
Recent Advances in Visible-Light-Mediated Amide Synthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
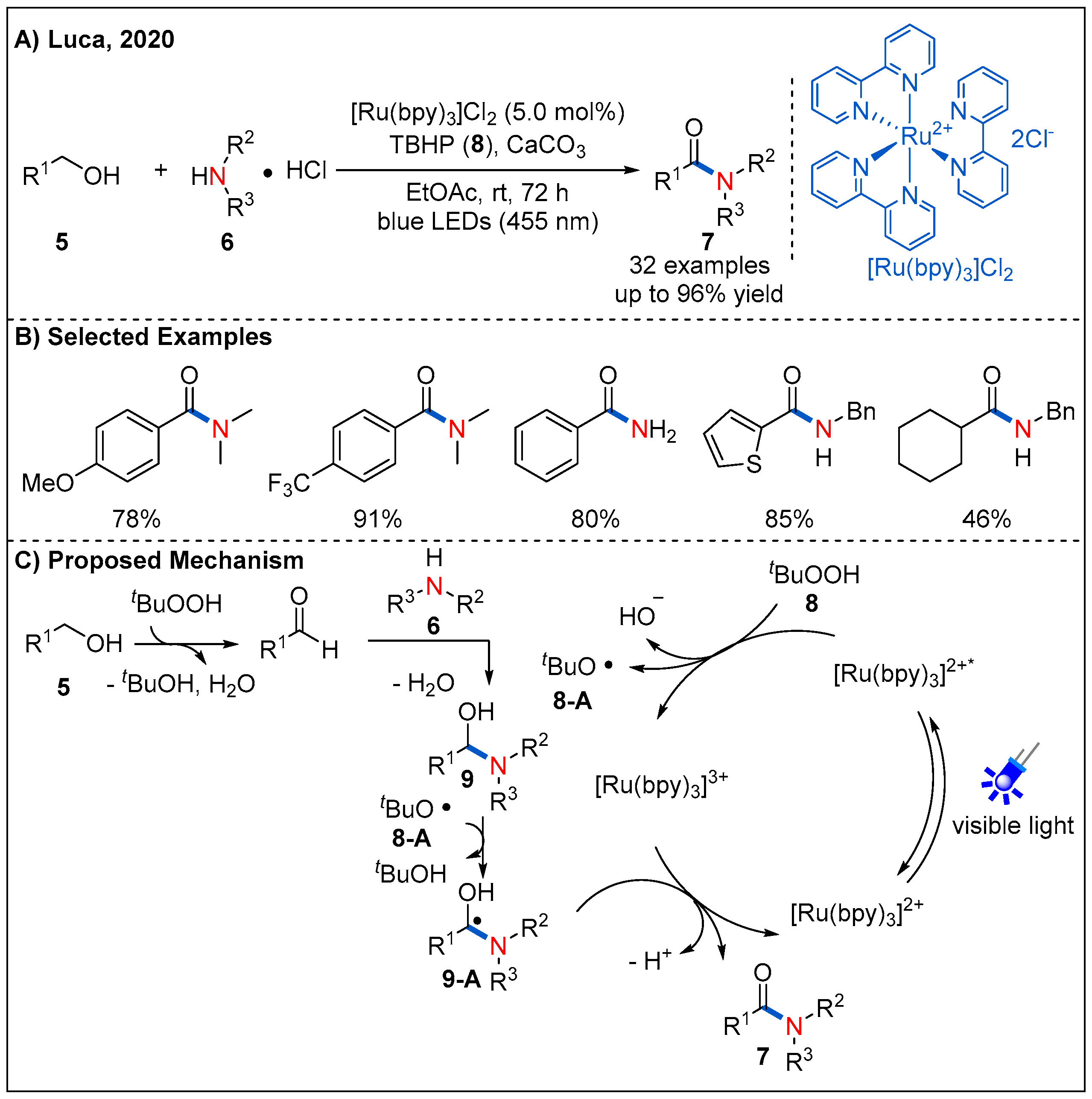{kind=link}
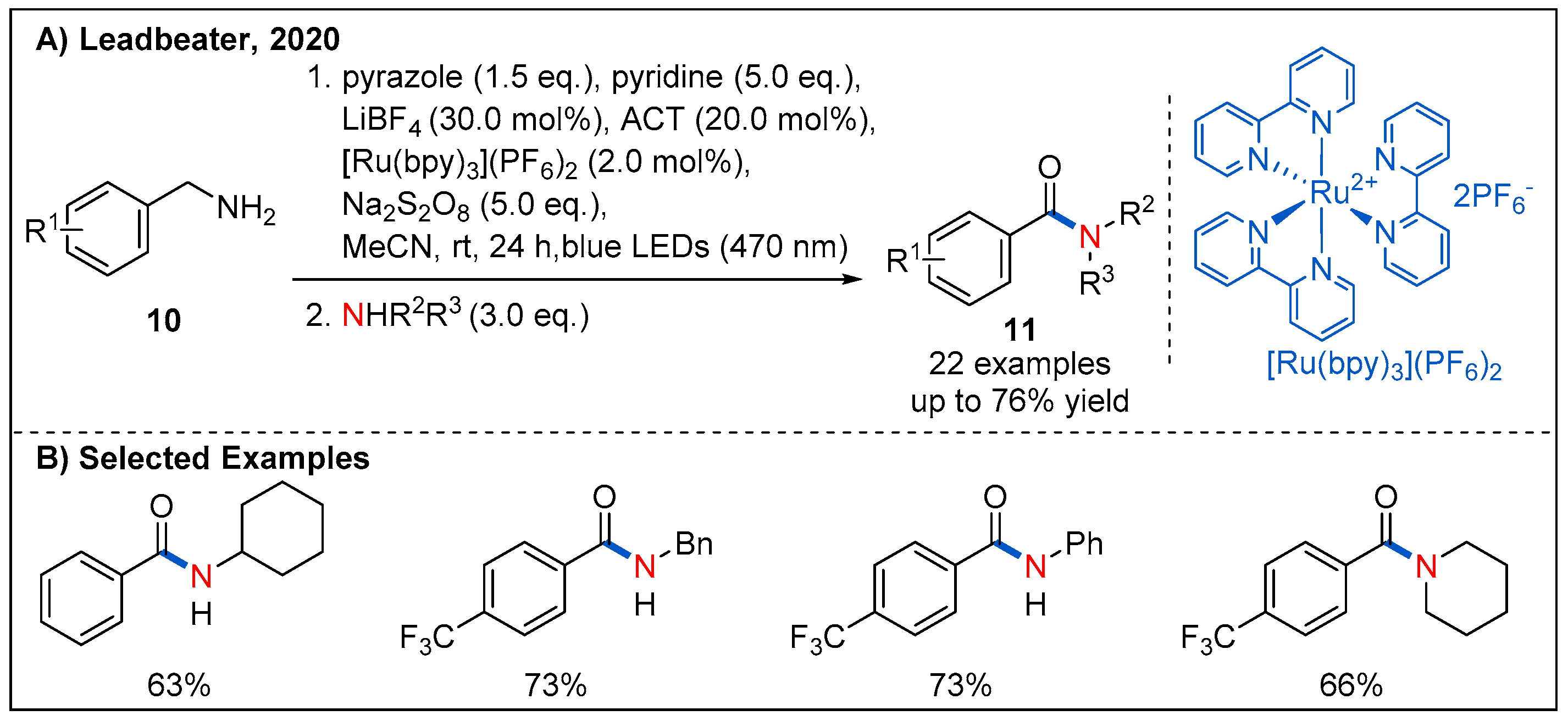{kind=link}
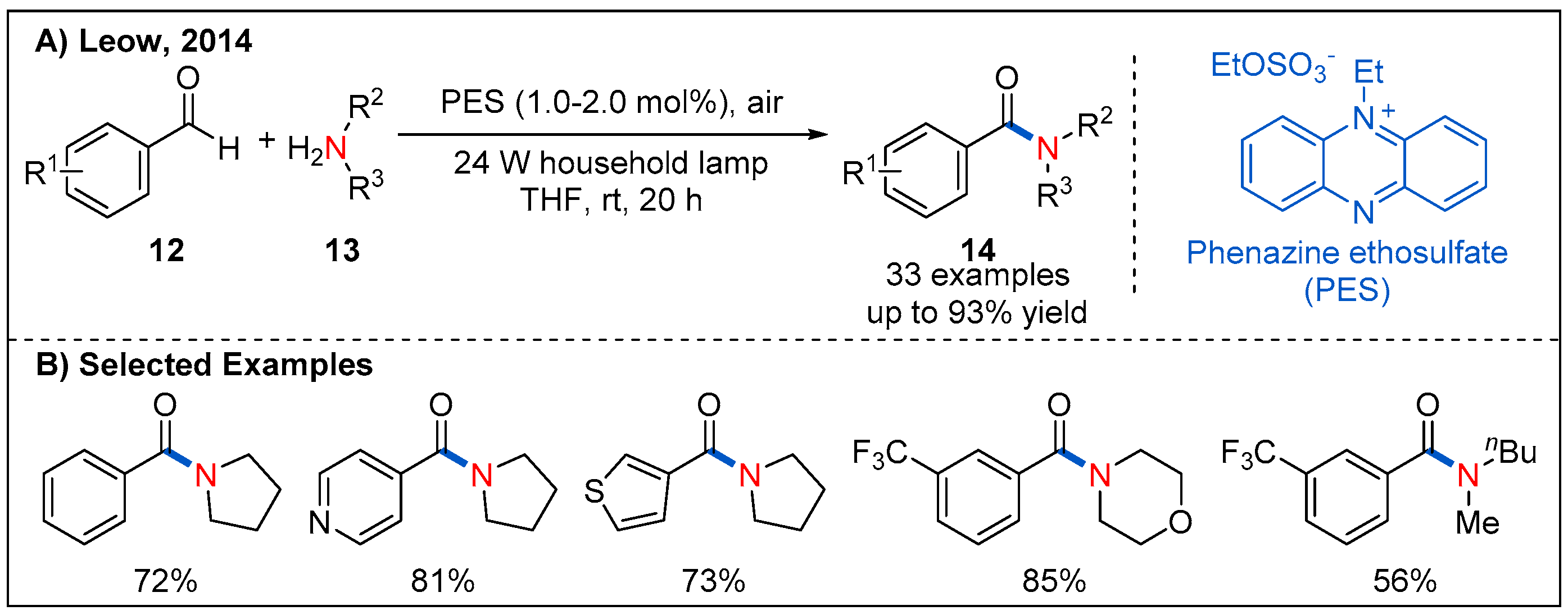{kind=link}
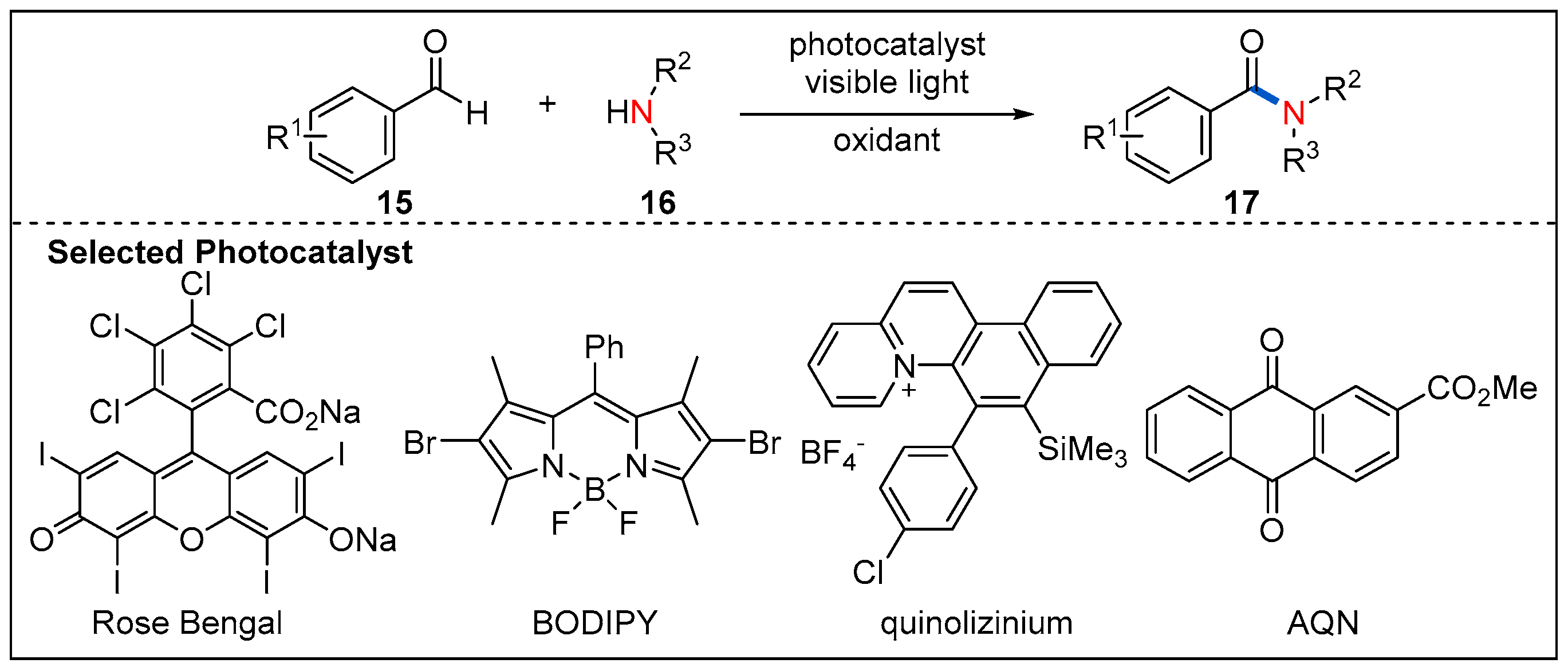{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
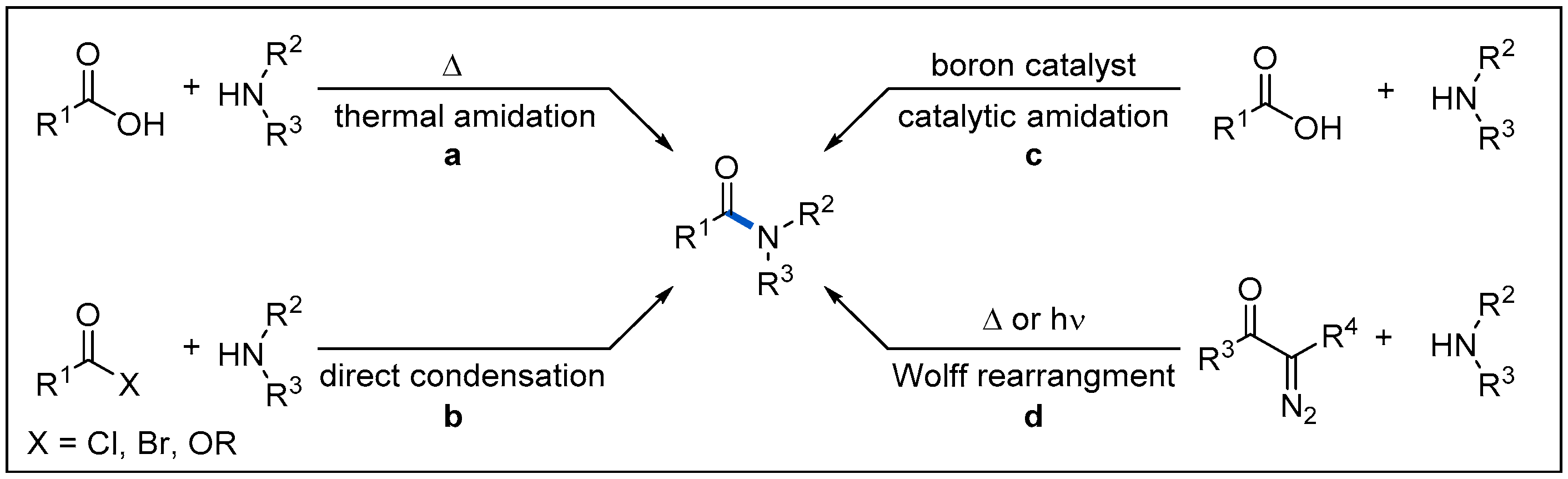
1.1. General Strategies for Amide Bond Formation
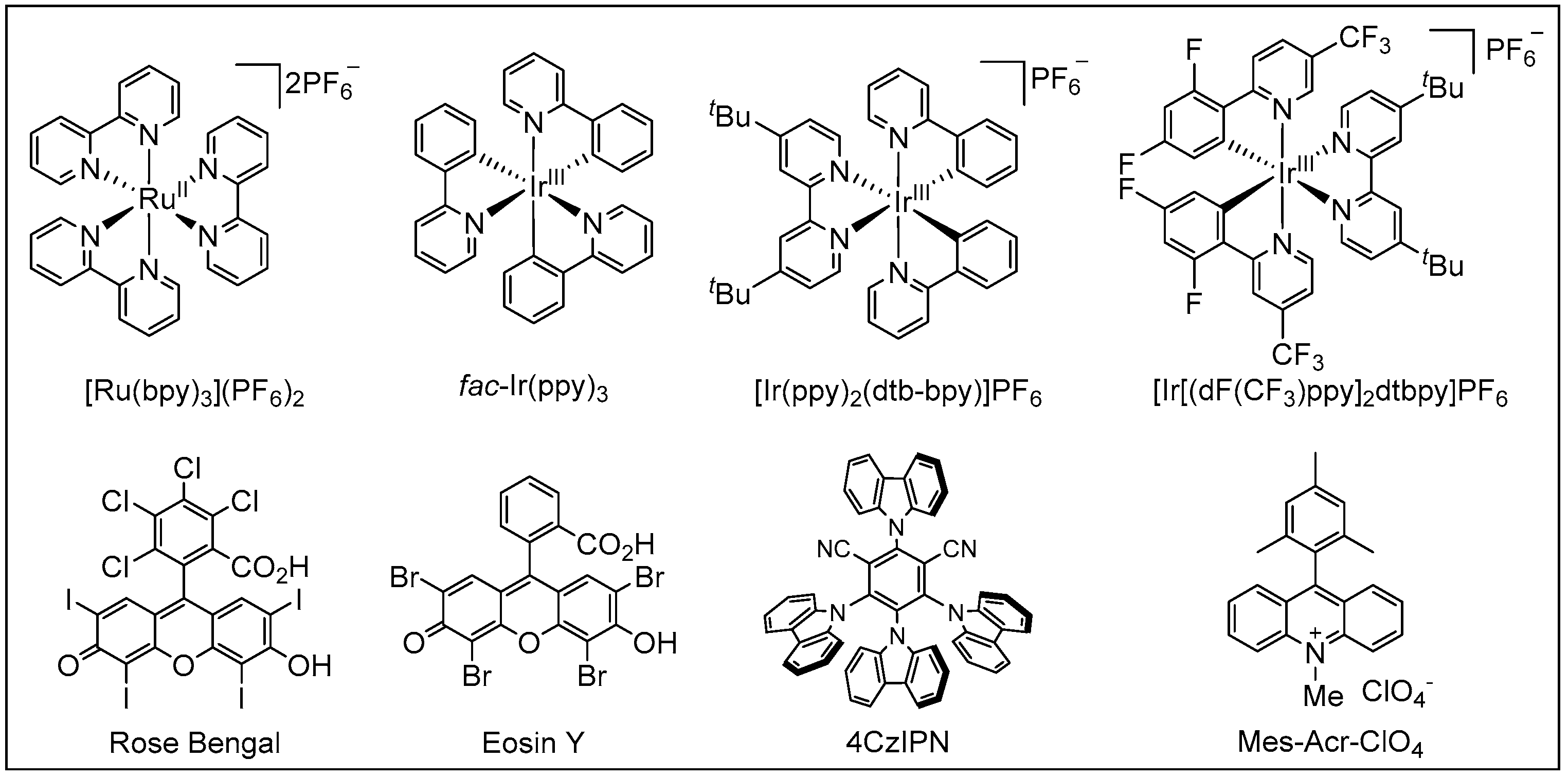
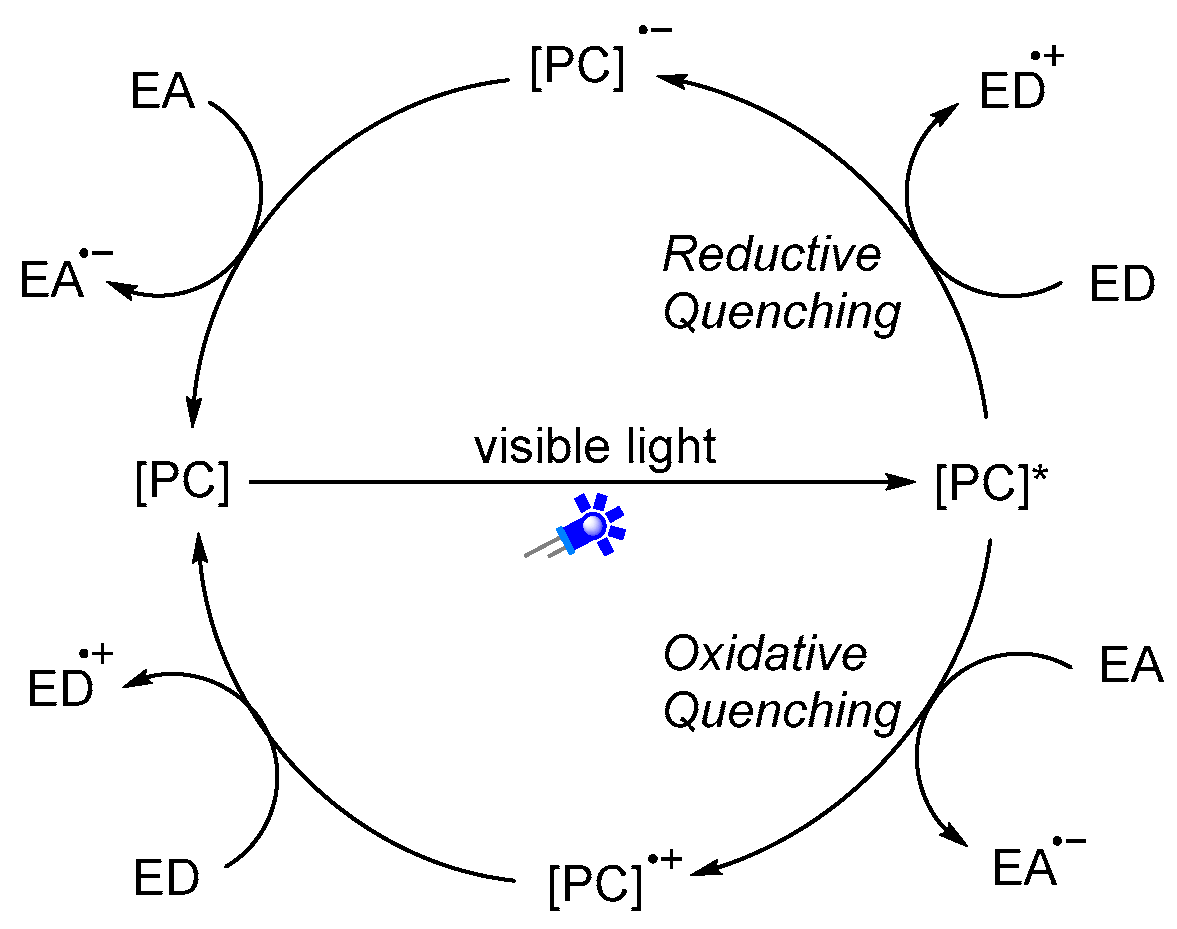
1.2. Visible-Light Photoredox Catalysis
2. Oxidative Amidations
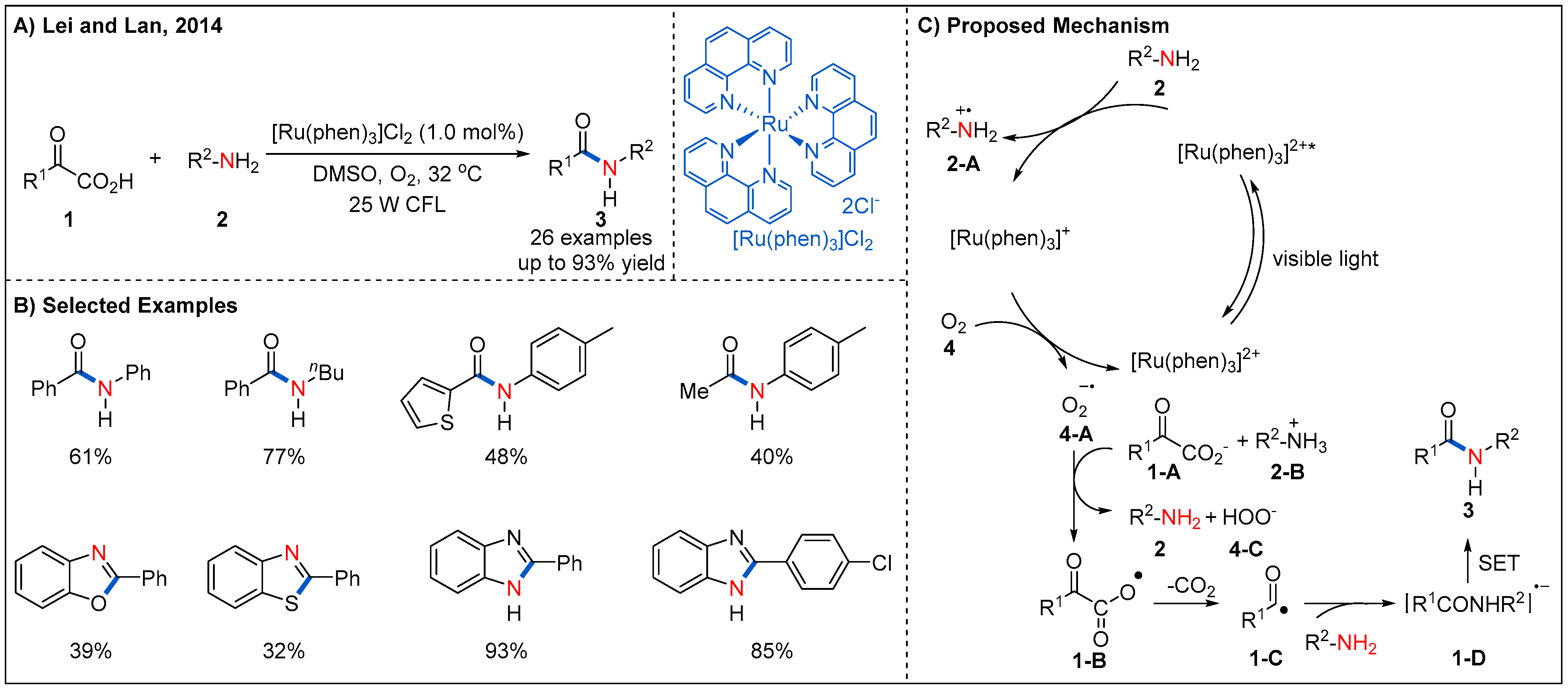
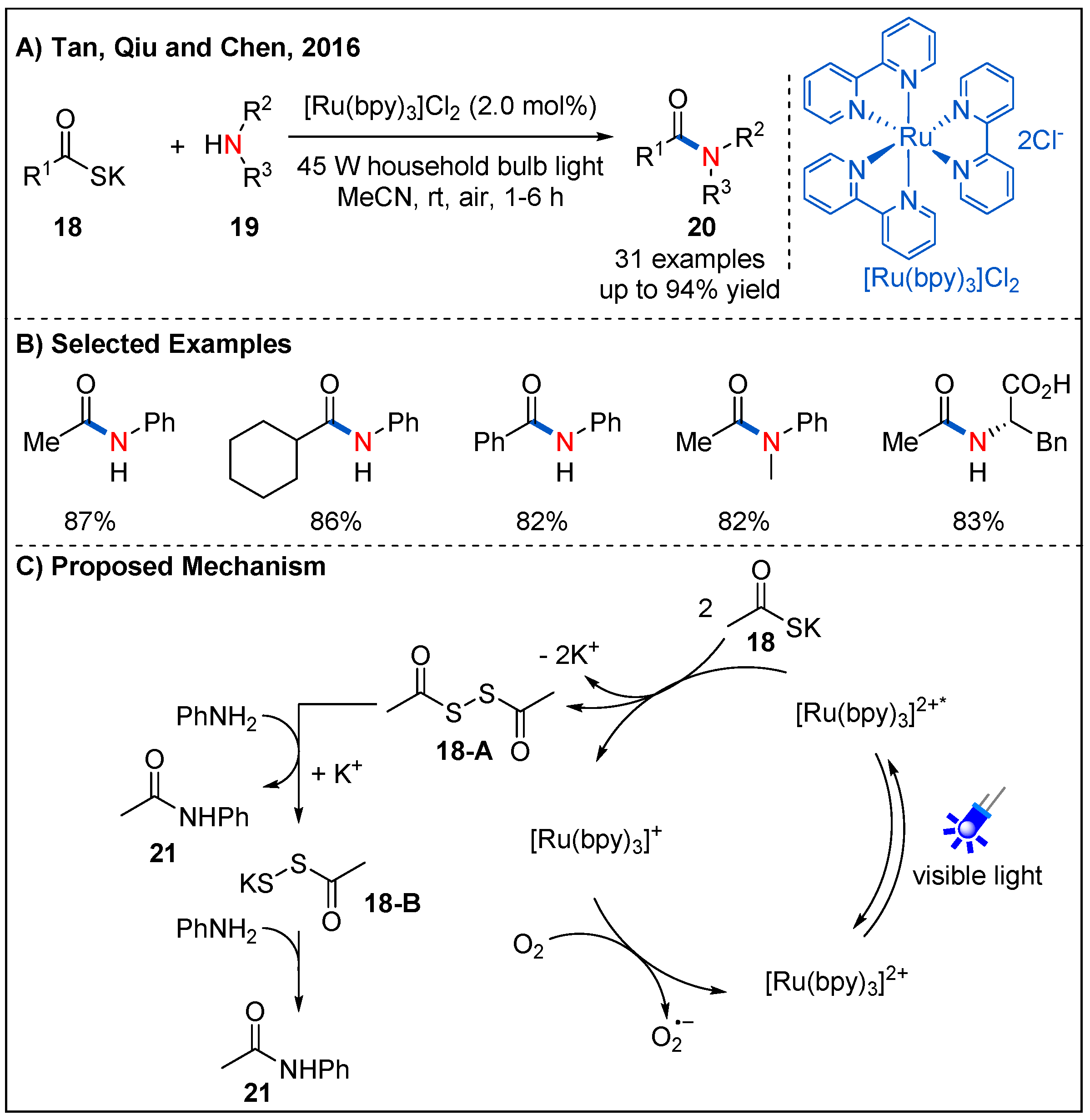
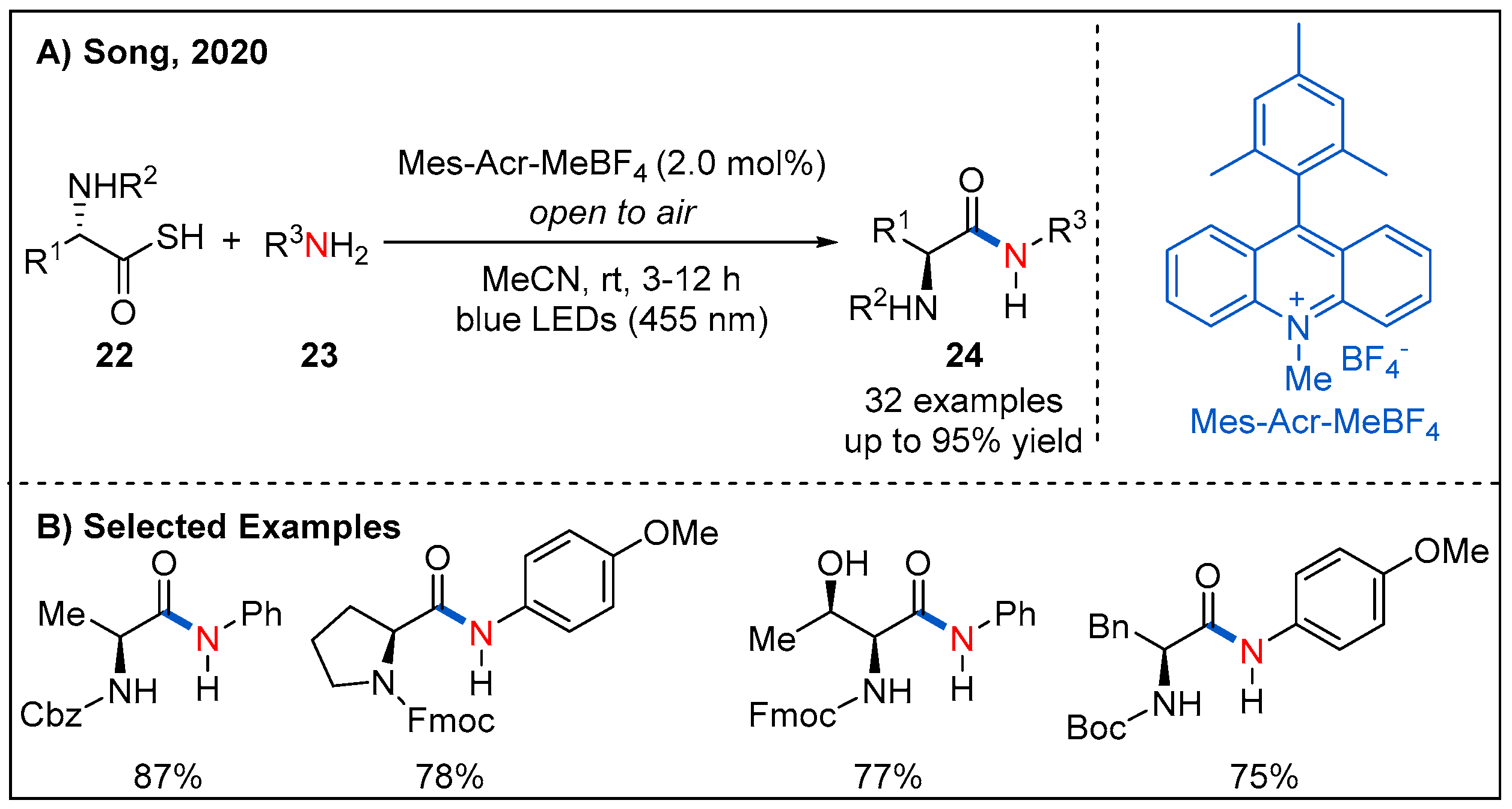
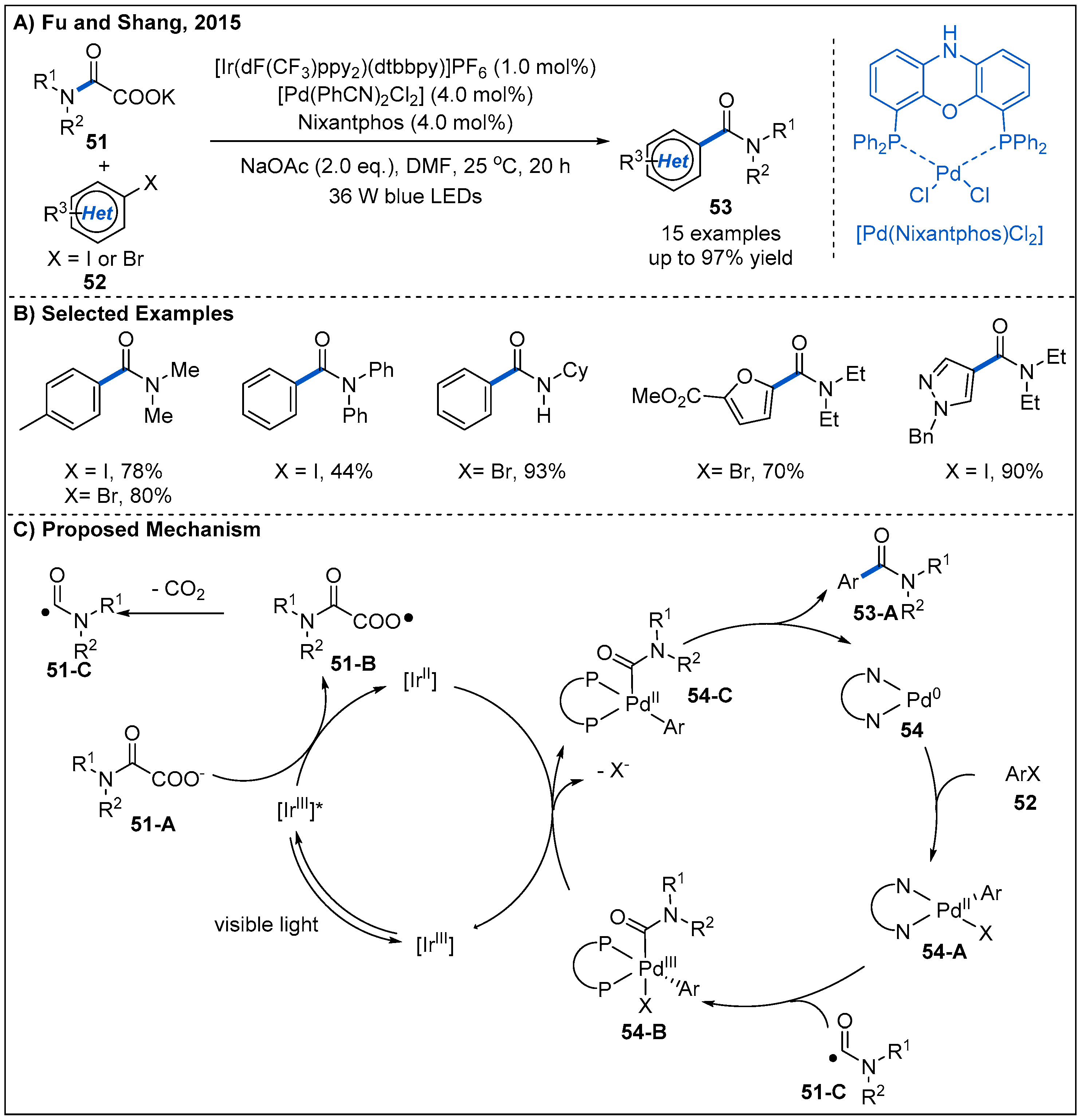
2.1. From Keto Acids
2.2. From Alcohols
2.3. From Amines
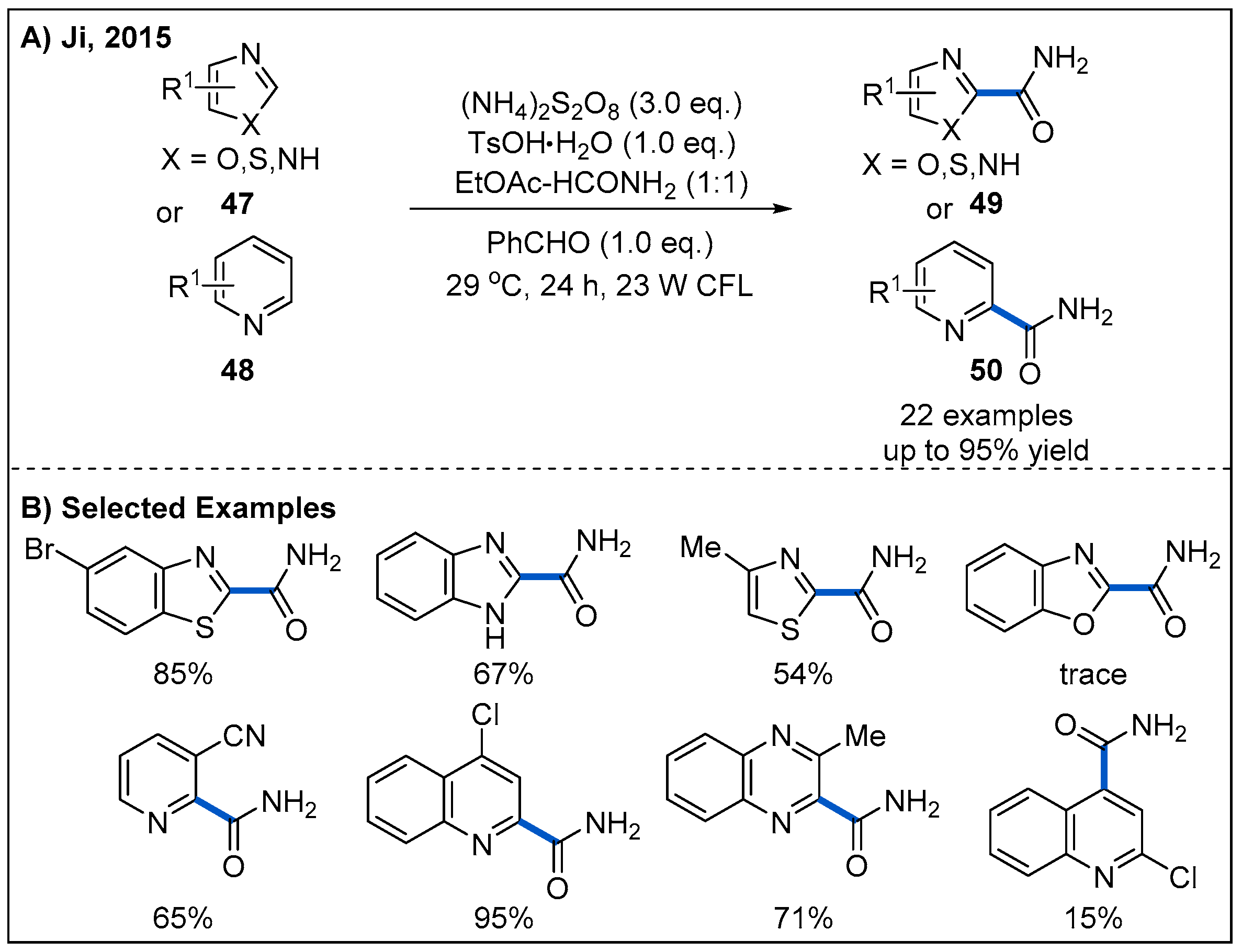
2.4. From Aldehydes
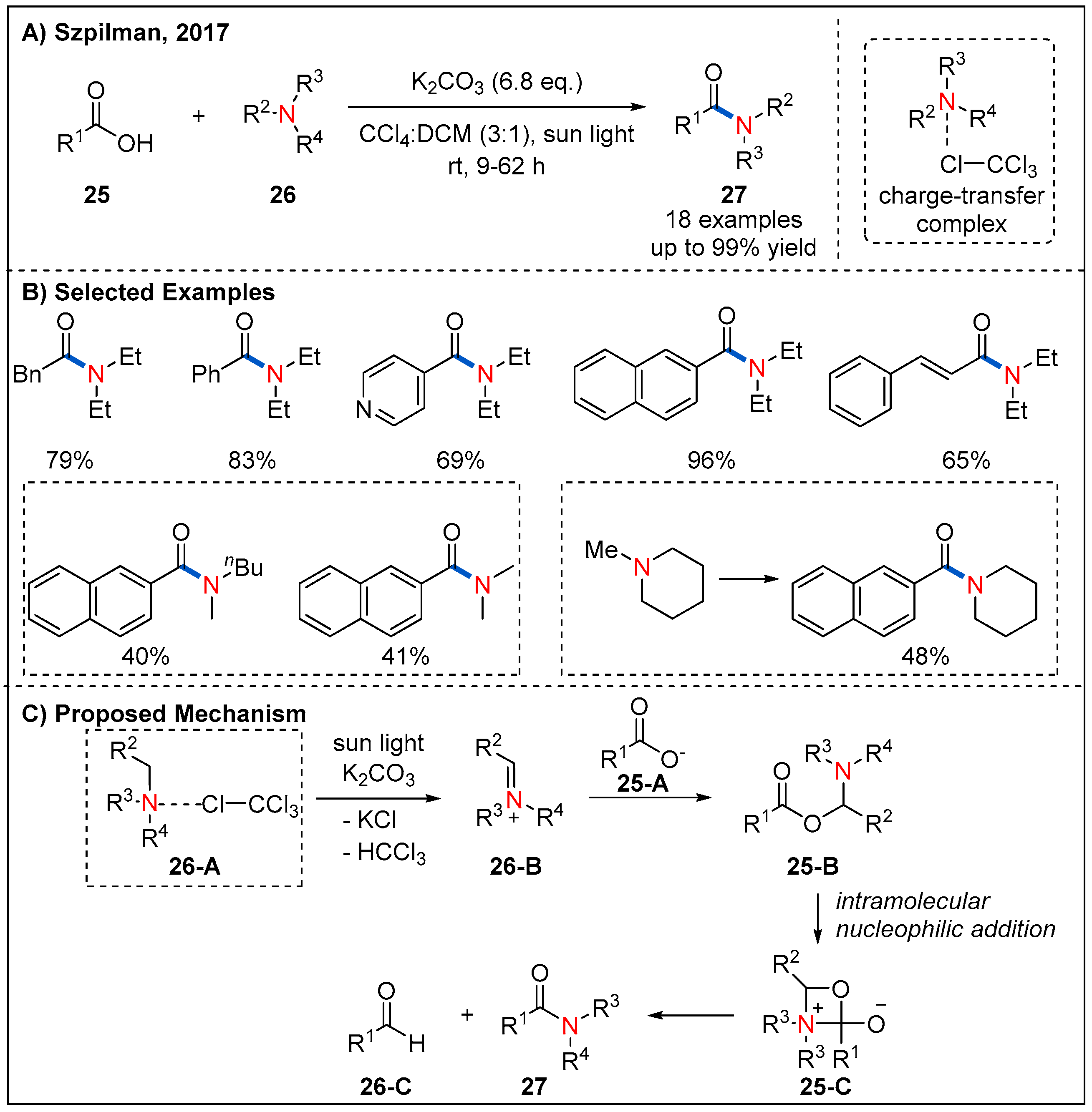
2.5. From Acids
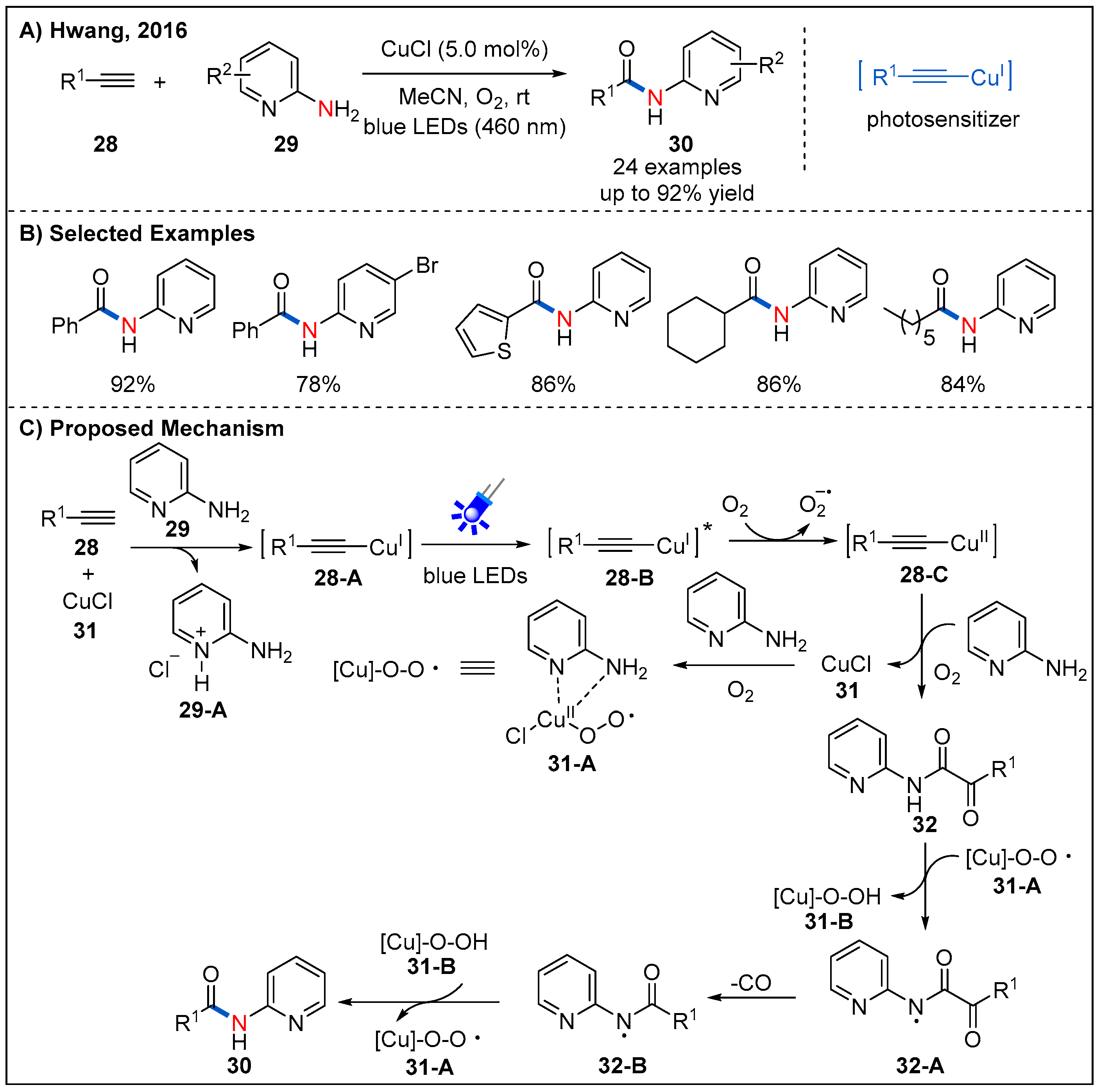
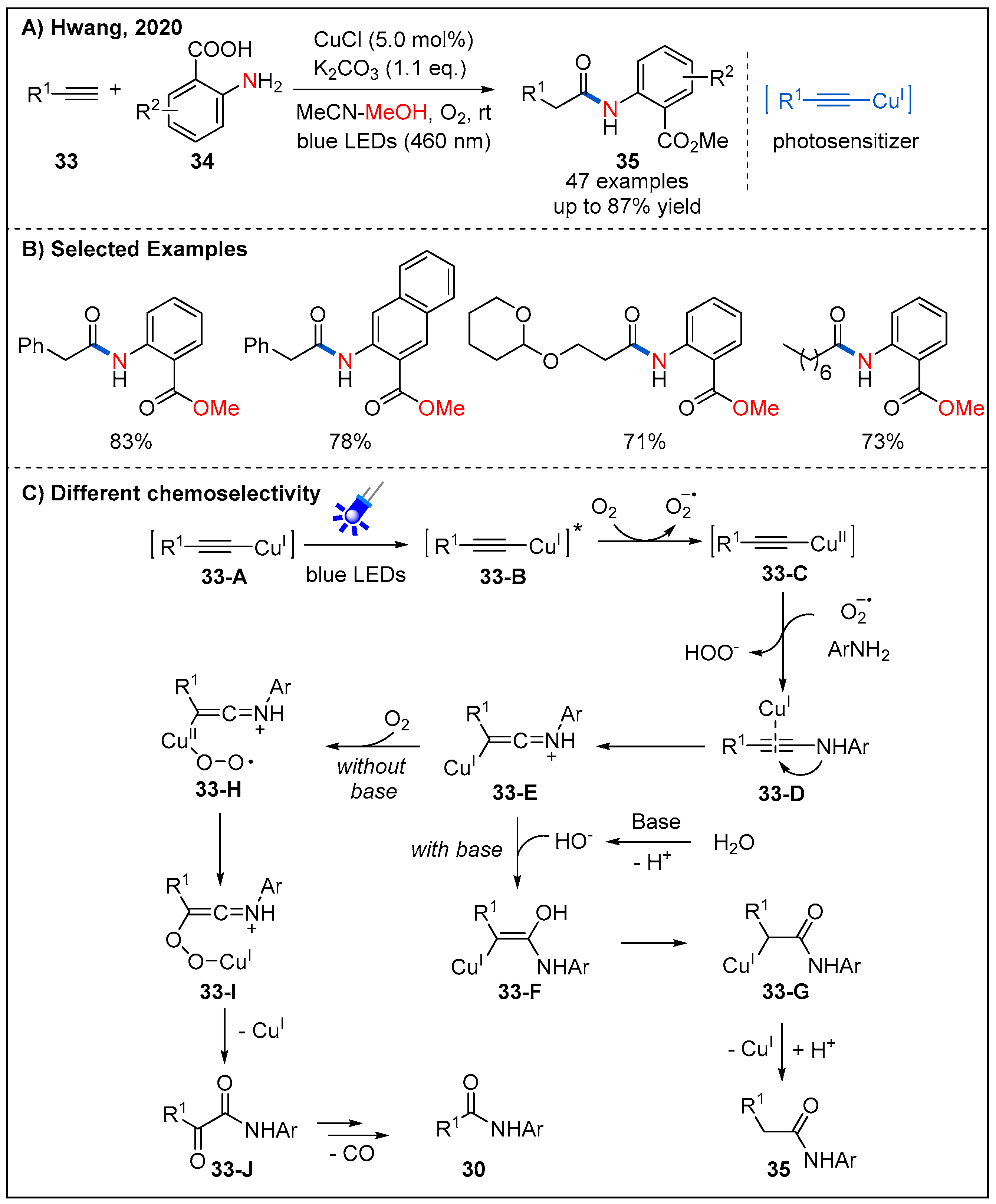
2.6. From Alkynes
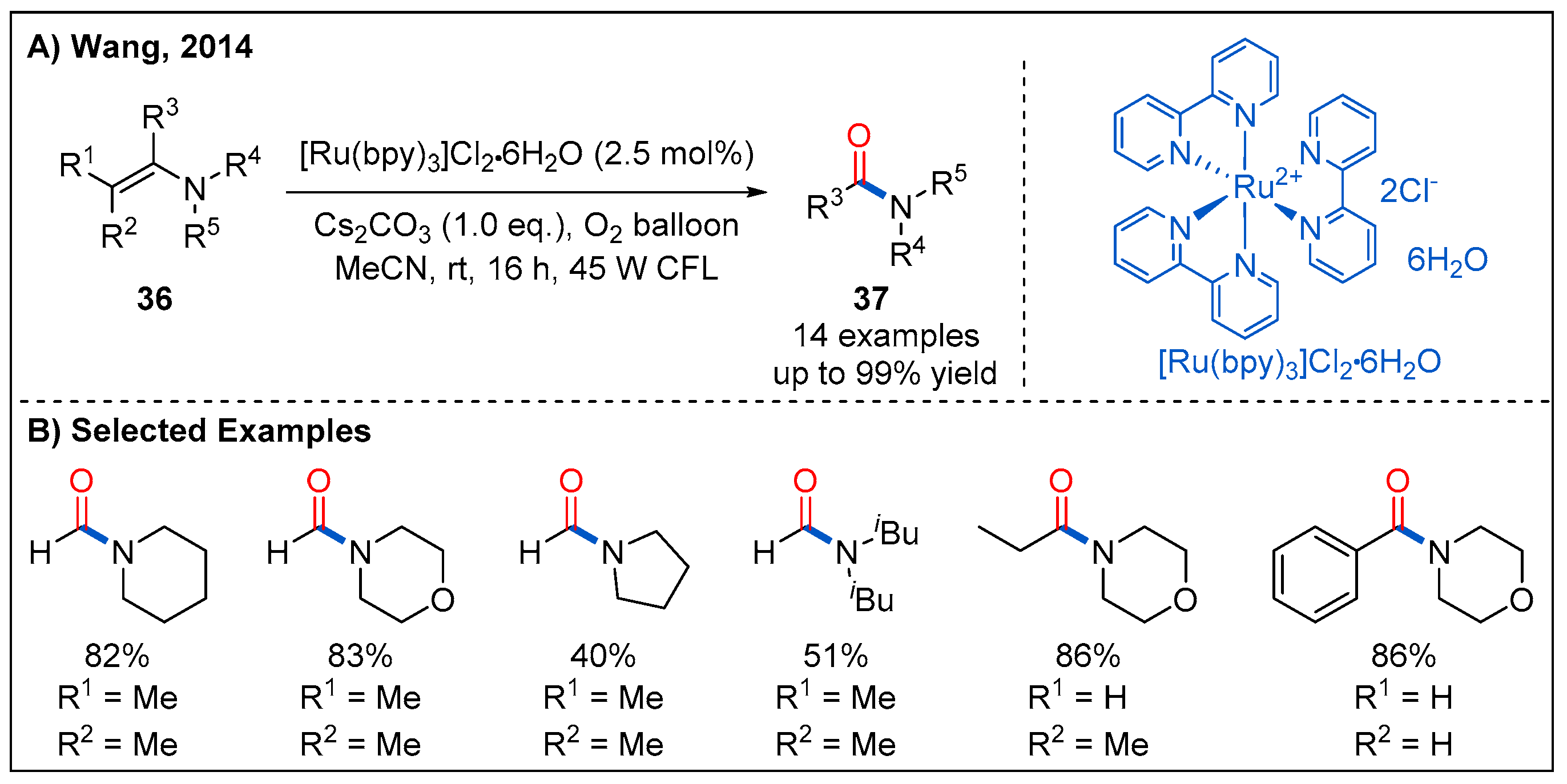
2.7. From Enamines
3. Ritter-Type Reactions
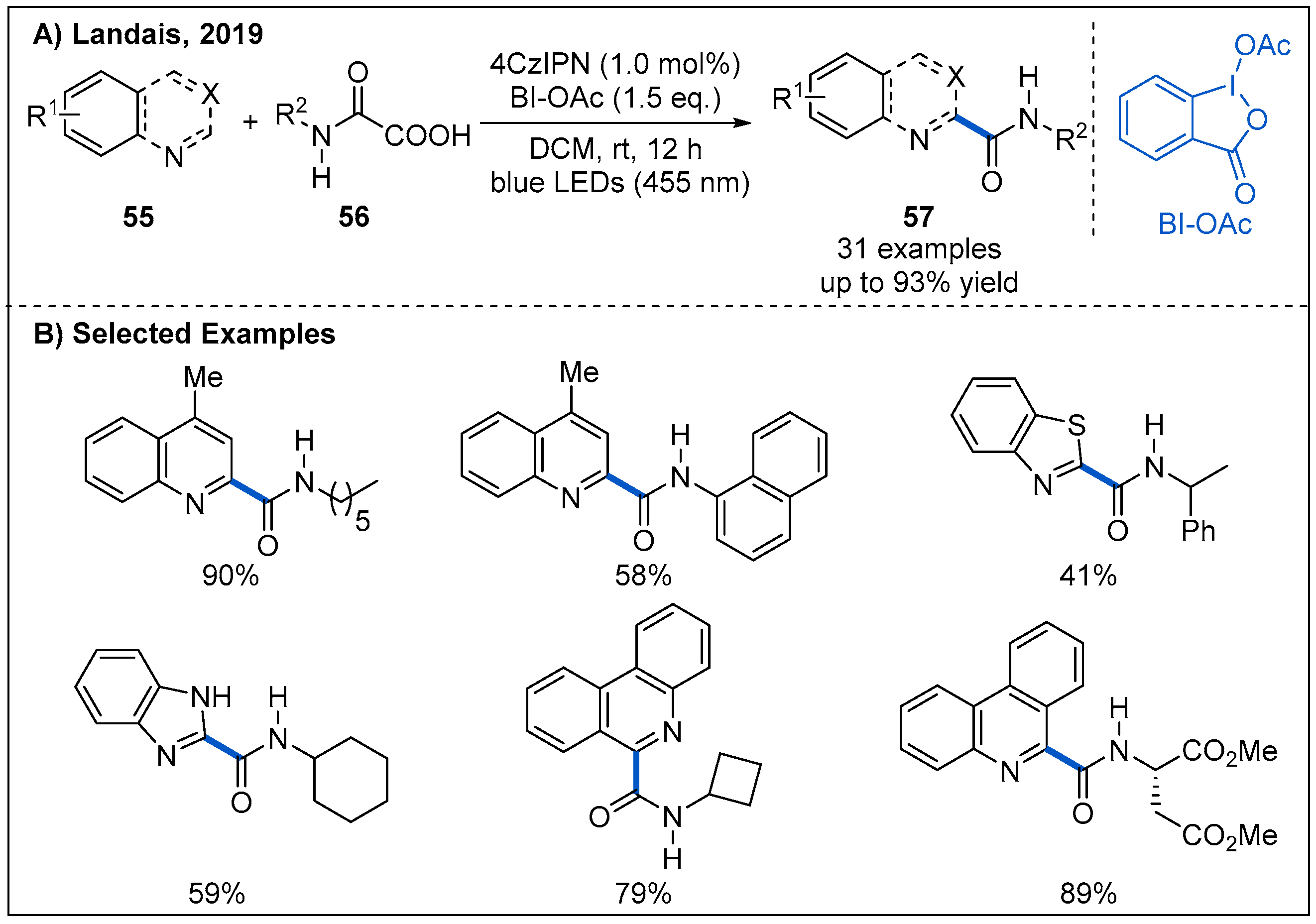
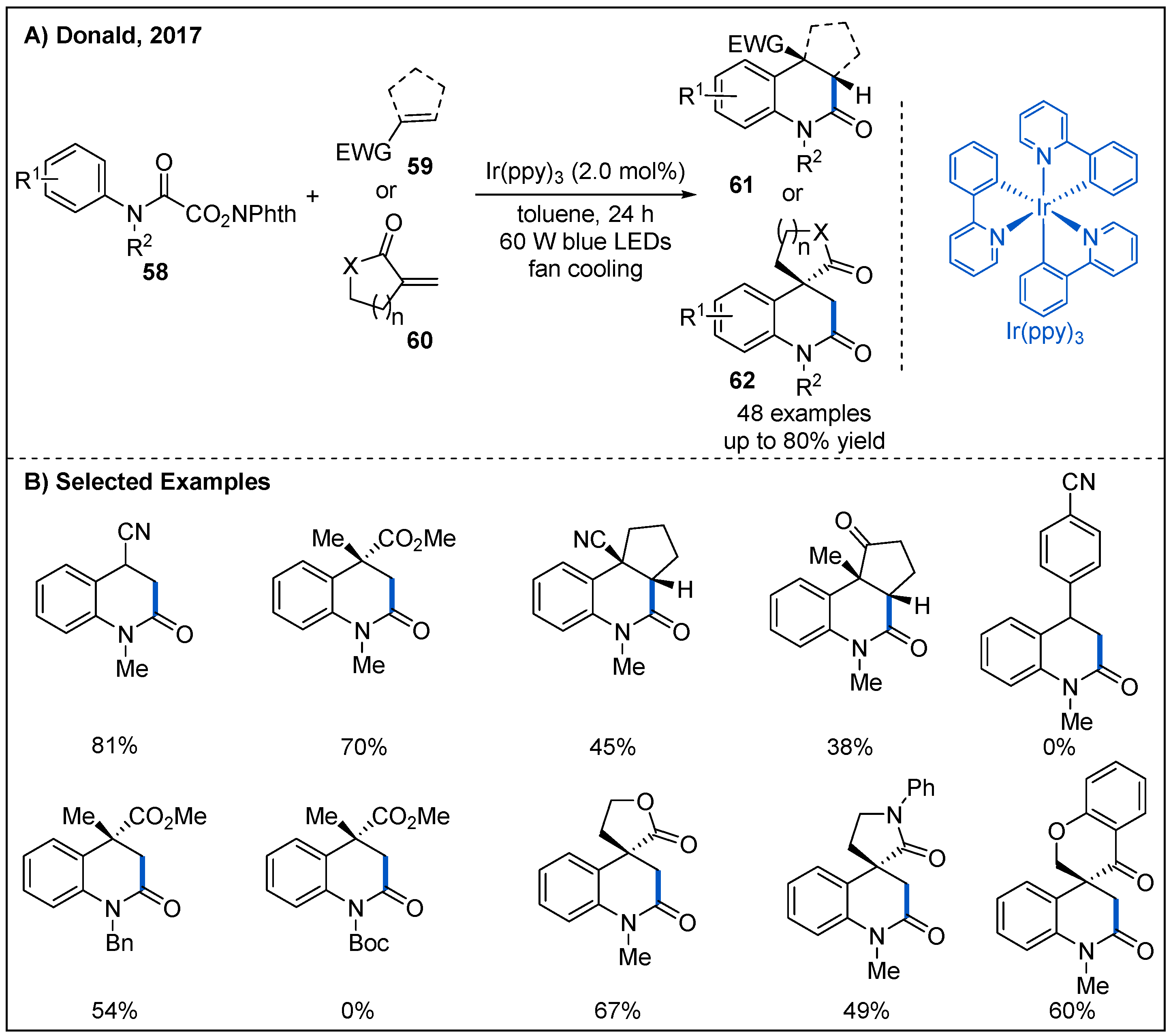
4. Carbamoylation
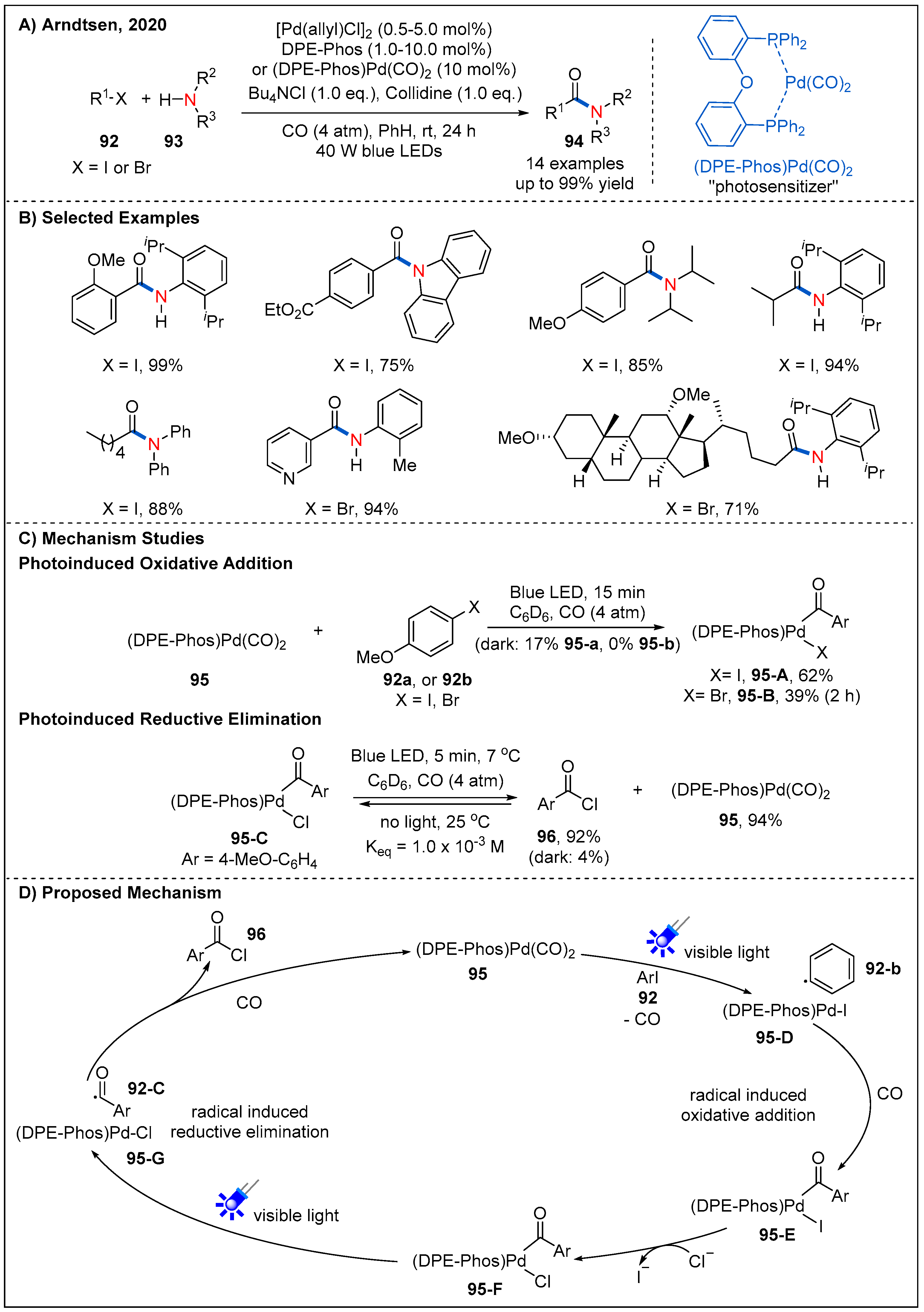
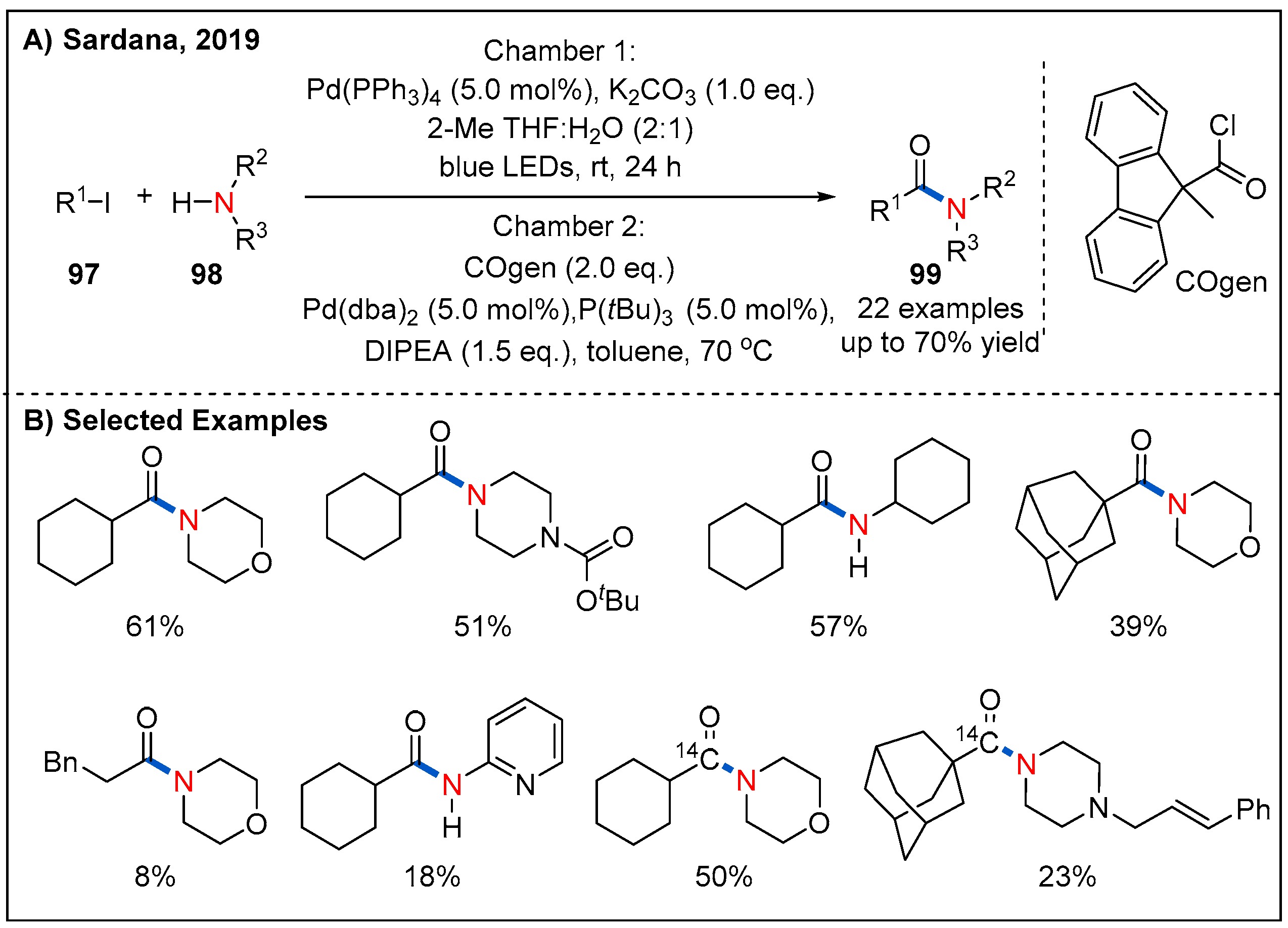
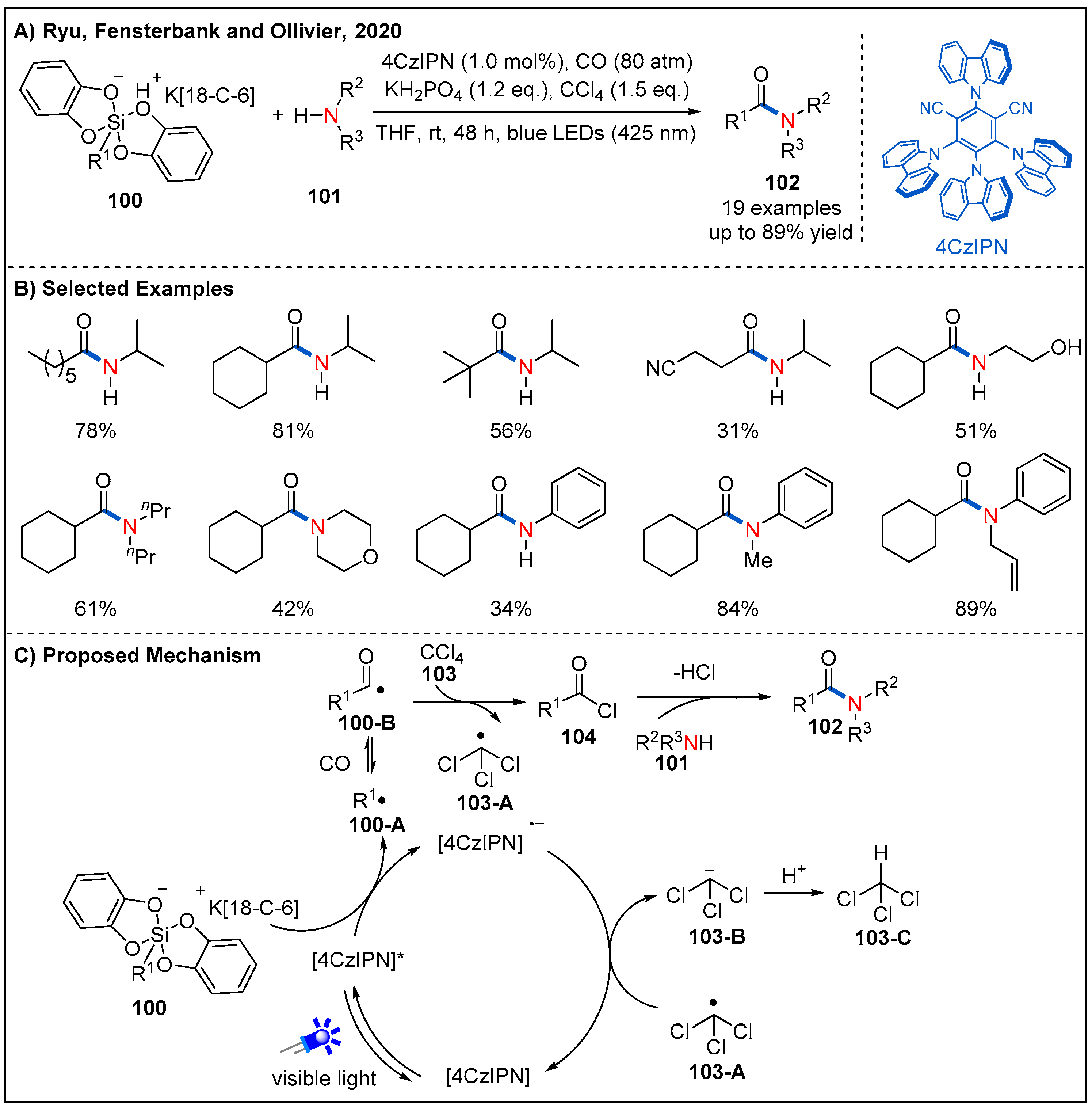
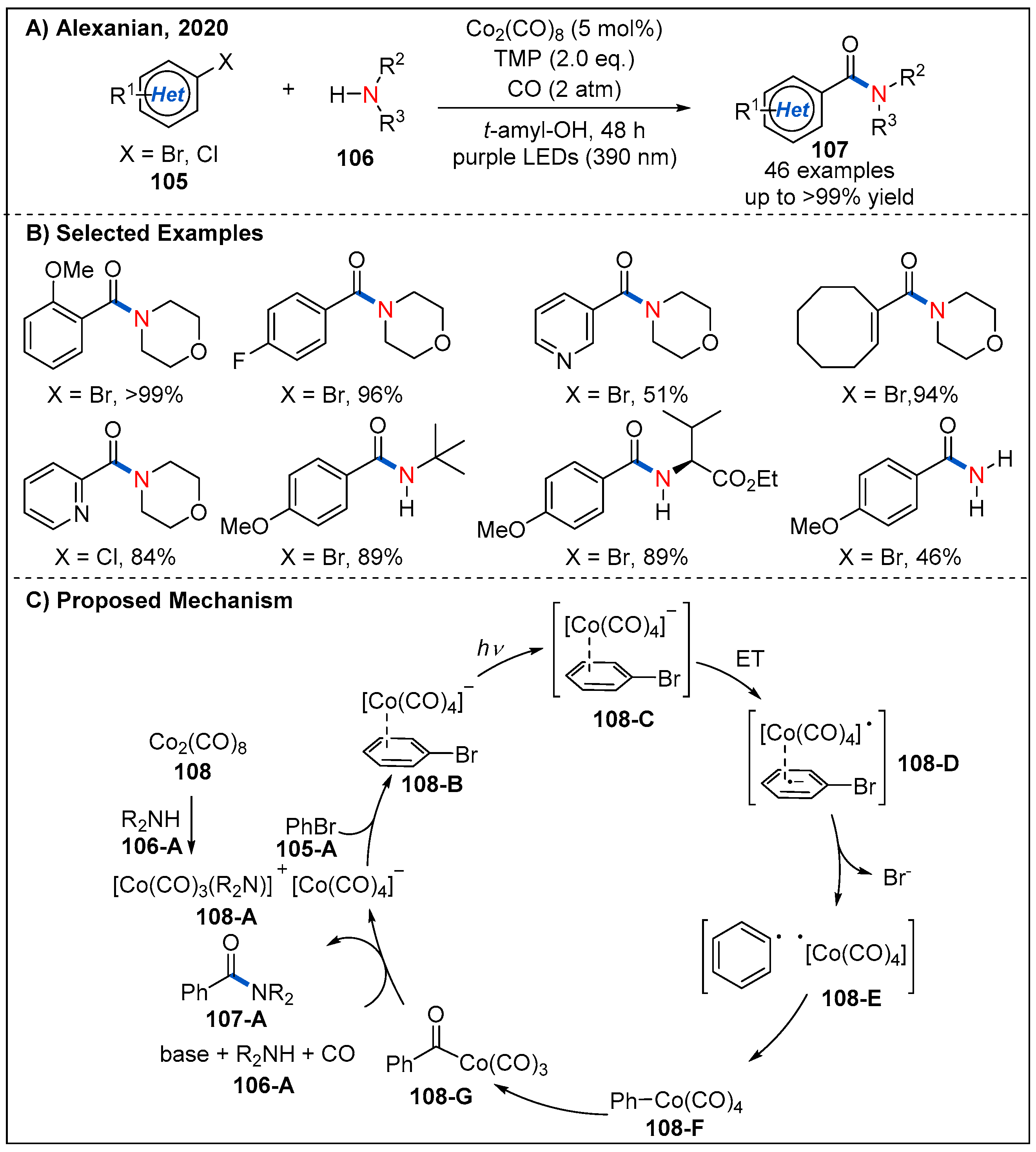
5. Radical Aminocarbonylation
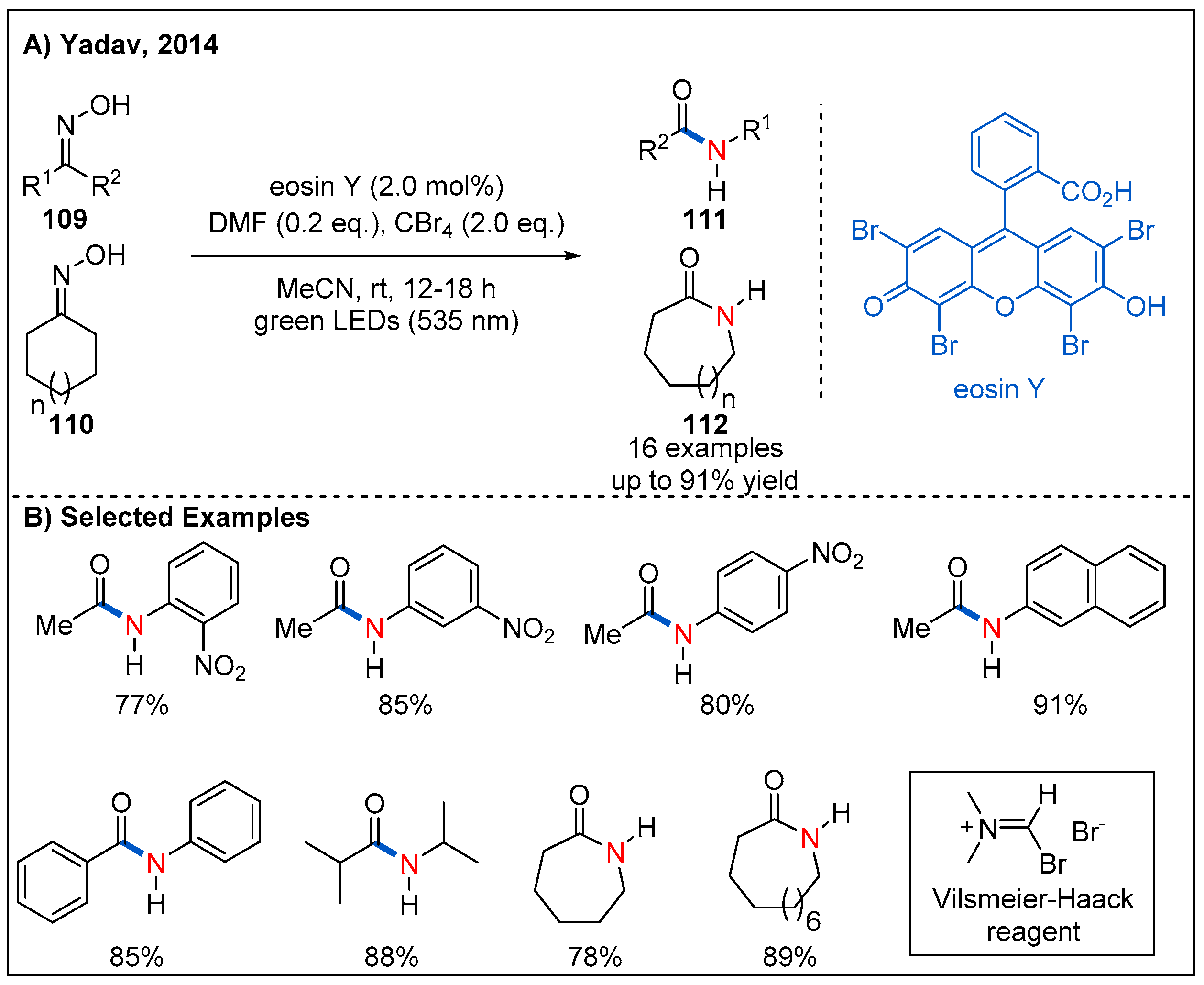
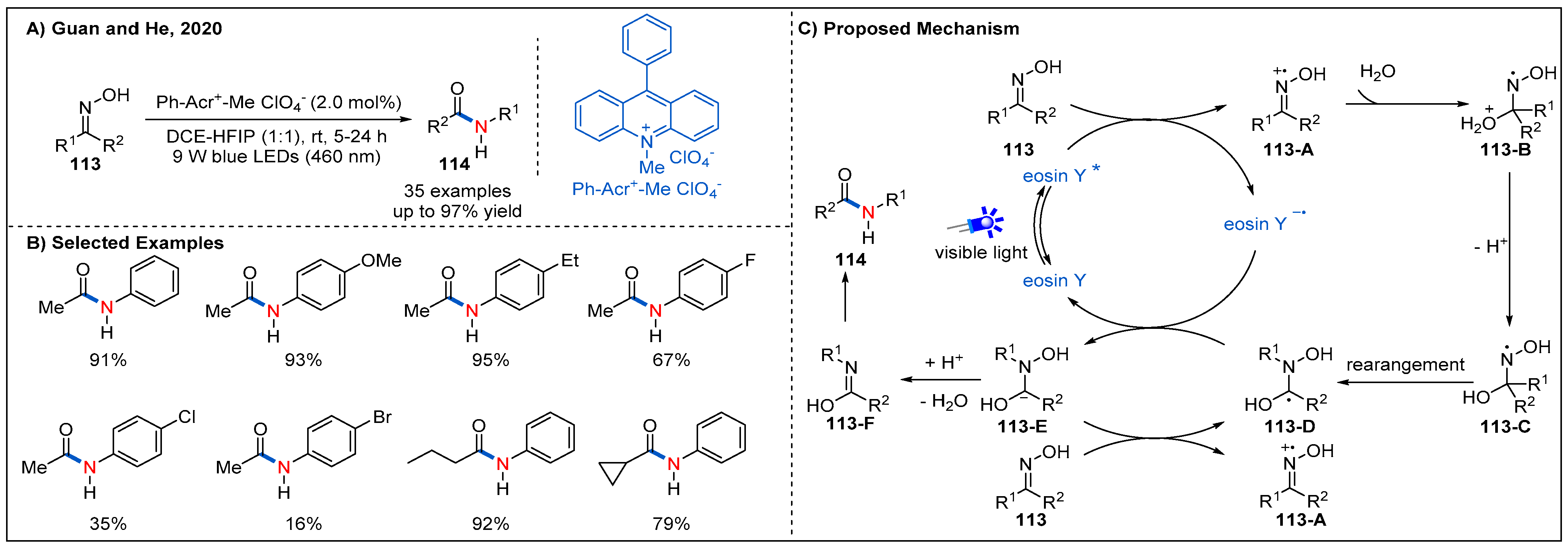
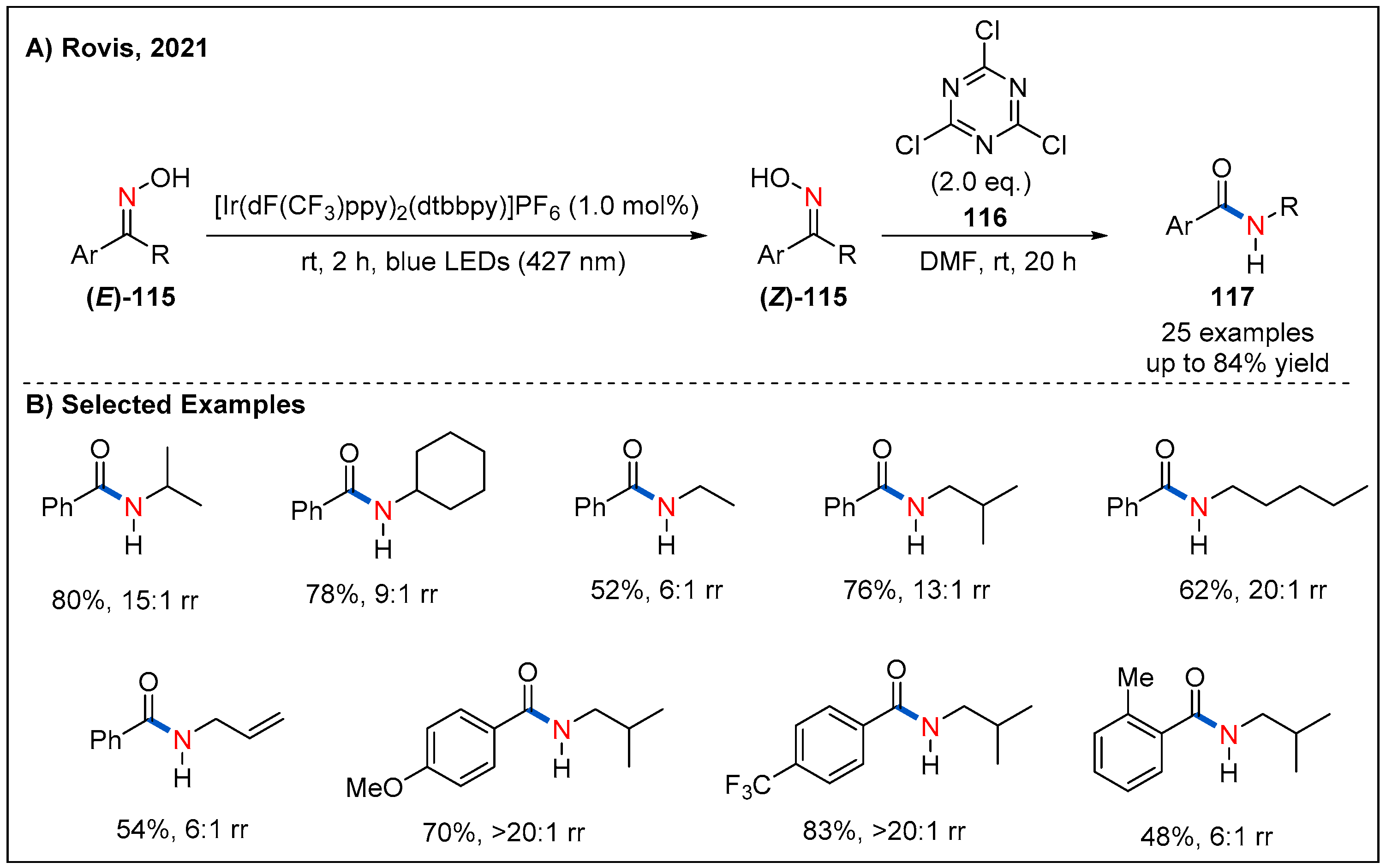
6. Beckmann Rearrangements
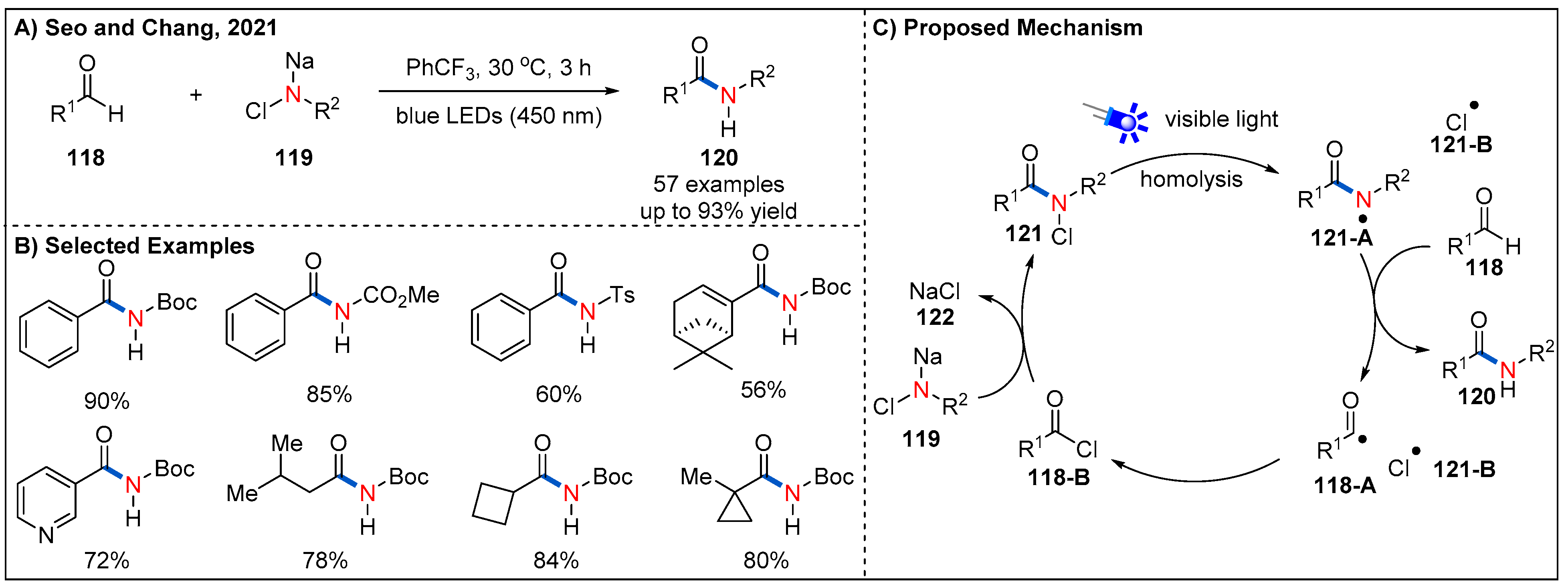
7. Miscellaneous Amidation Methods
8. Conclusions and Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Figueiredo, R.M.; Suppo, J.S.; Campagne, J.M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, C.; Wehl, I.; Jung, N.; Schepers, U.; Brase, S. The Staudinger Ligation. Chem. Rev. 2020, 120, 4301–4354. [Google Scholar] [CrossRef]
- Massolo, E.; Pirola, M.; Benaglia, M. Amide Bond Formation Strategies: Latest Advances on a Dateless Transformation. Eur. J. Org. Chem. 2020, 2020, 4641–4651. [Google Scholar] [CrossRef]
- Santos, A.S.; Silva, A.M.S.; Marques, M.M.B. Sustainable Amidation Reactions-Recent Advances. Eur. J. Org. Chem. 2020, 2020, 2501–2516. [Google Scholar] [CrossRef]
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 2014, 43, 2714–2742. [Google Scholar] [CrossRef] [Green Version]
- Carey, J.S.; Laffan, D.; Thomson, C.; Williams, M.T. Analysis of the Reactions Used for the Preparation of Drug Candidate Molecules. Org. Biomol. Chem. 2006, 4, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, M.; Perrin, D.M. Recent developments in catalytic amide bond formation. Pept. Sci. 2020, 112, e24210. [Google Scholar] [CrossRef]
- Sabatini, M.T.; Boulton, L.T.; Sneddon, H.F.; Sheppard, T.D. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2019, 2, 10–17. [Google Scholar] [CrossRef]
- Valeur, E.; Bradley, M. Amide bond formation: Beyond the myth of coupling reagents. Chem. Soc. Rev. 2009, 38, 606–631. [Google Scholar] [CrossRef] [PubMed]
