
Synthesis and Biological Activity of 3-(Heteroaryl)quinolin-2(1H)-ones Bis-Heterocycles as Potential Inhibitors of the Protein Folding Machinery Hsp90

 , ,
, ,
Abstract
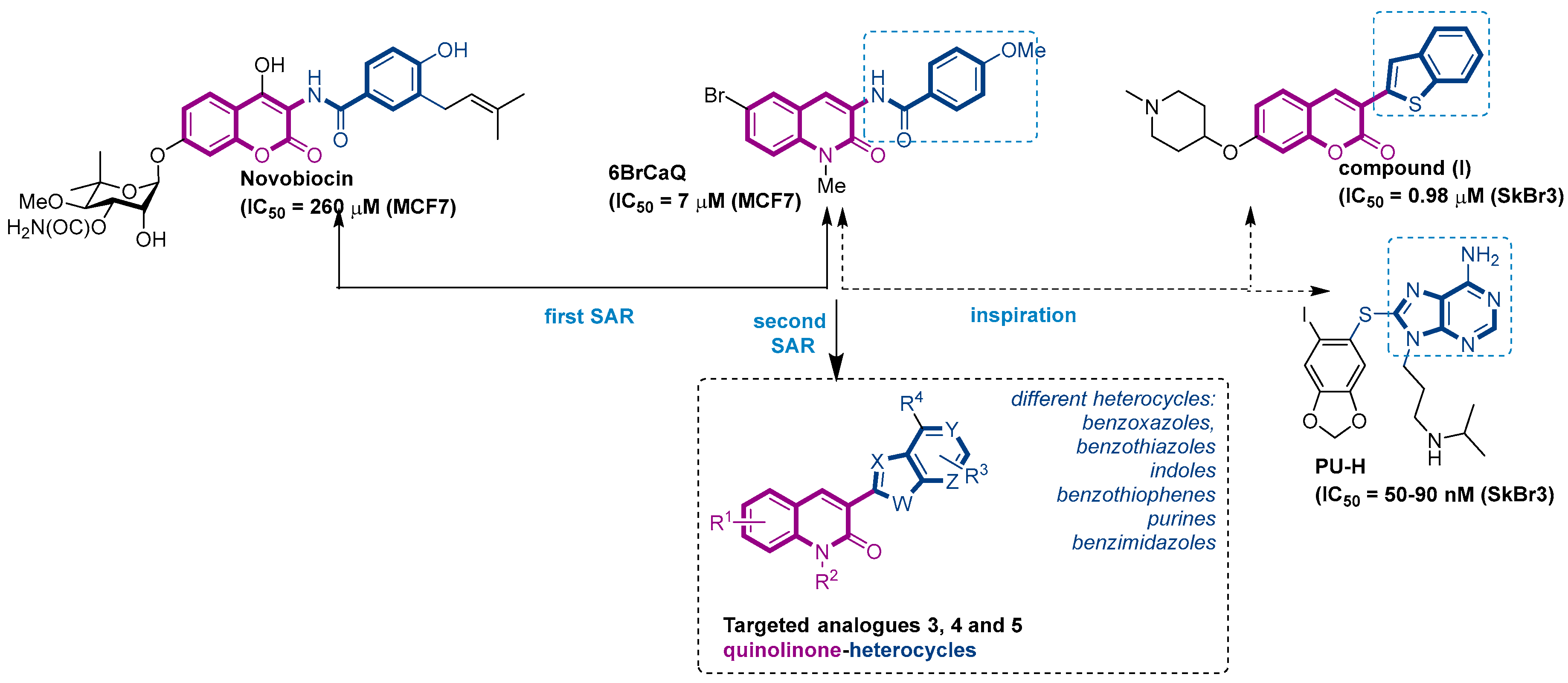
:1. Introduction
2. Results and Discussion
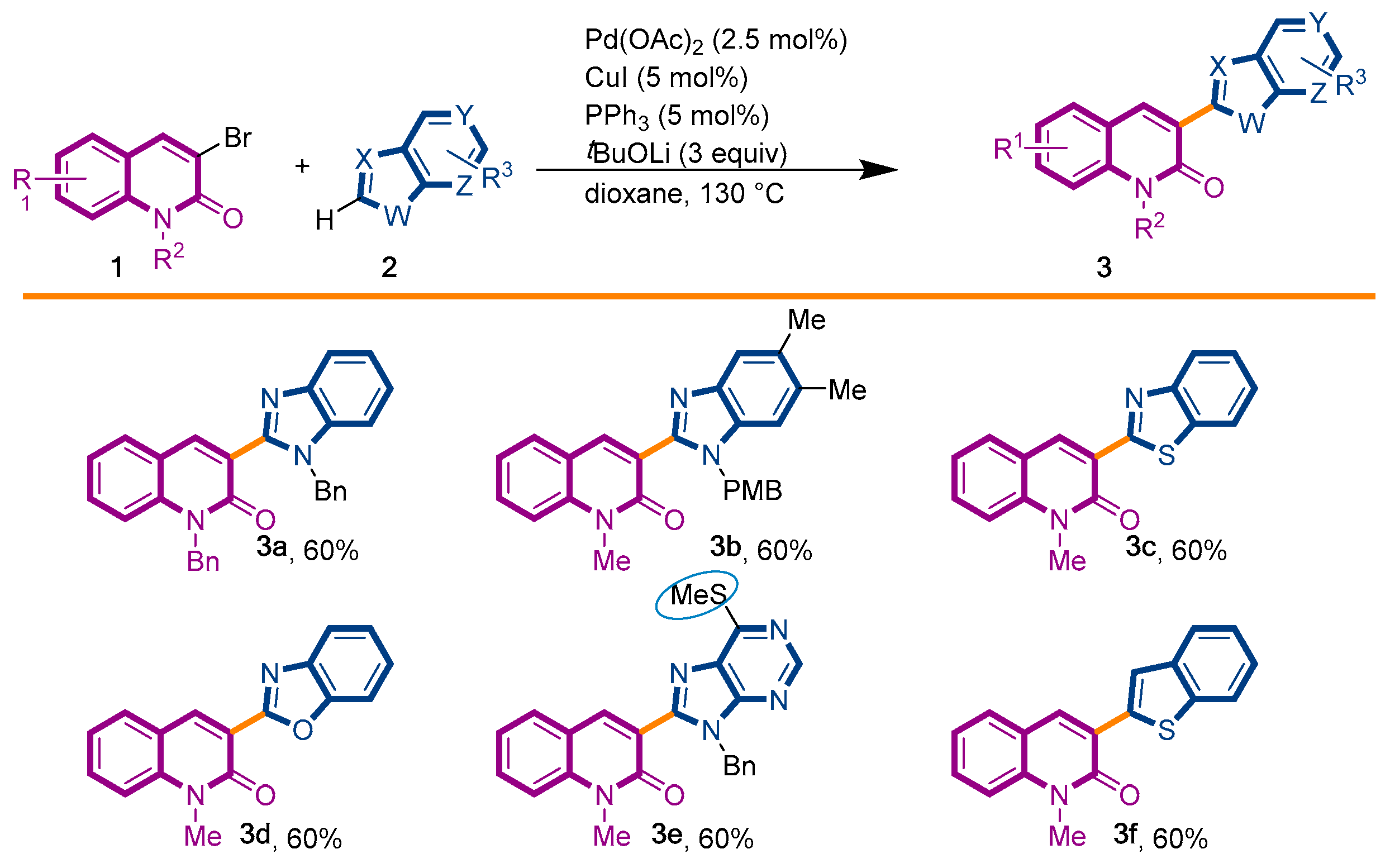
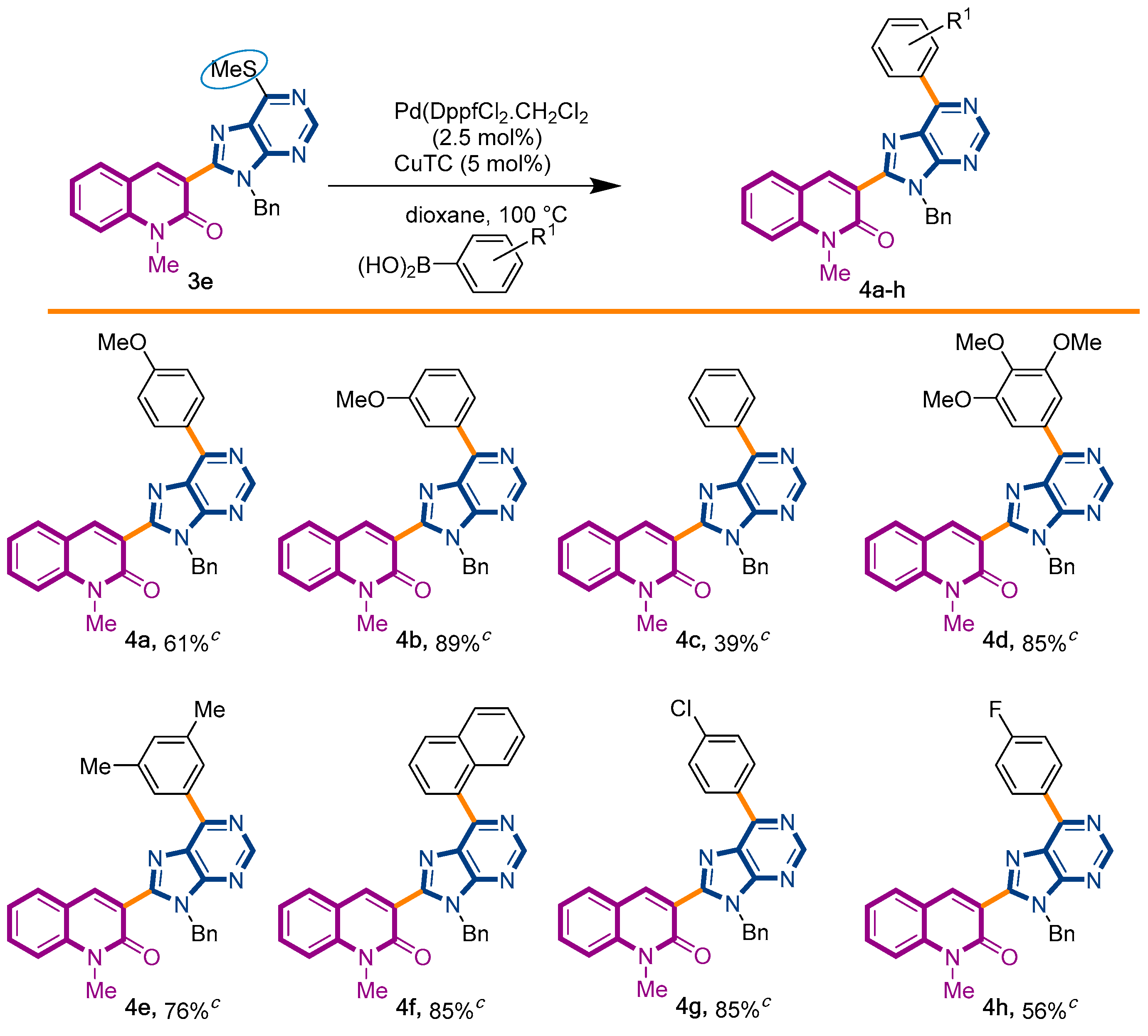
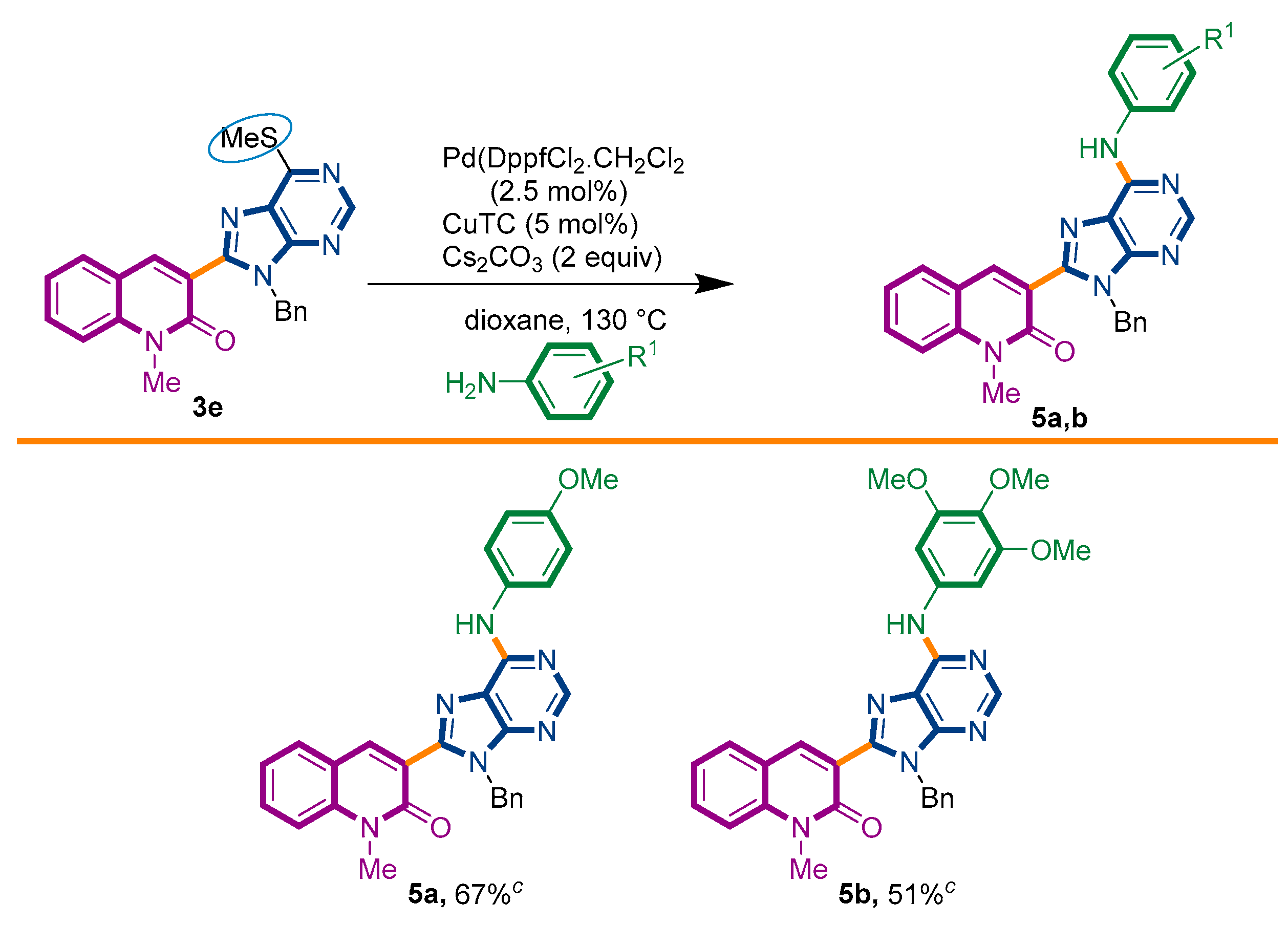
2.1. Chemistry
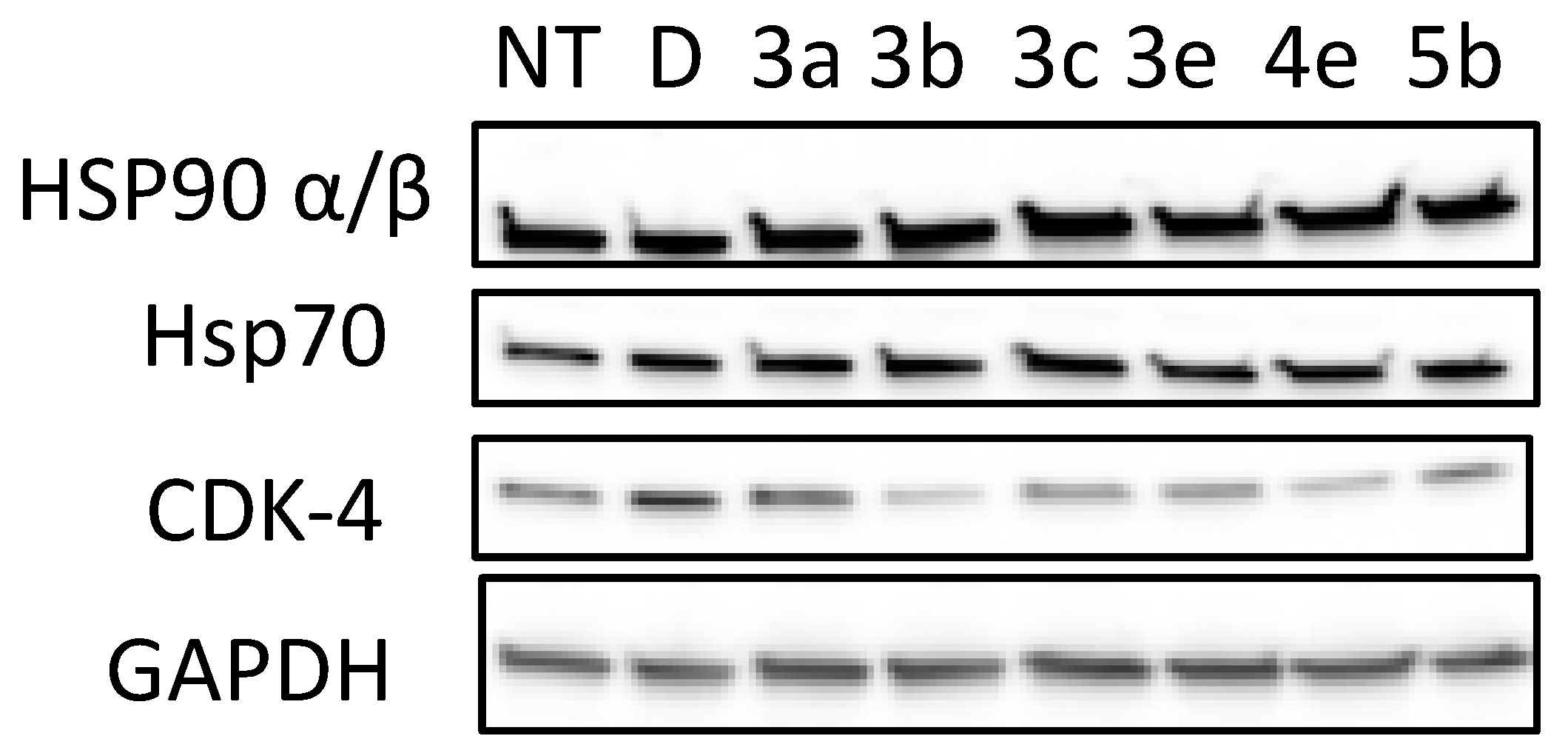
2.2. Biological Evaluation of Quinolones Analogues
Antiproliferative Activity
3. Conclusions
4. Materials and Methods
4.1. General Experimental Methods
4.2. General Procedure for the Liebeskind–Srogl Coupling of 3-(6-Methylthiopurine)-2-quinolone (3e) and Boronic Acids
4.3. Procedure for the Liebeskind–Srogl Coupling of 3-(6-Methylthiopurine)-2-quinolone (3e) and Anilines
4.4. Materials and Methods for Cell Culture and Western Blot Analysis
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Messaoudi, S.; Peyrat, J.-F.; Brion, J.-D.; Alami, M. Recent advances in Hsp90 inhibitors as antitumor agents. Anticancer Agents Med. Chem. 2008, 8, 761–782. [Google Scholar] [CrossRef]
- Sgobba, M.; Rastelli, G. Structure-based and in silico design of Hsp90 inhibitors. ChemMedChem. 2009, 4, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Janin, Y.L. ATPase inhibitors of heat-shock protein 90, second season. Drug Discov. Today 2010, 15, 342–353. [Google Scholar] [CrossRef]
- Messaoudi, S.; Peyrat, J.-F.; Brion, J.-D.; Alami, M. Heat-shock protein 90 inhibitors as antitumor agents: A survey of the literature from 2005 to 2010. Expert Opin. Ther. Pat. 2011, 21, 1501–1542. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, S.; Peyrat, J.-F.; Brion, J.-D.; Alami, M. Recent advances in hsp90 inhibitors as antitumor agents. In Advances in Anti-Cancer Agents in Medicinal Chemistry, 1st ed.; Prudhomme, M., Ed.; Bentham Sciences Publishers: Oak Park, IL, USA, 2013; Volume 1, pp. 107–183. [Google Scholar]
