; Wang, H.M.; Wenz, G.; Ambrus, R.; Bojin, F.; Anghel, M. Betulin complex in γ-cyclodextrin derivatives: Properties and antineoplasic activities in in vitro and in vivo tumor models. Int. J. Mol. Sci. 2012, 13, 14992–15011. [Google Scholar] [CrossRef]
- Hordyjewska, A.; Ostapiuk, A.; Horecka, A.; Kurzepa, J. Betulin and betulinic acid: Triterpenoids derivatives with a powerful biological potential. Phytochem. Rev. 2019, 18, 929–951. [Google Scholar] [CrossRef]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.-R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38, 107409. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti, S.; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Current perspectives on naturally occurring saponins as anticancer agents. Arch. Pharm. 2022, 355, 2100469. [Google Scholar] [CrossRef] [PubMed]
- Cmoch, P.; Korda, A.; Rárová, L.; Oklešťková, J.; Strnad, M.; Luboradzki, R.; Pakulski, Z. Synthesis and structure–activity relationship study of cytotoxic lupane-type 3β-O-monodesmosidic saponins with an extended C-28 side chain. Tetrahedron 2014, 70, 2717–2730. [Google Scholar] [CrossRef]
- Kuczynska, K.; Cmoch, P.; Rárová, L.; Oklešťková, J.; Korda, A.; Pakulski, Z.; Strnad, M. Influence of intramolecular hydrogen bonds on regioselectivity of glycosylation. Synthesis of lupane-type saponins bearing the OSW-1 saponin disaccharide unit and its isomers. Carbohydr. Res. 2016, 423, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Jastrzębska, M.; Marciniec, K.; Chrobak, E.; Bębenek, E.; Latocha, M.; Kuśmierz, D.; Boryczka, S. Design, synthesis and biological activity of 1,4-quinone moiety attached to betulin derivatives as potent DT-diaphorase substrate. Bioorg. Chem. 2021, 106, 104478. [Google Scholar] [CrossRef] [PubMed]
- Anh, D.T.T.; Giang, L.N.T.; Hien, N.T.; Cuc, D.T.; Thanh, N.H.; Ha, N.T.T.; Chinh, P.T.; Van Tuyen, N.; Van Kiem, P. Synthesis and Cytotoxic Evaluation of Betulin–Triazole–AZT Hybrids. Nat. Prod. Commun. 2017, 12, 1934578X1701201. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Semenova, A.A.; Nedopekina, D.A.; Davletshin, E.V.; Spivak, A.Y.; Belosludtsev, K.N. Effect of F16-Betulin Conjugate on Mitochondrial Membranes and Its Role in Cell Death Initiation. Membranes 2021, 11, 352. [Google Scholar] [CrossRef]
- Sousa, M.C.; Varandas, R.; Santos, R.C.; Santos-Rosa, M.; Alves, V.; Salvador, J.A.R. Antileishmanial Activity of Semisynthetic Lupane Triterpenoids Betulin and Betulinic Acid Derivatives: Synergistic Effects with Miltefosine. PLoS ONE 2014, 9, e89939. [Google Scholar] [CrossRef] [Green Version]
- Lugiņina, J.; Linden, M.; Bazulis, M.; Kumpiņš, V.; Mishnev, A.; Popov, S.A.; Golubeva, T.S.; Waldvogel, S.R.; Shults, E.E.; Turks, M. Electrosynthesis of Stable Betulin-Derived Nitrile Oxides and their Application in Synthesis of Cytostatic Lupane-Type Triterpenoid-Isoxazole Conjugates. Eur. J. Org. Chem. 2021, 2021, 2557–2577. [Google Scholar] [CrossRef]
- Karagöz, A.Ç.; Leidenberger, M.; Hahn, F.; Hampel, F.; Friedrich, O.; Marschall, M.; Kappes, B.; Tsogoeva, S.B. Synthesis of new betulinic acid/betulin-derived dimers and hybrids with potent antimalarial and antiviral activities. Bioorg. Med. Chem. 2019, 27, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, T.C.; Cunha-Oliveira, T.; Serafim, T.L.; Holy, J.; Krasutsky, D.; Kolomitsyna, O.; Krasutsky, P.; Moreno, A.M.; Oliveira, P.J. Dimethylaminopyridine derivatives of lupane triterpenoids cause mitochondrial disruption and induce the permeability transition. Bioorg. Med. Chem. 2013, 21, 7239–7249. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Semenova, A.A.; Ilzorkina, A.I.; Markelova, N.Y.; Penkov, N.V.; Shakurova, E.R.; Belosludtsev, K.N.; Parfenova, L.V. New quaternized pyridinium derivatives of betulin: Synthesis and evaluation of membranotropic properties on liposomes, pro- and eukaryotic cells, and isolated mitochondria. Chem. Biol. Interact. 2021, 349, 109678. [Google Scholar] [CrossRef] [PubMed]
