


Computational Insights into β-Carboline Inhibition of Monoamine Oxidase A

 , and
, and 
Abstract
:1. Introduction
2. Results
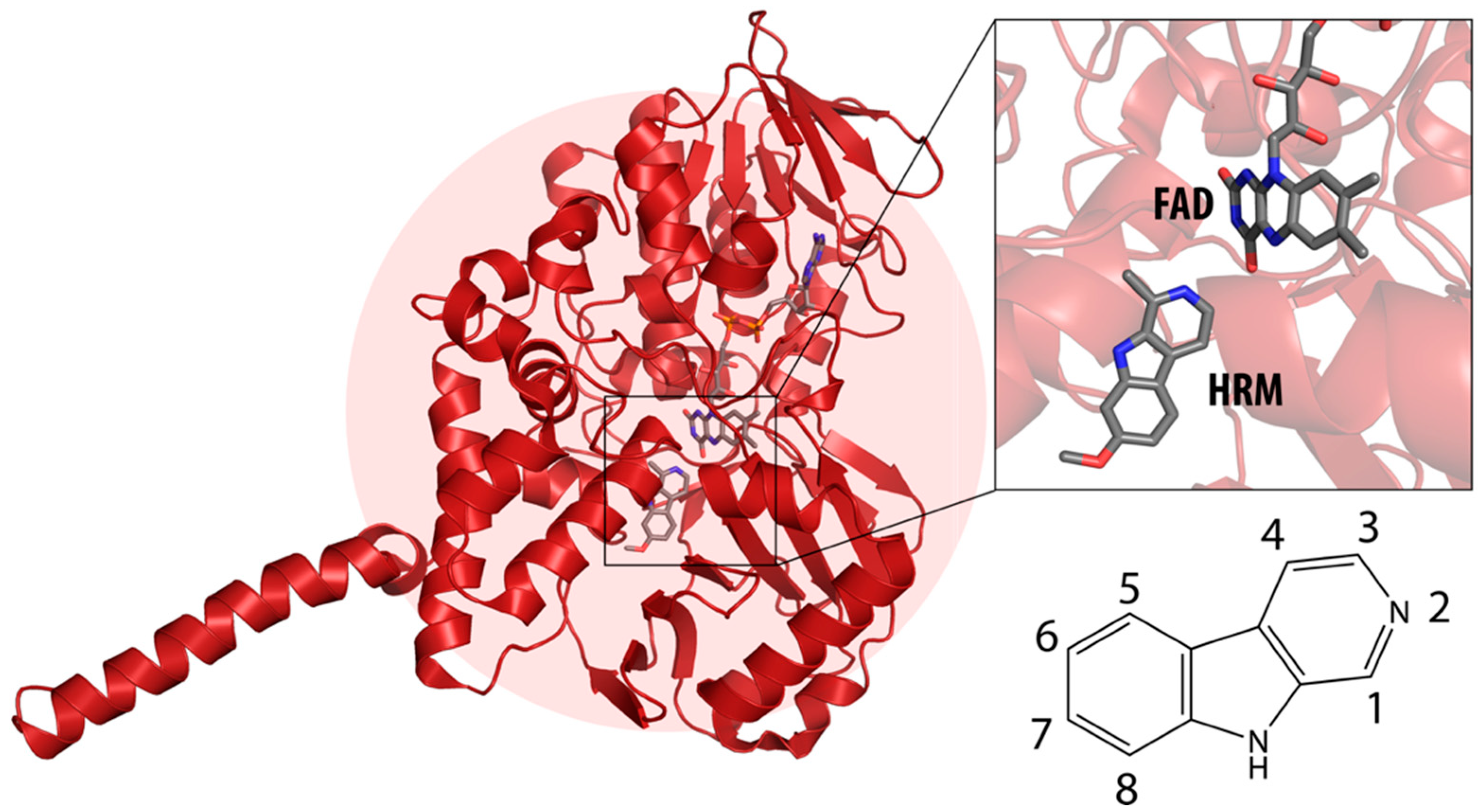
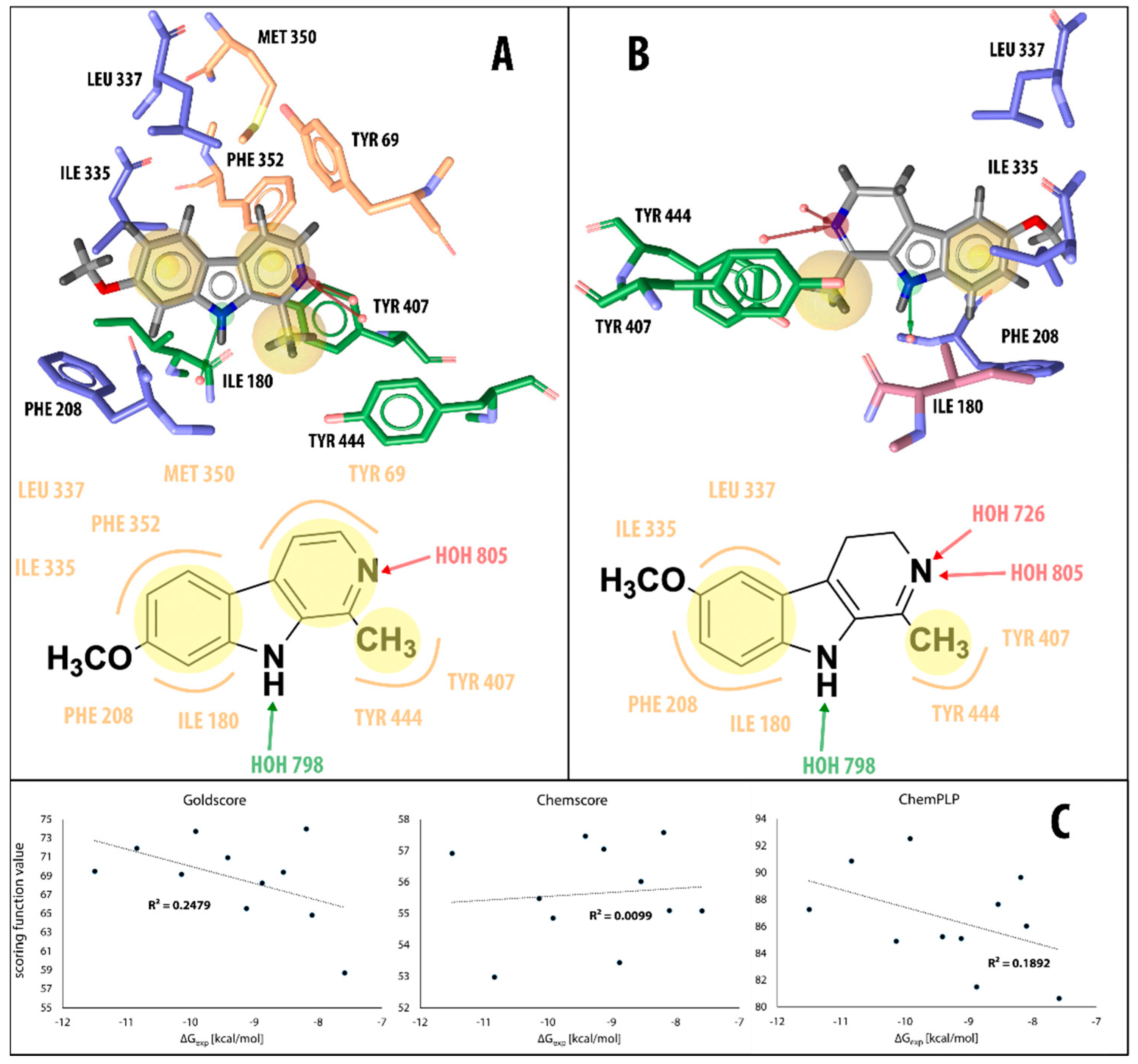
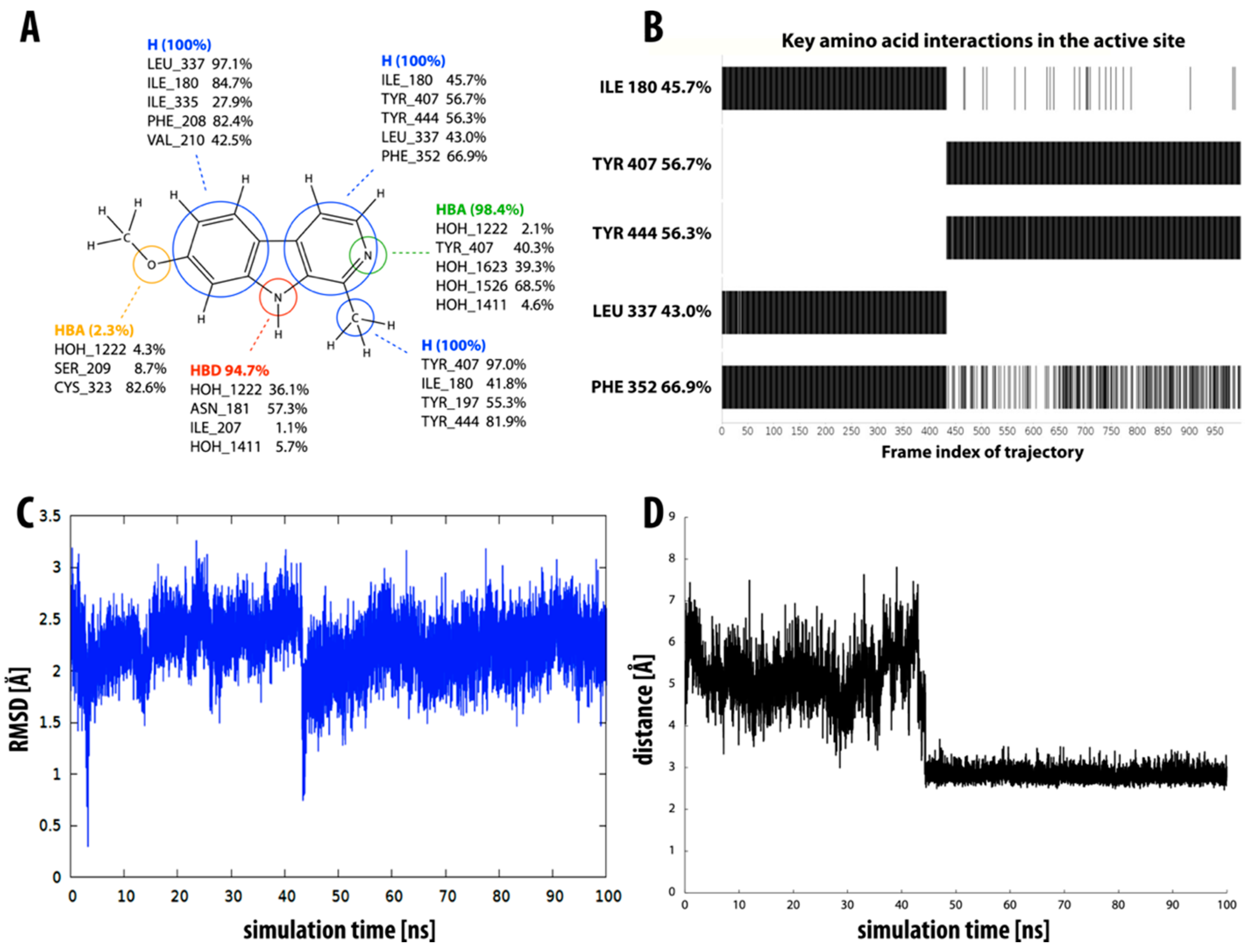
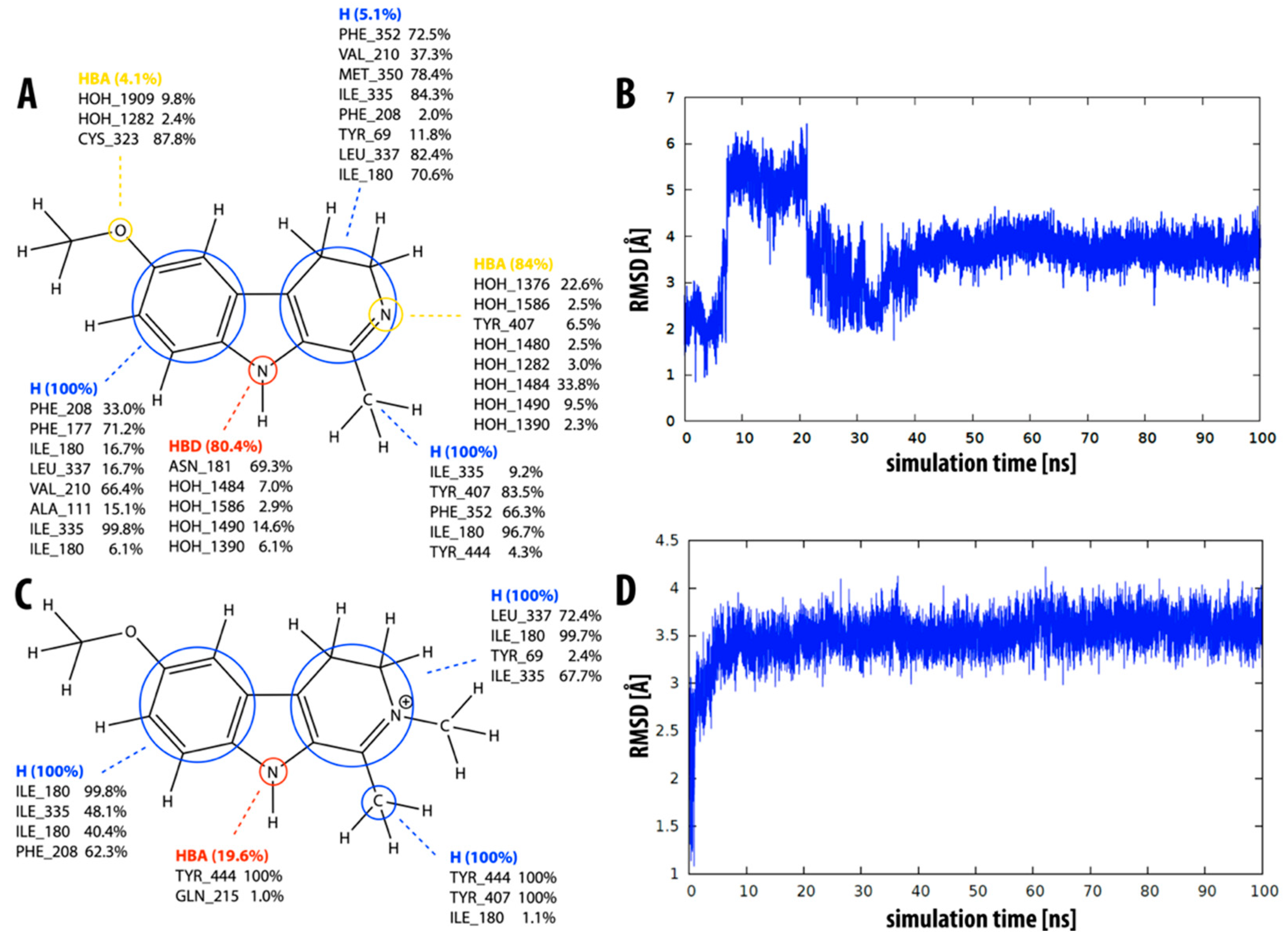
2.1. Static Insight into the β-Carboline–MAO-A Molecular Recognition
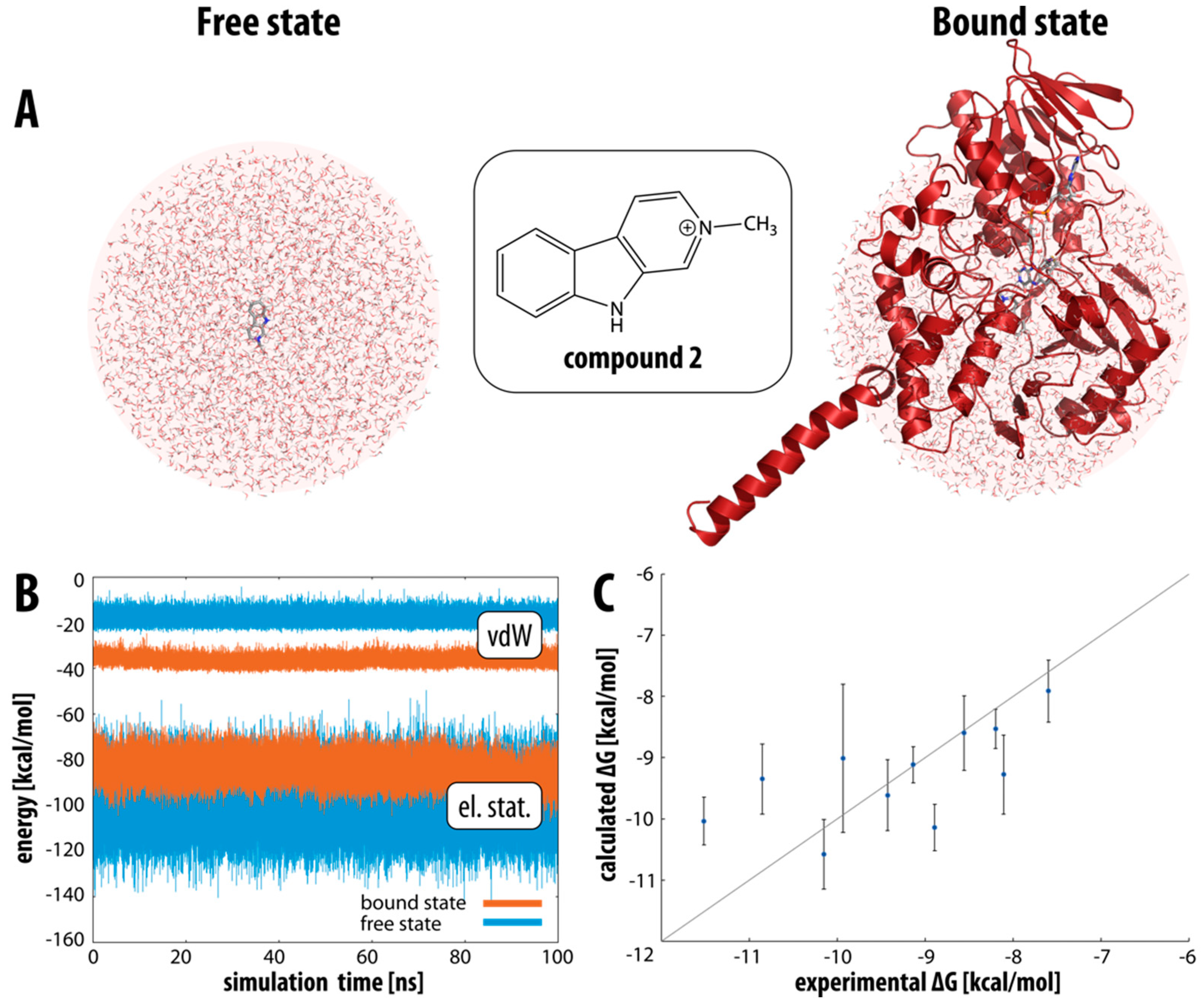
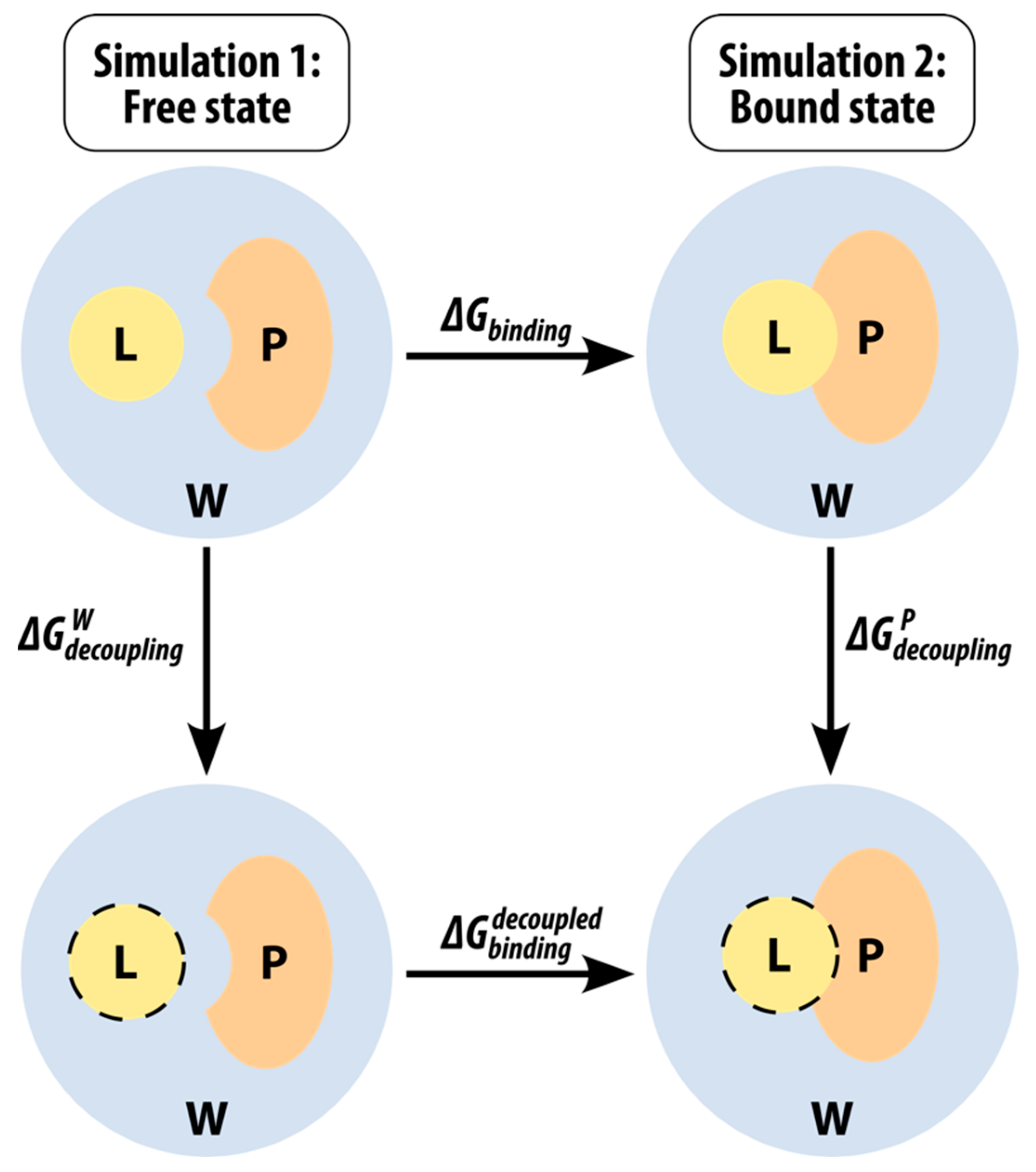
2.2. Binding Free Energies of β-Carbolines
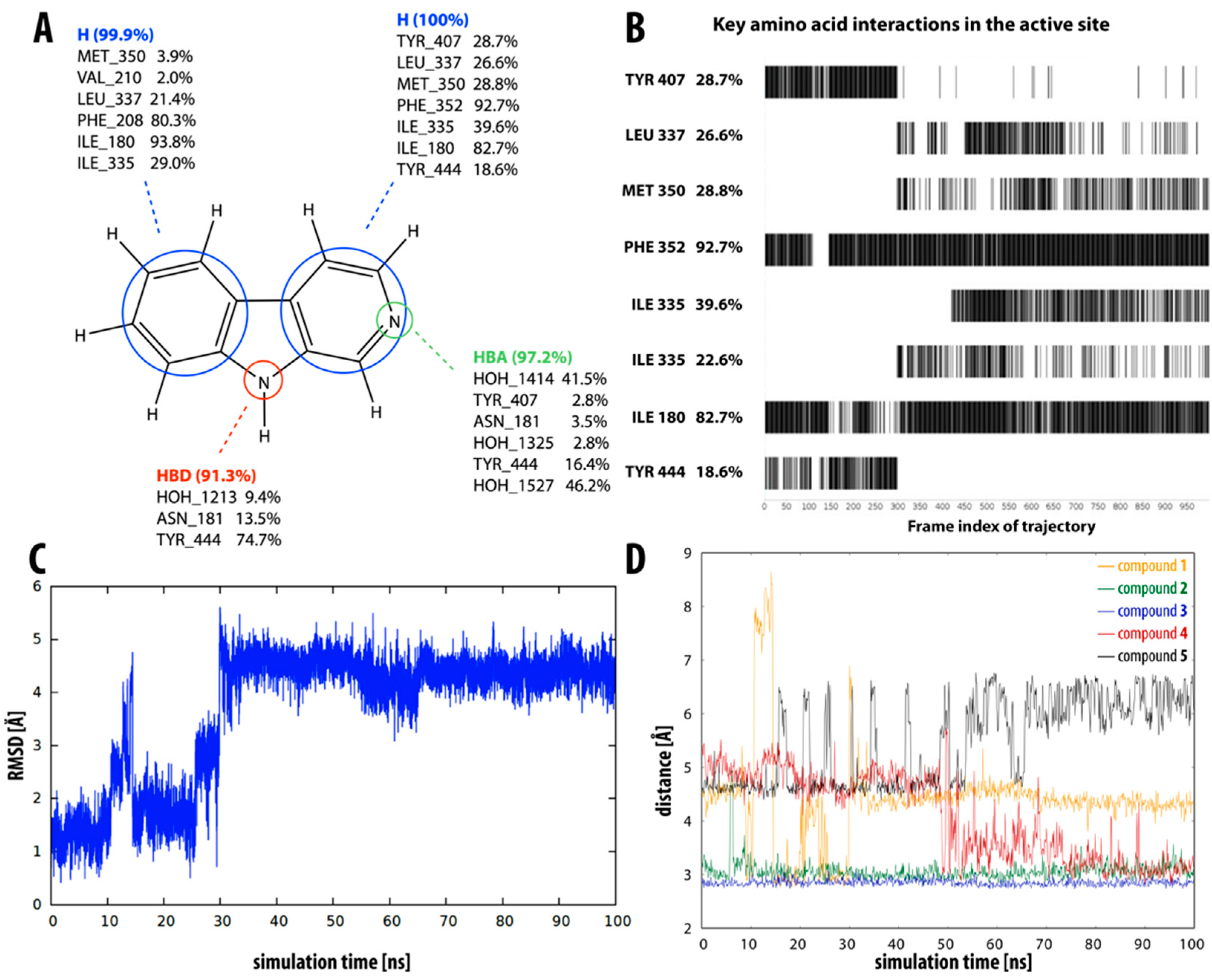
2.3. Dynamic Insight into the β-Carboline–MAO-A Molecular Recognition
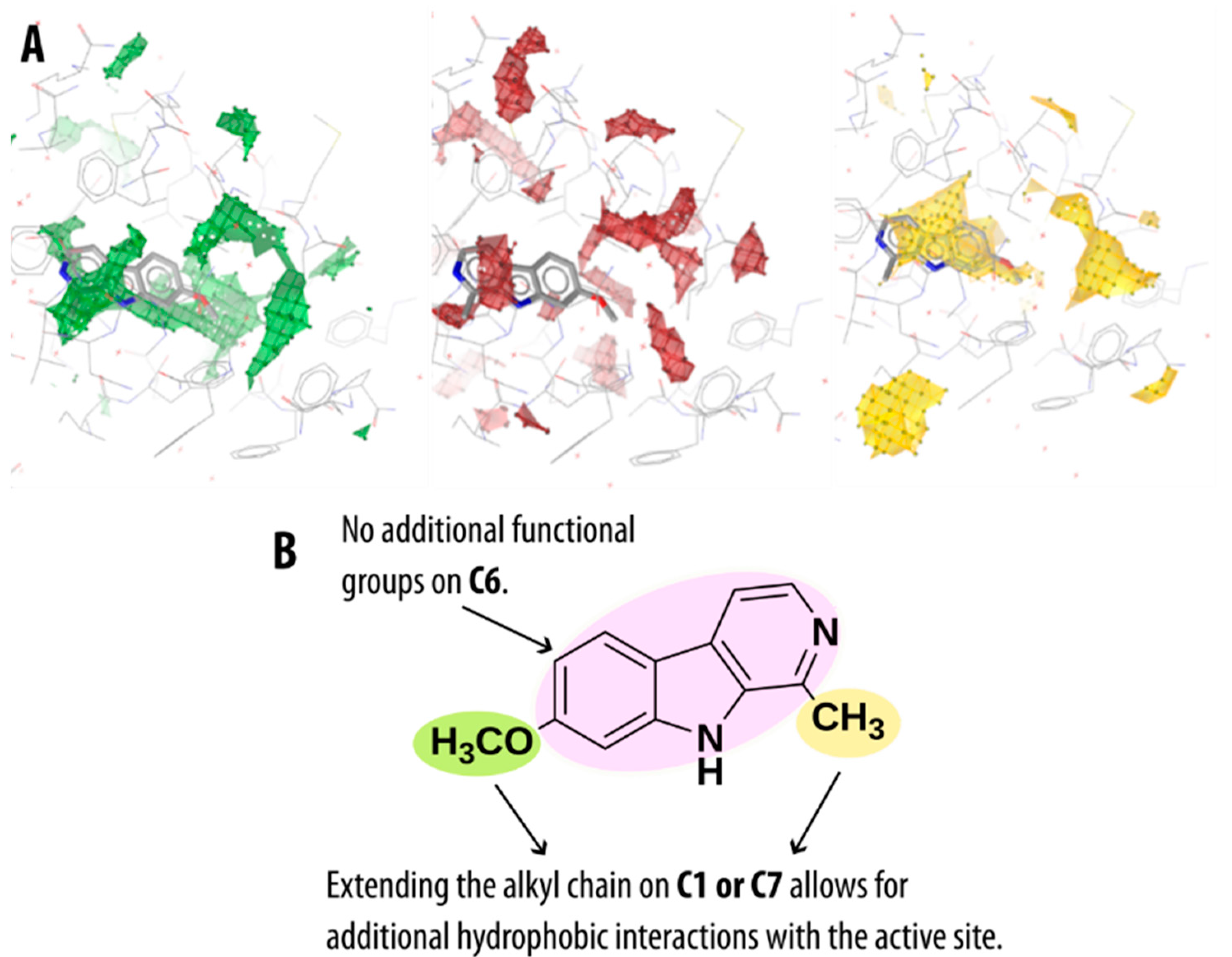
2.4. Guidelines for Further Optimization of β-Carbolines as Non-Covalent MAO-A Inhibitors
3. Discussion
4. Materials and Methods
4.1. Molecular Docking and Binding Site Analysis
4.2. Preparation of the Initial Structures for Molecular Dynamics Simulations
4.3. Molecular Dynamics Simulations
4.4. Generation of Dynamical Pharmacophore Models
4.5. Calculation of Binding Free Energies Using the Linear Interaction Energy Method
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.R.; Edmondson, D.E. Structure-activity relationships in the oxidation of para-substituted benzylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry 1999, 38, 13670–13683. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.B. Radical ideas about monoamine-oxidase. Acc. Chem. Res. 1995, 28, 335–342. [Google Scholar] [CrossRef]
- Abad, E.; Zenn, R.K.; Kästner, J. Reaction Mechanism of Monoamine Oxidase from QM/MM Calculations. J. Phys. Chem. B 2013, 117, 14238–14246. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Torres, G.; Fierro, A.; Barriga-Gonzalez, G.; Salgado, J.C.; Celis-Barros, C. Revealing monoamine oxidase B catalytic mechanisms by means of the quantum chemical cluster approach. J. Chem. Inf. Model. 2015, 55, 1349–1360. [Google Scholar] [CrossRef] [Green Version]
- Ralph, E.C.; Hirschi, J.S.; Anderson, M.A.; Cleland, W.W.; Singleton, D.A.; Fitzpatrick, P.F. Insights into the mechanism of flavoprotein-catalyzed amine oxidation from nitrogen isotope effects on the reaction of N-methyltryptophan oxidase. Biochemistry 2007, 46, 7655–7664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdem, S.S.; Karahan, O.; Yildiz, I.; Yelekci, K. A computational study on the amine-oxidation mechanism of monoamine oxidase: Insight into the polar nucleophilic mechanism. Org. Biomol. Chem. 2006, 4, 646–658. [Google Scholar] [CrossRef]