- Neckers, L. Heat shock protein 90: The cancer chaperone. J. Biosci. 2007, 32, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Amolins, M.W.; Blagg, B.S.J. Natural product inhibitors of Hsp90: Potential leads for drug discovery. Mini Rev. Med. Chem. 2009, 9, 140–152. [Google Scholar] [CrossRef] [Green Version]
- Maloney, A.; Workman, P. HSP90 as a new therapeutic target for cancer therapy: The story unfolds. Expert Opin. Biol. Ther. 2002, 2, 3–24. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.-L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer. 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Zhang, H.; Burrows, F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J. Mol. Med. 2004, 82, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Blagg, B.S.J.; Kerr, T.D. Hsp90 inhibitors: Small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med. Res. Rev. 2006, 26, 310–338. [Google Scholar] [CrossRef]
- Chène, P. ATPases as drug targets: Learning from their structure. Nat. Rev. Drug Discov. 2002, 1, 665–673. [Google Scholar] [CrossRef]
- Taldone, T.; Sun, W.; Chiosis, G. Discovery and development of heat shock protein 90 inhibitors. Bioorg. Med. Chem. 2009, 17, 2225–2235. [Google Scholar] [CrossRef] [Green Version]
- Taldone, T.; Gozman, A.; Maharaj, R.; Chiosis, G. Targeting Hsp90: Small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 2008, 8, 370–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting Hsp90 for cancer therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar] [CrossRef] [Green Version]
- Panaretou, B.; Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998, 17, 4829–4836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, P.A.; Hostein, I.; Banerji, U.; Stefano, F.D.; Maloney, A.; Walton, M.; Judson, I.; Workman, P. Gene expression profiling of human colon cancer cells following inhibition of signal transduction by 17-allylamino-17-demethoxygeldanamycin, an inhibitor of the Hsp90 molecular chaperone. Oncogene 2000, 19, 4125–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlichman, C. Tanespimycin: The opportunities and challenges of targeting heat shock protein 90. Expert Opin. Investig. Drugs 2009, 18, 861–868. [Google Scholar] [CrossRef]