- Shakurova, E.R.; Pozdnyakova, D.A.; Tretyakova, E.V.; Parfenova, L.V. One-pot Synthesis of Betulin Triterpenoid Quaternized Pyridine Derivatives and their Antimicrobial Activity. Lett. Drug Des. Discov. 2019, 17, 79–84. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Bębenek, E.; Chrobak, E.; Marciniec, K.; Latocha, M.; Kuśmierz, D.; Jastrzębska, M.; Boryczka, S. Betulin-1,4-quinone hybrids: Synthesis, anticancer activity and molecular docking study with NQO1 enzyme. Eur. J. Med. Chem. 2019, 177, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.; Hodoň, J.; Kazakova, A.; D’Acunto, C.W.; Kaňovský, P.; Urban, M.; Strnad, M. Novel pentacyclic triterpenes exhibiting strong neuroprotective activity in SH-SY5Y cells in salsolinol- and glutamate-induced neurodegeneration models. Eur. J. Med. Chem. 2021, 213, 113168. [Google Scholar] [CrossRef]
- Yamansarov, E.Y.; Lopatukhina, E.V.; Evteev, S.A.; Skvortsov, D.A.; Lopukhov, A.V.; Kovalev, S.V.; Vaneev, A.N.; Shkil’, D.O.; Akasov, R.A.; Lobov, A.N.; et al. Discovery of Bivalent GalNAc-Conjugated Betulin as a Potent ASGPR-Directed Agent against Hepatocellular Carcinoma. Bioconjug. Chem. 2021, 32, 763–781. [Google Scholar] [CrossRef]
- Grymel, M.; Pastuch-Gawołek, G.; Lalik, A.; Zawojak, M.; Boczek, S.; Krawczyk, M.; Erfurt, K. Glycoconjugation of Betulin Derivatives Using Copper-Catalyzed 1,3-Dipolar Azido-Alkyne Cycloaddition Reaction and a Preliminary Assay of Cytotoxicity of the Obtained Compounds. Molecules 2020, 25, 6019. [Google Scholar] [CrossRef]
- Bębenek, E.; Jastrzębska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolińska, K.; Latocha, M.; Mertas, A.; Czuba, Z.; Boryczka, S. Novel Triazole Hybrids of Betulin: Synthesis and Biological Activity Profile. Molecules 2017, 22, 1876. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Xu, X.; Liu, J.; Jia, Q.; Ke, C.; Zhang, H.; Xu, C.; Ou, E.; Tan, W.; Zhao, Y. Mitochondria-Targeted Triphenylphosphonium Conjugated C-3 Modified Betulin: Synthesis, Antitumor Properties and Mechanism of Action. ChemMedChem 2022, 17, e202100659. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Nemtarev, A.V.; Abdullin, T.I.; Grigor’eva, L.R.; Kuznetsova, E.V.; Akhmadishina, R.A.; Ziganshina, L.E.; Cong, H.H.; Mironov, V.F. Design, Synthesis, and Cancer Cell Growth Inhibitory Activity of Triphenylphosphonium Derivatives of the Triterpenoid Betulin. J. Nat. Prod. 2017, 80, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Csuk, R.; Sczepek, R.; Siewert, B.; Nitsche, C. Cytotoxic betulin-derived hydroxypropargylamines trigger apoptosis. Bioorg. Med. Chem. 2013, 21, 425–435. [Google Scholar] [CrossRef]
- Holy, J.; Kolomitsyna, O.; Krasutsky, D.; Oliveira, P.J.; Perkins, E.; Krasutsky, P.A. Dimethylaminopyridine derivatives of lupane triterpenoids are potent disruptors of mitochondrial structure and function. Bioorg. Med. Chem. 2010, 18, 6080–6088. [Google Scholar] [CrossRef] [PubMed]
- Myszka, H.; Grzywacz, D.; Zdrowowicz, M.; Spisz, P.; Butowska, K.; Rak, J.; Piosik, J.; Jaśkiewicz, M.; Kamysz, W.; Liberek, B. Design, synthesis and biological evaluation of betulin-3-yl 2-amino-2-deoxy-β-d-glycopyranosides. Bioorg. Chem. 2020, 96, 103568. [Google Scholar] [CrossRef]
- Korda, A.; Rárová, L.; Pakulski, Z.; Strnad, M.; Oklešťková, J.; Kuczynska, K.; Cmoch, P.; Gwardiak, K.; Karczewski, R. New lupane bidesmosides exhibiting strong cytotoxic activities in vitro. Bioorg. Chem. 2020, 100, 103868. [Google Scholar] [CrossRef] [PubMed]
- Mihoub, M.; Pichette, A.; Sylla, B.; Gauthier, C.; Legault, J. Bidesmosidic betulin saponin bearing L-rhamnopyranoside moieties induces apoptosis and inhibition of lung cancer cells growth in vitro and in vivo. PLoS ONE 2018, 13, e0193386. [Google Scholar] [CrossRef] [Green Version]
- Sidoryk, K.; Korda, A.; Rárová, L.; Oklešťková, J.; Strnad, M.; Cmoch, P.; Pakulski, Z.; Gwardiak, K.; Karczewski, R.; Luboradzki, R. Synthesis and biological activity of new homolupanes and homolupane saponins. Tetrahedron 2015, 71, 2004–2012. [Google Scholar] [CrossRef]
- Grishko, V.V.; Tolmacheva, I.A.; Nebogatikov, V.O.; Galaiko, N.V.; Nazarov, A.V.; Dmitriev, M.V.; Ivshina, I.B. Preparation of novel ring-A fused azole derivatives of betulin and evaluation of their cytotoxicity. Eur. J. Med. Chem. 2017, 125, 629–639. [Google Scholar] [CrossRef]
