
Discovery of Potential Antiviral Compounds against Hendra Virus by Targeting Its Receptor-Binding Protein (G) Using Computational Approaches

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Methodology
2.1. Drug Target Selection and Preparation
2.2. Small Molecule Preparation
2.3. Molecular Docking
2.4. Molecular Dynamics (MD) Simulation
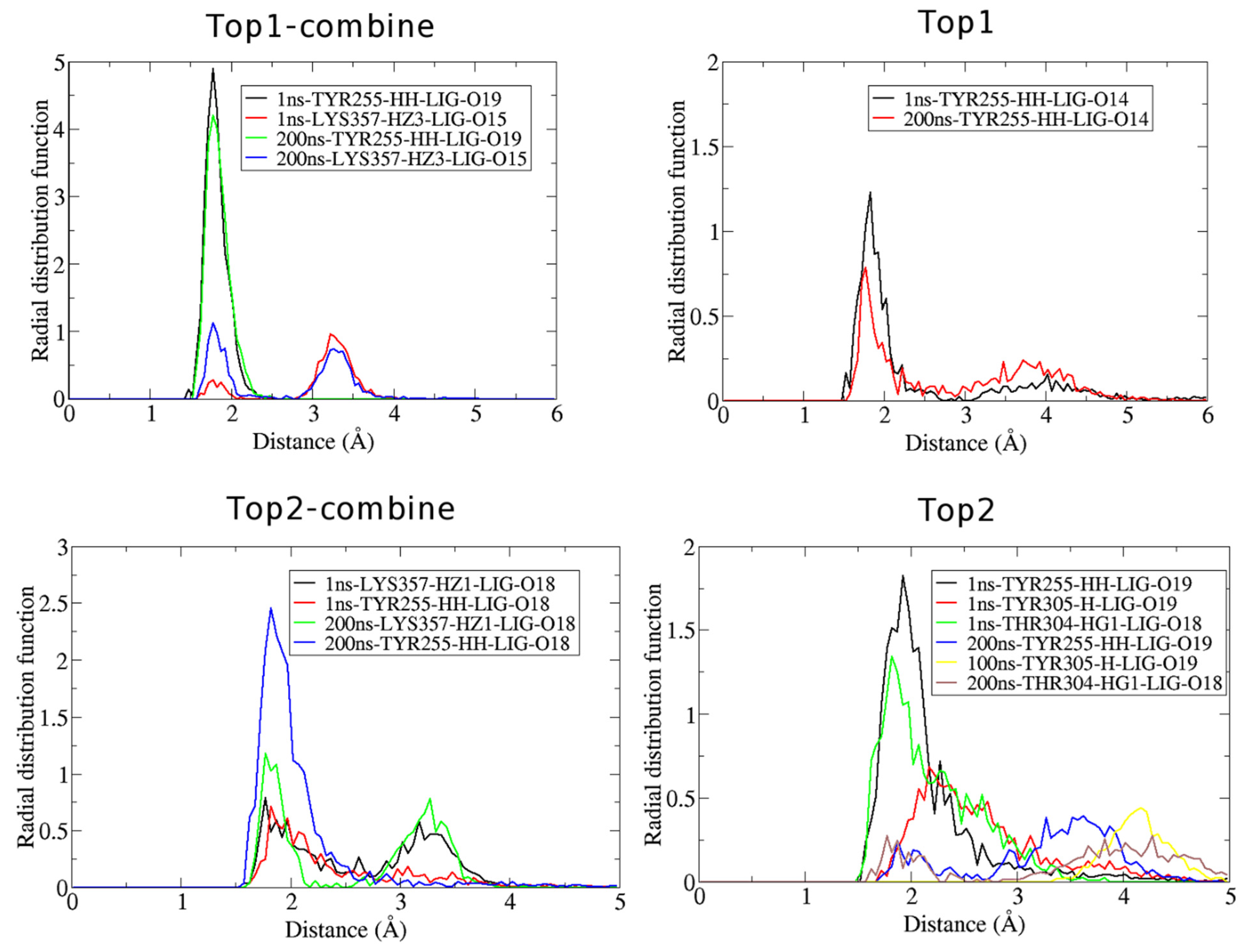
2.5. Radial Distribution Function
2.6. Binding Free Energy Calculations
2.7. WaterSwap Absolute Binding Free Energy Calculations
3. Results
3.1. Filtering Antiviral Chemical Libraries
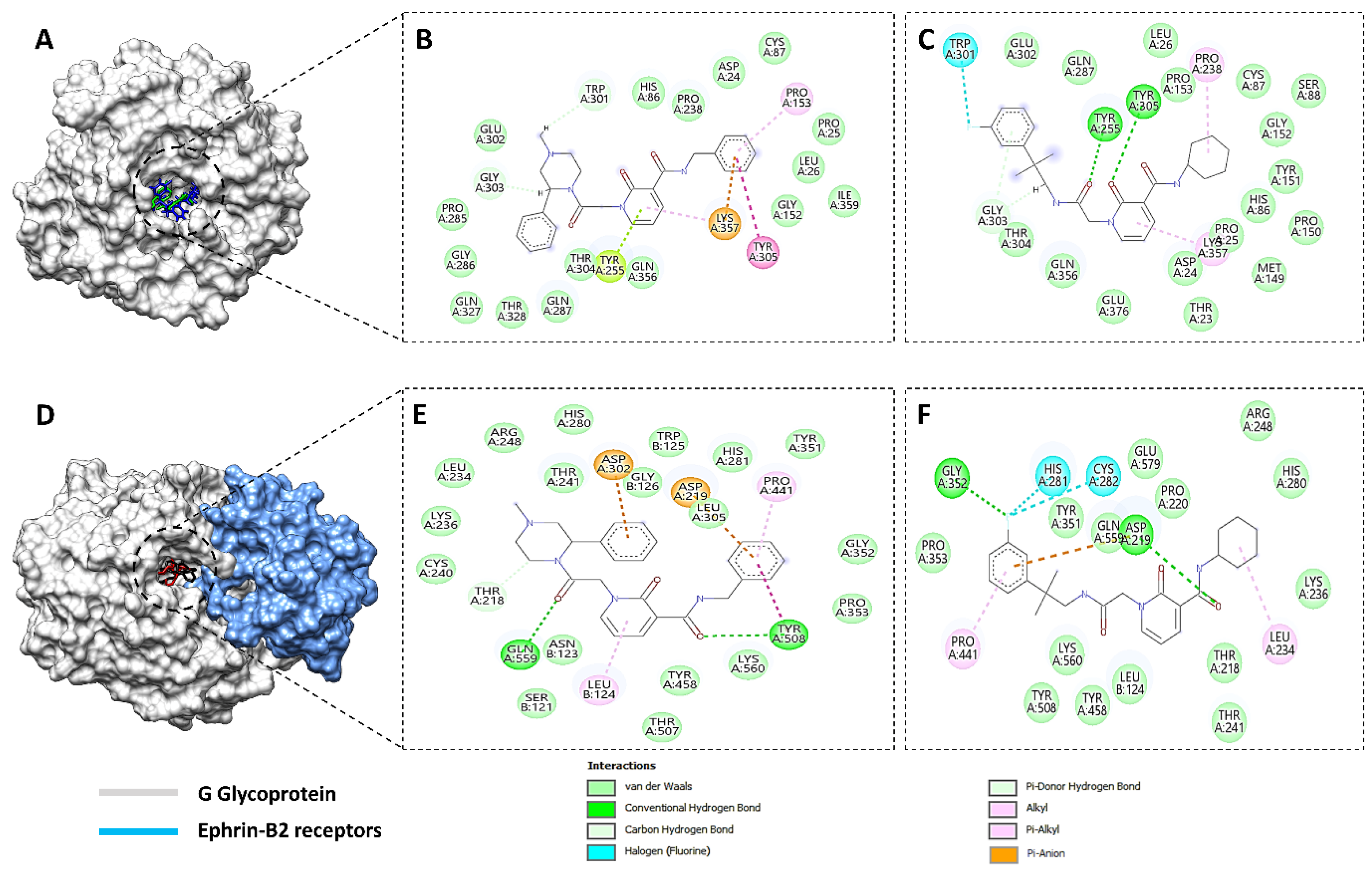
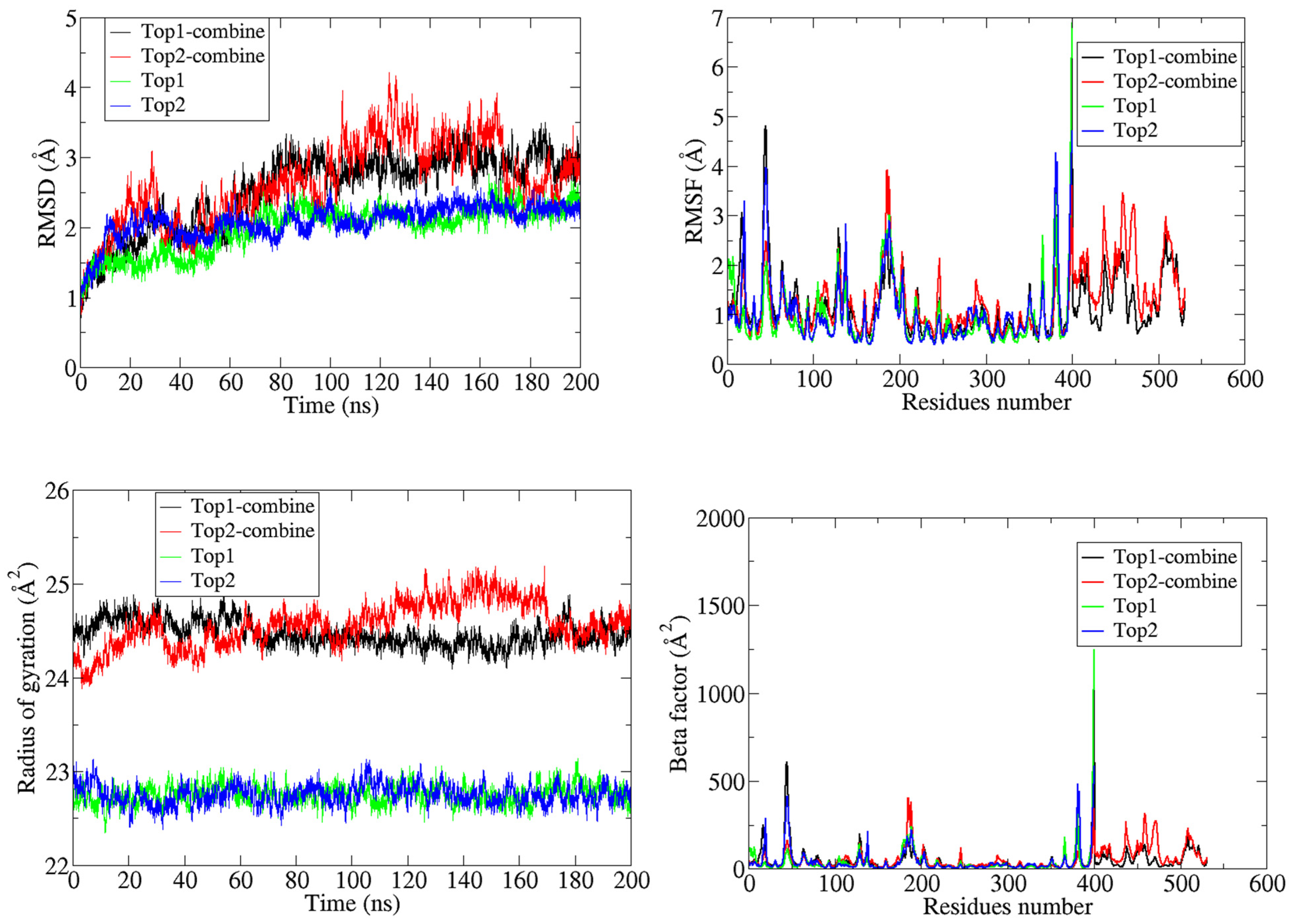
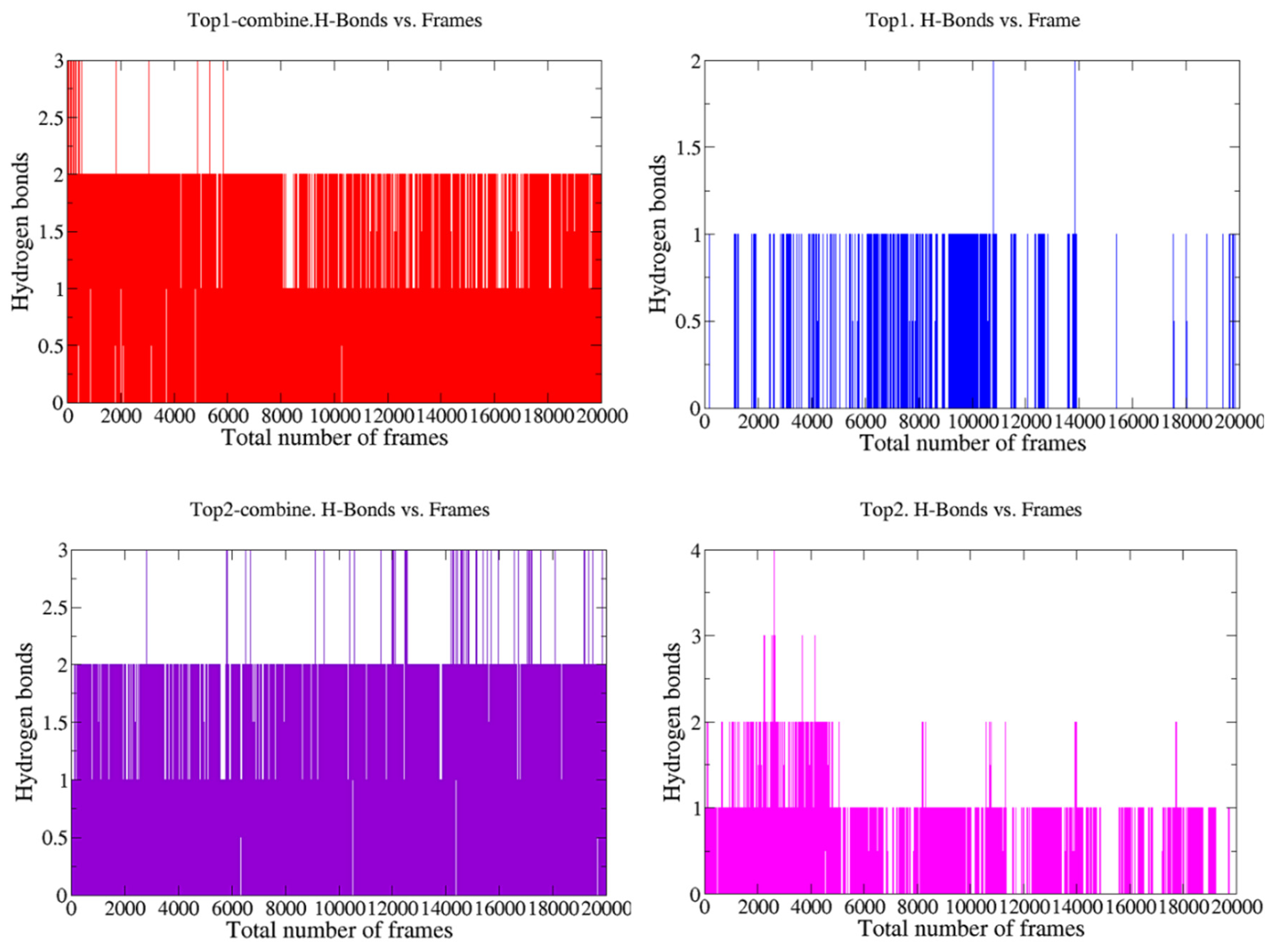
3.2. MD Simulations
3.2.1. Radial Distribution Function (RDF)
3.2.2. Binding Free Energy Calculation
3.3. WaterSwap Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eaton, B.T.; Broder, C.C.; Middleton, D.; Wang, L.-F. Hendra and Nipah viruses: Different and dangerous. Nat. Rev. Microbiol. 2006, 4, 23–35. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Lo Presti, A.; Cella, E.; Giovanetti, M.; Lai, A.; Angeletti, S.; Zehender, G.; Ciccozzi, M. Origin and evolution of Nipah virus. J. Med. Virol. 2016, 88, 380–388. [Google Scholar] [CrossRef]
- Wang, L.-F.; Yu, M.; Hansson, E.; Pritchard, L.I.; Shiell, B.; Michalski, W.P.; Eaton, B.T. The exceptionally large genome of Hendra virus: Support for creation of a new genus within the family Paramyxoviridae. J. Virol. 2000, 74, 9972–9979. [Google Scholar] [CrossRef] [Green Version]