- McCollum, A.K.; Teneyck, C.J.; Sauer, B.M.; Toft, D.O.; Erlichman, C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res. 2006, 66, 10967–10975. [Google Scholar] [CrossRef] [Green Version]
- Demidenko, Z.N.; Vivo, C.; Halicka, H.D.; Li, C.J.; Bhalla, K.; Broude, E.V.; Blagosklonny, M.V. Pharmacological induction of Hsp70 protects apoptosis-prone cells from doxorubicin: Comparison with caspase-inhibitor- and cycle-arrest- mediated cytoprotection. Cell. Death Differ. 2006, 13, 1434–1441. [Google Scholar] [CrossRef] [Green Version]
- Gabai, V.L.; Budagova, K.R.; Sherman, M.Y. Increased expression of the major heat shock protein Hsp72 in human prostate carcinoma cells is dispensable for their viability but confers resistance to a variety of anticancer agents. Oncogene 2005, 24, 3328–3338. [Google Scholar] [CrossRef] [Green Version]
- Pocaly, M.; Lagarde, V.; Etienne, G.; Ribeil, J.A.; Claverol, S.; Bonneu, M.; Moreau-Gaudry, F.; Guyonnet-Duperat, V.; Hermine, O.; Melo, J.V.; et al. Overexpression of the heat-shock protein 70 is associated to imatinib resistance in chronic myeloid leukemia. Leukemia 2007, 21, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.E. Targeting heat shock proteins 70/90 and proteasome for cancer therapy. Curr. Med. Chem. 2011, 18, 4250–4264. [Google Scholar] [CrossRef]
- Wang, H.; Tan, M.S.; Lu, R.C.; Yu, J.T.; Tan, L. Heat shock proteins at the crossroads between cancer and Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 239164. [Google Scholar] [CrossRef]
- Murphy, M.E. The Hsp70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, A.; Blagg, B.S.J. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem. 2008, 15, 2702–2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskew, J.D.; Sadikot, T.; Morales, P.; Duren, A.; Dunwiddie, I.; Swink, M.; Zhang, X.; Hembruff, S.; Donnelly, A.; Rajewski, R.A.; et al. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 2011, 11, 468. [Google Scholar] [CrossRef] [Green Version]