- Dunetz, J.R.; Magano, J.; Weisenburger, G.A. Large-Scale Applications of Amide Coupling Reagents for the Synthesis of Pharmaceuticals. Org. Process Res. Dev. 2016, 20, 140–177. [Google Scholar] [CrossRef]
- Sabatini, M.T.; Boulton, L.T.; Sheppard, T.D. Borate esters: Simple catalysts for the sustainable synthesis of complex amides. Sci. Adv. 2017, 3, e1701028. [Google Scholar] [CrossRef] [Green Version]
- Mylavarapu, R.K.; Gcm, K.; Kolla, N.; Veeramalla, R.; Koilkonda, P.; Bhattacharya, A.; Bandichhor, R. Boric Acid Catalyzed Amidation in the Synthesis of Active Pharmaceutical Ingredients. Org. Process Res. Dev. 2007, 11, 1065–1068. [Google Scholar] [CrossRef]
- Ciszewski, L.W.; Rybicka-Jasinska, K.; Gryko, D. Recent developments in photochemical reactions of diazo compounds. Org. Biomol. Chem. 2019, 17, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.; Miel, H.; Ring, A.; Slattery, C.N.; Maguire, A.R.; McKervey, M.A. Modern Organic Synthesis with alpha-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. [Google Scholar] [CrossRef]
- Hua, T.-B.; Yang, Q.-Q.; Zou, Y.-Q. Recent Advances in Enantioselective Photochemical Reactions of Stabilized Diazo Compounds. Molecules 2019, 24, 3191. [Google Scholar] [CrossRef] [Green Version]
- Kirmse, W. 100 Years of the Wolff Rearrangement. Eur. J. Org. Chem. 2002, 2002, 2193–2256. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [Green Version]
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Xuan, J.; Zhang, Z.-G.; Xiao, W.-J. Visible-Light-Induced Decarboxylative Functionalization of Carboxylic Acids and Their Derivatives. Angew. Chem. Int. Ed. 2015, 54, 15632–15641. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, L.-Q.; Yu, D.-G.; Zhu, C.-J.; Xiao, W.-J. Visible Light-driven Organic PhotoChemical Synthesis in China. Sci. China Chem. 2019, 62, 24–57. [Google Scholar] [CrossRef]
- Ren, L.; Ran, M.; He, J.; Qian, Y.; Yao, Q. Recent Advance in the Transition-Metal Free Coupling Reactions for the Construction of C–X Bonds Induced by Light. Chin. J. Org. Chem. 2019, 39, 1583–1595. [Google Scholar] [CrossRef]
- Wang, P.-Z.; Zhao, Q.-Q.; Xiao, W.-J.; Chen, J.-R. Recent advances in visible-light photoredox-catalyzed nitrogen radical cyclization. Green Synth. Catal. 2020, 1, 42–51. [Google Scholar] [CrossRef]
- Rao, M.; Wu, W.-H.; Yang, C. Recent progress on the enantioselective excited-state photoreactions by pre-arrangement of photosubstrate(s). Green Synth. Catal. 2021, 2, 131–144. [Google Scholar] [CrossRef]
- Zhao, S.N.; Wang, G.; Poelman, D.; Van Der Voort, P. Metal Organic Frameworks Based Materials for Heterogeneous Photocatalysis. Molecules 2018, 23, 2947. [Google Scholar] [CrossRef] [Green Version]
- Pawlowski, R.; Stanek, F.; Stodulski, M. Recent Advances on Metal-Free, Visible-Light- Induced Catalysis for Assembling Nitrogen- and Oxygen-Based Heterocyclic Scaffolds. Molecules 2019, 24, 1533. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L. Recent Advances in C-F Bond Cleavage Enabled by Visible Light Photoredox Catalysis. Molecules 2021, 26, 7051. [Google Scholar] [CrossRef] [PubMed]