- Harcourt, B.H.; Tamin, A.; Halpin, K.; Ksiazek, T.G.; Rollin, P.E.; Bellini, W.J.; Rota, P.A. Molecular characterization of the polymerase gene and genomic termini of Nipah virus. Virology 2001, 287, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Marsh, G.A.; Wang, L.-F. Hendra and Nipah viruses: Why are they so deadly? Curr. Opin. Virol. 2012, 2, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M. WHO’s blueprint list of priority diseases. JAMA 2018, 319, 1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, K.Y.; Fraser, N.S.; Henning, J.; Halpin, K.; Gibson, J.S.; Betzien, L. Hendra virus: Epidemiology dynamics in relation to climate change, diagnostic tests and control measures. One Health 2021, 12, 100207. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.M.; Plemper, R.K. Structure and organization of paramyxovirus particles. Curr. Opin. Virol. 2017, 24, 105–114. [Google Scholar] [CrossRef]
- Rizzardini, G.; Saporito, T.; Visconti, A. What is new in infectious diseases: Nipah virus, MERS-CoV and the blueprint list of the World Health Organization. Infez. Med. 2018, 26, 195–198. [Google Scholar]
- Pal, M. Hendra virus disease: A highly infectious emerging anthropozoonosis. Acta Sci. Microbiol. 2021, 4, 29–30. [Google Scholar] [CrossRef]
- Wong, K.; Ong, K. Pathology of acute henipavirus infection in humans and animals. Pathol. Res. Int. 2011, 2011, 567248. [Google Scholar] [CrossRef] [Green Version]
- Playford, E.G.; McCall, B.; Smith, G.; Slinko, V.; Allen, G.; Smith, I.; Moore, F.; Taylor, C.; Kung, Y.-H.; Field, H. Human Hendra virus encephalitis associated with equine outbreak, Australia, 2008. Emerg. Infect. Dis. 2010, 16, 219. [Google Scholar] [CrossRef]