- Buckton, K.; Wahyudi, H.; McAlpine, S.R. The first report of direct inhibitors that target the C-terminal MEEVD region on heat shock protein 90 L. Chem. Commun. 2016, 52, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Burlison, J.A.; Avila, C.; Vielhauer, G.; Lubbers, D.J.; Holzbeierlein, J.; Blagg, B.S.J. Development of novobiocin analogues that manifest anti-proliferative activity against several cancer cell lines. J. Org. Chem. 2008, 73, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, H.K.; Koay, Y.C.; Irani, S.; Das, R.; Nassar, Z.D.; Australian Prostate Cancer BioResource; Selth, L.A.; Centenera, M.M.; McAlpine, S.R.; Butler, L.M. A novel class of Hsp90 C-Terminal modulators have pre-clinical efficacy in prostate tumor cells without induction of a heat shock response. Prostate 2016, 76, 1546–1559. [Google Scholar] [CrossRef] [PubMed]
- Moses, M.A.; Henry, E.C.; Ricke, W.A.; Gasiewicz, T.A. The heat shock protein 90 inhibitor, (-)-epigallocatechin gallate, has anticancer activity in a novel human prostate cancer progression model. Cancer Prev. Res. (Phila.) 2015, 8, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Le Bras, G.; Radanyi, C.; Peyrat, J.-F.; Brion, J.-D.; Alami, M.; Marsaud, V.; Stella, B.; Renoir, J.-M. New novobiocin analogues as antiproliferative agents in breast cancer cells and potential inhibitors of heat shock protein 90. J. Med. Chem. 2007, 50, 6189–6200. [Google Scholar] [CrossRef] [PubMed]