- Luu, T.G.; Jung, Y.; Kim, H.K. Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst. Molecules 2021, 26, 7380. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Q.; Yi, H.; Qin, C.; Bai, R.; Qi, X.; Lan, Y.; Lei, A. Visible-light-mediated decarboxylation/oxidative amidation of alpha-keto acids with amines under mild reaction conditions using O2. Angew. Chem. Int. Ed. 2014, 53, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Gaspa, S.; Farina, A.; Tilocca, M.; Porcheddu, A.; Pisano, L.; Carraro, M.; Azzena, U.; De Luca, L. Visible-Light Photoredox-Catalyzed Amidation of Benzylic Alcohols. J. Org. Chem. 2020, 85, 11679–11687. [Google Scholar] [CrossRef]
- Singha, K.; Ghosh, S.C.; Panda, A.B. Visible Light-Driven Efficient Synthesis of Amides from Alcohols using Cu−N−TiO2 Heterogeneous Photocatalyst. Eur. J. Org. Chem. 2021, 4, 657–662. [Google Scholar] [CrossRef]
- Nandi, J.; Vaughan, M.Z.; Sandoval, A.L.; Paolillo, J.M.; Leadbeater, N.E. Oxidative Amidation of Amines in Tandem with Transamidation: A Route to Amides Using Visible-Light Energy. J. Org. Chem. 2020, 85, 9219–9229. [Google Scholar] [CrossRef] [PubMed]
- Leow, D. Phenazinium Salt-catalyzed Aerobic Oxidative Amidation of Aromatic Aldehydes. Org. Lett. 2014, 16, 5812–5815. [Google Scholar] [CrossRef] [PubMed]
- Leung, F.K.-C.; Cui, J.-F.; Hui, T.-W.; Kung, K.K.-Y.; Wong, M.-K. Photooxidative Amidation of Aldehydes with Amines Catalyzed by Rose Bengal. Asian J. Org. Chem. 2015, 4, 533–536. [Google Scholar] [CrossRef]
- Wang, X.-F.; Yu, S.-S.; Wang, C.; Xue, D.; Xiao, J. BODIPY Catalyzed Amide Synthesis Promoted by BHT and Air under Visible Light. Org. Biomol. Chem. 2016, 14, 7028–7037. [Google Scholar] [CrossRef]
- Deng, J.-R.; Chan, W.-C.; Chun-Him Lai, N.; Yang, B.; Tsang, C.S.; Chi-Bun Ko, B.; Lai-Fung Chan, S.; Wong, M.K. Photosensitizer-free Visible Light-mediated Gold-catalysed cis-Difunctionalization of Silyl-substituted Alkynes. Chem. Sci. 2017, 8, 7537–7544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhao, L.; Yuan, Y.; Xu, Z.; Chen, K.; Qiu, S.; Tan, H. Potassium Thioacids Mediated Selective Amide and Peptide Constructions Enabled by Visible Light Photoredox Catalysis. ACS Catal. 2016, 6, 1732–1736. [Google Scholar] [CrossRef]
- Song, W.; Dong, K.; Li, M. Visible Light-Induced Amide Bond Formation. Org. Lett. 2020, 22, 371–375. [Google Scholar] [CrossRef]
- Cohen, I.; Mishra, A.K.; Parvari, G.; Edrei, R.; Dantus, M.; Eichen, Y.; Szpilman, A.M. Sunlight Assisted Direct Amide Formation via A Charge-transfer Complex. Chem. Commun. 2017, 53, 10128–10131. [Google Scholar] [CrossRef]
- Ragupathi, A.; Sagadevan, A.; Lin, C.C.; Hwu, J.R.; Hwang, K.C. Copper(I)-catalysed Oxidative C-N Coupling of 2-Aminopyridine with Terminal Alkynes Featuring a C≡C Bond Cleavage Promoted by Visible Light. Chem. Commun. 2016, 52, 11756–11759. [Google Scholar] [CrossRef] [PubMed]
- Pampana, V.K.K.; Sagadevan, A.; Ragupathi, A.; Hwang, K.C. Visible Light-promoted Copper Catalyzed Regioselective Acetamidation of Terminal Alkynes by Arylamines. Green Chem. 2020, 22, 1164–1170. [Google Scholar] [CrossRef]