- Bowden, T.A.; Aricescu, A.R.; Gilbert, R.J.; Grimes, J.M.; Jones, E.Y.; Stuart, D.I. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat. Struct. Mol. Biol. 2008, 15, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Bonaparte, M.I.; Dimitrov, A.S.; Bossart, K.N.; Crameri, G.; Mungall, B.A.; Bishop, K.A.; Choudhry, V.; Dimitrov, D.S.; Wang, L.-F.; Eaton, B.T. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10652–10657. [Google Scholar] [CrossRef] [Green Version]
- Shehroz, M.; Zaheer, T.; Hussain, T. Computer-aided drug design against spike glycoprotein of SARS-CoV-2 to aid COVID-19 treatment. Heliyon 2020, 6, e05278. [Google Scholar] [CrossRef]
- Anasir, M.I.; Ramanathan, B.; Poh, C.L. Structure-based design of antivirals against envelope glycoprotein of dengue virus. Viruses 2020, 12, 367. [Google Scholar] [CrossRef] [Green Version]
- Dash, R.; Das, R.; Junaid, M.; Akash, M.F.C.; Islam, A.; Hosen, S.Z. In silico-based vaccine design against Ebola virus glycoprotein. Adv. Appl. Bioinform. Chem. AABC 2017, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Kraljevic, S.; Stambrook, P.J.; Pavelic, K. Accelerating drug discovery: Although the evolution of ‘-omics’ methodologies is still in its infancy, both the pharmaceutical industry and patients could benefit from their implementation in the drug development process. EMBO Rep. 2004, 5, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studio, D. Discovery Studio; Accelrys: San Diego, CA, USA, 2008. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Tabachnick, B.G.; Fidell, L.S.; Ullman, J.B. Using Multivariate Statistics; Pearson: Boston, MA, USA, 2007; Volume 5. [Google Scholar]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, Y.; Isomura, T.; Kashima, H. Wisdom of crowds for synthetic accessibility evaluation. J. Mol. Graph. Model. 