- Cakir, K.; Erdem, S.S.; Atalay, V.E. ONIOM calculations on serotonin degradation by monoamine oxidase B: Insight into the oxidation mechanism and covalent reversible inhibition. Org. Biomol. Chem. 2016, 14, 9239–9252. [Google Scholar] [CrossRef]
- Akyuz, M.A.; Erdem, S.S. Computational modeling of the direct hydride transfer mechanism for the MAO catalyzed oxidation of phenethylamine and benzylamine: ONIOM (QM/QM) calculations. J. Neural Transm. 2013, 120, 937–945. [Google Scholar] [CrossRef]
- Atalay, V.E.; Erdem, S.S. A comparative computational investigation on the proton and hydride transfer mechanisms of monoamine oxidase using model molecules. Comput. Biol. Chem. 2013, 47, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, K.A.; Rishavy, M.A.; Cleland, W.W.; Fitzpatrick, P.F. Nitrogen isotope effects as probes of the mechanism of D-amino acid oxidase. J. Am. Chem. Soc. 2000, 122, 12896–12897. [Google Scholar] [CrossRef]
- Fitzpatrick, P.F. Oxidation of amines by flavoproteins. Arch. Biochem. Biophys. 2010, 493, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Vianello, R.; Repič, M.; Mavri, J. How are biogenic amines metabolized by monoamine oxidases? Eur. J. Org. Chem. 2012, 36, 7057–7065. [Google Scholar] [CrossRef]
- Fowler, J.S.; Volkow, N.D.; Wang, G.J.; Pappas, N.; Logan, J.; Shea, C.; Alexoff, D.; MacGregor, R.R.; Schlyer, D.J.; Zezulkova, I.; et al. Brain monoamine oxidase A inhibition in cigarette smokers. Proc. Natl. Acad. Sci. USA 1996, 93, 14065–14069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, J.S.; Volkow, N.D.; Wang, G.J.; Pappas, N.; Logan, J.; MacGregor, R.; Alexoff, D.; Shea, C.; Schlyer, D.; Wolf, A.P.; et al. Inhibition of monoamine oxidase B in the brains of smokers. Nature 1996, 379, 733–736. [Google Scholar] [CrossRef]
- Castagnoli, K.; Murugesan, T. Tobacco Leaf, Smoke and Smoking, MAO Inhibitors, Parkinson’s Disease and Neuroprotection; Are There Links? Neurotoxicology 2004, 25, 279–291. [Google Scholar] [CrossRef]