- Wang, D.; Li, J.; Cai, S.; Chen, J.; Zhao, Y. Visible Light Induced Photocatalytic Conversion of Enamines into Amides. Synlett 2014, 25, 1626–1628. [Google Scholar] [CrossRef]
- Guerinot, A.; Reymond, S.; Cossy, J.; Jiang, D.; He, T.; Ma, L.; Wang, Z.; Bolsakova, J.; Jirgensons, A. Ritter Reaction: Recent Catalytic Developments. Eur. J. Org. Chem. 2012, 1, 19–28. [Google Scholar] [CrossRef]
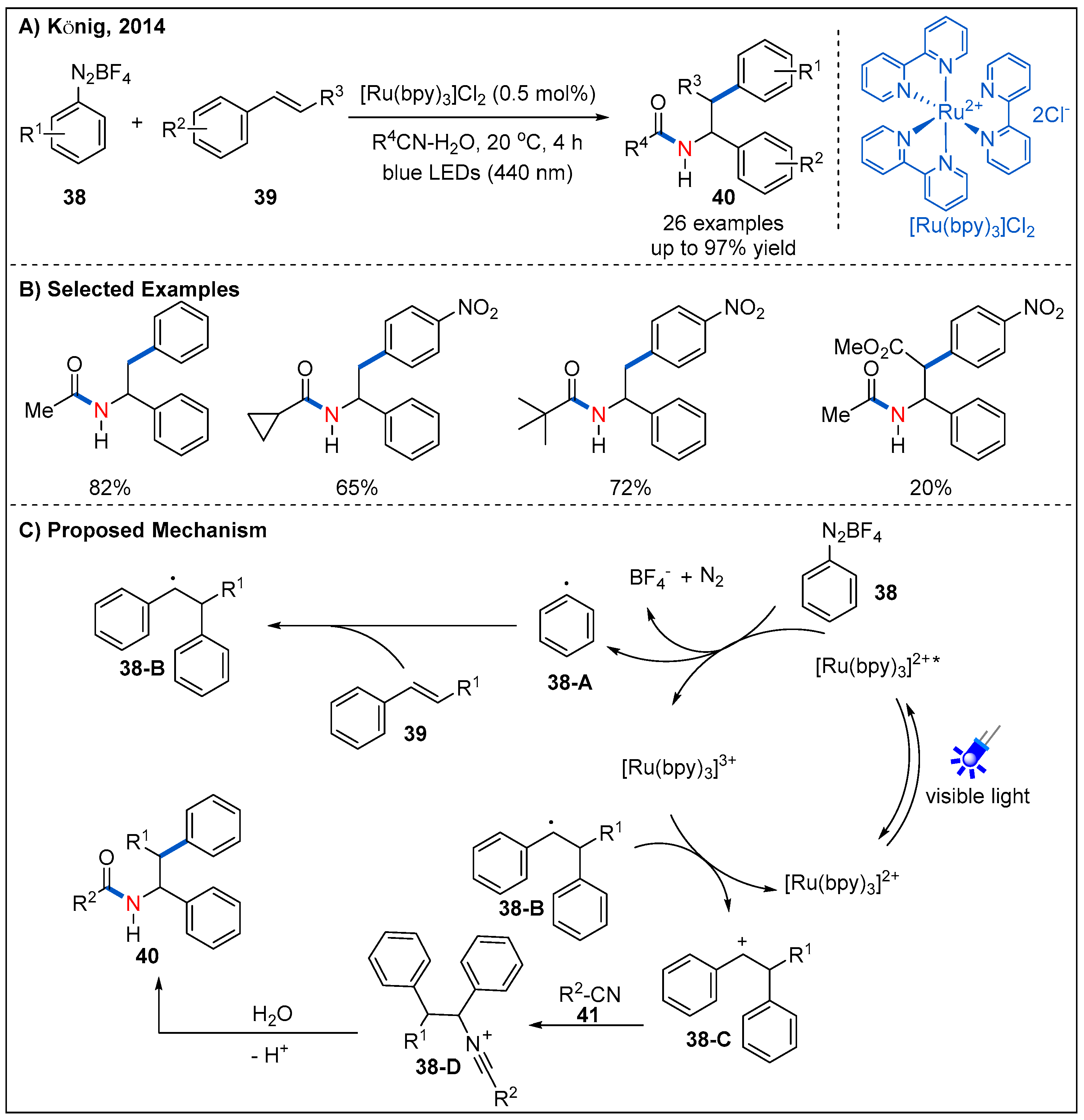
- Hari, D.P.; Hering, T.; Konig, B. The Photoredox-catalyzed Meerwein Addition Reaction: Intermolecular Amino-arylation of Alkenes. Angew. Chem. Int. Ed. 2014, 53, 725–728. [Google Scholar] [CrossRef]
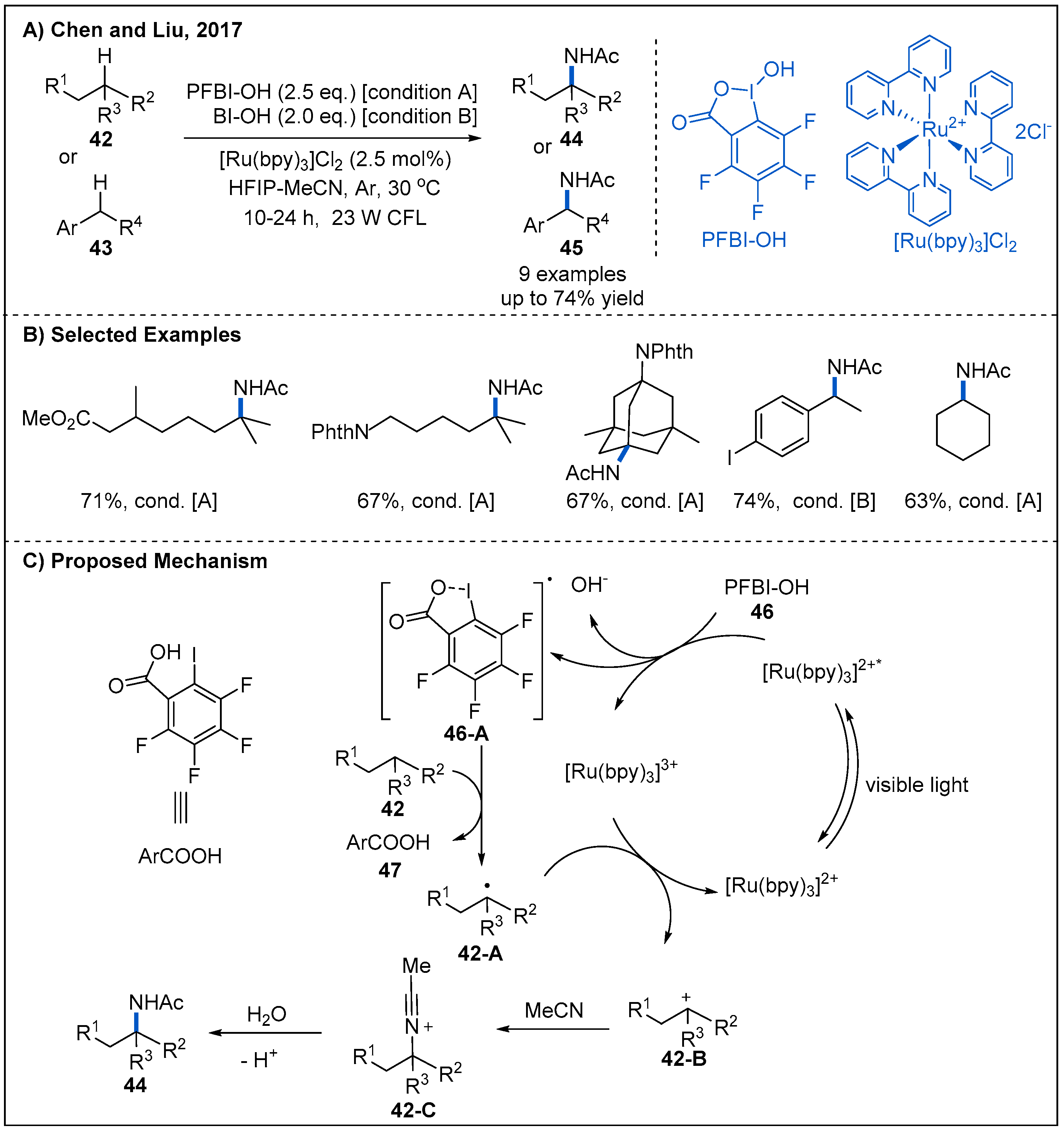
- Li, G.X.; Morales-Rivera, C.A.; Gao, F.; Wang, Y.; He, G.; Liu, P.; Chen, G. A Unified Photoredox-catalysis Strategy for C(sp3)-H Hydroxylation and Amidation Using Hypervalent Iodine. Chem. Sci. 2017, 8, 7180–7185. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Teuscher, K.B.; Ji, H. Direct α-heteroarylation of Amides (α to nitrogen) And Ethers through A Benzaldehyde-mediated Photoredox Reaction. Chem. Sci. 2016, 7, 2111–2118. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.-M.; Shang, R.; Yu, H.Z.; Fu, Y. Room-Temperature Decarboxylative Couplings of α-Oxocarboxylates with Aryl Halides by Merging Photoredox with Palladium Catalysis. Chem. Eur. J. 2015, 21, 13191–13195. [Google Scholar] [CrossRef]
- Shang, R.; Fu, Y.; Li, J.-B.; Zhang, S.-L.; Guo, Q.-X.; Liu, L. Synthesis of Aromatic Esters via Pd-catalyzed Decarboxylative Coupling of Potassium Oxalate Monoesters with Aryl Bromides and Chlorides. J. Am. Chem. Soc. 2009, 131, 5738–5739. [Google Scholar] [CrossRef]
- Jatoi, A.H.; Pawar, G.G.; Robert, F.; Landais, Y. Visible-light Mediated Carbamoyl Radical Addition to Heteroarenes. Chem. Commun. 2019, 55, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Tlili, A.; Lakhdar, S. Acridinium Salts and Cyanoarenes as Powerful Photocatalysts: Opportunities in Organic Synthesis. Angew. Chem. Int. Ed. 2021, 60, 19526–19549. [Google Scholar] [CrossRef] [PubMed]
- Shang, T.Y.; Lu, L.H.; Cao, Z.; Liu, Y.; He, W.M.; Yu, B. Recent advances of 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) in photocatalytic transformations. Chem. Commun. 2019, 55, 5408–5419. [Google Scholar] [CrossRef]
- Petersen, W.F.; Taylor, R.J.K.; Donald, J.R. Photoredox-catalyzed Procedure for Carbamoyl Radical Generation: 3,4-Dihydroquinolin-2-one And Quinolin-2-one Synthesis. Org. Biomol. Chem. 2017, 15, 5831–5845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.-Z.; Chen, J.-R.; Xiao, W.-J. Hantzsch esters: An emerging versatile class of reagents in photoredox catalyzed organic synthesis. Org. Biomol. Chem. 2019, 17, 6936–6951. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Huang, W. Hantzsch Esters as Multifunctional Reagents in Visible-Light Photoredox Catalysis. Synlett 2016, 28, 148–158. [Google Scholar] [CrossRef]
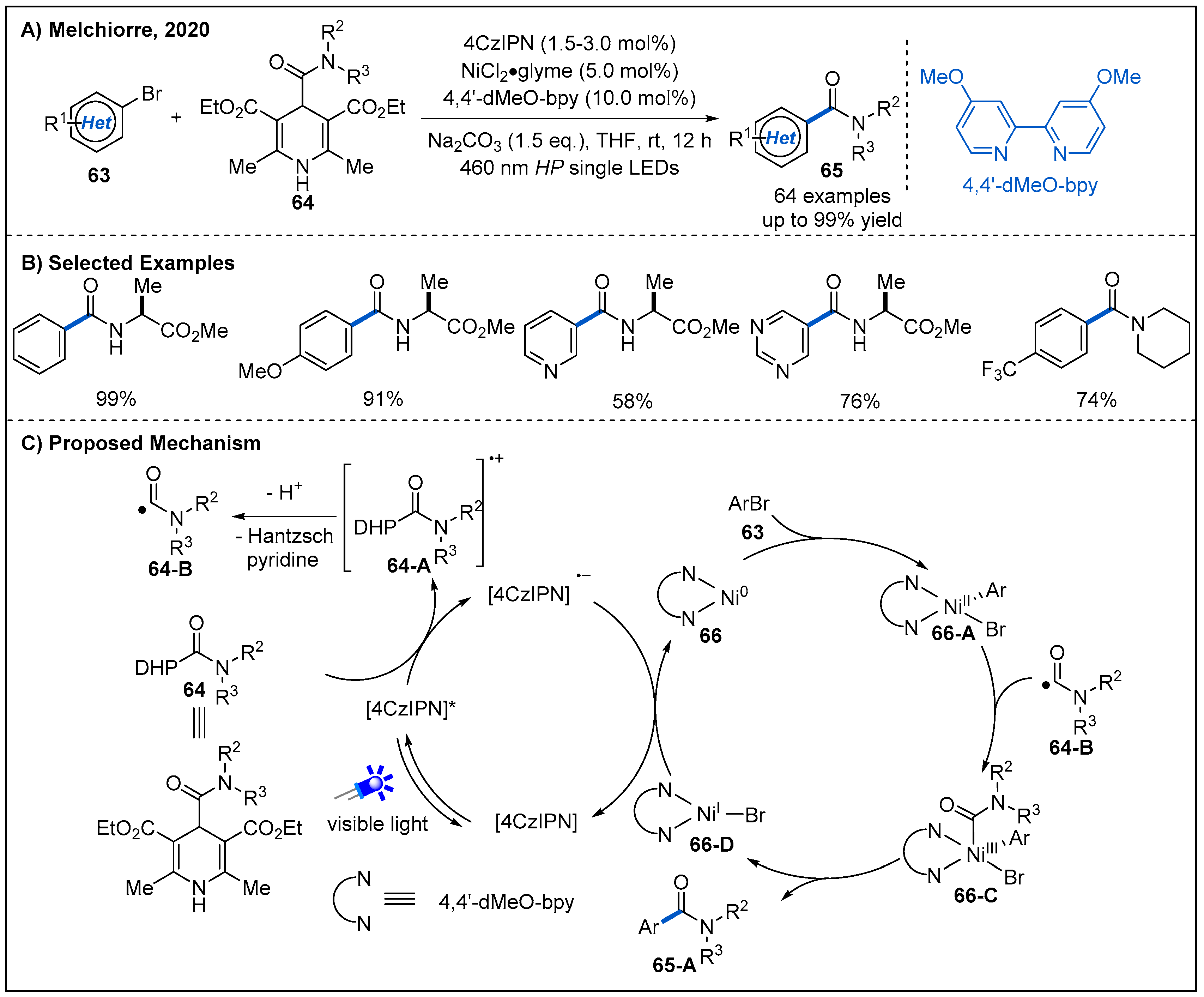