- Radanyi, C.; Le Bras, G.; Marsaud, V.; Peyrat, J.-F.; Messaoudi, S.; Catelli, M.G.; Brion, J.-D.; Alami, M.; Renoir, J.-M. Antiproliferative and apoptotic activities of tosylcyclonovobiocic acids as potent heat shock protein 90 inhibitors in human cancer cells. Cancer Lett. 2008, 274, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Radanyi, C.; Le Bras, G.; Bouclier, C.; Messaoudi, S.; Peyrat, J.-F.; Brion, J.-D.; Alami, M.; Renoir, J.-M. Tosylcyclonovobiocic acids promote cleavage of the hsp90-associated cochaperone p23. Biochem. Biophys. Res. Commun. 2009, 379, 514–518. [Google Scholar] [CrossRef]
- Audisio, D.; Methy-Gonnot, D.; Radanyi, C.; Renoir, J.-M.; Denis, S.; Sauvage, F.; Vergnaud-Gauduchon, J.; Brion, J.-D.; Messaoudi, S.; Alami, M. synthesis and antiproliferative activity of novobiocin analogues as potential Hsp90 inhibitors. Eur. J. Med. Chem. 2014, 83, 498–507. [Google Scholar] [CrossRef]
- Radanyi, C.; Le Bras, G.; Messaoudi, S.; Bouclier, C.; Peyrat, J.-F.; Brion, J.-D.; Marsaud, V.; Renoir, J.-M.; Alami, M. Synthesis and biological activity of simplified denoviose-coumarins related to novobiocin as potent inhibitors of heat-shock protein 90 (hsp90). Bioorg. Med. Chem. Lett. 2008, 18, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Audisio, D.; Messaoudi, S.; Cegielkowski, L.; Peyrat, J.-F.; Brion, J.-D.; Methy-Gonnot, D.; Radanyi, C.; Renoir, J.-M.; Alami, M. Discovery and biological activity of 6brcaq as an inhibitor of the Hsp90 protein folding machinery. ChemMedChem 2011, 6, 804–815. [Google Scholar] [CrossRef]
- Messaoudi, S.; Audisio, D.; Brion, J.-D.; Alami, M. Rapid access to 3-(N-substituted)-aminoquinolin-2(1H)-ones using palladium-catalyzed C–N bond coupling reaction. Tetrahedron 2007, 63, 10202–10210. [Google Scholar] [CrossRef]
- Sauvage, F.; Fattal, E.; Al-Shaer, W.; Denis, S.; Brotin, E.; Denoyelle, C.; Blanc-Fournier, C.; Toussaint, B.; Messaoudi, S.; Alami, M.; et al. Antitumor activity of nanoliposomes encapsulating the novobiocin analog 6brcaq in a triple-negative breast cancer model in mice. Cancer Lett. 2018, 432, 103–111. [Google Scholar] [CrossRef]