- Herraiz, T. Relative exposure to beta-carbolines norharman and harman from foods and tobacco smoke. Food Addit. Contam. 2004, 21, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, T.; Chaparro, C. Human monoamine oxidase is inhibited by tobacco smoke: Beta-carboline alkaloids act as potent and reversible inhibitors. Biochem. Biophys. Res. Commun. 2005, 326, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Piechowska, P.; Zawirska-Wojtasiak, R.; Mildner-Szkudlarz, S. Bioactive beta-Carbolines in Food: A Review. Nutrients 2019, 11, 814. [Google Scholar] [CrossRef] [Green Version]
- Moura, D.J.; Richter, M.F.; Boeira, J.M.; Pegas Henriques, J.A.; Saffi, J. Antioxidant properties of beta-carboline alkaloids are related to their antimutagenic and antigenotoxic activities. Mutagenesis 2007, 22, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Freedland, C.S.; Mansbach, R.S. Behavioral profile of constituents in ayahuasca, an Amazonian psychoactive plant mixture. Drug Alcohol Depend. 1999, 54, 183–194. [Google Scholar] [CrossRef]
- Boeira, J.M.; Viana, A.F.; Picada, J.N.; Henriques, J.A.P. Genotoxic and recombinogenic activities of the two β-carboline alkaloids harman and harmine in Saccharomyces cerevisiae. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2002, 500, 39–48. [Google Scholar] [CrossRef]
- Husbands, S.M.; Glennon, R.A.; Gorgerat, S.; Gough, R.; Tyacke, R.; Crosby, J.; Nutt, D.J.; Lewis, J.W.; Hudson, A.L. β-carboline binding to imidazoline receptors. Drug Alcohol Depend. 2001, 64, 203–208. [Google Scholar] [CrossRef]
- Squires, P.E.; Hills, C.E.; Rogers, G.J.; Garland, P.; Farley, S.R.; Morgan, N.G. The putative imidazoline receptor agonist, harmane, promotes intracellular calcium mobilisation in pancreatic beta-cells. Eur. J. Pharmacol. 2004, 501, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Grella, B.; Dukat, M.; Young, R.; Teitler, M.; Herrick-Davis, K.; Gauthier, C.B.; Glennon, R.A. Investigation of hallucinogenic and related β-carbolines. Drug Alcohol Depend. 1998, 50, 99–107. [Google Scholar] [CrossRef]
- Loew, G.H.; Nienow, J.; Lawson, J.A.; Toll, L.; Uyeno, E.T. Theoretical structure-activity studies of beta-carboline analogs. Requirements for benzodiazepine receptor affinity and antagonist activity. Mol. Pharmacol. 1985, 28, 17–31. [Google Scholar]