- Laavola, M.; Haavikko, R.; Hämäläinen, M.; Leppänen, T.; Nieminen, R.; Alakurtti, S.; Moreira, V.M.; Yli-Kauhaluoma, J.; Moilanen, E. Betulin Derivatives Effectively Suppress Inflammation in Vitro and in Vivo. J. Nat. Prod. 2016, 79, 274–280. [Google Scholar] [CrossRef]
- Oliveira-Costa, J.F.; Meira, C.S.; das Neves, M.V.G.; Dos Reis, B.P.Z.C.; Soares, M.B.P. Anti-Inflammatory Activities of Betulinic Acid: A Review. Front. Pharmacol. 2022, 13, 883857. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Liang, N.; Liu, Y.; Cheng, M.-S. Recent progress on betulinic acid and its derivatives as antitumor agents: A mini review. Chin. J. Nat. Med. 2021, 19, 641–647. [Google Scholar] [CrossRef]
- Ulici, A.; Milan, A.; Mioc, M.; Ghiulai, R.; Racoviceanu, R.; Șoica, C. Ring-Modified Triterpene Derivatives as Potential Pharmacological Active Compounds. Timisoara Med. J. 2021, 2020, 7. [Google Scholar] [CrossRef]
- Wang, Q.; Li, Y.; Zheng, L.; Huang, X.; Wang, Y.; Chen, C.-H.; Cheng, Y.-Y.; Morris-Natschke, S.L.; Lee, K.-H. Novel Betulinic Acid–Nucleoside Hybrids with Potent Anti-HIV Activity. ACS Med. Chem. Lett. 2020, 11, 2290–2293. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zou, X.; Wang, Y.; Zou, M.; Ma, F.; Wang, N.; Li, J.; Wang, M.; Hung, H.-Y.; Wang, Q. Betulinic acid-nucleoside hybrid prevents acute alcohol -induced liver damage by promoting anti-oxidative stress and autophagy. Eur. J. Pharmacol. 2022, 914, 174686. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.-W.; He, Y.; Wang, J.; Gao, W.; Liu, T.; Qin, M.; Wang, X.; Gao, C.; Wang, Y.; Liu, M.-Y.; et al. Synthesis of heterocycle-modified betulinic acid derivatives as antitumor agents. Eur. J. Med. Chem. 2015, 95, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Bildziukevich, U.; Rárová, L.; Šaman, D.; Wimmer, Z. Picolyl amides of betulinic acid as antitumor agents causing tumor cell apoptosis. Eur. J. Med. Chem. 2018, 145, 41–50. [Google Scholar] [CrossRef]
- Sylla, B.; Lavoie, S.; Legault, J.; Gauthier, C.; Pichette, A. Synthesis, cytotoxicity and anti-inflammatory activity of rhamnose-containing ursolic and betulinic acid saponins. RSC Adv. 2019, 9, 39743–39757. [Google Scholar] [CrossRef] [Green Version]
- Özdemir, Z.; Rybková, M.; Vlk, M.; Šaman, D.; Rárová, L.; Wimmer, Z. Synthesis and Pharmacological Effects of Diosgenin–Betulinic Acid Conjugates. Molecules 2020, 25, 3546. [Google Scholar] [CrossRef]
- Meira, C.S.; Barbosa-Filho, J.M.; Lanfredi-Rangel, A.; Guimarães, E.T.; Moreira, D.R.M.; Soares, M.B.P. Antiparasitic evaluation of betulinic acid derivatives reveals effective and selective anti-Trypanosoma cruzi inhibitors. Exp. Parasitol. 2016, 166, 108–115. [Google Scholar] [CrossRef]
- Ibezim, A.; Nwodo, N.J.; Nnaji, N.J.N.; Ujam, O.T.; Olubiyi, O.O.; Mba, C.J. In silico investigation of morpholines as novel class of trypanosomal triosephosphate isomerase inhibitors. Med. Chem. Res. 2017, 26, 180–189. [Google Scholar] [CrossRef]
- Yang, Y.; Xie, T.; Tian, X.; Han, N.; Liu, X.; Chen, H.; Qi, J.; Gao, F.; Li, W.; Wu, Q.; et al. Betulinic Acid-Nitrogen Heterocyclic Derivatives: Design, Synthesis, and Antitumor Evaluation in Vitro. Molecules 2020, 25, 948. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hou, S.; Li, B.; Pan, J.; Jiang, L.; Zhou, G.; Gu, H.; Zhao, C.; Lu, H.; Ma, F. Combination of betulinic acid with diazen-1-ium-1,2-diolate nitric oxide moiety donating a novel anticancer candidate. OncoTargets Ther. 2018, 11, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodr, D.; Stanková, J.; Rumlová, M.; Džubák, P.; Řehulka, J.; Zimmermann, T.; Křížová, I.; Gurská, S.; Hajdúch, M.; Drašar, P.B.; et al. Betulinic Acid Decorated with Polar Groups and Blue Emitting BODIPY Dye: Synthesis, Cytotoxicity, Cell-Cycle Analysis and Anti-HIV Profiling. Biomedicines 2021, 9, 1104. [Google Scholar] [CrossRef]