2018, 80, 217–223. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. Chem. Med. Chem. 2008, 3, 435. [Google Scholar] [CrossRef] [PubMed]
- Tabeshpour, J.; Sahebkar, A.; Zirak, M.R.; Zeinali, M.; Hashemzaei, M.; Rakhshani, S.; Rakhshani, S. Computer-aided drug design and drug pharmacokinetic prediction: A mini-review. Curr. Pharm. Des. 2018, 24, 3014–3019. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem. Med. Chem. 2016, 11, 1117. [Google Scholar] [CrossRef] [Green Version]
- Dressman, J.; Vertzoni, M.; Goumas, K.; Reppas, C. Estimating drug solubility in the gastrointestinal tract. Adv. Drug Deliv. Rev. 2007, 59, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Arnautova, Y.A.; Abagyan, R.A.; Totrov, M. Development of a new physics-based internal coordinate mechanics force field and its application to protein loop modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 477–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teodoro, M.L.; Phillips, G.N.; Kavraki, L.E. Molecular docking: A problem with thousands of degrees of freedom. In Proceedings of the 2001 ICRA. IEEE International Conference on Robotics and Automation (Cat. No. 01CH37164), Seoul, Korea, 21–26 May 2001; IEEE: Piscataway, NJ, USA, 2001; pp. 960–965. [Google Scholar]
- Ahmad, S.; Waheed, Y.; Ismail, S.; Najmi, M.H.; Ansari, J.K. Rational design of potent anti-COVID-19 main protease drugs: An extensive multi-spectrum in silico approach. J. Mol. Liq. 2021, 330, 115636. [Google Scholar] [CrossRef] [PubMed]