- Alandini, N.; Buzzetti, L.; Favi, G.; Schulte, T.; Candish, L.; Collins, K.D.; Melchiorre, P. Amide Synthesis by Nickel/Photoredox-Catalyzed Direct Carbamoylation of (Hetero)Aryl Bromides. Angew. Chem. Int. Ed. 2020, 59, 5248–5253. [Google Scholar] [CrossRef] [Green Version]
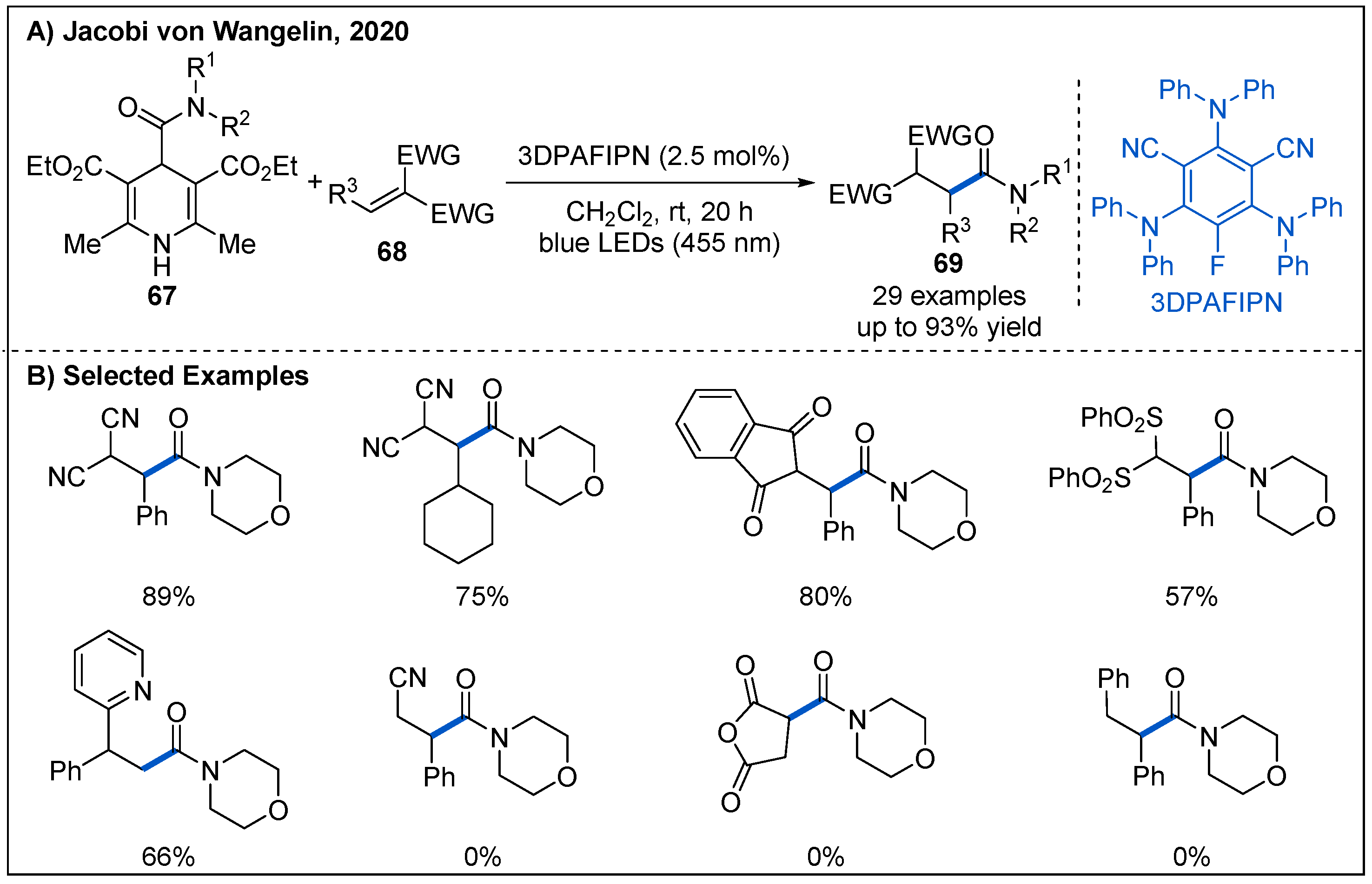
- Cardinale, L.; Konev, M.O.; Jacobi von Wangelin, A. Photoredox-Catalyzed Addition of Carbamoyl Radicals to Olefins: A 1,4-Dihydropyridine Approach. Chem. Eur. J. 2020, 26, 8239–8243. [Google Scholar] [CrossRef]
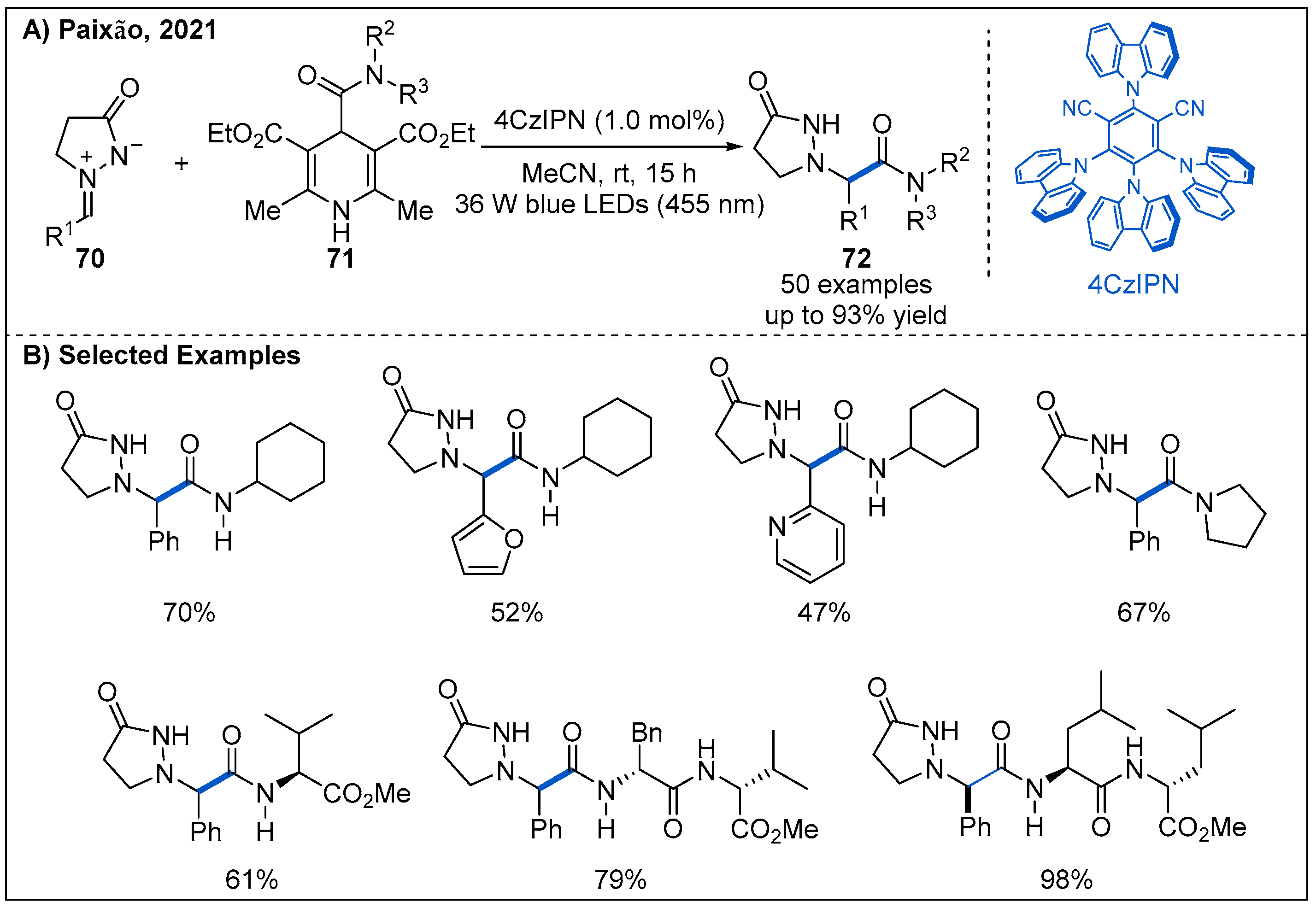
- Matsuo, B.T.; Oliveira, P.H.R.; Correia, J.T.M.; Paixao, M.W. Carbamoylation of Azomethine Imines via Visible-Light Photoredox Catalysis. Org. Lett. 2021, 23, 6775–6779. [Google Scholar] [CrossRef]
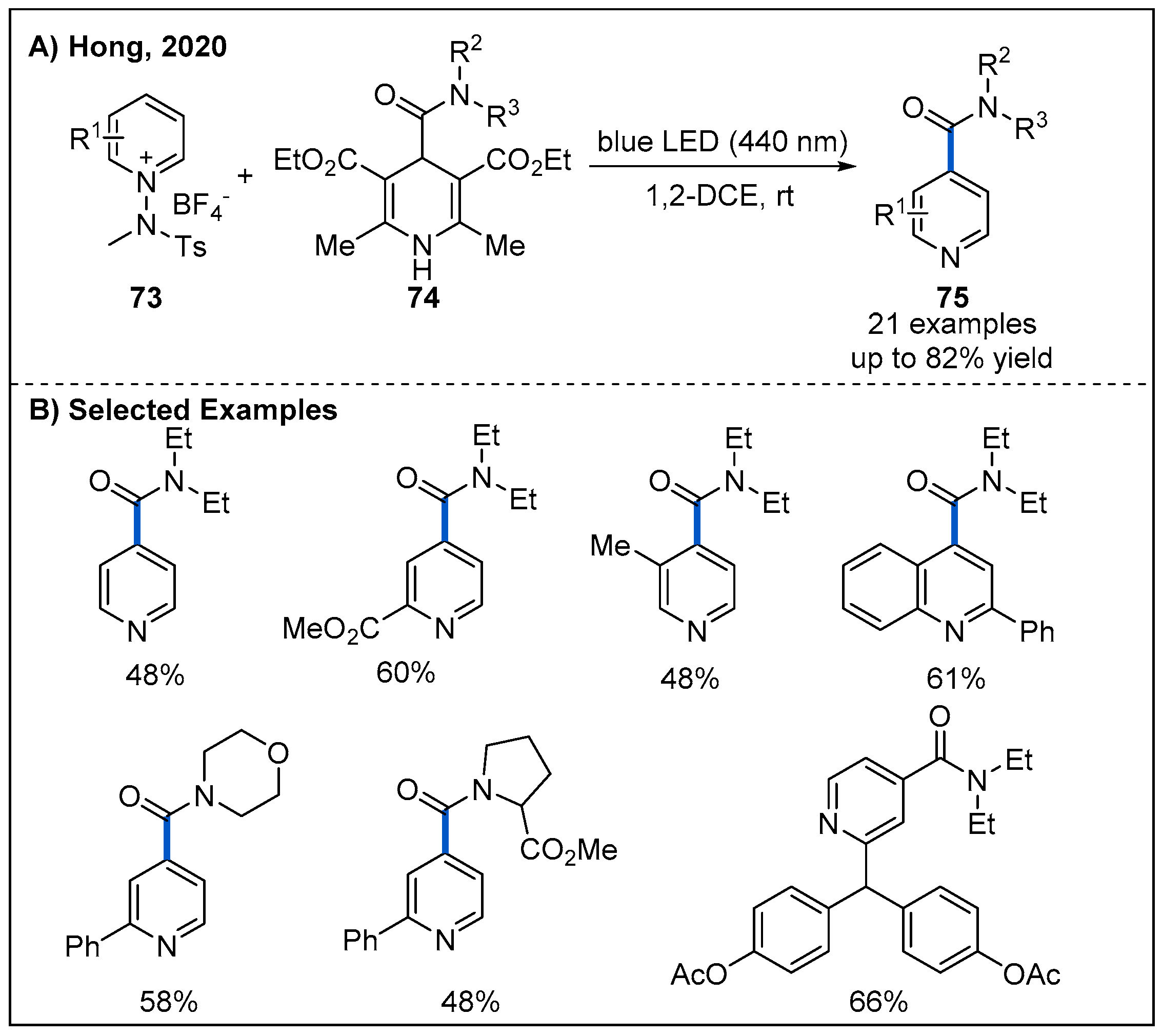
- Kim, I.; Park, S.; Hong, S. Functionalization of Pyridinium Derivatives with 1,4-Dihydropyridines Enabled by Photoinduced Charge Transfer. Org. Lett. 2020, 22, 8730–8734. [Google Scholar] [CrossRef]
- Ryu, I.; Sonoda, N. Free-Radical Carbonylations: Then and Now. Angew. Chem. Int. Ed. 1996, 35, 1050–1066. [Google Scholar] [CrossRef]
- Ryu, I.; Sonoda, N.; Curran, D.P. Tandem Radical Reactions of Carbon Monoxide, Isonitriles, and Other Reagent Equivalents of the Geminal Radical Acceptor/Radical Precursor Synthon. Chem. Rev. 1996, 96, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Crich, D.; Komatsu, M.; Ryu, I. Chemistry of Acyl Radicals. Chem. Rev. 1999, 99, 1991–2070. [Google Scholar] [CrossRef]
- Sumino, S.; Fusano, A.; Fukuyama, T.; Ryu, I. Carbonylation Reactions of Alkyl Iodides through the Interplay of Carbon Radicals and Pd Catalysts. Acc. Chem. Res. 2014, 47, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.-B.; Geng, H.-Q.; Wu, X.-F. The Chemistry of CO: Carbonylation. J. Chem. 2019, 5, 526–552. [Google Scholar] [CrossRef] [Green Version]
- Brennfuhrer, A.; Neumann, H.; Beller, M. Palladium-Catalyzed Carbonylation Reactions of Aryl Halides and Related Com-pounds. Angew. Chem. Int. Ed. 2009, 48, 4114–4133. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-F.; Neumann, H.; Beller, M. Synthesis of heterocycles via palladium-catalyzed carbonylations. Chem. Rev. 2013, 113, 1–35. [Google Scholar] [CrossRef]
- Peng, J.-B.; Wu, F.-P.; Wu, X.-F. First-Row Transition-Metal-Catalyzed Carbonylative Transformations of Carbon Electrophiles. Chem. Rev. 2019, 119, 2090–2127. [Google Scholar] [CrossRef]
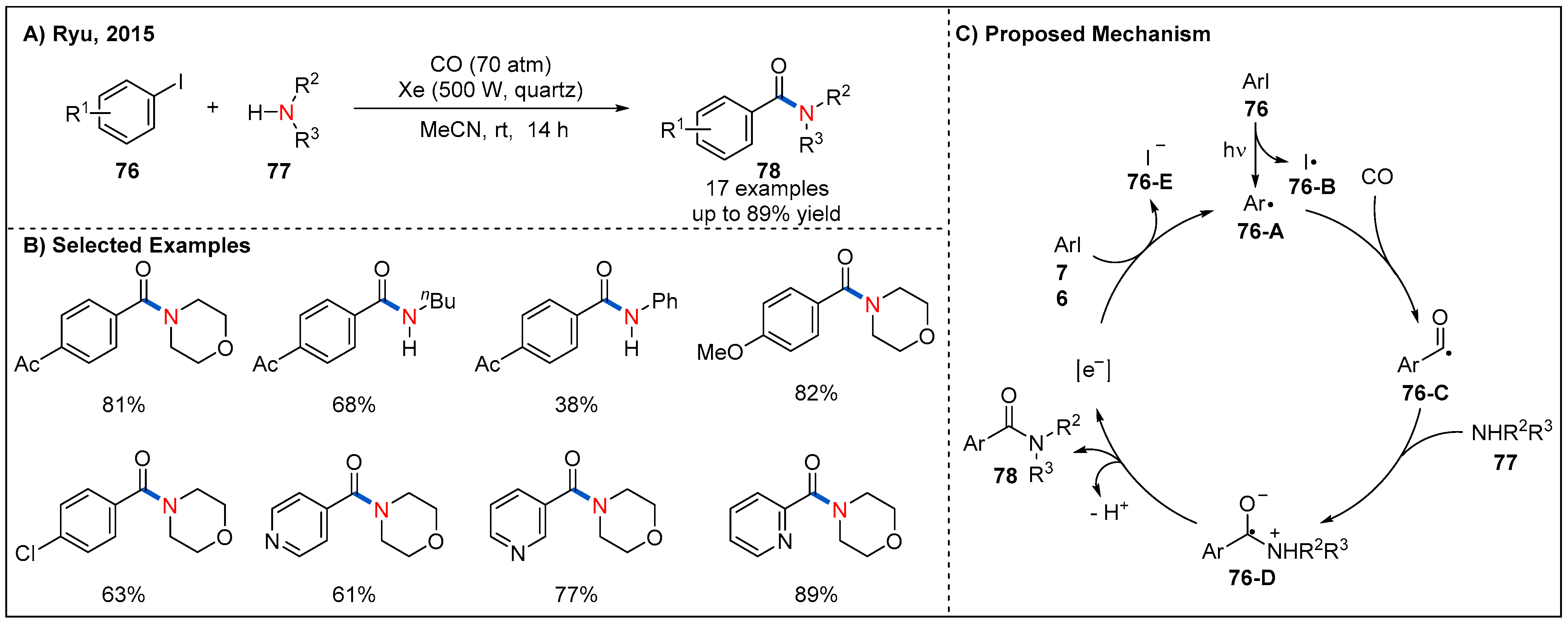
- Kawamoto, T.; Sato, A.; Ryu, I. Photoinduced Aminocarbonylation of Aryl Iodides. Chem. Eur. J. 2015, 21, 14764–14767. [Google Scholar] [CrossRef]
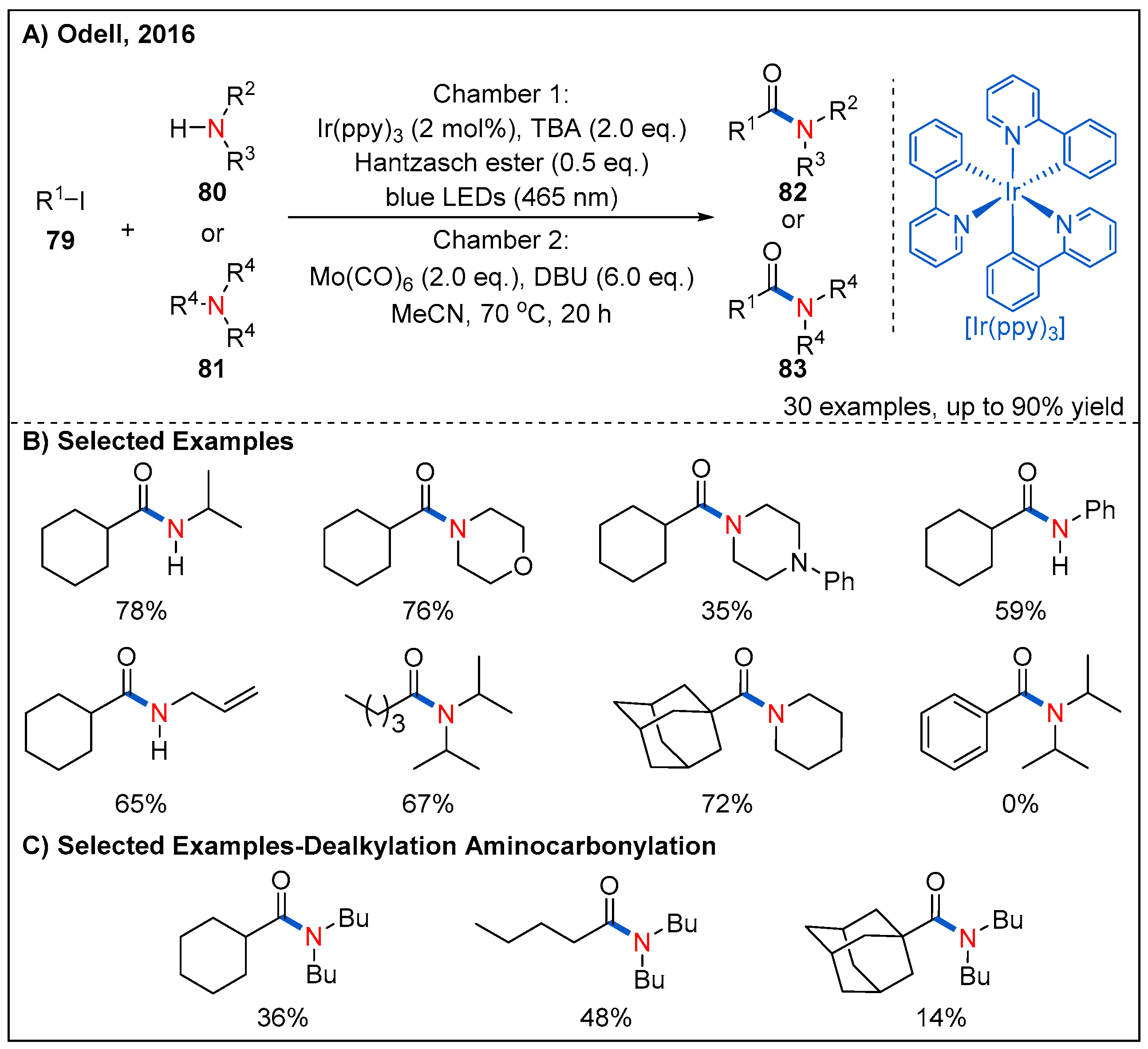
- Chow, S.Y.; Stevens, M.Y.; Akerbladh, L.; Bergman, S.; Odell, L.R. Mild and Low-Pressure fac-Ir(ppy)3-Mediated Radical Ami-nocarbonylation of Unactivated Alkyl Iodides through Visible-Light Photoredox Catalysis. Chem. Eur. J. 2016, 22, 9155–9161. [Google Scholar] [CrossRef]
- Zhao, S.; Mankad, N.P. Metal-catalysed radical carbonylation reactions. Catal. Sci. Technol. 2019, 9, 3603–3613. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, S.; Sharma, A. Photocatalytic Carbonylation Strategies: A Recent Trend in Organic Synthesis. J. Org. Chem. 2021, 86, 24–48. [Google Scholar] [CrossRef] [PubMed]