- Glennon, R.A.; Dukat, M.; Grella, B.; Hong, S.-S.; Costantino, L.; Teitler, M.; Smith, C.; Egan, C.; Davis, K.; Mattson, M.V. Binding of β-carbolines and related agents at serotonin (5-HT2 and 5-HT1A), dopamine (D2) and benzodiazepine receptors. Drug Alcohol Depend. 2000, 60, 121–132. [Google Scholar] [CrossRef]
- Pimpinella, G.; Palmery, M. Interaction of β-carbolines with central dopaminergic transmission in mice: Structure-activity relationships. Neurosci. Lett. 1995, 189, 121–124. [Google Scholar] [CrossRef]
- Matsubara, K.; Gonda, T.; Sawada, H.; Uezono, T.; Kobayashi, Y.; Kawamura, T.; Ohtaki, K.; Kimura, K.; Akaike, A. Endogenously occurring beta-carboline induces parkinsonism in nonprimate animals: A possible causative protoxin in idiopathic Parkinson’s disease. J. Neurochem. 1998, 70, 727–735. [Google Scholar] [CrossRef]
- Hayashi, K.; Nagao, M.; Sugimura, T. Interactions of norharman and harman with DNA. Nucleic Acids Res. 1977, 4, 3679–3685. [Google Scholar] [CrossRef] [Green Version]
- Funayama, Y.; Nishio, K.; Wakabayashi, K.; Nagao, M.; Shimoi, K.; Ohira, T.; Hasegawa, S.; Saijo, N. Effects of β- and γ-carboline derivatives on DNA topoisomerase activities. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1996, 349, 183–191. [Google Scholar] [CrossRef]
- Balón, M.; Muñoz, M.A.; Carmona, C.; Guardado, P.; Galán, M. A fluorescence study of the molecular interactions of harmane with the nucleobases, their nucleosides and mononucleotides. Biophys. Chem. 1999, 80, 41–52. [Google Scholar] [CrossRef]
- Pari, K.; Sundari, C.S.; Chandani, S.; Balasubramanian, D. Beta-carbolines that accumulate in human tissues may serve a protective role against oxidative stress. J. Biol. Chem. 2000, 275, 2455–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, H.; Sato, M.; Watanabe, I. Antiplatelet activity of soy sauce as functional seasoning. J. Agric. Food. Chem. 1999, 47, 4167–4174. [Google Scholar] [CrossRef] [PubMed]
- Ancolio, C.; Azas, N.; Mahiou, V.; Ollivier, E.; Di Giorgio, C.; Keita, A.; Timon-David, P.; Balansard, G. Antimalarial activity of extracts and alkaloids isolated from six plants used in traditional medicine in Mali and Sao Tome. Phytother. Res. 2002, 16, 646–649. [Google Scholar] [CrossRef]