- ul Qamar, M.T.; Ahmad, S.; Khan, A.; Mirza, M.U.; Ahmad, S.; Abro, A.; Chen, L.-L.; Almatroudi, A.; Wei, D.-Q. Structural probing of HapR to identify potent phytochemicals to control Vibrio cholera through integrated computational approaches. Comput. Biol. Med. 2021, 138, 104929. [Google Scholar] [CrossRef]
- Ahmad, S.; Waheed, Y.; Ismail, S.; Bhatti, S.; Abbasi, S.W.; Muhammad, K. Structure-based virtual screening identifies multiple stable binding sites at the RecA domains of SARS-CoV-2 helicase enzyme. Molecules 2021, 26, 1446. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Weiner, P.K.; Kollman, P.A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. [Google Scholar] [CrossRef]
- Turner, P. XMGRACE; Version 5.1. 19; Weizmann Institute of Science: Rehovot, Israel, 2005. [Google Scholar]
- Ahmad, S.; Waheed, Y.; Abro, A.; Abbasi, S.W.; Ismail, S. Molecular screening of glycyrrhizin-based inhibitors against ACE2 host receptor of SARS-CoV-2. J. Mol. Modeling 2021, 27, 206. [Google Scholar] [CrossRef]
- Abbasi, S.; Raza, S.; Azam, S.S.; Liedl, K.R.; Fuchs, J.E. Interaction mechanisms of a melatonergic inhibitor in the melatonin synthesis pathway. J. Mol. Liq. 2016, 221, 507–517. [Google Scholar] [CrossRef]
- Abro, A.; Azam, S.S. Binding free energy based analysis of arsenic (+3 oxidation state) methyltransferase with S-adenosylmethionine. J. Mol. Liq. 2016, 220, 375–382. [Google Scholar] [CrossRef]
- Woods, C.J.; Malaisree, M.; Michel, J.; Long, B.; McIntosh-Smith, S.; Mulholland, A.J. Rapid decomposition and visualisation of protein–ligand binding free energies by residue and by water. Faraday Discuss. 2014, 169, 477–499. [Google Scholar] [CrossRef] [Green Version]