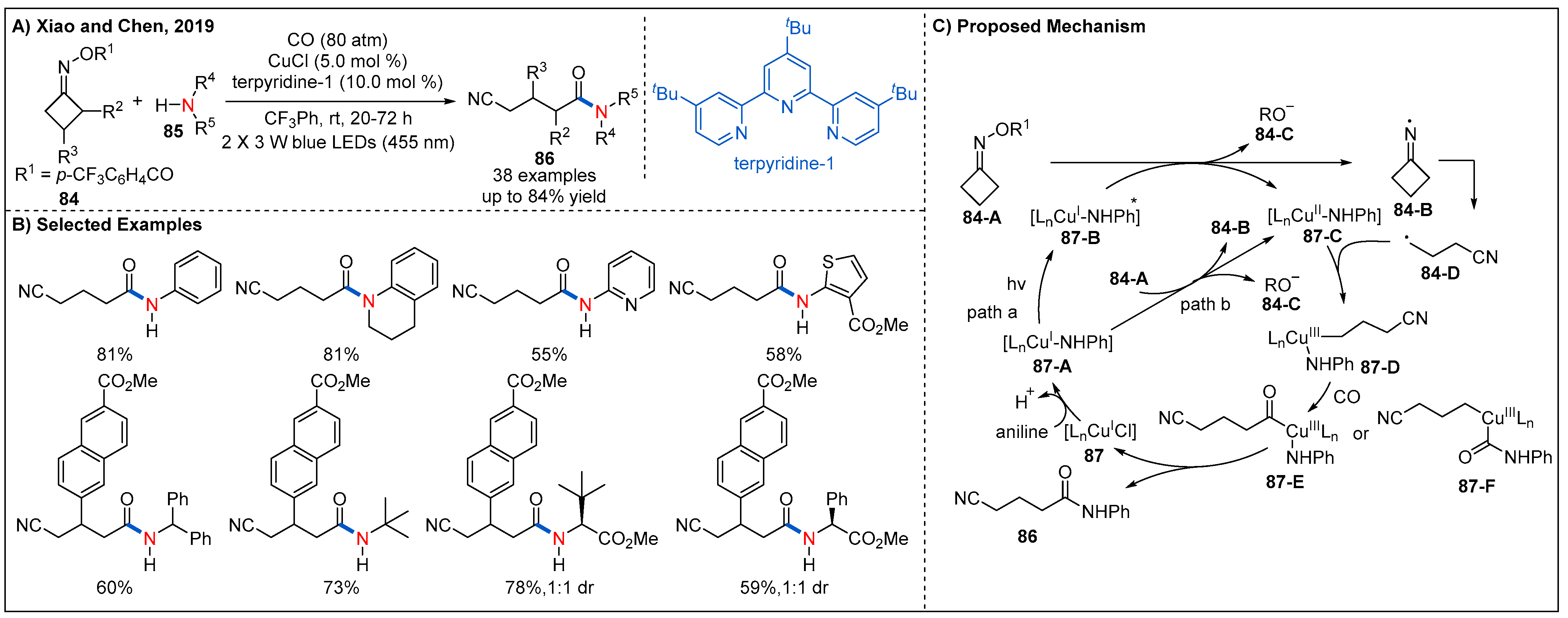
- Lu, B.; Cheng, Y.; Chen, L.-Y.; Chen, J.-R.; Xiao, W.-J. Photoinduced Copper-Catalyzed Radical Aminocarbonylation of Cycloketone Oxime Esters. ACS Catal. 2019, 9, 8159–8164. [Google Scholar] [CrossRef]
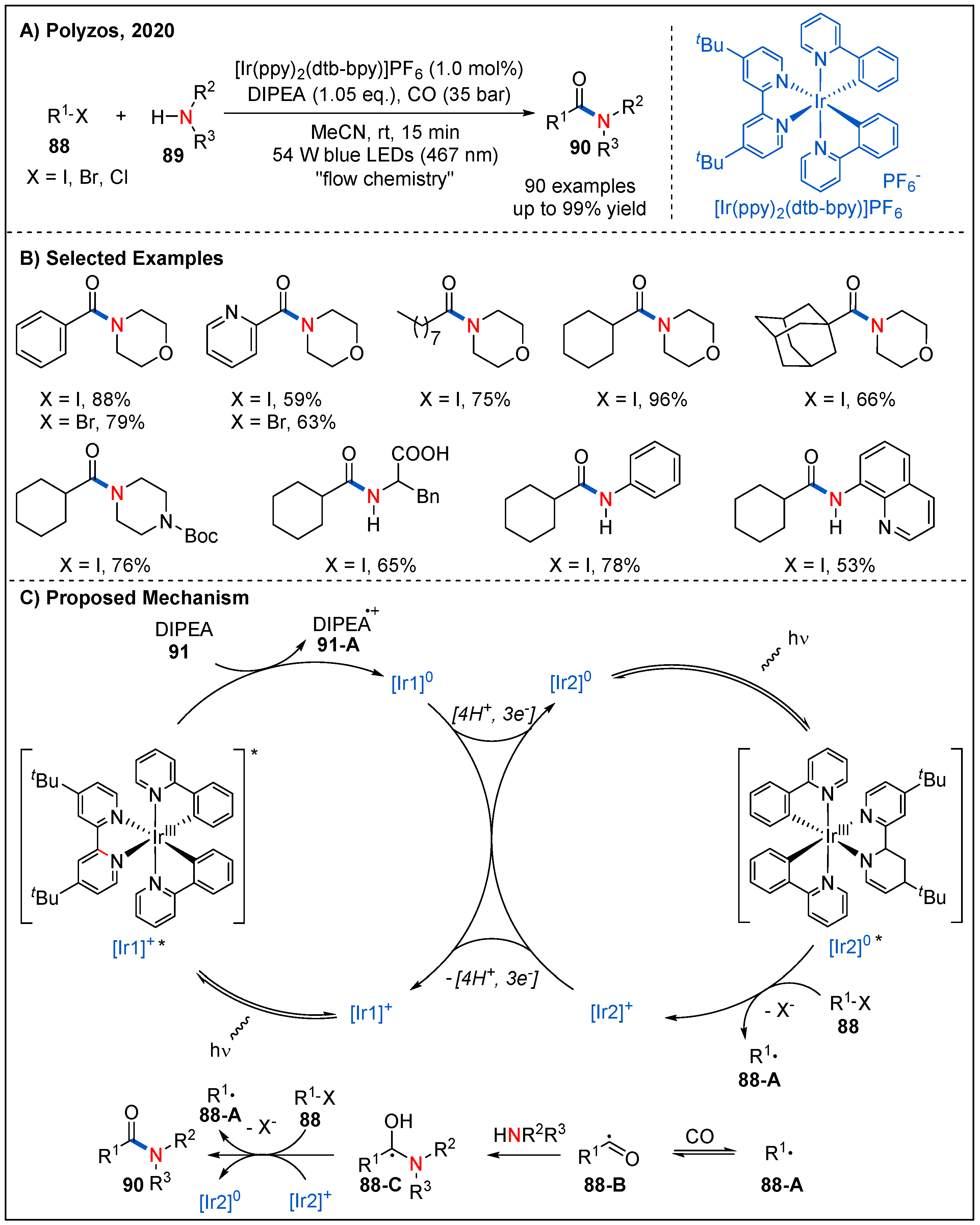
- Forni, J.A.; Micic, N.; Connell, T.U.; Weragoda, G.; Polyzos, A. Tandem Photoredox Catalysis: Enabling Carbonylative Amidation of Aryl and Alkylhalides. Angew. Chem. Int. Ed. 2020, 59, 18646–18654. [Google Scholar] [CrossRef]
- Kwiatkowski, M.R.; Alexanian, E.J. Transition-Metal (Pd, Ni, Mn)-Catalyzed C-C Bond Constructions Involving Unactivated Alkyl Halides and Fundamental Synthetic Building Blocks. Acc. Chem. Res. 2019, 52, 1134–1144. [Google Scholar] [CrossRef]
- Torres, G.M.; Liu, Y.; Arndtsen, B.A. A dual light-driven palladium catalyst: Breaking the barriers in carbonylation reactions. Science 2020, 368, 318–323. [Google Scholar] [CrossRef]
- Kathe, P.; Fleischer, I. Light expands a catalyst‘s repertoire. Science 2020, 368, 242–243. [Google Scholar] [CrossRef]
- Sardana, M.; Bergman, J.; Ericsson, C.; Kingston, L.P.; Schou, M.; Dugave, C.; Audisio, D.; Elmore, C.S. Visible-Light-Enabled Aminocarbonylation of Unactivated Alkyl Iodides with Stoichiometric Carbon Monoxide for Application on Late-Stage Carbon Isotope Labeling. J. Org. Chem. 2019, 84, 16076–16085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartier, A.; Levernier, E.; Dhimane, A.L.; Fukuyama, T.; Ollivier, C.; Ryu, I.; Fensterbank, L. Synthesis of Aliphatic Amides through a Photoredox Catalyzed Radical Carbonylation Involving Organosilicates as Alkyl Radical Precursors. Adv. Synth. Catal. 2020, 362, 2254–2259. [Google Scholar] [CrossRef]
- Cartier, A.; Levernier, E.; Corce, V.; Fukuyama, T.; Dhimane, A.L.; Ollivier, C.; Ryu, I.; Fensterbank, L. Carbonylation of Alkyl Radicals Derived from Organosilicates through Visible-Light Photoredox Catalysis. Angew. Chem. Int. Ed. 2019, 58, 1789–1793. [Google Scholar] [CrossRef]
- Veatch, A.M.; Alexanian, E.J. Cobalt-catalyzed Aminocarbonylation of (Hetero)Aryl Halides Promoted by Visible Light. Chem. Sci. 2020, 11, 7210–7213. [Google Scholar] [CrossRef]
- Debnath, P. Recent Advances in the Synthesis of Amides via Oxime Rearrangements and its Applications. Curr. Org. Synth. 2018, 15, 666–706. [Google Scholar] [CrossRef]
- Amin, J.H.; de Mayo, P. The irradiation of aryl aldoximes. Tetrahedron Lett. 1963, 4, 1585–1589. [Google Scholar] [CrossRef]
- Yadav, L.; Srivastava, V.; Yadav, A. The Beckmann Rearrangement Executed by Visible-Light-Driven Generation of Vilsmeier–Haack Reagent. Synlett 2014, 25, 665–670. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, V.; Singh, P.P. Eosin Y catalysed photoredox synthesis: A review. RSC Adv. 2017, 7, 31377–31392. [Google Scholar] [CrossRef]
- Tang, L.; Wang, Z.-L.; Wan, H.-L.; He, Y.-H.; Guan, Z. Visible-Light-Induced Beckmann Rearrangement by Organic Photoredox Catalysis. Org. Lett. 2020, 22, 6182–6186. [Google Scholar] [CrossRef]
