
The Discovery of Small Allosteric and Active Site Inhibitors of the SARS-CoV-2 Main Protease via Structure-Based Virtual Screening and Biological Evaluation

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
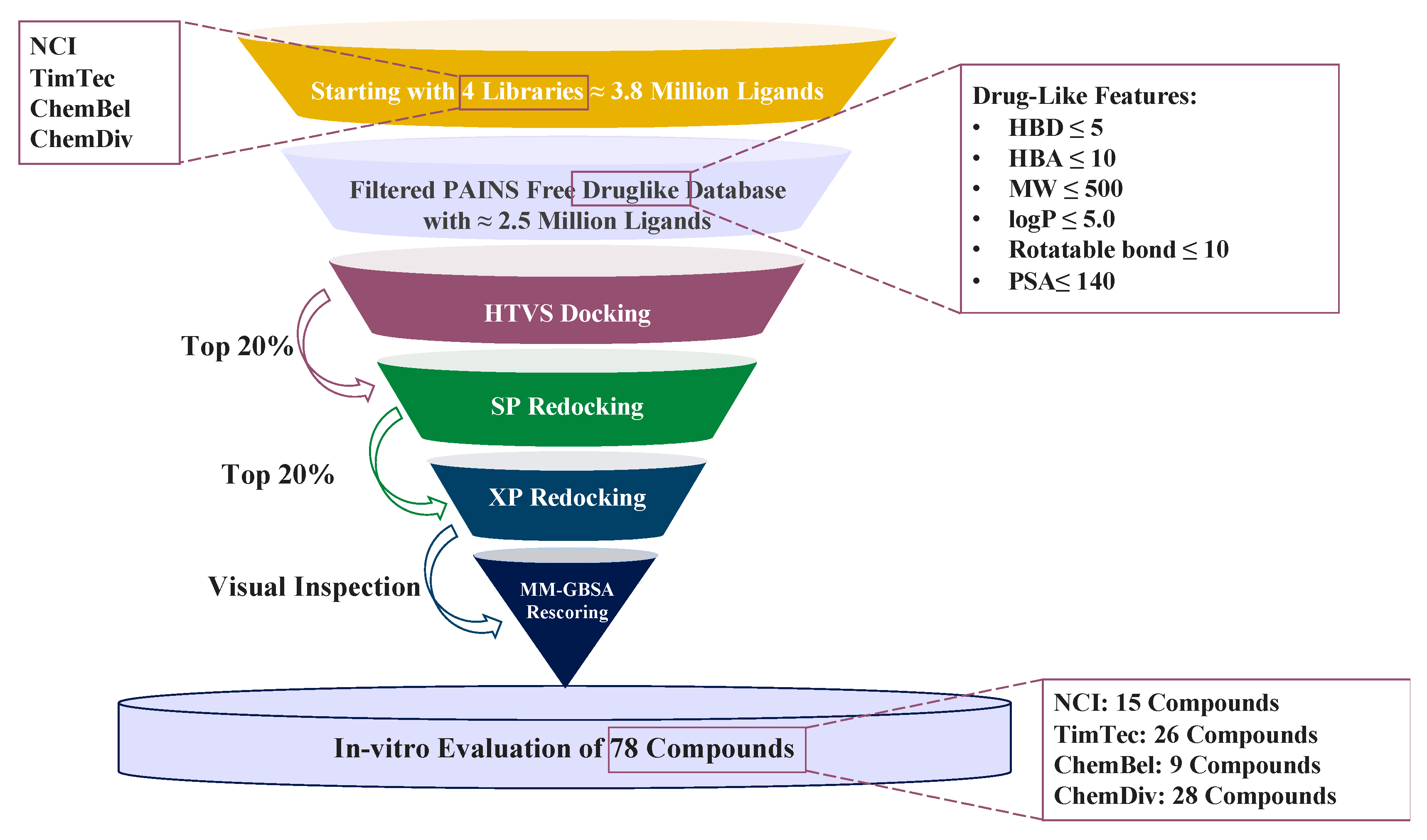
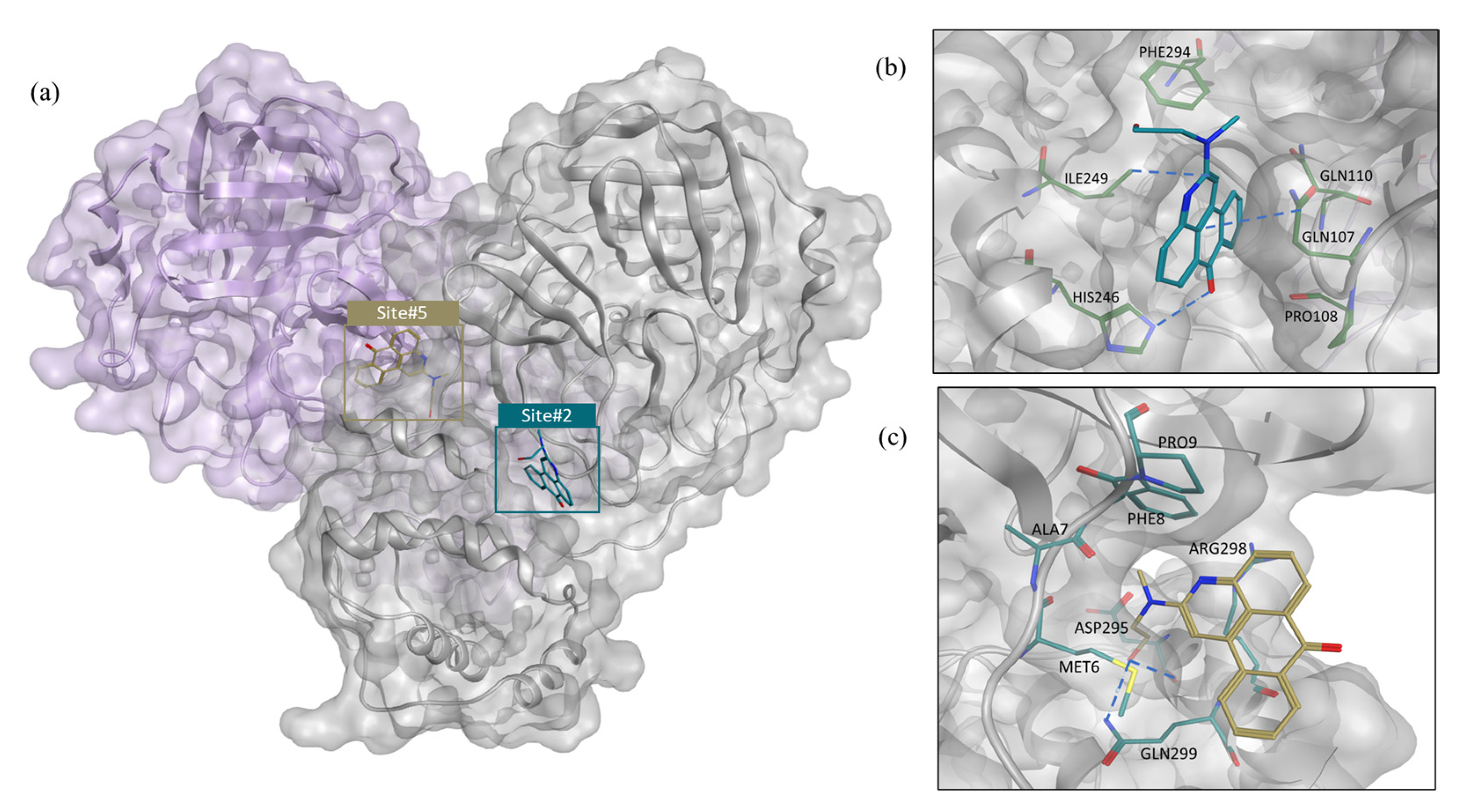
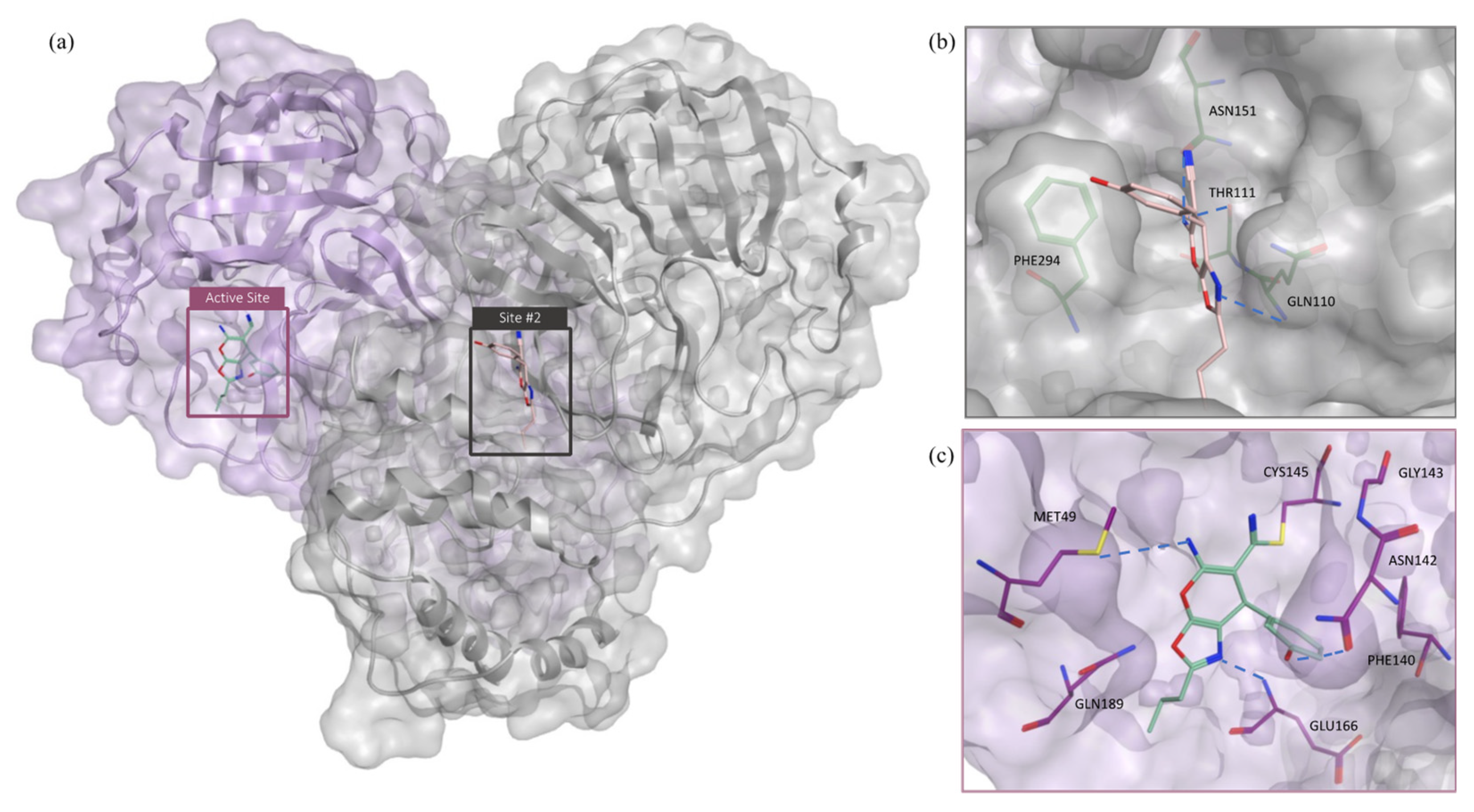