- Sauvage, F.; Franzè, S.; Bruneau, A.; Alami, M.; Denis, S.; Nicolas, V.; Lesieur, S.; Legrand, F.-X.; Barratt, G.; Messaoudi, S.; et al. Formulation and in vitro efficacy of liposomes containing the Hsp90 inhibitor 6BrCaQ in prostate cancer cells. Int. J. Pharm. 2016, 499, 101–109. [Google Scholar] [CrossRef]
- Mathieu, C.; Chamayou, Q.; Luong, T.T.H.; Naud, D.; Mahuteau, F.; Alami, M.; Fattal, E.; Messaoudi, S.; Vergnaud-Gauduchon, J. Synthesis and antiproliferative activity of 6BrCaQ-TPP conjugates for targeting the mitochondrial heat shock protein TRAP1. Eur. J. Med. Chem. 2022, 229, 114052–114065. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, B.; Peterson, L.B.; Blagg, B.S.J. 3-Arylcoumarin derivatives manifest anti-proliferative activity through hsp90 inhibition. ACS Med. Chem. Lett. 2012, 3, 327–331. [Google Scholar] [CrossRef]
- Bruneau, A.; Brion, J.-B.; Messaoudi, S.; Alami, M. A General Pd/Cu-catalyzed C-H heteroarylation of 3-bromoquinolin-2(1H)-ones. Org. Biomol. Chem. 2014, 12, 8533–8541. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, W.; Yu, Z. Transition-metal mediated carbon–sulfur bond activation and transformations. Chem. Soc. Rev. 2013, 42, 599–621. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Shi, Z.-J. Recent advances in transition-metal-catalyzed C–S activation: From thioester to (hetero)aryl thioether. ACS Catal. 2014, 4, 280–288. [Google Scholar] [CrossRef]
- Prokopcová, H.; Kappe, C.O. The Liebeskind–Srogl C-C cross-coupling reaction. Angew. Chem. Int. Ed. 2009, 48, 2276–2286. [Google Scholar] [CrossRef]
- Vabre, R. Fonctionnalisation Directe de Liaisons C-H et Couplages Croisés pour la Formation de Liaisons C-C et C-N: Synthèse de Purines 6,8,9-trisubstituées. Ph.D. Thesis, Université Paris Sud—Paris XI, Paris, France, 2013. [Google Scholar]
- Vabre, R.; Chevot, F.; Legraverend, M.; Piguel, S. Microwave-assisted Pd/Cu-catalyzed C-8 direct alkenylation of purines and related azoles: An alternative access to 6, 8, 9-trisubstituted purines. J. Org. Chem. 2011, 76, 9542–9547. [Google Scholar] [CrossRef]