- Krajcovicova, S.; Stankova, J.; Dzubak, P.; Hajduch, M.; Soural, M.; Urban, M. A Synthetic Approach for the Rapid Preparation of BODIPY Conjugates and their use in Imaging of Cellular Drug Uptake and Distribution. Chem. A Eur. J. 2018, 24, 4957–4966. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Ganguly, A.; Chowdhuri, S.; Yousuf, M.; Ghosh, A.; Barui, A.K.; Kotcherlakota, R.; Adhikari, S.; Banerjee, R. Bis -Arylidene Oxindole–Betulinic Acid Conjugate: A Fluorescent Cancer Cell Detector with Potent Anticancer Activity. ACS Med. Chem. Lett. 2015, 6, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suman, P.; Patel, A.; Solano, L.; Jampana, G.; Gardner, Z.S.; Holt, C.M.; Jonnalagadda, S.C. Synthesis and cytotoxicity of Baylis-Hillman template derived betulinic acid-triazole conjugates. Tetrahedron 2017, 73, 4214–4226. [Google Scholar] [CrossRef]
- Santos, R.C.; Salvador, J.A.R.; Marín, S.; Cascante, M.; Moreira, J.N.; Dinis, T.C.P. Synthesis and structure–activity relationship study of novel cytotoxic carbamate and N-acylheterocyclic bearing derivatives of betulin and betulinic acid. Bioorg. Med. Chem. 2010, 18, 4385–4396. [Google Scholar] [CrossRef]
- Nedopekina, D.A.; Gubaidullin, R.R.; Odinokov, V.N.; Maximchik, P.V.; Zhivotovsky, B.; Bel’skii, Y.P.; Khazanov, V.A.; Manuylova, A.V.; Gogvadze, V.; Spivak, A.Y. Mitochondria-targeted betulinic and ursolic acid derivatives: Synthesis and anticancer activity. MedChemComm 2017, 8, 1934–1945. [Google Scholar] [CrossRef] [Green Version]
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Bel’skii, Y.P.; Bel’skaya, N.V.; Khazanov, V.A. Triphenylphosphonium cations of betulinic acid derivatives: Synthesis and antitumor activity. Med. Chem. Res. 2017, 26, 518–531. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Davletshin, E.V.; Tukhbatullin, A.A.; D’yakonov, V.A.; Yunusbaeva, M.M.; Dzhemileva, L.U.; Dzhemilev, U.M. Pentacyclic triterpene acid conjugated with mitochondria-targeting cation F16: Synthesis and evaluation of cytotoxic activities. Med. Chem. Res. 2021, 30, 940–951. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Semenova, A.A.; Ilzorkina, A.I.; Penkov, N.V.; Nedopekina, D.A.; Sharapov, V.A.; Khoroshavina, E.I.; Davletshin, E.V.; Belosludtseva, N.V.; Spivak, A.Y.; et al. Mitochondria-targeted prooxidant effects of betulinic acid conjugated with delocalized lipophilic cation F16. Free Radic. Biol. Med. 2021, 168, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Tsepaeva, O.V.; Nemtarev, A.V.; Salikhova, T.I.; Abdullin, T.I.; Grigor‘eva, L.R.; Khozyainova, S.A.; Mironov, V.F. Synthesis, Anticancer, and Antibacterial Activity of Betulinic and Betulonic Acid C-28-Triphenylphosphonium Conjugates with Variable Alkyl Linker Length. Anticancer Agents Med. Chem. 2020, 20, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Kraft, O.; Kozubek, M.; Hoenke, S.; Serbian, I.; Major, D.; Csuk, R. Cytotoxic triterpenoid–safirinium conjugates target the endoplasmic reticulum. Eur. J. Med. Chem. 2021, 209, 112920. [Google Scholar] [CrossRef]
- Bori, I.D.; Hung, H.-Y.; Qian, K.; Chen, C.-H.; Morris-Natschke, S.L.; Lee, K.-H. Anti-AIDS agents 88. Anti-HIV conjugates of betulin and betulinic acid with AZT prepared via click chemistry. Tetrahedron Lett. 2012, 53, 1987–1989. [Google Scholar] [CrossRef] [Green Version]
- Thi, T.A.D.; Tuyet, N.T.K.; The, C.P.; Nguyen, H.T.; Thi, C.B.; Duy, T.D.; D’Hooghe, M.; Van Nguyen, T. Synthesis and cytotoxic evaluation of novel ester-triazole-linked triterpenoid–AZT conjugates. Bioorg. Med. Chem. Lett. 2014, 24, 5190–5194. [Google Scholar] [CrossRef]
- Thi, T.A.D.; Tuyet, N.T.K.; The, C.P.; Nguyen, H.T.; Thi, C.B.; Phuong, H.T.; Van Boi, L.; Van Nguyen, T.; D’Hooghe, M. Synthesis and cytotoxic evaluation of novel amide–triazole-linked triterpenoid–AZT conjugates. Tetrahedron Lett. 2015, 56, 218–224. [Google Scholar] [CrossRef]
- Thurnher, D.; Turhani, D.; Pelzmann, M.; Wannemacher, B.; Knerer, B.; Formanek, M.; Wacheck, V.; Selzer, E. Betulinic acid: A new cytotoxic compound against malignant head and neck cancer cells. Head Neck 2003, 25, 732–740. [Google Scholar] [CrossRef]
- Eder-Czembirek, C.; Czembirek, C.; Erovic, B.; Selzer, E.; Turhani, D.; Vormittag, L.; Thurnher, D. Combination of betulinic acid with cisplatin—different cytotoxic effects in two head and neck cancer cell lines. Oncol. Rep. 2005, 14, 667–671. [Google Scholar] [CrossRef]