- Woods, C.J.; Malaisree, M.; Hannongbua, S.; Mulholland, A.J. A water-swap reaction coordinate for the calculation of absolute protein–ligand binding free energies. J. Chem. Phys. 2011, 134, 054114. [Google Scholar] [CrossRef] [Green Version]
- Nutho, B.; Pengthaisong, S.; Tankrathok, A.; Lee, V.S.; Ketudat Cairns, J.R.; Rungrotmongkol, T.; Hannongbua, S. Structural basis of specific glucoimidazole and mannoimidazole binding by Os3BGlu7. Biomolecules 2020, 10, 907. [Google Scholar] [CrossRef]
- Tariq, M.H. Computational Screening of Phytochemicals against Munc13-1, a Promising target to treat Alcoholism. NUST J. Nat. Sci. 2020, 5. [Google Scholar] [CrossRef]
- Hussain, G.; Ashfaq, U.A.; Masoud, M.S.; Nahid, N.; Bhinder, M.A.; Aslam, N.; Yousaf, N.; Ahmed, U.; Qasim, M. Computational screening of phytochemicals against survivin protein: A potent target for cancer. Pak. J. Pharm. Sci. 2019, 32, 1145–1154. [Google Scholar] [PubMed]
- ul Qamar, M.T.; Maryam, A.; Muneer, I.; Xing, F.; Ashfaq, U.A.; Khan, F.A.; Anwar, F.; Geesi, M.H.; Khalid, R.R.; Rauf, S.A. Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Sci. Rep. 2019, 9, 1433. [Google Scholar] [CrossRef]
- Lim, S.; Othman, R.; Yusof, R.; Heh, C. Rational drug discovery of HCV helicase inhibitor: Improved docking accuracy with multiple seedings of Autodock Vina and in situ minimization. Curr. Comput. Aided Drug Des. 2017, 31, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Vernès, L.; Cadoret, J.-P. Docking and in silico toxicity assessment of Arthrospira compounds as potential antiviral agents against SARS-CoV-2. J. Appl. Phycol. 2021, 33, 1579–1602. [Google Scholar] [CrossRef] [PubMed]