- Benner, S.A. Understanding nucleic acids using synthetic chemistry. Acc. Chem. Res. 2004, 37, 784–797. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef]
- Sugahara, T.; Murakami, K.; Yorimitsu, H.; Osuka, A. Palladium-catalyzed amination of aryl sulfides with anilines. Angew. Chem. Int. Ed. 2014, 53, 9329–9333. [Google Scholar] [CrossRef] [Green Version]
- Pellegatti, L.; Vedrenne, E.; Leger, J.-M.; Jarry, C.; Routier, S. First efficient palladium-catalyzed aminations of pyrimidines, 1, 2, 4-triazines and tetrazines by original methyl sulfur release. Synlett 2009, 13, 2137–2142. [Google Scholar] [CrossRef]
- Bruneau, L.A.; Roche, M.; Alami, M.; Messaoudi, S. 2-Aminobiphenyl Palladacycles: The “Most Powerful” Precatalysts in C–C and C–Heteroatom Cross-Couplings. ACS Catal. 2015, 5, 1386–1396. [Google Scholar] [CrossRef]
- Liu, J.; Robins, M.J. Fluoro, alkylsulfanyl, and alkylsulfonyl leaving groups in Suzuki cross-coupling reactions of purine 2′-deoxynucleosides and nucleosides. Org. Lett. 2005, 7, 1149–1151. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  |  |  |  |  | |||||||||||||
| Cell viability [%] [a] | ||||||||||||||||||
| MDA-MB-231 | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | ||
| 47 | 47 | 36 | 60 | 60 | 23 | 55 | 72 | 65 | 98 | 73 | 70 | 50 | 98 | |||||
| PC-3 (15 µM) | 70 | 61 | 61 | ND | 61 | ND | ||||||||||||
| MRC-5 (15 µM) | 80 | 95 | 88 | ND | 88 | ND | ||||||||||||
 |  |  |  |  |  | |||||||||||||
| Cell viability [%] [a] | ||||||||||||||||||
| MDA-MB-231 | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM |
| 66 | 81 | 87 | 70 | 69 | 61 | 71 | 62 | 35 | 60 | 47 | 55 | 57 | 62 | 50 | 73 | 81 | 92 | |
| PC-3 (15 µM) | 73 | 106 | 97 | 89 | 56 | 88 | ||||||||||||
| MRC-5 (15 µM) | 94 | 92 | 82 | 70 | 71 | 98 | ||||||||||||
 |  |  |  |  | DMSO | |||||||||||||
| Cell viability [%] [a] | ||||||||||||||||||
| MDA-MB-231 | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | 15 µM | 25 µM | 10 µM | 100 | ||||