- Nevesely, T.; Wienhold, M.; Molloy, J.J.; Gilmour, R. Advances in the E → Z Isomerization of Alkenes Using Small Molecule Photocatalysts. Chem. Rev. 2021. [Google Scholar] [CrossRef]
- Yuan, P.-F.; Huang, T.; He, J.; Huang, X.-T.; Jin, X.-L.; Sun, C.; Wu, L.-Z.; Liu, Q. Controllable Z/E-selective synthesis of α-amino-ketoximes from N-nitrososulfonamides and aryl alkenes under neutral conditions. Org. Chem. Front. 2021, 8, 5785–5792. [Google Scholar] [CrossRef]
- Zhang, X.; Rovis, T. Photocatalyzed Triplet Sensitization of Oximes Using Visible Light Provides a Route to Nonclassical Beckmann Rearrangement Products. J. Am. Chem. Soc. 2021, 143, 21211–21217. [Google Scholar] [CrossRef]
- Lowry, M.S.; Goldsmith, J.I.; Slinker, J.D.; Rohl, R.; Pascal, R.A.; Malliaras, G.G.; Bernhard, S. Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex. Chem. Mater. 2005, 17, 5712–5719. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.; Xiao, W.-J.; Chen, J.-R. When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts. Acc. Chem. Res. 2020, 53, 1066–1083. [Google Scholar] [CrossRef]
- Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Visible Light Photoredox-Controlled Reactions of N-Radicals and Radical Ions. Chem. Soc. Rev. 2016, 45, 2044–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Xia, W. Recent Advances in Radical-Based C-N Bond Formation via Photo-/Electrochemistry. Chem. Soc. Rev. 2018, 47, 2591–2608. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Jeon, H.J.; Jung, H.; Kim, D.; Seo, S.; Chang, S. Controlled Relay Process to Access N-Centered Radicals for Catalyst-free Amidation of Aldehydes under Visible Light. J. Chem. 2021, 7, 495–508. [Google Scholar] [CrossRef]
- Jeon, H.J.; Lee, W.; Seo, S.; Chang, S. N-Chloro-N-sodio-carbamates as a Practical Amidating Reagent for Scalable and Sustainable Amidation of Aldehydes under Visible Light. Org. Process Res. Dev. 2021, 25, 1176–1183. [Google Scholar] [CrossRef]
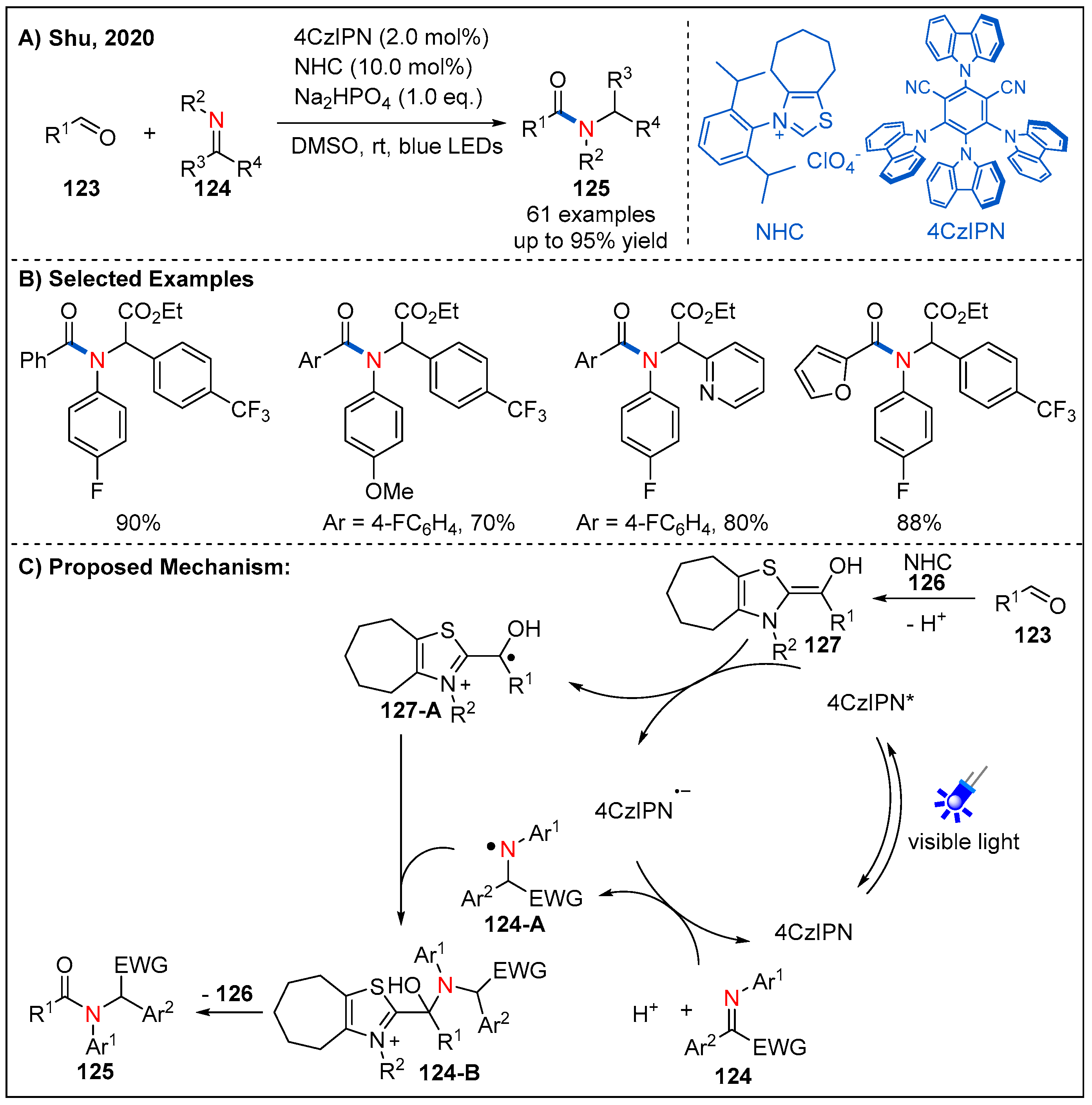
- Liu, M.-S.; Shu, W. Catalytic, Metal-Free Amide Synthesis from Aldehydes and Imines Enabled by a Dual-Catalyzed Umpolung Strategy under Redox-Neutral Conditions. ACS Catal. 2020, 10, 12960–12966. [Google Scholar] [CrossRef]
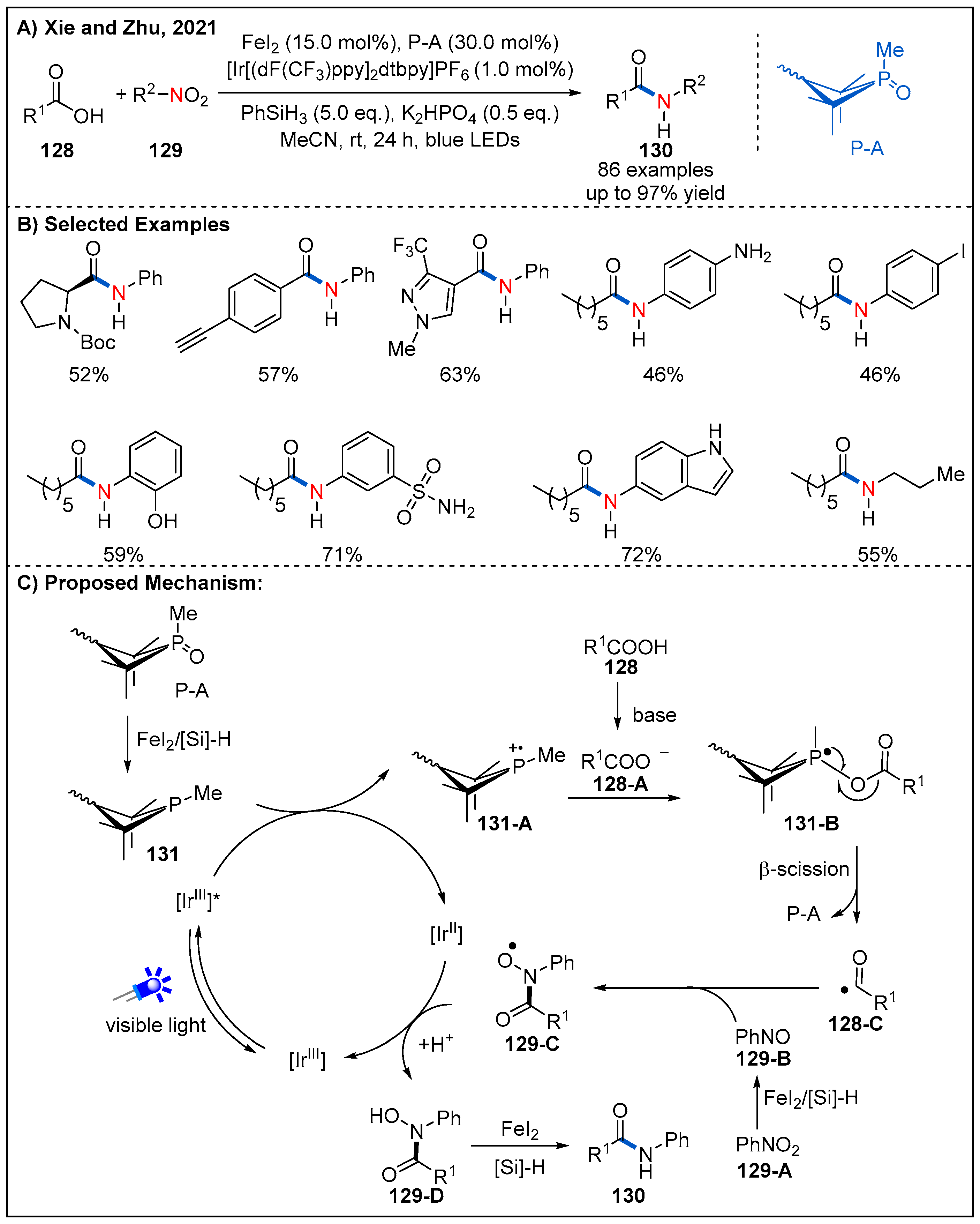
- Ning, Y.; Wang, S.; Li, M.; Han, J.; Zhu, C.; Xie, J. Site-specific Umpolung amidation of carboxylic acids via triplet synergistic catalysis. Nat. Commun. 2021, 12, 4637. [Google Scholar] [CrossRef]
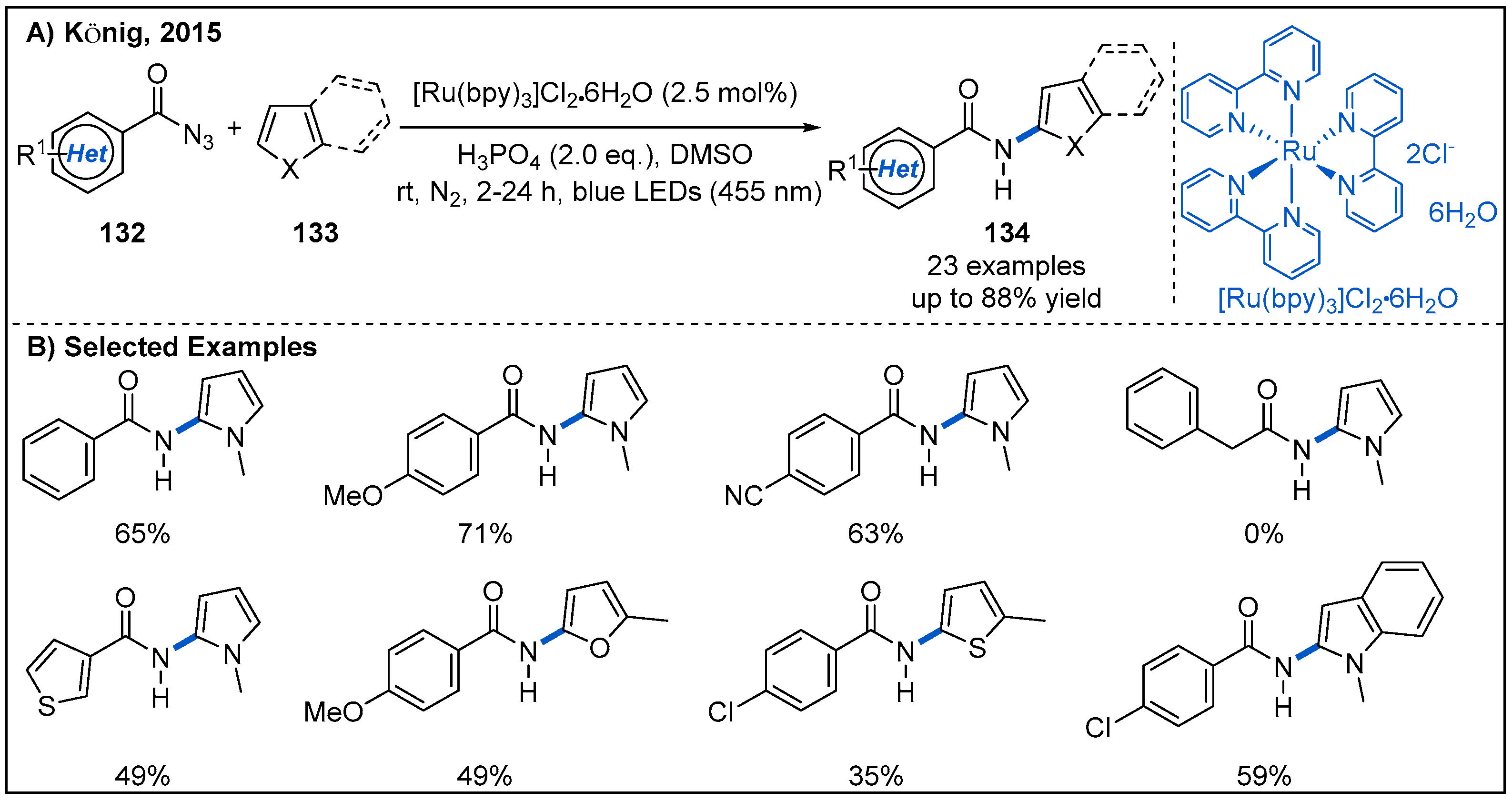
- Brachet, E.; Ghosh, T.; Ghosh, I.; Konig, B. Visible Light C-H Amidation of Heteroarenes with Benzoyl Azides. Chem. Sci. 2015, 6, 987–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
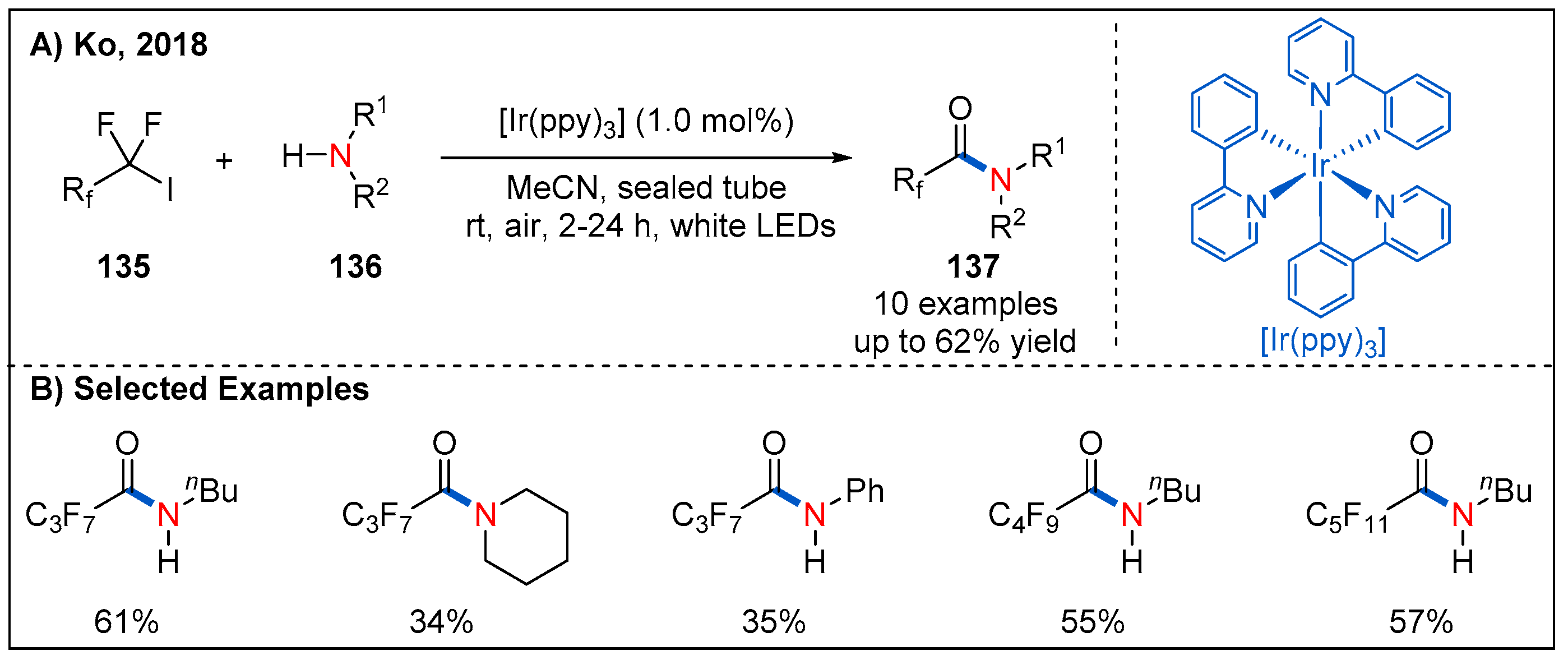
- Ng, C.-O.; Feng, H.; Cheng, S.-C.; Xiao, Y.; Lo, L.T.-L.; Ko, C.-C. Photoredox Catalysis of Cyclometalated IrIII Complex for the Conversion of Amines to Fluorinated Alkyl Amides. Asian J. Org. Chem. 2018, 7, 1587–1590. [Google Scholar] [CrossRef]
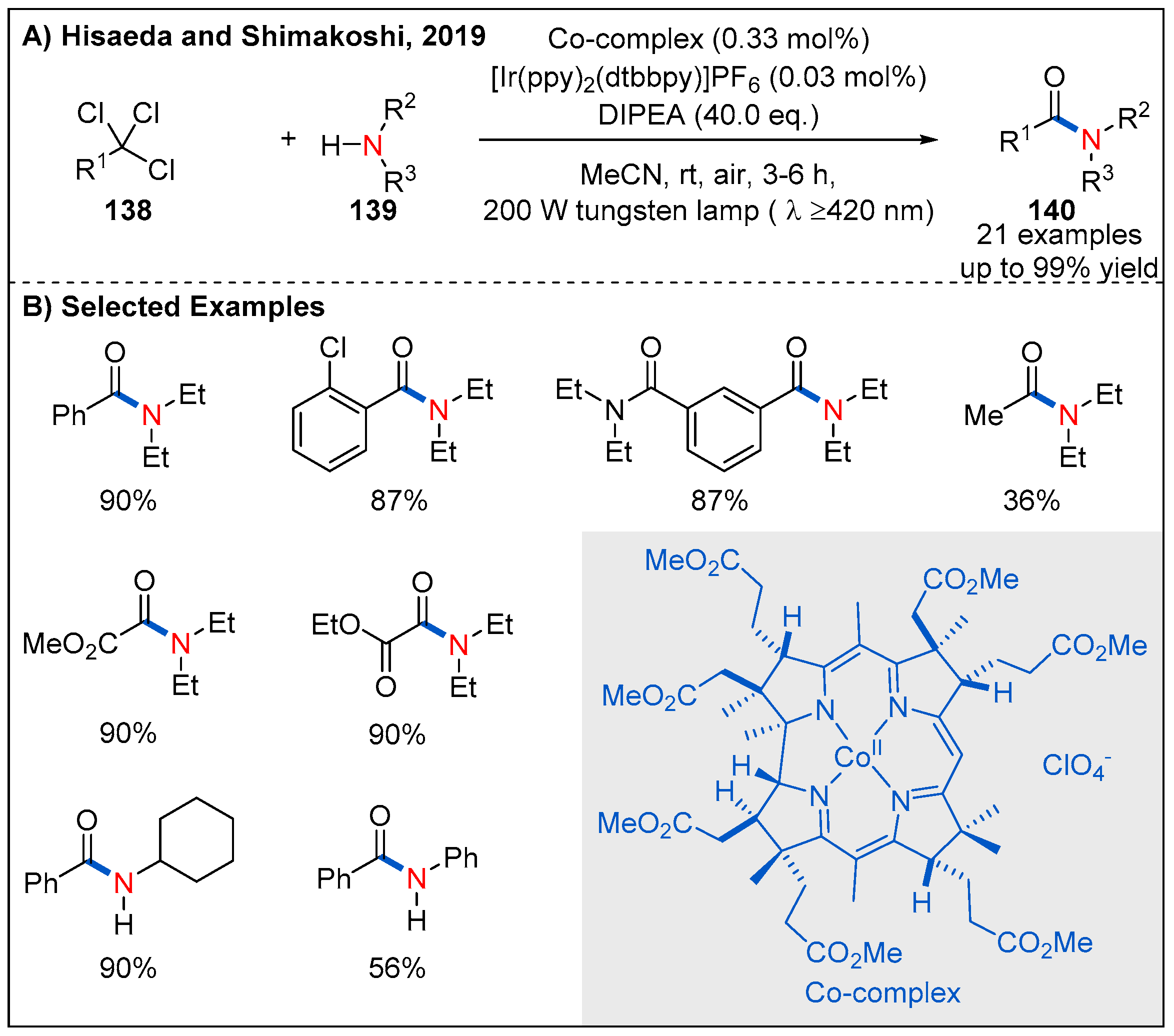