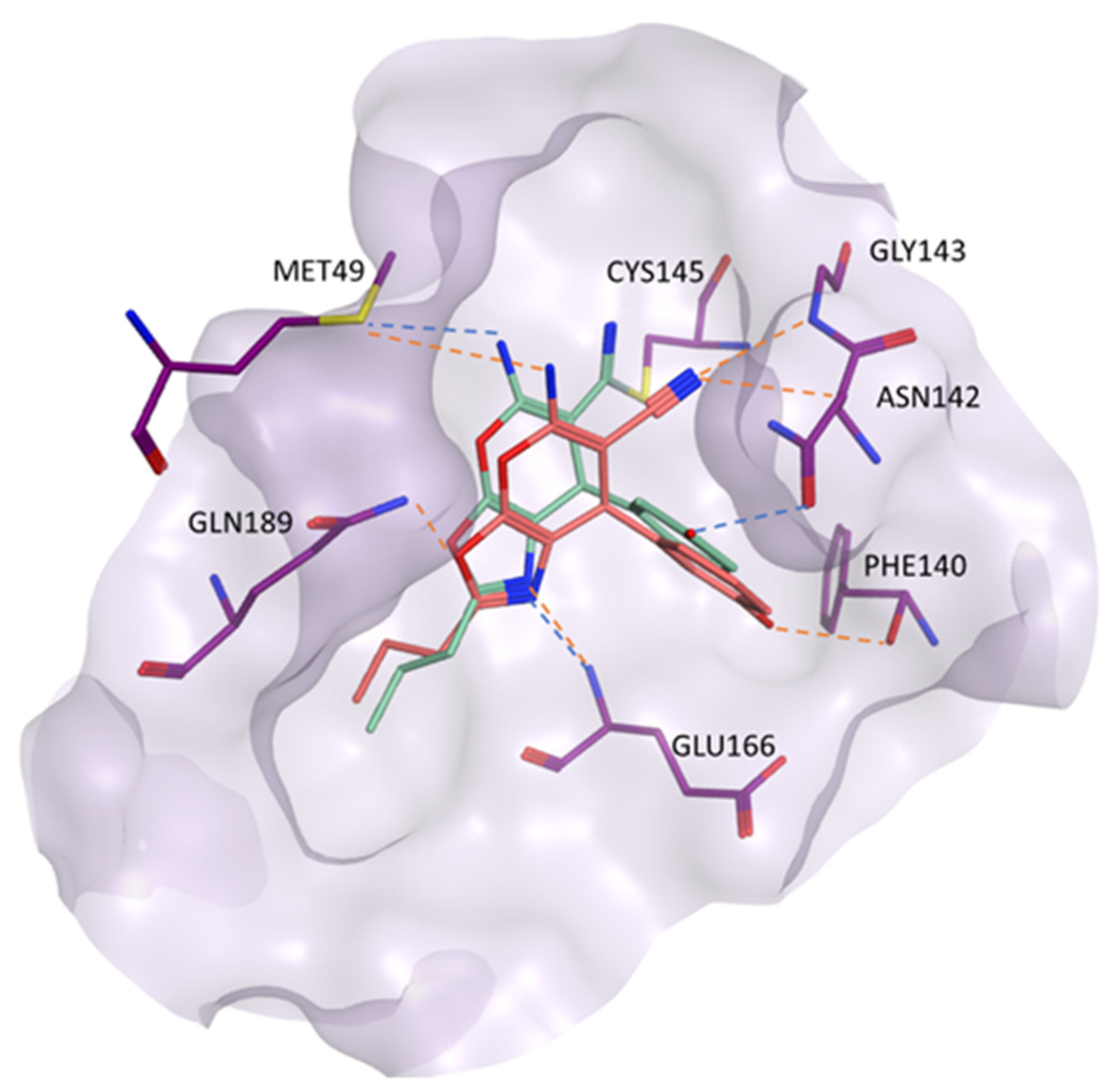
2.1. Structure-Based Virtual Screening
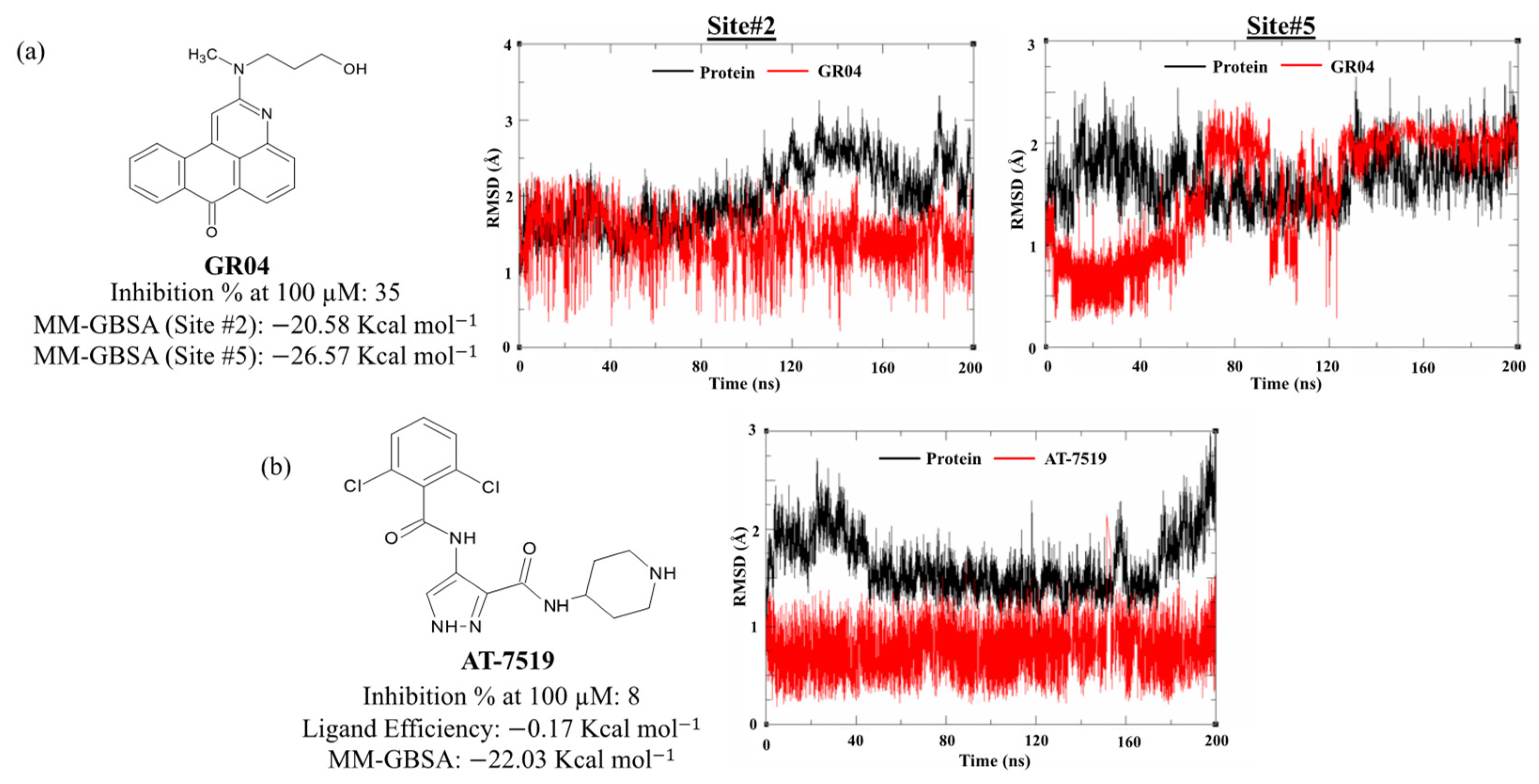
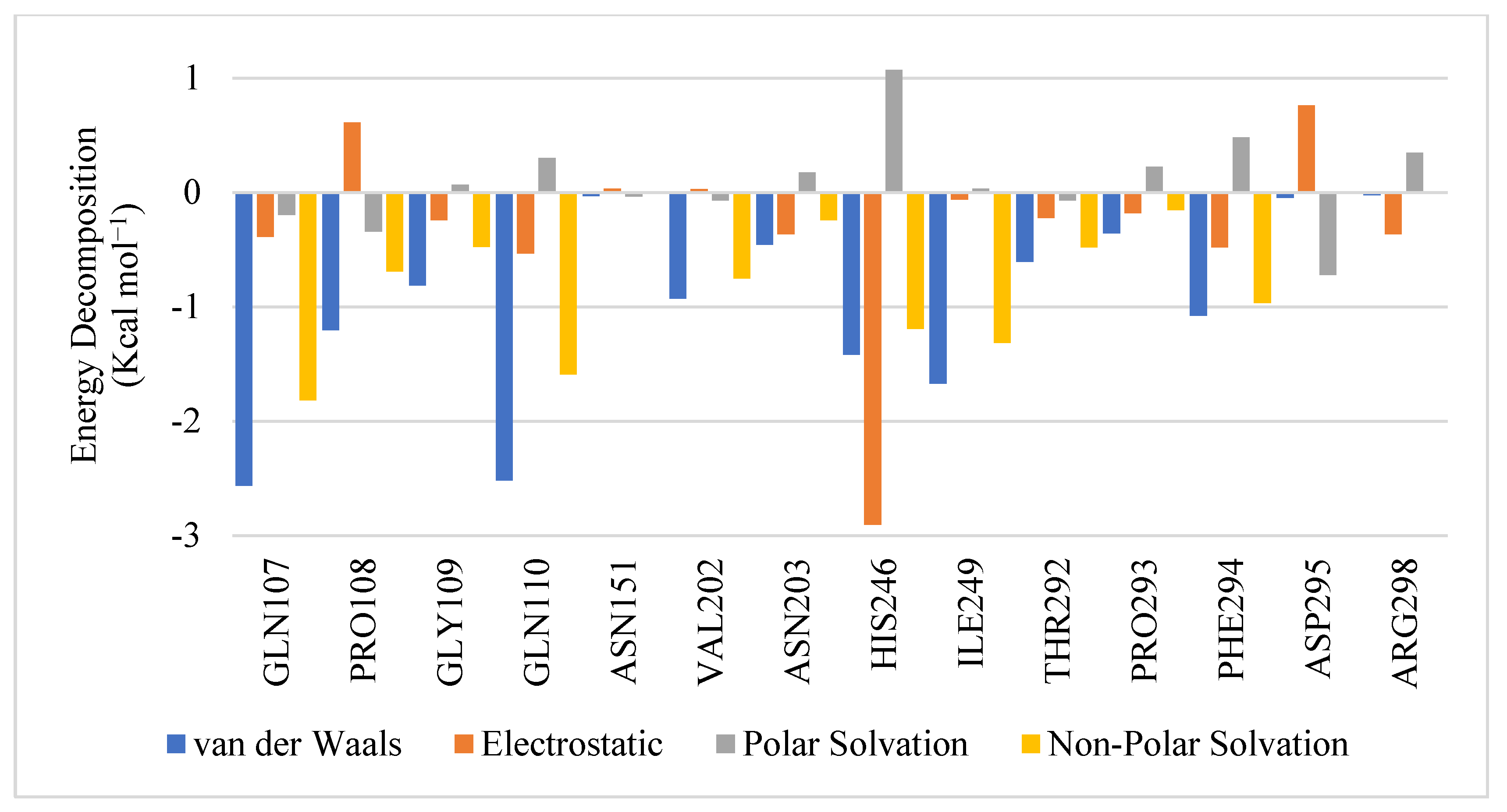
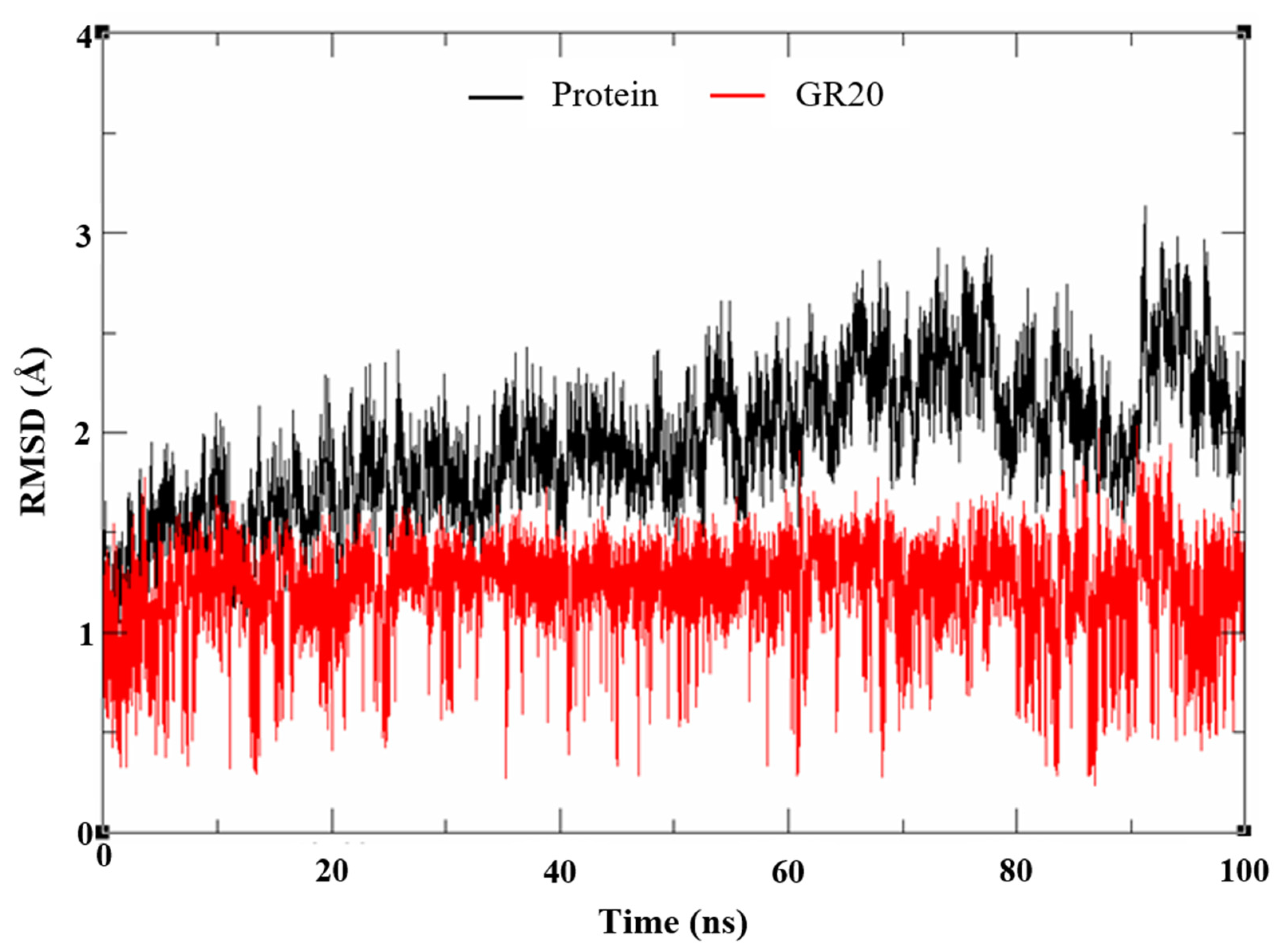
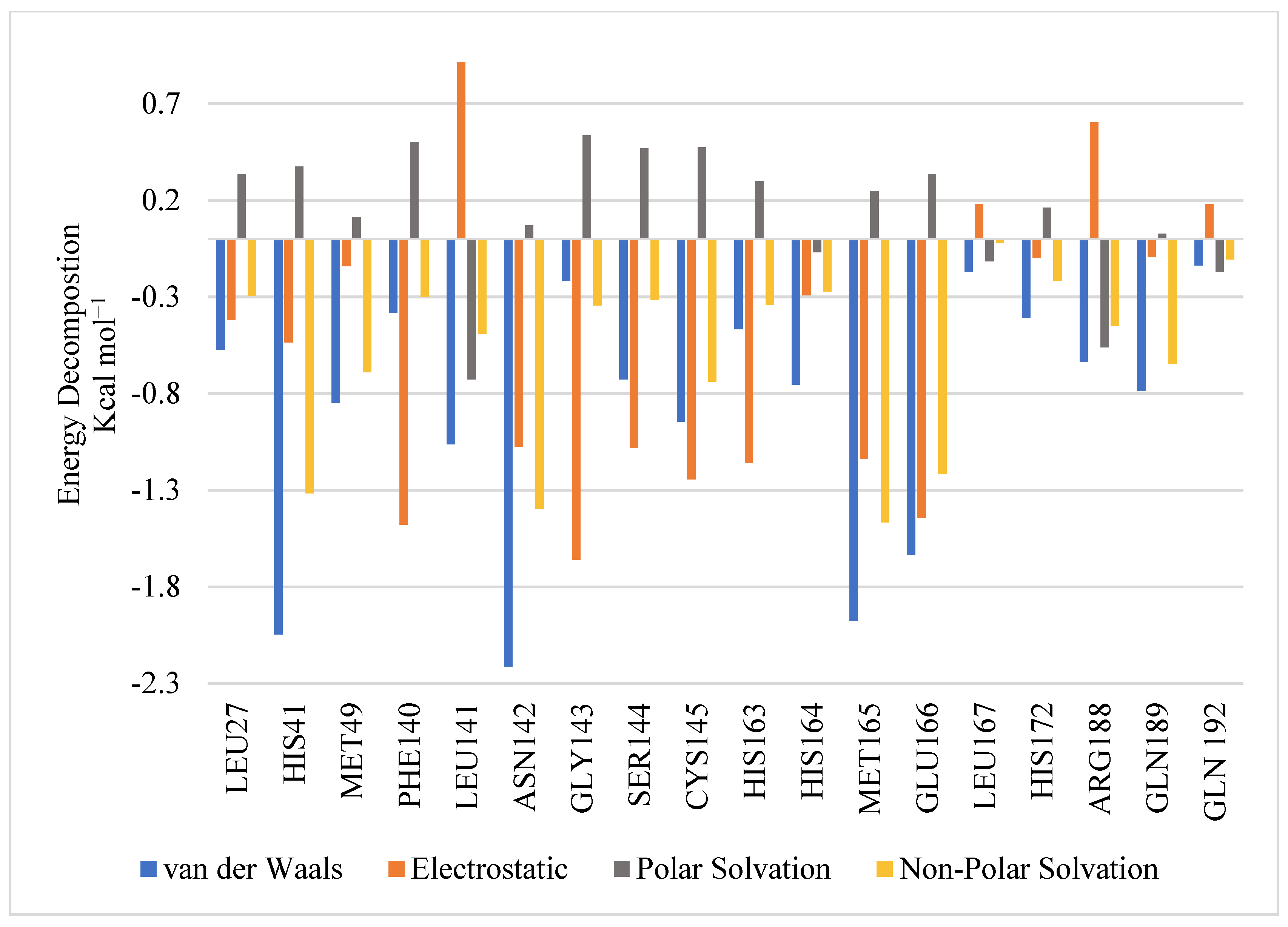
2.2. MD Simulation and MM-GBSA Scoring
2.3. ADME Profile of Top Hits
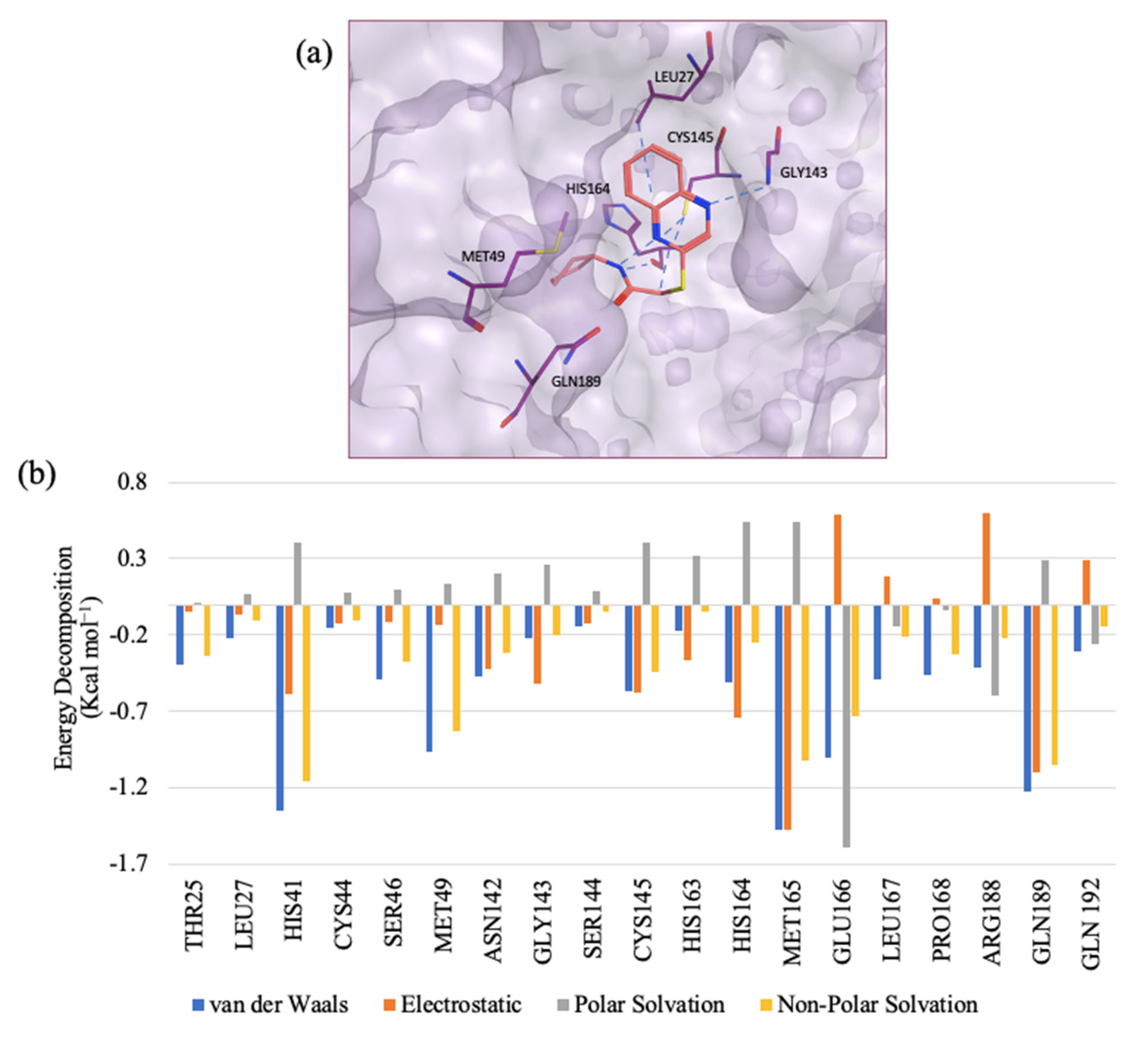