- Emmerich, D.; Vanchanagiri, K.; Baratto, L.C.; Schmidt, H.; Paschke, R. Synthesis and studies of anticancer properties of lupane-type triterpenoid derivatives containing a cisplatin fragment. Eur. J. Med. Chem. 2014, 75, 460–466. [Google Scholar] [CrossRef]
- Bache, M.; Hein, A.; Petrenko, M.; Güttler, A.; Keßler, J.; Wichmann, H.; Kappler, M.; Emmerich, D.; Paschke, R.; Vordermark, D. Evaluation of the Betulinic Acid–Cisplatin conjugate APC and its precursor DE9B for the treatment of human malignant glioma. Chem. Biol. Interact. 2019, 314, 108841. [Google Scholar] [CrossRef] [PubMed]
- Prodea, A.; Mioc, A.; Banciu, C.; Trandafirescu, C.; Milan, A.; Racoviceanu, R.; Ghiulai, R.; Mioc, M.; Soica, C. The Role of Cyclodextrins in the Design and Development of Triterpene-Based Therapeutic Agents. Int. J. Mol. Sci. 2022, 23, 736. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ding, X.; Fu, Y.; Xiang, C.; Yuan, Y.; Zhang, Y.; Yu, P. Cyclodextrins based delivery systems for macro biomolecules. Eur. J. Med. Chem. 2021, 212, 113105. [Google Scholar] [CrossRef] [PubMed]
- Lihumis, H.; Talebi, Z.; Shanan, S. Review on role alkyne and azide building blocks for click chemistry in organic synthesis and their application. Asian J. Green Chem. 2022, 6, 68–87. [Google Scholar]
- Chen, Y.; Wang, X.; Zhu, Y.; Si, L.; Zhang, B.; Zhang, Y.; Zhang, L.; Zhou, D.; Xiao, S. Synthesis of a Hexavalent Betulinic Acid Derivative as a Hemagglutinin-Targeted Influenza Virus Entry Inhibitor; ACS Publications: Washington, DC, USA, 2020; Volume 17, ISBN 8610828050. [Google Scholar]
- Chen, Y.; Wang, X.; Ma, X.; Liang, S.; Gao, Q.; Tretyakova, E.V.; Zhang, Y.; Zhou, D.; Xiao, S. Facial Synthesis and Bioevaluation of Well-Defined OEGylated Betulinic Acid-Cyclodextrin Conjugates for Inhibition of Influenza Infection. Molecules 2022, 27, 1163. [Google Scholar] [CrossRef]
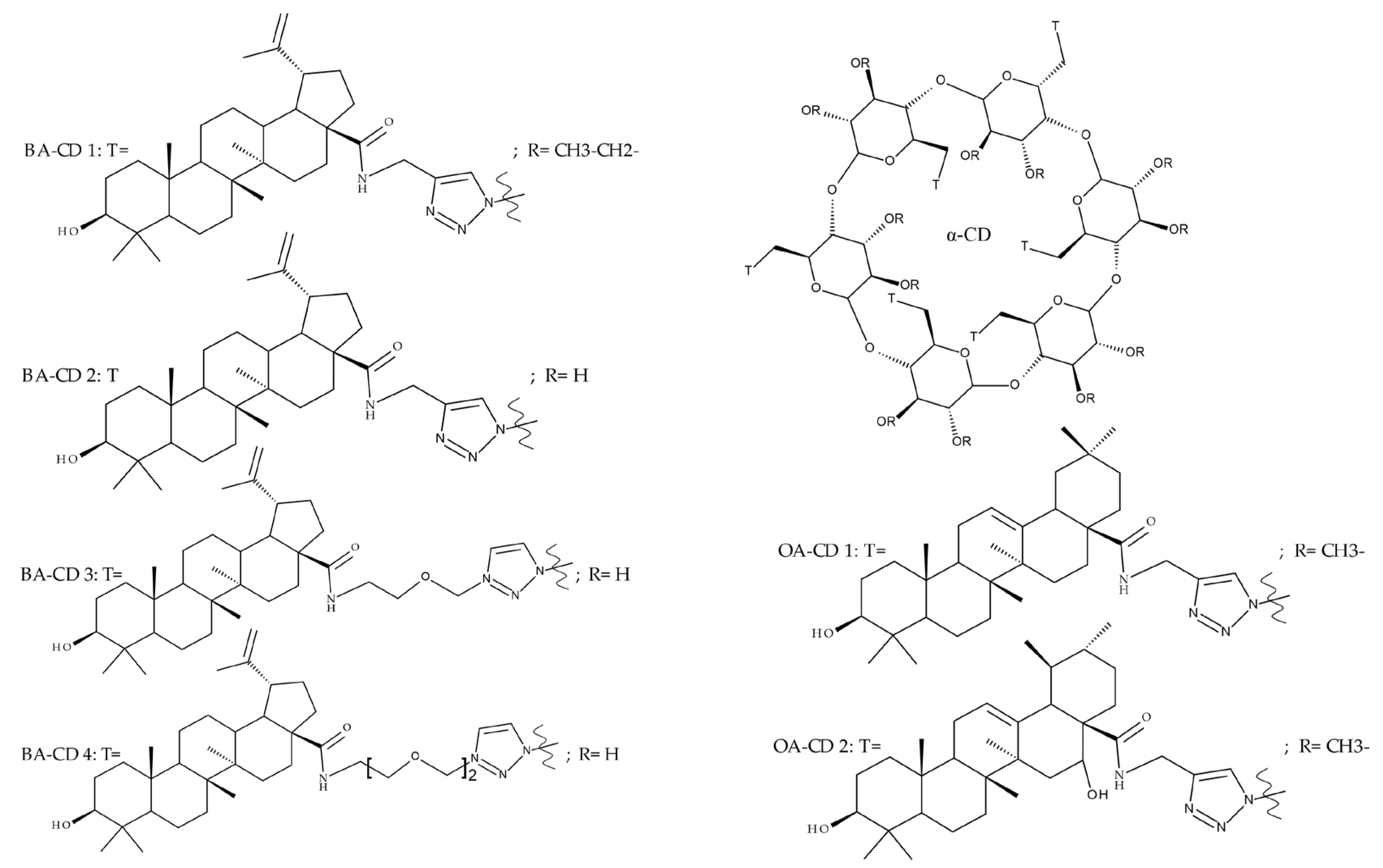
- Tian, Z.; Si, L.; Meng, K.; Zhou, X.; Zhang, Y.; Zhou, D.; Xiao, S. Inhibition of influenza virus infection by multivalent pentacyclic triterpene-functionalized per-O-methylated cyclodextrin conjugates. Eur. J. Med. Chem. 2017, 134, 133–139. [Google Scholar] [CrossRef]
- Khlebnikova, T.S.; Piven’, Y.A.; Lakhvich, F.A.; Frolova, T.S.; Sorokina, I.V.; Tolstikova, T.G. Synthesis and Cytotoxicity of Conjugates of Betulinic Acid and F-Containing 2-Acylcycloalkane-1,3-Diones. Chem. Nat. Compd. 2018, 54, 1100–1105. [Google Scholar] [CrossRef]
- Tajudin Mohd Ali, M.; Zahari, H.; Aliasak, A.; Meng Lim, S.; Ramasamy, K.; Patrick, G.; Macabeo, A. Synthesis, Characterization and Cytotoxic Activity of Betulinic Acid and Seco-Betulinic Acid Derivatives against Human Colorectal Carcinoma. Orient. J. Chem. 2017, 33, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Sidova, V.; Zoufaly, P.; Pokorny, J.; Dzubak, P.; Hajduch, M.; Popa, I.; Urban, M. Cytotoxic conjugates of betulinic acid and substituted triazoles prepared by Huisgen Cycloaddition from 30-azidoderivatives. PLoS ONE 2017, 12, e0171621. [Google Scholar] [CrossRef] [Green Version]
- Shakurova, E.R.; Parfenova, L.V. Synthesis of N-Heterocyclic Analogues of 28-O-Methyl Betulinate, and Their Antibacterial and Antifungal Properties. Molbank 2019, 2020, M1100. [Google Scholar] [CrossRef] [Green Version]
- Yamansarov, E.Y.; Skvortsov, D.A.; Lopukhov, A.V.; Kovalev, S.V.; Evteev, S.A.; Petrov, R.A.; Klyachko, N.L.; Zyk, N.V.; Beloglazkina, E.K.; Ivanenkov, Y.A.; et al. New ASGPR-targeted ligands based on glycoconjugated natural triterpenoids. Russ. Chem. Bull. 2019, 68, 2331–2338. [Google Scholar] [CrossRef]
- Olshanova, A.S.; Yamansarov, E.Y.; Seleznev, E.I.; Kovalev, S.V.; Lopuhov, A.V.; Skvortsov, D.A.; Evteev, S.A.; Klyachko, N.L.; Beloglazkina, E.K.; Ivanenkov, Y.A.; et al. Synthesis of a new betulinic acid glycoconjugate with N-acetyl-d-galactosamine for the targeted delivery to hepatocellular carcinoma cells. Russ. Chem. Bull. 2020, 69, 158–163. [Google Scholar] [CrossRef]