- Sadati, S.M.; Gheibi, N.; Ranjbar, S.; Hashemzadeh, M.S. Docking study of flavonoid derivatives as potent inhibitors of influenza H1N1 virus neuraminidase. Biomed. Rep. 2019, 10, 33–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Huang, D. Hydrogen bonding penalty upon ligand binding. PLoS ONE 2011, 6, e19923. [Google Scholar] [CrossRef]
- Naz, S.; Farooq, U.; Khan, S.; Sarwar, R.; Mabkhot, Y.N.; Saeed, M.; Alsayari, A.; Muhsinah, A.B.; Ul-Haq, Z. Pharmacophore model-based virtual screening, docking, biological evaluation and molecular dynamics simulations for inhibitors discovery against α-tryptophan synthase from Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2021, 39, 610–620. [Google Scholar] [CrossRef]
- Nirwan, S.; Chahal, V.; Kakkar, R. Structure-based virtual screening, free energy of binding and molecular dynamics simulations to propose novel inhibitors of Mtb-MurB oxidoreductase enzyme. J. Biomol. Struct. Dyn. 2021, 39, 656–671. [Google Scholar] [CrossRef]
- Nagar, P.R.; Gajjar, N.D.; Dhameliya, T.M. In search of SARS CoV-2 replication inhibitors: Virtual screening, molecular dynamics simulations and ADMET analysis. J. Mol. Struct. 2021, 1246, 131190. [Google Scholar] [CrossRef]
- Kitamura, K.; Tamura, Y.; Ueki, T.; Ogata, K.; Noda, S.; Himeno, R.; Chuman, H. Binding free-energy calculation is a powerful tool for drug optimization: Calculation and measurement of binding free energy for 7-azaindole derivatives to glycogen synthase kinase-3β. J. Chem. Inf. Modeling 2014, 54, 1653–1660. [Google Scholar] [CrossRef]