- Ferraz, C.A.A.; de Oliveira Junior, R.G.; Picot, L.; da Silva Almeida, J.R.G.; Nunes, X.P. Pre-clinical investigations of beta-carboline alkaloids as antidepressant agents: A systematic review. Fitoterapia 2019, 137, 104196. [Google Scholar] [CrossRef]
- Aricioglu, F.; Altunbas, H. Harmane induces anxiolysis and antidepressant-like effects in rats. Ann. N. Y. Acad. Sci. 2003, 1009, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.L.; Ford, G.K.; Jessop, D.S.; Finn, D.P. Behavioural, neurochemical and neuroendocrine effects of the endogenous beta-carboline harmane in fear-conditioned rats. J. Psychopharm. 2013, 27, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Farzin, D.; Mansouri, N. Antidepressant-like effect of harmane and other beta-carbolines in the mouse forced swim test. Eur. Neuropsychopharmacol. 2006, 16, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Adell, A.; Biggs, T.A.; Myers, R.D. Action of harman (1-methyl-β-carboline) on the brain: Body temperature and in vivo efflux of 5-HT from hippocampus of the rat. Neuropharmacology 1996, 35, 1101–1107. [Google Scholar] [CrossRef]
- Fortunato, J.J.; Reus, G.Z.; Kirsch, T.R.; Stringari, R.B.; Stertz, L.; Kapczinski, F.; Pinto, J.P.; Hallak, J.E.; Zuardi, A.W.; Crippa, J.A.; et al. Acute harmine administration induces antidepressive-like effects and increases BDNF levels in the rat hippocampus. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2009, 33, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, J.J.; Reus, G.Z.; Kirsch, T.R.; Stringari, R.B.; Fries, G.R.; Kapczinski, F.; Hallak, J.E.; Zuardi, A.W.; Crippa, J.A.; Quevedo, J. Effects of beta-carboline harmine on behavioral and physiological parameters observed in the chronic mild stress model: Further evidence of antidepressant properties. Brain Res. Bull. 2010, 81, 491–496. [Google Scholar] [CrossRef]
- WHO. Depression. Available online: https://www.who.int/news-room/fact-sheets/detail/depression (accessed on 13 September 2022).
- Cao, R.; Peng, W.; Wang, Z.; Xu, A. Beta-Carboline alkaloids: Biochemical and pharmacological functions. Curr. Med. Chem. 2007, 14, 479–500. [Google Scholar] [CrossRef] [PubMed]
- Varela, A.P.; Burrows, H.D.; Douglas, P.; da Graça Miguel, M. Triplet state studies of β-carbolines. J. Photochem. Photobiol. A Chem. 2001, 146, 29–36. [Google Scholar] [CrossRef]