- Xu, J.; Li, Z.; Luo, J.; Yang, F.; Liu, T.; Liu, M.; Qiu, W.-W.; Tang, J. Synthesis and Biological Evaluation of Heterocyclic Ring-Fused Betulinic Acid Derivatives as Novel Inhibitors of Osteoclast Differentiation and Bone Resorption. J. Med. Chem. 2012, 55, 3122–3134. [Google Scholar] [CrossRef] [PubMed]
- Soural, M.; Hodon, J.; Dickinson, N.J.; Sidova, V.; Gurska, S.; Dzubak, P.; Hajduch, M.; Sarek, J.; Urban, M. Preparation of Conjugates of Cytotoxic Lupane Triterpenes with Biotin. Bioconjug. Chem. 2015, 26, 2563–2570. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Lv, S.; Wu, Z.; Qian, M.; Xu, Y.; Gao, X.; Wang, T.; Guo, W.; Hou, T.; Li, X.; et al. Role of betulinic acid derivative SH-479 in triple negative breast cancer and bone microenvironment. Oncol. Lett. 2021, 22, 605. [Google Scholar] [CrossRef]
- Li, J.; Jing, J.; Bai, Y.; Li, Z.; Xing, R.; Tan, B.; Ma, X.; Qiu, W.-W.; Du, C.; Du, B.; et al. SH479, a Betulinic Acid Derivative, Ameliorates Experimental Autoimmune Encephalomyelitis by Regulating the T Helper 17/Regulatory T Cell Balance. Mol. Pharmacol. 2017, 91, 464–474. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Bai, Y.; Li, Z.; Jia, K.; Jin, Y.; He, B.; Qiu, W.-W.; Du, C.; Siwko, S.; Chen, H.; et al. A betulinic acid derivative SH479 inhibits collagen-induced arthritis by modulating T cell differentiation and cytokine balance. Biochem. Pharmacol. 2017, 126, 69–78. [Google Scholar] [CrossRef]
- Zhang, H.; Li, F.; Zhu, P.; Liu, J.; Yao, H.; Jiang, J.; Ye, W.; Wu, X.; Xu, J. Synthesis and Biological Evaluation of Oxygen-containing Heterocyclic Ring-fused 23-Hydroxybetulinic Acid Derivatives as Antitumor Agents. Chem. Biol. Drug Des. 2015, 86, 424–431. [Google Scholar] [CrossRef]
- Lombrea, A.; Scurtu, A.D.; Avram, S.; Pavel, I.Z.; Turks, M.; Lugiņina, J.; Peipiņš, U.; Dehelean, C.A.; Soica, C.; Danciu, C. Anticancer potential of betulonic acid derivatives. Int. J. Mol. Sci. 2021, 22, 3676. [Google Scholar] [CrossRef]
- Sorokina, I.V.; Baev, D.S.; Zhukova, N.A.; Tolstikova, T.G.; Antimonova, A.N.; Petrenko, N.I.; Shults, E.E.; Grigor’ev, I.A. Hepatoprotective activity of betulonic acid amides containing piperidine or pyrrolidine nitroxide moieties. Russ. J. Bioorg. Chem. 2013, 39, 668–670. [Google Scholar] [CrossRef]
- Khlebnicova, T.S.; Piven, Y.A.; Baranovsky, A.V.; Lakhvich, F.A.; Sorokina, I.V.; Tolstikova, T.G. Fluorine-containing lupane triterpenoid acid derivatives: Design, synthesis and biological evaluation as potential anti-inflammatory agents. Steroids 2019, 147, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.A.; Semenova, M.D.; Baev, D.S.; Sorokina, I.V.; Zhukova, N.A.; Frolova, T.S.; Tolstikova, T.G.; Shults, E.E.; Turks, M. Lupane-type conjugates with aminoacids, 1,3,4- oxadiazole and 1,2,5-oxadiazole-2-oxide derivatives: Synthesis, anti-inflammatory activity and in silico evaluation of target affinity. Steroids 2019, 150, 108443. [Google Scholar] [CrossRef] [PubMed]
- Vasilevsky, S.F.; Govdi, A.I.; Sorokina, I.V.; Tolstikova, T.G.; Baev, D.S.; Tolstikov, G.A.; Mamatuyk, V.I.; Alabugin, I.V. Rapid access to new bioconjugates of betulonic acid via click chemistry. Bioorg. Med. Chem. Lett. 2011, 21, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Lipeeva, A.V.; Dolgikh, M.P.; Tolstikova, T.G.; Shults, E.E. A Study of Plant Coumarins. 18. Conjugates of Coumarins with Lupane Triterpenoids and 1,2,3-Triazoles: Synthesis and Anti-Inflammatory Activity. Russ. J. Bioorg. Chem. 2020, 46, 125–132. [Google Scholar] [CrossRef]
- Bębenek, E.; Kadela-Tomanek, M.; Chrobak, E.; Latocha, M.; Boryczka, S. Novel triazoles of 3-acetylbetulin and betulone as anticancer agents. Med. Chem. Res. 2018, 27, 2051–2061. [Google Scholar] [CrossRef] [Green Version]