2.4. Enzyme Assay and IC50 Determination
2.5. Promiscuity Tests
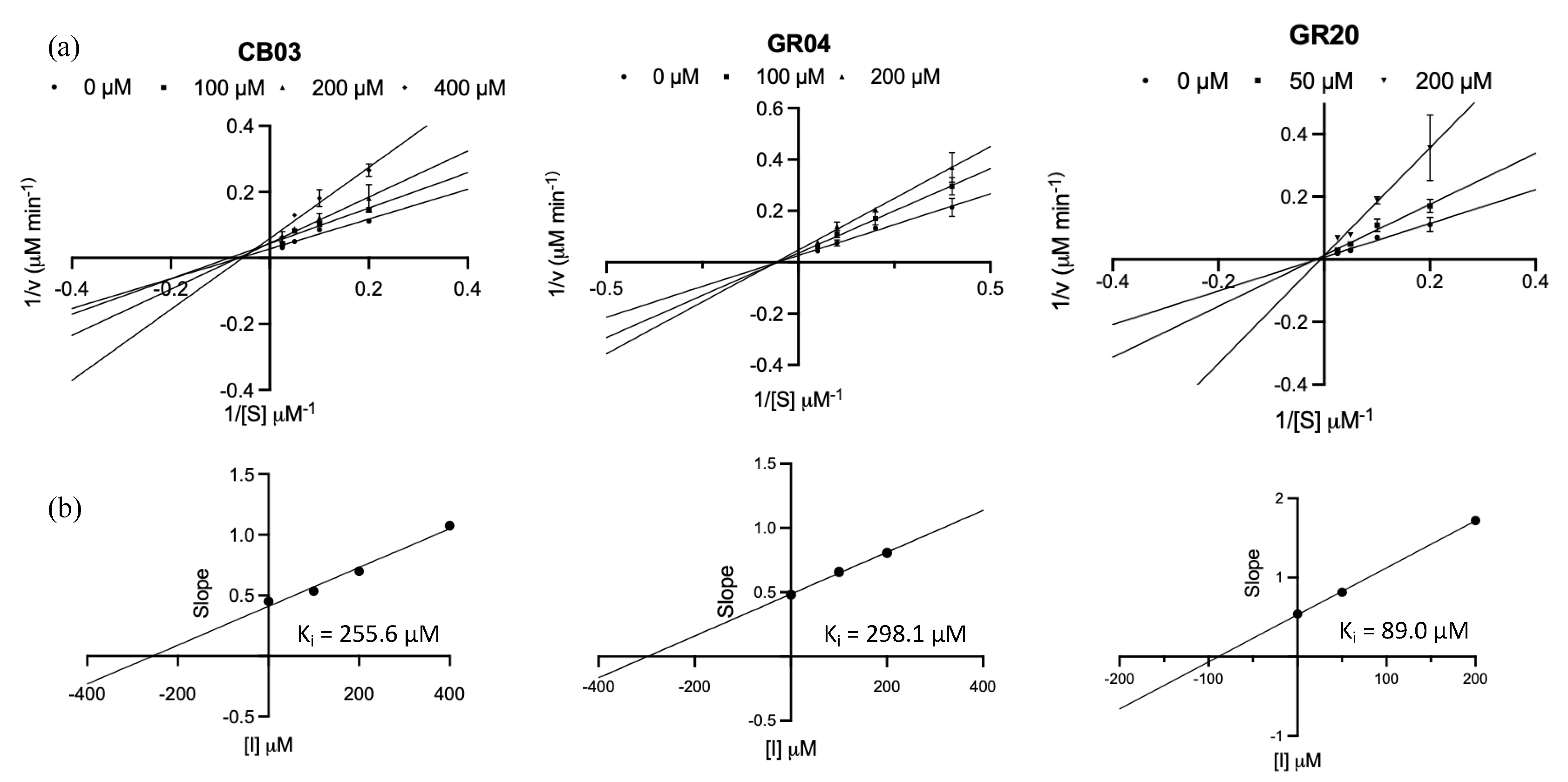
2.6. Inhibition Kinetic Study of CB03, GR04, and GR20
2.6.1. CB03 as a Competitive Inhibitor
2.6.2. GR04 as a Noncompetitive Inhibitor
2.6.3. GR20 as an Irreversible vs. Noncompetitive Inhibitor
3. Materials and Methods
3.1. Protein Preparation and Grid Generation
3.2. Ligand Filtration and Preparation
3.3. Structure-Based Virtual Screening
3.4. MD Simulation and MM-GBSA Scoring
3.5. Covalent Docking of GR20
3.6. Expression and Purification of Mpro
3.7. ADME Screening
3.8. Enzyme Assay and IC50 Determination
3.9. Promiscuity Tests (BSA and Increasing Enzyme Concentration)
3.10. Kinetic Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lam, S.; Lombardi, A.; Ouanounou, A. COVID-19: A Review of the Proposed Pharmacological Treatments. Eur. J. Pharmacol. 2020, 886, 173451. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.W.; Li, K.S.M.; To, K.K.W.; Cheng, V.C.C.; Chen, H.; Yuen, K.Y. Is the Discovery of the Novel Human Betacoronavirus 2c EMC/2012 (HCoV-EMC) the Beginning of Another SARS-like Pandemic? J. Infect. 2012, 65, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO Coronavirus (COVID-19) Dashboard|WHO Coronavirus (COVID-19) Dashboard with Vaccination Data. Available online: https://covid19.who.int/table (accessed on 13 March 2022).