| 56 | 57 | 27 | 55 | 42 | 17 | 62 | 63 | 69 | 44 | 55 | 61 | 76 | ||||||
| PC-3 (15 µM) | 91 | 97 | 87 | 88 | ND | 100 | ||||||||||||
| MRC-5 (15 µM) | 79 | 84 | 91 | 81 | ND | 100 | ||||||||||||
| Compound | PC-3 |
|---|---|
| 6Br-CaQ | 10 |
| 3a | 48 |
| 3b | 28 |
| 3c | 37 |
| 3e | 38 |
| Primary Antibody | Dilution | Secondary Antibody (from Santa-Cruz) | Dilution |
|---|---|---|---|
| Anti-Hsp90 α/β (H-114) (Santa-Cruz) | 1/500 | Anti-rabbit | 1/10,000 |
| Anti-Hsp70 (Santa-Cruz) | 1/500 | Anti-mouse | 1/3000 |
| Anti-CDK-4 (C-22) (Santa-Cruz) | 1/500 | Anti-rabbit | 1/10,000 |
| Anti-GAPDH (Sigma Aldrich) | 1/5000 | Anti-rabbit | 1/10,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larghi, E.L.; Bruneau, A.; Sauvage, F.; Alami, M.; Vergnaud-Gauduchon, J.; Messaoudi, S. Synthesis and Biological Activity of 3-(Heteroaryl)quinolin-2(1H)-ones Bis-Heterocycles as Potential Inhibitors of the Protein Folding Machinery Hsp90. Molecules 2022, 27, 412. https://doi.org/10.3390/molecules27020412
Larghi EL, Bruneau A, Sauvage F, Alami M, Vergnaud-Gauduchon J, Messaoudi S. Synthesis and Biological Activity of 3-(Heteroaryl)quinolin-2(1H)-ones Bis-Heterocycles as Potential Inhibitors of the Protein Folding Machinery Hsp90. Molecules. 2022; 27(2):412. https://doi.org/10.3390/molecules27020412
Chicago/Turabian StyleLarghi, Enrique L., Alexandre Bruneau, Félix Sauvage, Mouad Alami, Juliette Vergnaud-Gauduchon, and Samir Messaoudi. 2022. "Synthesis and Biological Activity of 3-(Heteroaryl)quinolin-2(1H)-ones Bis-Heterocycles as Potential Inhibitors of the Protein Folding Machinery Hsp90" Molecules 27, no. 2: 412. https://doi.org/10.3390/molecules27020412
APA StyleLarghi, E. L., Bruneau, A., Sauvage, F., Alami, M., Vergnaud-Gauduchon, J., & Messaoudi, S. (2022). Synthesis and Biological Activity of 3-(Heteroaryl)quinolin-2(1H)-ones Bis-Heterocycles as Potential Inhibitors of the Protein Folding Machinery Hsp90. Molecules, 27(2), 412. https://doi.org/10.3390/molecules27020412