- May, T.; Rommelspacher, H.; Pawlik, M. [3H]harman binding experiments. I: A reversible and selective radioligand for monoamine oxidase subtype A in the CNS of the rat. J. Neurochem. 1991, 56, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Son, S.Y.; Ma, A.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-angstrom resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Sablin, S.O.; Ramsay, R.R. Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch. Biochem. Biophys. 1997, 337, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef] [PubMed]
- King, E.; Aitchison, E.; Li, H.; Luo, R. Recent Developments in Free Energy Calculations for Drug Discovery. Front. Mol. Biosci. 2021, 8, 712085. [Google Scholar] [CrossRef] [PubMed]
- van Lipzig, M.M.; ter Laak, A.M.; Jongejan, A.; Vermeulen, N.P.; Wamelink, M.; Geerke, D.; Meerman, J.H. Prediction of ligand binding affinity and orientation of xenoestrogens to the estrogen receptor by molecular dynamics simulations and the linear interaction energy method. J. Med. Chem. 2004, 47, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Stjernschantz, E.; Oostenbrink, C. Improved ligand-protein binding affinity predictions using multiple binding modes. Biophys. J. 2010, 98, 2682–2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, J.; Kollman, P.A. What determines the van der Waals coefficient in the LIE (linear interaction energy) method to estimate binding free energies using molecular dynamics simulations? Proteins Struct. Funct. Genet. 1999, 34, 395–402. [Google Scholar] [CrossRef]
- Bock, A.; Bermudez, M.; Krebs, F.; Matera, C.; Chirinda, B.; Sydow, D.; Dallanoce, C.; Holzgrabe, U.; De Amici, M.; Lohse, M.J.; et al. Ligand Binding Ensembles Determine Graded Agonist Efficacies at a G Protein-coupled Receptor. J. Biol. Chem. 2016, 291, 16375–16389. [Google Scholar] [CrossRef] [Green Version]
- Bermudez, M.; Rakers, C.; Wolber, G. Structural Characteristics of the Allosteric Binding Site Represent a Key to Subtype Selective Modulators of Muscarinic Acetylcholine Receptors. Mol. Inform. 2015, 34, 526–530. [Google Scholar] [CrossRef]
- Sydow, D. Dynophores: Novel Dynamic Pharmacophores; Lebenswissenschaftliche Fakultät: Berlin, Germany, 2015. [Google Scholar]