- Tian, H.; Shimakoshi, H.; Ono, T.; Hisaeda, Y. Visible Light-Driven, One-pot Amide Synthesis Catalyzed by the B12 Model Complex under Aerobic Conditions. Chempluschem 2019, 84, 237–240. [Google Scholar] [CrossRef]
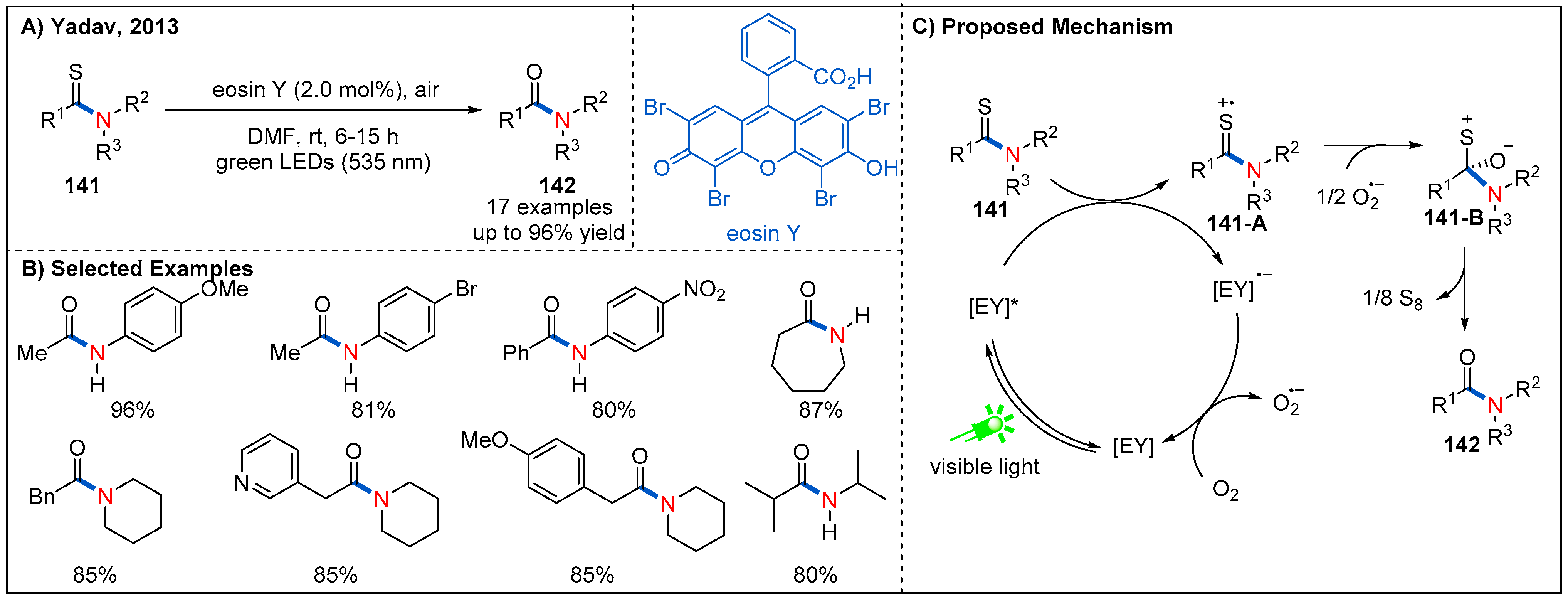
- Yadav, A.K.; Srivastava, V.P.; Yadav, L.D.S. Visible-light-mediated eosin Y catalyzed aerobic desulfurization of thioamides into amides. New J. Chem. 2013, 37, 4119–4124. [Google Scholar] [CrossRef]
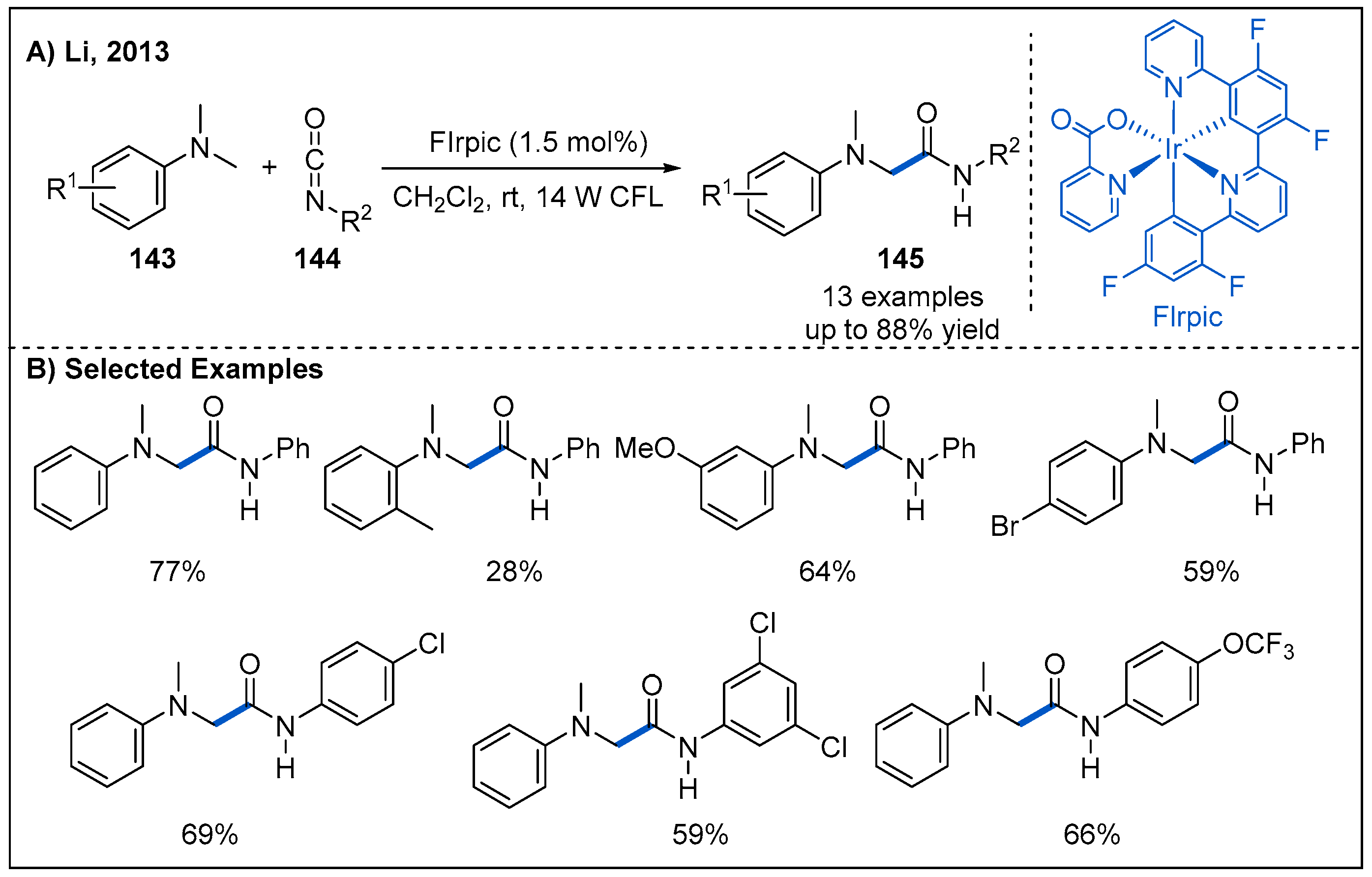
- Zhou, H.; Lu, P.; Gu, X.; Li, P. Visible-light-mediated nucleophilic addition of an alpha-aminoalkyl radical to isocyanate or isothiocyanate. Org. Lett. 2013, 15, 5646–5649. [Google Scholar] [CrossRef] [PubMed]
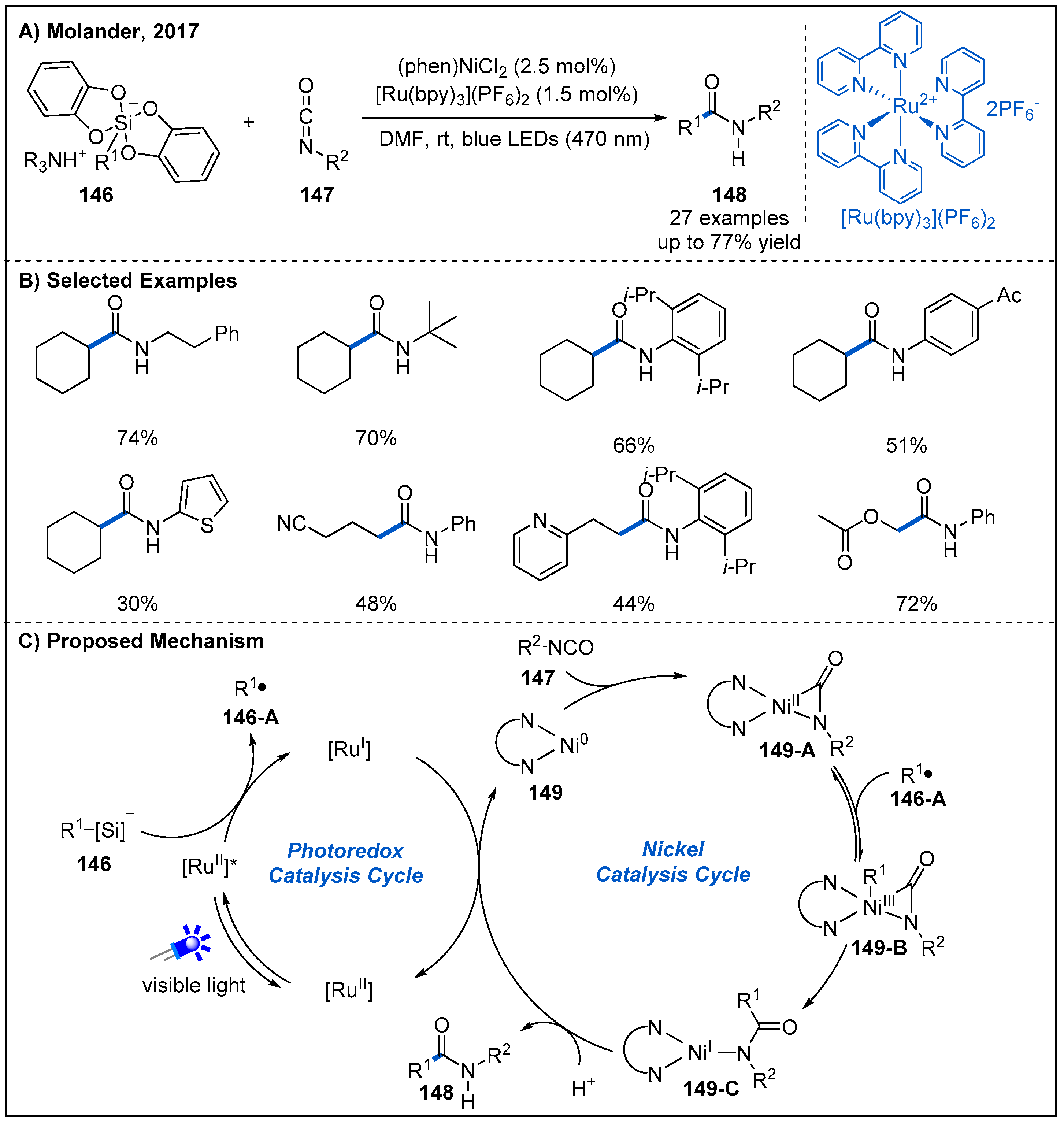
- Zheng, S.; Primer, D.N.; Molander, G.A. Nickel/Photoredox-Catalyzed Amidation via Alkylsilicates and Isocyanates. ACS Catal. 2017, 7, 7957–7961. [Google Scholar] [CrossRef] [PubMed]
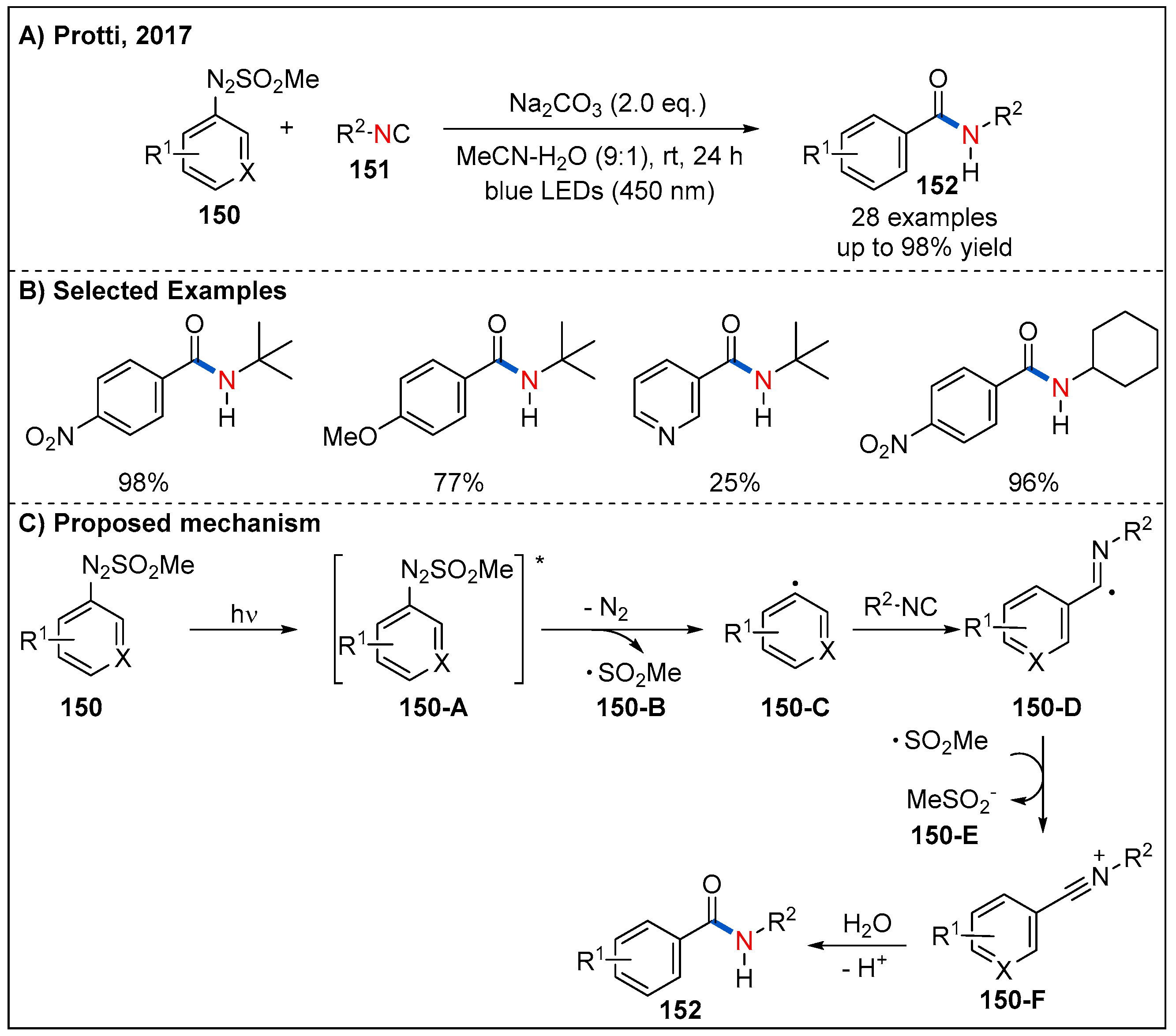
- Malacarne, M.; Protti, S.; Fagnoni, M. A Visible-Light-Driven, Metal-free Route to Aromatic Amides via Radical Arylation of Isonitriles. Adv. Synth. Catal. 2017, 359, 3826–3830. [Google Scholar] [CrossRef]
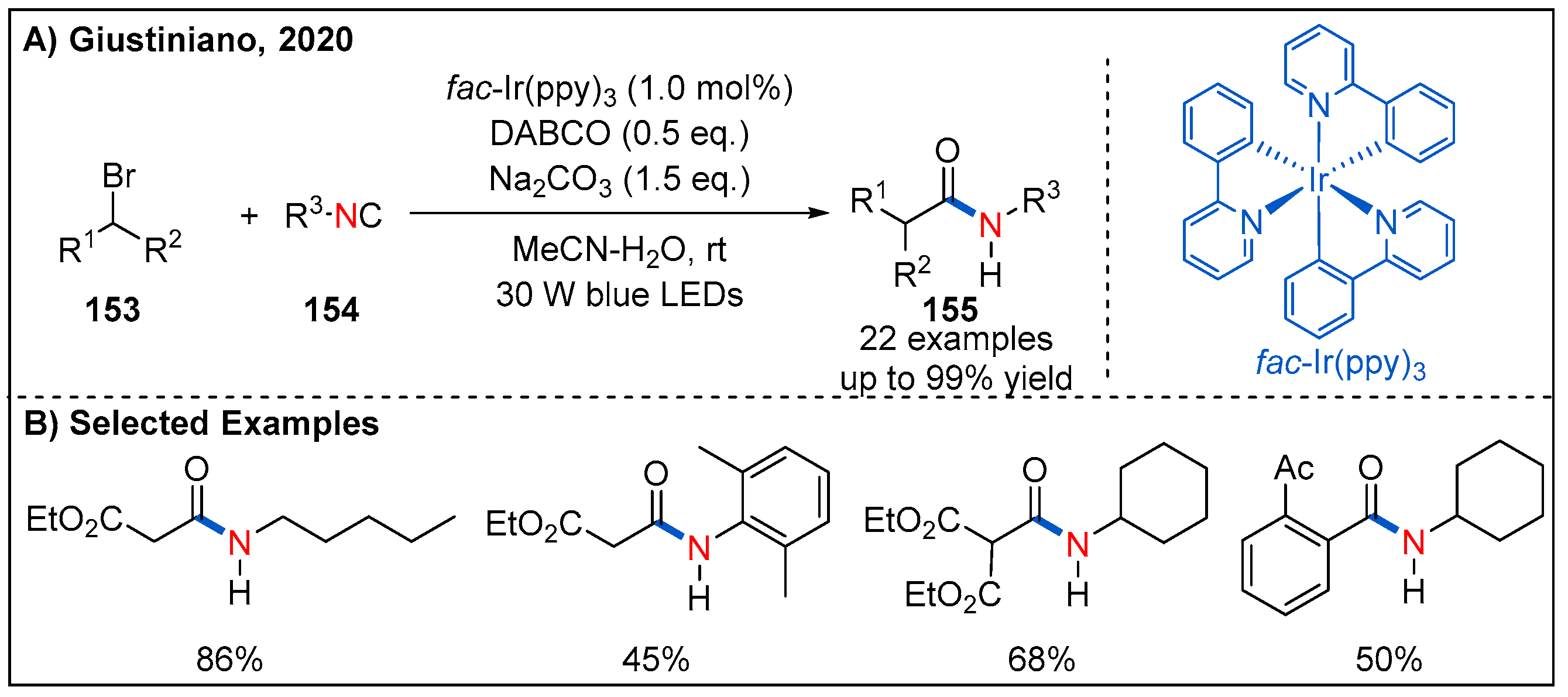
- Cannalire, R.; Amato, J.; Summa, V.; Novellino, E.; Tron, G.C.; Giustiniano, M. Visible-Light Photocatalytic Functionalization of Isocyanides for the Synthesis of Secondary Amides and Ketene Aminals. J. Org. Chem. 2020, 85, 14077–14086. [Google Scholar] [CrossRef]
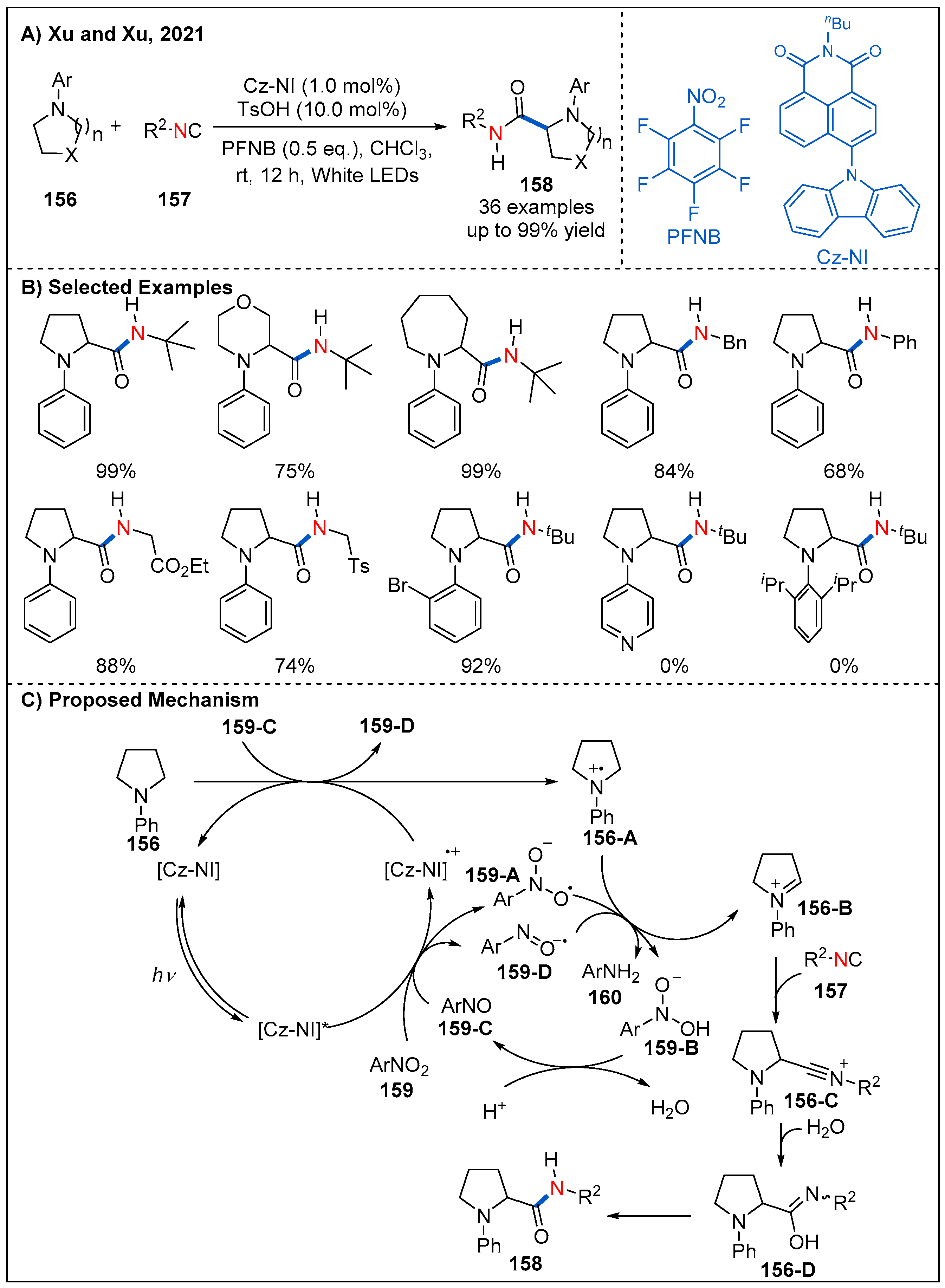
- Yi, M.-J.; Zhang, H.-X.; Xiao, T.-F.; Zhang, J.-H.; Feng, Z.-T.; Wei, L.-P.; Xu, G.-Q.; Xu, P.-F. Photoinduced Metal-Free α-C(sp3)–H Carbamoylation of Saturated Aza-Heterocycles via Rationally Designed Organic Photocatalyst. ACS Catal. 2021, 11, 3466–3472. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Liao, G.; Shi, B.-F. Stereoselective construction of atropisomers featuring a C–N chiral axis. Green Synth. Catal. 2022. [Google Scholar] [CrossRef]
- Cambié, D.; Bottecchia, C.; Straathof, N.J.W.; Hessel, V.; Noël, T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. [Google Scholar] [CrossRef]
- Peng, J.-B.; Liu, X.-L.; Li, L.; Wu, X.-F. Palladium-catalyzed enantioselective carbonylation reactions. Sci. China Chem. 2022, in press. [CrossRef]















































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, B.; Xiao, W.-J.; Chen, J.-R. Recent Advances in Visible-Light-Mediated Amide Synthesis. Molecules 2022, 27, 517. https://doi.org/10.3390/molecules27020517
Lu B, Xiao W-J, Chen J-R. Recent Advances in Visible-Light-Mediated Amide Synthesis. Molecules. 2022; 27(2):517. https://doi.org/10.3390/molecules27020517
Chicago/Turabian StyleLu, Bin, Wen-Jing Xiao, and Jia-Rong Chen. 2022. "Recent Advances in Visible-Light-Mediated Amide Synthesis" Molecules 27, no. 2: 517. https://doi.org/10.3390/molecules27020517
APA StyleLu, B., Xiao, W. -J., & Chen, J. -R. (2022). Recent Advances in Visible-Light-Mediated Amide Synthesis. Molecules, 27(2), 517. https://doi.org/10.3390/molecules27020517