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Rox, K.; Hilgenfeld, R. X-Ray Structure of Main Protease of the Novel Coronavirus SARS-CoV-2 Enables Design of α-Ketoamide Inhibitors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 7. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Song, J. The Catalysis of the SARS 3C-like Protease Is under Extensive Regulation by Its Extra Domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of Two Coronavirus Main Proteases: Implications for Substrate Binding and Antiviral Drug Design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Pan, Y.; Huang, N.; Cho, S.; MacKerell, A.D. Consideration of Molecular Weight during Compound Selection in Virtual Target-Based Database Screening. J. Chem. Inf. Comput. Sci. 2003, 43, 267–272. [Google Scholar] [CrossRef]
- Guimarães, C.R.W.; Cardozo, M. MM-GB/SA Rescoring of Docking Poses in Structure-Based Lead Optimization. J. Chem. Inf. Model. 2008, 48, 958–970. [Google Scholar] [CrossRef]
- Montanari, F.; Ecker, G.F. Prediction of Drug-ABC-Transporter Interaction—Recent Advances and Future Challenges. In Advanced Drug Delivery Reviews; Elsevier: Amsterdam, The Netherlands, 2015; pp. 17–26. [Google Scholar] [CrossRef] [Green Version]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Hugh Gilbert, I.; Frearson, J.; Graham Wyatt, P. Lessons Learnt from Assembling Screening Libraries for Drug Discovery for Neglected Diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Aljafari, A.A. Kinetics for the Inhibition of Acetylcholinesterase from Human Erythrocyte by Cisplatin. Int. J. Biochem. Cell Biol. 1995, 27, 965–970. [Google Scholar] [CrossRef]
- Dinos, G.P.; Coutsogeorgopoulos, C. Kinetic Study of Irreversible Inhibition of an Enzyme Consumed in the Reaction It Catalyses. Application to the Inhibition of the Puromycin Reaction by Spiramycin and Hydroxylamine. J. Enzym. Inhib. 1997, 12, 79–99. [Google Scholar] [CrossRef]
- Oballa, R.M.; Truchon, J.F.; Bayly, C.I.; Chauret, N.; Day, S.; Crane, S.; Berthelette, C. A Generally Applicable Method for Assessing the Electrophilicity and Reactivity of Diverse Nitrile-Containing Compounds. Bioorg. Med. Chem. Lett. 2007, 17, 998–1002. [Google Scholar] [CrossRef]
- Schirmeister, T.; Schmitz, J.; Jung, S.; Schmenger, T.; Krauth-Siegel, R.L.; Gütschow, M. Evaluation of Dipeptide Nitriles as Inhibitors of Rhodesain, a Major Cysteine Protease of Trypanosoma Brucei. Bioorg. Med. Chem. Lett. 2017, 27, 45–50. [Google Scholar] [CrossRef]
- Löser, R.; Gütschow, M. Dipeptide-Derived Nitriles Containing Additional Electrophilic Sites: Potentially Irreversible Inhibitors of Cysteine Proteases. J. Enzyme Inhib. Med. Chem. 2009, 24, 1245–1252. [Google Scholar] [CrossRef] [Green Version]
- Tahir, T.; Tabassum, R.; Javed, Q.; Ali, A.; Ashfaq, M.; Shahzad, M.I. Synthesis, Kinetics, Structure-Activity Relationship and in Silico ADME Studies of New Diazenyl Azo-Phenol Derivatives against Urease, SARS-CoV-2 Main Protease (Mpro) and Ribosomal Protein S1 (RpsA) of Mycobacterium Tuberculosis. J. Mol. Struct. 2022, 1254, 132336. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yiu, C.-P.B.; Wong, K.-Y. Prediction of the SARS-CoV-2 (2019-NCoV) 3C-like Protease (3CLpro) Structure: Virtual Screening Reveals Velpatasvir, Ledipasvir, and Other Drug Repurposing Candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montana, M.; Montero, V.; Khoumeri, O.; Vanelle, P. Quinoxaline Derivatives as Antiviral Agents: A Systematic Review. Molecules 2020, 25, 2784. [Google Scholar] [CrossRef]
- Kleim, J.P.; Bender, R.; Billhardt, U.M.; Meichsner, C.; Riess, G.; Rosner, M.; Winkler, I.; Paessens, A. Activity of a Novel Quinoxaline Derivative against Human Immunodeficiency Virus Type 1 Reverse Transcriptase and Viral Replication. Antimicrob. Agents Chemother. 1993, 37, 1659–1664. [Google Scholar] [CrossRef] [Green Version]
- Günther, S.; Reinke, P.Y.A.; Fernández-Garciá, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-Ray Screening Identifies Active Site and Allosteric Inhibitors of SARS-CoV-2 Main Protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and Electrophilic Fragment Screening of the SARS-CoV-2 Main Protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef]
- Sztain, T.; Amaro, R.; McCammon, J.A. Elucidation of Cryptic and Allosteric Pockets within the SARS-CoV-2 Main Protease. J. Chem. Inf. Model. 2021, 61, 3495–3501. [Google Scholar] [CrossRef]
- Goyal, B.; Goyal, D. Targeting the Dimerization of the Main Protease of Coronaviruses: A Potential Broad-Spectrum Therapeutic Strategy. In ACS Combinatorial Science; American Chemical Society: Washington, DC, USA, 2020; pp. 297–305. [Google Scholar] [CrossRef]
- SiteMap, Schrödinger, LLC, New York, NY, 2021. Available online: http://www.schrodinger.com (accessed on 2 June 2022).
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Yang, W.-H.; Yang, C.-S.; Hou, M.-H.; Tsai, C.-L.; Chou, Y.-Z.; Hung, M.-C.; Chen, Y. Structural Basis of SARS-CoV-2 Main Protease Inhibition by a Broad-Spectrum Anti-Coronaviral Drug. Am. J. Cancer Res. 2020, 10, 2535–2545. [Google Scholar]
- Prince, G.A. An Update on Respiratory Syncytial Virus Antiviral Agents. Expert Opin. Investig. Drugs 2001, 10, 297–308. [Google Scholar] [CrossRef]
- Krilov, L.R. Recent Developments in the Treatment and Prevention of Respiratory Syncytial Virus Infection. Expert Opin. Ther. Pat. 2002, 12, 441–449. [Google Scholar] [CrossRef]
- Alamri, M.A.; Tahir ul Qamar, M.; Afzal, O.; Alabbas, A.B.; Riadi, Y.; Alqahtani, S.M. Discovery of Anti-MERS-CoV Small Covalent Inhibitors through Pharmacophore Modeling, Covalent Docking and Molecular Dynamics Simulation. J. Mol. Liq. 2021, 330, 115699. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2022.02 Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- NCI Database Download Page. Available online: https://cactus.nci.nih.gov/download/nci/ (accessed on 11 February 2021).