- Akyüz, M.A.; Erdem, S.S.; Edmondson, D.E. The aromatic cage in the active site of monoamine oxidase B: Effect on the structural and electronic properties of bound benzylamine and p-nitrobenzylamine. J. Neural Transm. 2007, 114, 693–698. [Google Scholar] [CrossRef]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T.; The International Natural Product Sciences Taskforce. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Reniers, J.; Robert, S.; Frederick, R.; Masereel, B.; Vincent, S.; Wouters, J. Synthesis and evaluation of beta-carboline derivatives as potential monoamine oxidase inhibitors. Bioorg. Med. Chem. 2011, 19, 134–144. [Google Scholar] [CrossRef]
- Aqvist, J.; Medina, C.; Samuelsson, J.E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Perrin, D.D.; Dempsey, B.; Serjeant, E.P. pKa Prediction for Organic Acids and Bases; Chapman and Hall: London, UK; New York, NY, USA, 1981; 146p. [Google Scholar]
- Poberžnik, M.; Purg, M.; Repič, M.; Mavri, J.; Vianello, R. Empirical valence bond simulations of the hydride-transfer step in the monoamine oxidase A catalyzed metabolism of noradrenaline. J. Phys. Chem. B 2016, 120, 11419–11427. [Google Scholar] [CrossRef]
- Marelius, J.; Kolmodin, K.; Feierberg, I.; Aqvist, J. Q: A molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 1998, 16, 213–225, 261. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Modell. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Aqvist, J.; Luzhkov, V.B.; Brandsdal, B.O. Ligand binding affinities from MD simulations. Acc. Chem. Res. 2002, 35, 358–365. [Google Scholar] [CrossRef]
- Perdih, A.; Bren, U.; Solmajer, T. Binding free energy calculations of N-sulphonyl-glutamic acid inhibitors of MurD ligase. J. Mol. Model. 2009, 15, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Aqvist, J.; Marelius, J. The linear interaction energy method for predicting ligand binding free energies. Comb. Chem. High Throughput Screen. 2001, 4, 613–626. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ||||||
|---|---|---|---|---|---|---|
| 1 | −28.4 ± 0.5 | −15.4 ± 0.1 | −13.1 ± 0.5 | −15.1 ± 1.6 | −22.2 ± 0.1 | +7.1 ± 1.6 |
| 2 * | −36.7 ± 0.5 | −17.9 ± 0.1 | −18.9 ± 0.5 | −84.1 ± 2.2 | −98.0 ± 0.2 | +13.9 ± 2.2 |
| 3 | −32.5 ± 0.3 | −16.8 ± 0.1 | −15.7 ± 0.3 | −15.7 ± 0.9 | −23.6 ± 0.1 | +7.9 ± 0.9 |
| 4 * | −38.2 ± 0.8 | −19.4 ± 0.1 | −17.7 ± 0.8 | −82.9 ± 1.6 | −96.9 ± 0.1 | +14.0 ± 1.6 |