- Borková, L.; Frydrych, I.; Jakubcová, N.; Adámek, R.; Lišková, B.; Gurská, S.; Medvedíková, M.; Hajdúch, M.; Urban, M. Synthesis and biological evaluation of triterpenoid thiazoles derived from betulonic acid, dihydrobetulonic acid, and ursonic acid. Eur. J. Med. Chem. 2020, 185, 111806. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Smirnova, I.E.; Kazakova, O.B. Synthesis and Cytotoxicity of 28-Oxo-Allobetulone Derivatives. Chem. Nat. Compd. 2020, 56, 465–471. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Giniyatullina, G.V.; Mustafin, A.G.; Babkov, D.A.; Sokolova, E.V.; Spasov, A.A. Evaluation of Cytotoxicity and α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids. Molecules 2020, 25, 4833. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Smirnova, I.E.; Kazakova, O.B.; Petrova, A.V.; Ha, N.T.T.; Viet, D.Q. Synthesis and evaluation of 2,3-indolotriterpenoids as new α-glucosidase inhibitors. Med. Chem. Res. 2017, 26, 2737–2742. [Google Scholar] [CrossRef]
- Cargnin, S.T.; Gnoatto, S.B. Ursolic acid from apple pomace and traditional plants: A valuable triterpenoid with functional properties. Food Chem. 2017, 220, 477–489. [Google Scholar] [CrossRef]
- Khwaza, V.; Oyedeji, O.O.; Aderibigbe, B.A. Ursolic Acid-Based Derivatives as Potential Anti-Cancer Agents: An Update. Int. J. Mol. Sci. 2020, 21, 5920. [Google Scholar] [CrossRef]
- Popov, S.A.; Semenova, M.D.; Baev, D.S.; Frolova, T.S.; Shestopalov, M.A.; Wang, C.; Qi, Z.; Shults, E.E.; Turks, M. Synthesis and cytotoxicity of hybrids of 1,3,4- or 1,2,5-oxadiazoles tethered from ursane and lupane core with 1,2,3-triazole. Steroids 2020, 162, 108698. [Google Scholar] [CrossRef] [PubMed]
- Brandes, B.; Hoenke, S.; Fischer, L.; Csuk, R. Design, synthesis and cytotoxicity of BODIPY FL labelled triterpenoids. Eur. J. Med. Chem. 2020, 185, 111858. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Ozbey, U.; Sabitaliyevich, U.Y.; Attar, R.; Ozcelik, B.; Zhang, Y.; Guo, M.; Liu, M.; Alhewairini, S.S.; Farooqi, A.A. Maslinic acid as an effective anticancer agent. Cell. Mol. Biol. 2018, 64, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, G.; Vail, D.R.; Nair, V.; Medina-Bolivar, F.; Lay, J.O. Plant-based corosolic acid: Future anti-diabetic drug? Biotechnol. J. 2009, 4, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Assefa, S.T.; Yang, E.-Y.; Chae, S.-Y.; Song, M.; Lee, J.; Cho, M.-C.; Jang, S. Alpha Glucosidase Inhibitory Activities of Plants with Focus on Common Vegetables. Plants 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zang, X.; Yin, X.; Yang, W.; Huang, J.; Huang, J.; Yu, C.; Ke, C.; Hong, Y. Semi-synthesis of C28-modified triterpene acid derivatives from maslinic acid or corosolic acid as potential α-glucosidase inhibitors. Bioorg. Chem. 2020, 97, 103694. [Google Scholar] [CrossRef]
- Pollier, J.; Goossens, A. Oleanolic acid. Phytochemistry 2012, 77, 10–15. [Google Scholar] [CrossRef]
- Gupta, N. A Review on Recent Developments in the Anticancer Potential of Oleanolic Acid and its Analogs (2017-2020). Mini Rev. Med. Chem. 2022, 22, 600–616. [Google Scholar] [CrossRef]
- Medvedeva, N.I.; Kazakova, O.B. Synthesis and Cytotoxicity of Lupaneand Oleanane-Type Triterpenoid Conjugates with 1,3,4-Oxadiazole. Chem. Nat. Compd. 2021, 57, 485–490. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
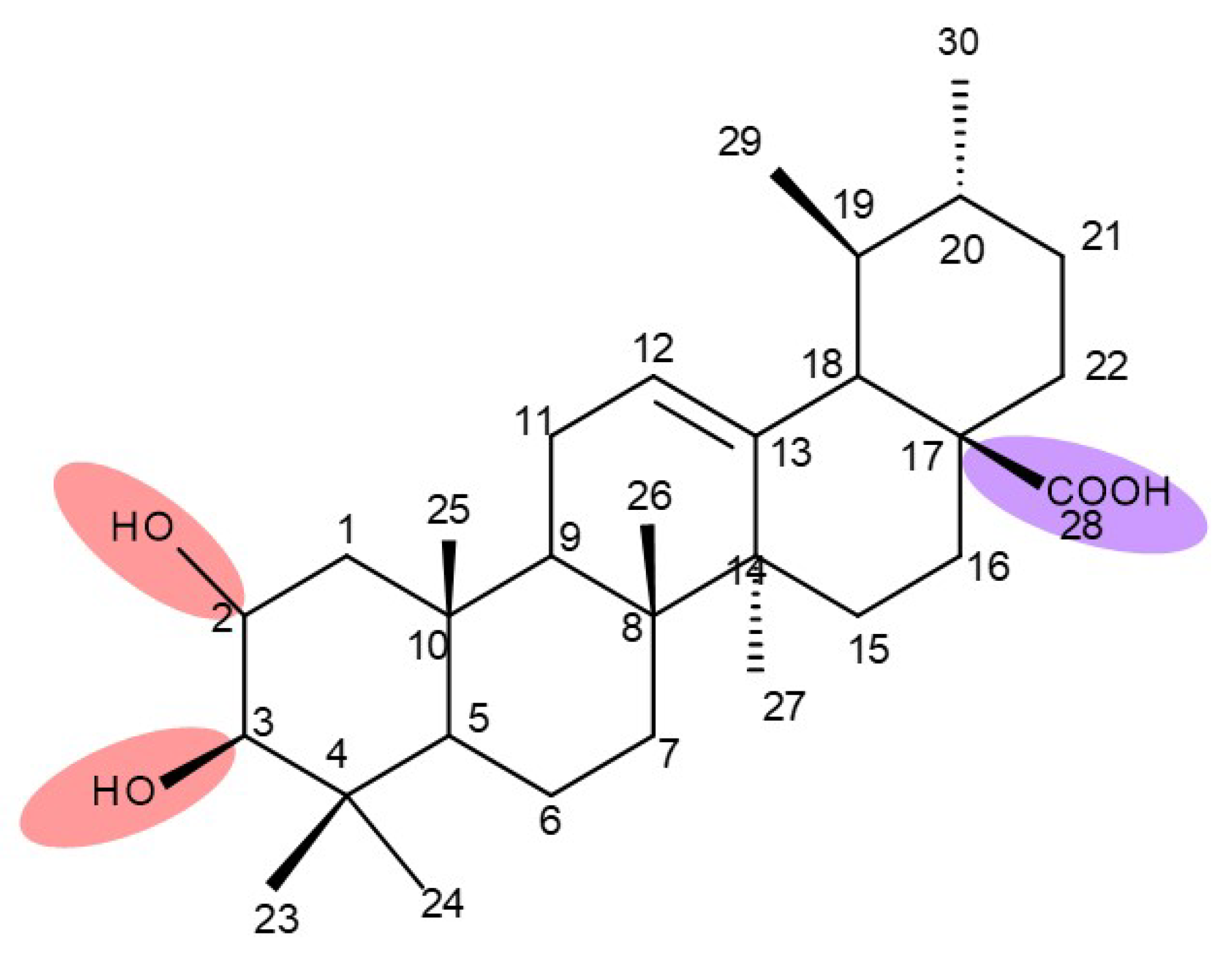{kind=link}
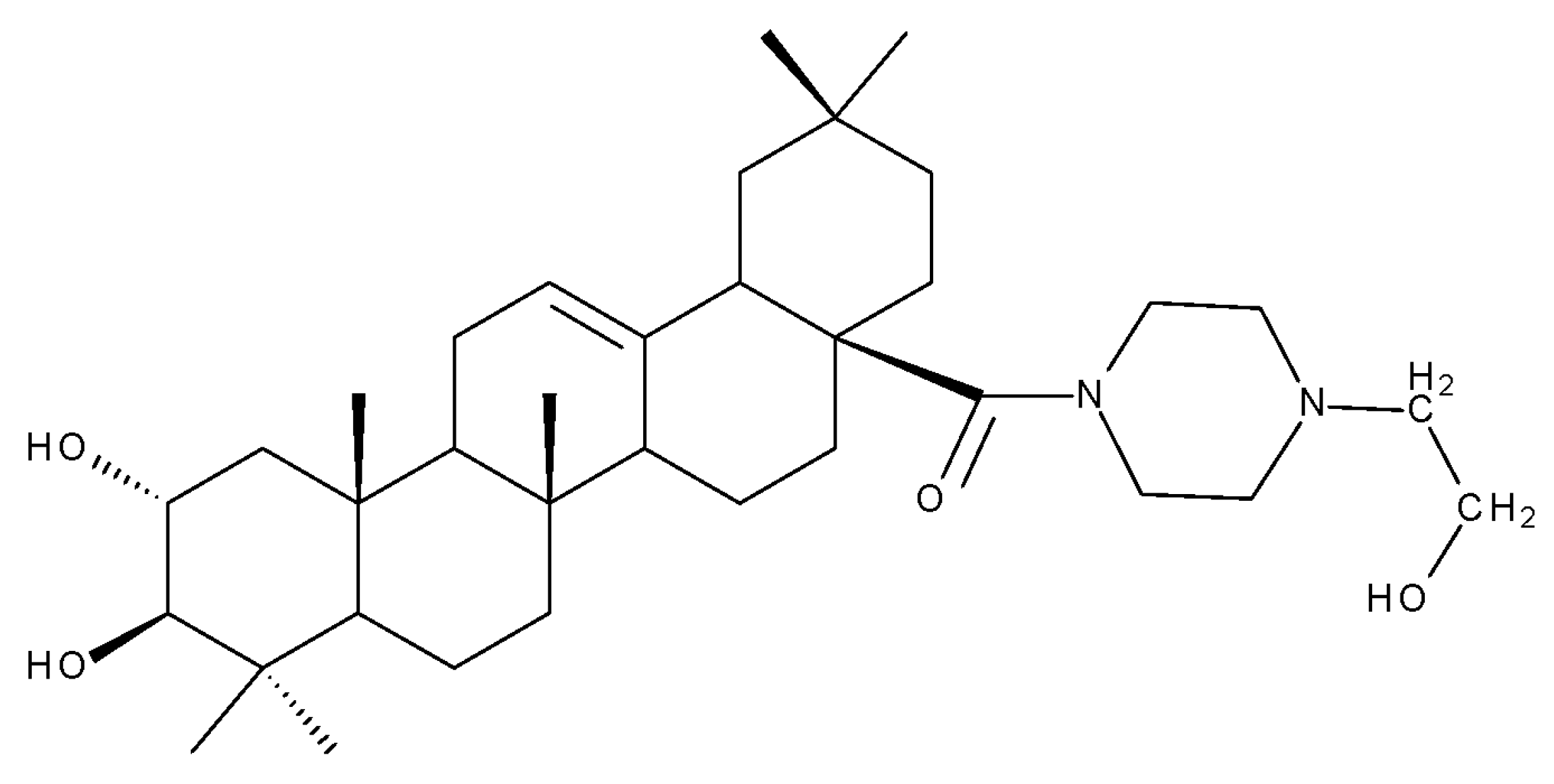{kind=link}
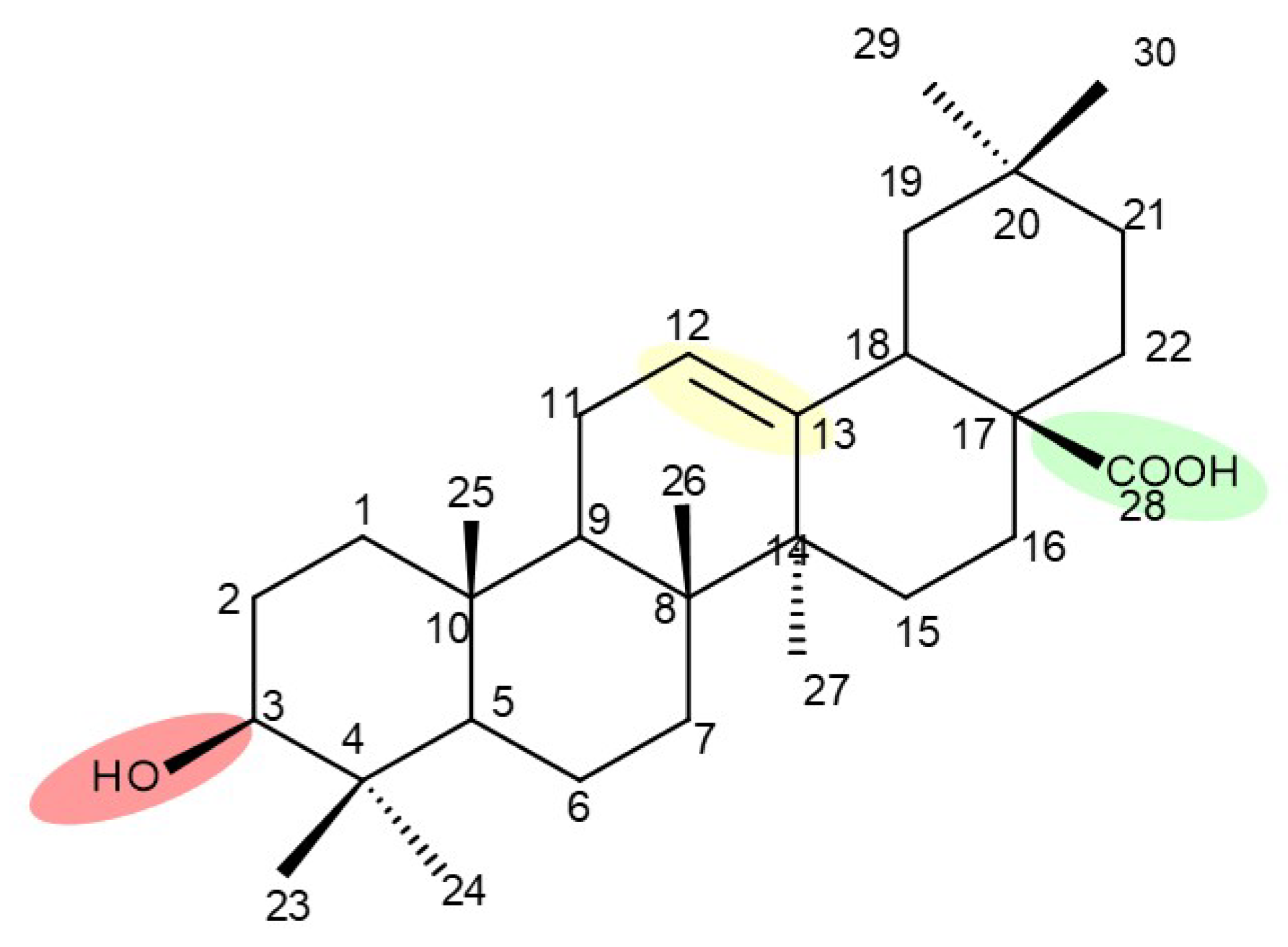{kind=link}
| Compound | Structure | Substituents | Reference |
|---|---|---|---|
| Lup 1–3 |  |  | [15] |
| Lup 4–7 |  |  | [15] |
| Lup 8–9 |  |  | [16] |
| Lup 10–11 |  |  | [16] |
| Lup 12–13 |  |  | [18] |
| Compound | Structure | Substituents | Reference |
|---|---|---|---|
| Bet 1 |  | Ac: -C=O-CH3 | [27] |
| Bet 2–5 |  | Bet 2 R: -OH Bet-3 R: =O Bet-4 R: =OC(O)CH3 Bet-5 R: =OC(O)CH2CH3 | [28] |
| Bet 6 |  | [29] | |
| Bet 7 |  | [30] | |
| Bet 8 |  | [31] | |
| Bet 9–10 |  | Bet-9 R:  Bet-10 R:  | [32] |
| Bet 11 |  | ||
| Bet 12 |  | ||
| Bet 13 |  | Ac: -C=O-CH3 | |
| Bet 14 |  | ||
| Bet 15 |  | [33] | |
| Bet 16 |  | [35] | |
| Bet 17 |  | Ac: -C=O-CH3 | [36] |
| Bet 18–20 |  | Bet 18 R2: -C(O)C≡CH Y:H, X:N Bet 19 R2: =C(O)CH3 Y:H, X: CH Bet 20 R2: -C(O)C≡CH Y:H, X:CH | [37] |
| Bet 21 |  | ||
| Bet 22 |  | ||
| Bet 23 |  | Ac: -C=O-CH3 | [38] |
| Bet 24 |  | ||
| Bet 25 |  | Ac: -C=O-CH3 | [39] |
| Bet 26 |  | [40] | |
| Bet 27 |  | [41] | |
| Bet 28 |  | Ac: -C=O-CH3 | |
| Bet 29 |  | [42] | |
| Bet 30 |  | [43] | |