- Screening-Compound-Libraries. Available online: https://www.timtec.net/screening-compound-libraries.html (accessed on 11 February 2021).
- ChEMBL. Available online: https://www.ebi.ac.uk/chembl/g/#browse/compounds (accessed on 11 February 2021).
- Screening Libraries. Available online: https://www.chemdiv.com/catalog/screening-libraries/ (accessed on 11 February 2021).
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58, 7076–7087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, S.M.; Bednar, D.; Damborsky, J. Computational Study of Protein-Ligand Unbinding for Enzyme Engineering. Front. Chem. 2019, 7, 650. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach to Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.C.; Fadl, S.; Ilter, M.; Pekel, H.; Rezgui, R.; Sensoy, O.; Rabeh, W.M. Dimethyl Sulfoxide Reduces the Stability but Enhances Catalytic Activity of the Main SARS-CoV-2 Protease 3CLpro. FASEB J. 2021, 35, e21774. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CB03 | CD06 | GR04 | GR20 | ||
|---|---|---|---|---|---|
| Pharmacokinetics | GI absorption | High | High | High | High |
| BBB permeant | No | Yes | Yes | No | |
| P-gp substrate | No | No | Yes | Yes | |
| CYP1A2 inhibitor | Yes | No | Yes | Yes | |
| CYP2C19 inhibitor | Yes | Yes | No | Yes | |
| CYP2C9 inhibitor | Yes | Yes | Yes | Yes | |
| CYP2D6 inhibitor | Yes | Yes | Yes | No | |
| CYP3A4 inhibitor | Yes | No | Yes | Yes | |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | |
| Drug-likeness | Lipinski | Yes | Yes | Yes | Yes |
| Ghose | Yes | Yes | Yes | Yes | |
| Veber | Yes | Yes | Yes | Yes | |
| Egan | Yes | Yes | Yes | Yes | |
| Muegge | Yes | No | Yes | Yes | |
| LogP > 3.5 | |||||
| Medicinal Chemistry | PAINS | 0 | 1 alert | 0 | 0 |
| Mannich in structure | |||||
| Brenk | 0 | 0 | 0 | 0 | |
| Lead-likeness | Yes | LogP > 3.5 MW > 350 | Yes | Yes | |
| Compound ID | Structure | Molecular Weight | MM-GBSA Score (kcal mol−1) | IC50 (μM) |
|---|---|---|---|---|
| CB03 |  | 304.4 | −22.7 ± 3.3 | 301.0 ± 10.0 |
| GR04 |  | 287.4 | −29.8 ± 3.5 | 346.0 ± 10.0 |
| GR20 |  | 297.3 | −31.5 ± 4.0 | 91.8 ± 4.34 |
| CD06 |  | 412.5 | −34.8 ± 5.1 | 226.2 ± 17.5 |
| Pre-Incubation Test | ||
| Inhibition % at 100 μM without pre-incubation | Inhibition % at 100 μM with 15 min pre-incubation | |
| CB03 | 15.1 | 14.2 |
| GR04 | 35.4 | 34.3 |
| GR20 | NT | NT |
| 10-Fold Enzyme Concentration Increase | ||
| Inhibition % at 100 μM at 4.6 μM enzyme concentration | Inhibition % at 100 μM at 46 μM enzyme concentration | |
| CB03 | 15.0 | 13.1 |
| GR04 | 33.5 | 33.9 |
| GR20 | 48.6 | 48.0 |
| BSA Buffer | ||
| Inhibition % at 100 μM without BSA in assay buffer | Inhibition % at 100 μM with 1g/mL BSA in assay buffer | |
| CB03 | 15.5 | 17.3 |
| GR04 | 33.0 | 35.0 |
| GR20 | 50.0 | 51.4 |
| Allosteric Site Number | PDB Code | Dscore (Site Map) | GR04 Docking Score (kcal mol−1) | GR20 Docking Score (kcal mol−1) |
|---|---|---|---|---|
| Site #1 | 5REC [29] | ND | −3.76 | −3.55 |
| Site #2 | 7AGA [28] | 1.02 | −5.31 | −5.53 |
| Site #3 | 7AXM [28] | 0.49 | −3.68 | −2.90 |
| Site #4 | 5RGJ [29] | ND | ND | −1.94 |
| Site #5 | 5RFA [29] | 0.83 | −4.72 | −4.90 |
| Site #6 | 5RF0 [29] | ND | ND | −2.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahgoub, R.E.; Mohamed, F.E.; Alzyoud, L.; Ali, B.R.; Ferreira, J.; Rabeh, W.M.; AlNeyadi, S.S.; Atatreh, N.; Ghattas, M.A. The Discovery of Small Allosteric and Active Site Inhibitors of the SARS-CoV-2 Main Protease via Structure-Based Virtual Screening and Biological Evaluation. Molecules 2022, 27, 6710. https://doi.org/10.3390/molecules27196710
Mahgoub RE, Mohamed FE, Alzyoud L, Ali BR, Ferreira J, Rabeh WM, AlNeyadi SS, Atatreh N, Ghattas MA. The Discovery of Small Allosteric and Active Site Inhibitors of the SARS-CoV-2 Main Protease via Structure-Based Virtual Screening and Biological Evaluation. Molecules. 2022; 27(19):6710. https://doi.org/10.3390/molecules27196710
Chicago/Turabian StyleMahgoub, Radwa E., Feda E. Mohamed, Lara Alzyoud, Bassam R. Ali, Juliana Ferreira, Wael M. Rabeh, Shaikha S. AlNeyadi, Noor Atatreh, and Mohammad A. Ghattas. 2022. "The Discovery of Small Allosteric and Active Site Inhibitors of the SARS-CoV-2 Main Protease via Structure-Based Virtual Screening and Biological Evaluation" Molecules 27, no. 19: 6710. https://doi.org/10.3390/molecules27196710
APA StyleMahgoub, R. E., Mohamed, F. E., Alzyoud, L., Ali, B. R., Ferreira, J., Rabeh, W. M., AlNeyadi, S. S., Atatreh, N., & Ghattas, M. A. (2022). The Discovery of Small Allosteric and Active Site Inhibitors of the SARS-CoV-2 Main Protease via Structure-Based Virtual Screening and Biological Evaluation. Molecules, 27(19), 6710. https://doi.org/10.3390/molecules27196710