| 5 * | −39.8 ± 1.0 | −22.7 ± 0.1 | −17.1 ± 1.0 | −77.6 ± 0.7 | −86.5 ± 0.2 | +8.9 ± 0.7 |
| 6 | −36.9 ± 0.4 | −19.6 ± 0.1 | −17.2 ± 0.4 | −17.0 ± 1.2 | −24.5 ± 0.1 | +7.2 ± 1.2 |
| 7 * | −42.3 ± 1.0 | −22.0 ± 0.1 | −20.3 ± 1.0 | −78.9 ± 4.1 | −96.7 ± 0.2 | +17.8 ± 4.1 |
| 8 * | −43.9 ± 0.7 | −25.3 ± 0.1 | −18.6 ± 0.7 | −74.5 ± 1.6 | −87.5 ± 0.2 | +12.9 ± 1.6 |
| 9 | −41.9 ± 1.0 | −20.6 ± 0.1 | −21.2 ± 1.0 | −9.7 ± 0.6 | −23.4 ± 0.1 | +13.7 ± 0.6 |
| 10 | −39.5 ± 0.6 | −20.7 ± 0.1 | −18.8 ± 0.6 | −12.9 ± 0.7 | −23.3 ± 0.1 | +10.4 ± 0.7 |
| 11 * | −41.0 ± 0.2 | −22.6 ± 0.1 | −18.4 ± 0.2 | −81.1 ± 1.1 | −96.8 ± 0.2 | +15.7 ± 1.1 |
| Compound | Structure | RMSD [Å] | |||
|---|---|---|---|---|---|
| 1 |  | 3.34 ± 0.10 | −7.6 ± 0.02 | −7.9 ± 0.5 | 3.7 |
| 2 |  | 1.43 ± 0.09 | −8.1 ± 0.04 | −9.3 ± 0.6 | 1.1 |
| 3 |  | 0.26 ± 0.024 | −9.1 ± 0.06 | −9.1 ± 0.3 | 1.9 |
| 4 |  | 0.68 ± 0.026 | −8.6 ± 0.02 | −8.6 ± 0.6 | 1.5 |
| 5 |  | 0.16 ± 0.036 | −9.4 ± 0.14 | −9.6 ± 0.6 | 1.6 |
| 6 |  | 0.005 ± 0.0002 | −11.5 ± 0.02 | −10.0 ± 0.4 | 2.3 |
| 7 |  | 0.069 ± 0.008 | −9.9 ± 0.07 | −9.0 ± 1.2 | 1.7 |
| 8 |  | 0.015 ± 0.0008 | −10.9 ± 0.03 | −9.4 ± 0.6 | 1.4 |
| 9 |  | 0.048 ± 0.007 | −10.2 ± 0.09 | −10.6 ± 0.6 | 1.9 |
| 10 |  | 0.39 ± 0.052 | −8.9 ± 0.08 | −10.1 ± 0.4 | 3.7 |
| 11 |  | 1.23 ± 0.27 | −8.2 ± 0.01 | −8.5 ± 0.3 | 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prah, A.; Gavranić, T.; Perdih, A.; Sollner Dolenc, M.; Mavri, J. Computational Insights into β-Carboline Inhibition of Monoamine Oxidase A. Molecules 2022, 27, 6711. https://doi.org/10.3390/molecules27196711
Prah A, Gavranić T, Perdih A, Sollner Dolenc M, Mavri J. Computational Insights into β-Carboline Inhibition of Monoamine Oxidase A. Molecules. 2022; 27(19):6711. https://doi.org/10.3390/molecules27196711
Chicago/Turabian StylePrah, Alja, Tanja Gavranić, Andrej Perdih, Marija Sollner Dolenc, and Janez Mavri. 2022. "Computational Insights into β-Carboline Inhibition of Monoamine Oxidase A" Molecules 27, no. 19: 6711. https://doi.org/10.3390/molecules27196711
APA StylePrah, A., Gavranić, T., Perdih, A., Sollner Dolenc, M., & Mavri, J. (2022). Computational Insights into β-Carboline Inhibition of Monoamine Oxidase A. Molecules, 27(19), 6711. https://doi.org/10.3390/molecules27196711