| Bet 31 |  | [34] | |
| Bet 32 |  | ||
| Bet 33 |  | ||
| Bet 34–36 |  | Bet 36 R1: -H, R2: -OH, R3: -OCOCH3 Bet 37 R1: -H R2 = R3: -OH Bet 38 R1: -OH R2: -H R3: -OH | [46] |
| Bet 37 |  | [47] | |
| Bet 38 |  | ||
| Bet 39 |  | [48] | |
| Bet 40 |  | [49] | |
| Bet 41 |  | [50] | |
| Bet 42 |  | [51] |
| Cell Line | Type of Cells Cancer/Non-Cancer | BA and BA Derivatives | ||||
|---|---|---|---|---|---|---|
| BA | Beviramat | BA 27 | BA 28 | BA 29 | ||
| CCRF-CEM | Leukemia | >50 | 12.82 | 0.29 | 1.55 | 2.92 |
| CEM-DNR | Leukemia | 23.05 | 22.17 | 0.35 | 11.53 | 7.00 |
| K562 | Leukemia | >50 | 23.60 | 0.40 | 5.25 | 23.19 |
| K562-Tax | Leukemia | >50 | 22.03 | 0.52 | 31.80 | 12.21 |
| A549 | Lung adenocarcinoma | 22.68 | 23.06 | 1.26 | 6.65 | 13.55 |
| HCT116 | Colorectal adenocarcinoma | >50 | 14.17 | 0.39 | 3.85 | 7.92 |
| HCT116P53−/− | Colorectal adenocarcinoma | >50 | 18.20 | 0.44 | 3.39 | 8.80 |
| U2OS | Osteosarcoma | 29.69 | 27.63 | 0.42 | 5.00 | 12.38 |
| MRC-5 | Human fibroblasts | >50 | >50 | 1.58 | 8.07 | 14.12 |
| BJ | Human fibroblasts | >50 | >50 | 1.59 | 8.37 | 15.49 |
| Compound | Structure | Substituents | Reference |
| BA 1 |  | - | [56] |
| BA 2–3 |  |  | [57] |
| BA 4–6 |  | - | [23] |
| BA 7–9 |  | - | [23] |
| BA 10 |  | - | [59] |
| BA 11–12 |  |  | [60] |
| BA 13 |  | - | [31] |
| BA 14–21 |  |  | [61] |
| BA 22 |  | - | [63] |
| BA 23–25 |  |  | [63] |
| BA 26 |  | [64] | |
| Beviramat |  | [65] | |
| BA 27 |  | [65] | |
| BA 28 |  | [65] | |
| BA 29 |  | [65] | |
| BA 30 |  | [67] | |
| BA 31–32 |  |  | [68] |
| BA 33–34 |  |  | [68] |
| BA 35 |  | - | [69] |
| BA 36 |  | - | [70] |
| BA 37–40 |  |  | [70] |
| BA 41–42 |  |  | [73] |
| BA 43 |  | [73] | |
| BA 44 |  | - | [74] |
| BA 45 |  | R = hexyl | [75] |
| BA 46 |  | [76] | |
| BA 47 |  | [77] | |
| BA 48–49 |  |  | [77] |
| BA 50 |  | - | [77] |
| BA 51 |  | - | [33] |
| BA 52 |  | [82] | |
| BA 53 |  | - | [91] |
| BA 54–55 |  |  | [92] |
| BA 56–57 |  | BA 56: R1= CHR-CH2-; R2= -COOH BA 57: R1= H; R2= -CH2-OH | [93] |
| BA 58 |  | - | [94] |
| SH479 |  | - | [95] |
| Compound | Structure | Substituents | Reference |
| BoA 1–2 |  |  | [102] |
| BoA 3 |  | - | [103] |
| BoA 4–5 |  |  | [104] |
| BoA 6 |  | - | [105] |
| BoA 7–8 |  |  | [106] |
| BoA 9 |  | - | [107] |
| BoA 10 |  | - | [108] |
| BoA 11 |  | [109] | |
| BoA 12 |  | [109] | |
| BoA 13 |  | - | [110] |
| BoA 14 |  | - | [111] |
| Compounds | Structure | Substituents | Reference |
|---|---|---|---|
| UA 1–4 |  | UA 1 R: -Rha  UA 2 R: -diRha UA 3 R: -triRha UA 4 R: -tetraRha | [59] |
| UA 5–6 |  | UA 5 R: -H UA 6 R: -C=O-CH3 | [114] |
| UA 7 |  | - | [115] |
| Number | Structure | Substituents | Reference |
| OA 1 |  | [122] | |
| OA 2 |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nistor, G.; Trandafirescu, C.; Prodea, A.; Milan, A.; Cristea, A.; Ghiulai, R.; Racoviceanu, R.; Mioc, A.; Mioc, M.; Ivan, V.; et al. Semisynthetic Derivatives of Pentacyclic Triterpenes Bearing Heterocyclic Moieties with Therapeutic Potential. Molecules 2022, 27, 6552. https://doi.org/10.3390/molecules27196552
Nistor G, Trandafirescu C, Prodea A, Milan A, Cristea A, Ghiulai R, Racoviceanu R, Mioc A, Mioc M, Ivan V, et al. Semisynthetic Derivatives of Pentacyclic Triterpenes Bearing Heterocyclic Moieties with Therapeutic Potential. Molecules. 2022; 27(19):6552. https://doi.org/10.3390/molecules27196552
Chicago/Turabian StyleNistor, Gabriela, Cristina Trandafirescu, Alexandra Prodea, Andreea Milan, Andreea Cristea, Roxana Ghiulai, Roxana Racoviceanu, Alexandra Mioc, Marius Mioc, Viviana Ivan, and et al. 2022. "Semisynthetic Derivatives of Pentacyclic Triterpenes Bearing Heterocyclic Moieties with Therapeutic Potential" Molecules 27, no. 19: 6552. https://doi.org/10.3390/molecules27196552
APA StyleNistor, G., Trandafirescu, C., Prodea, A., Milan, A., Cristea, A., Ghiulai, R., Racoviceanu, R., Mioc, A., Mioc, M., Ivan, V., & Șoica, C. (2022). Semisynthetic Derivatives of Pentacyclic Triterpenes Bearing Heterocyclic Moieties with Therapeutic Potential. Molecules, 27(19), 6552. https://doi.org/10.3390/molecules27196552