- ADuan, L.; Liu, X.; Zhang, J.Z. Interaction entropy: A new paradigm for highly efficient and reliable computation of protein–ligand binding free energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar]
- Ibrahim, M.A.; Abdelrahman, A.H.; Hussien, T.A.; Badr, E.A.; Mohamed, T.A.; El-Seedi, H.R.; Pare, P.W.; Efferth, T.; Hegazy, M.-E.F. In silico drug discovery of major metabolites from spices as SARS-CoV-2 main protease inhibitors. Comput. Biol. Med. 2020, 126, 104046. [Google Scholar] [CrossRef]
- Mirza, S.B.; Salmas, R.E.; Fatmi, M.Q.; Durdagi, S. Virtual screening of eighteen million compounds against dengue virus: Combined molecular docking and molecular dynamics simulations study. J. Mol. Graph. Model. 2016, 66, 99–107. [Google Scholar] [CrossRef]
- Ropón-Palacios, G.; Chenet-Zuta, M.E.; Olivos-Ramirez, G.E.; Otazu, K.; Acurio-Saavedra, J.; Camps, I. Potential novel inhibitors against emerging zoonotic pathogen Nipah virus: A virtual screening and molecular dynamics approach. J. Biomol. Struct. Dyn. 2020, 38, 3225–3234. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Bobb, K.; Borisevich, V.; Geisbert, J.B.; Agans, K.N.; Cross, R.W.; Prasad, A.N.; Fenton, K.A.; Yu, H.; Fouts, T.R. A single dose investigational subunit vaccine for human use against Nipah virus and Hendra virus. NPJ Vaccines 2021, 6, 23. [Google Scholar] [CrossRef]
- Kamthania, M.; Srivastava, S.; Desai, M.; Jain, A.; Shrivastav, A.; Sharma, D. Immunoinformatics Approach to design T-cell epitope-based vaccine against hendra virus. Int. J. Pept. Res. Ther. 2019, 25, 1627–1637. [Google Scholar] [CrossRef]
- Hossan, M.I.; Chowdhury, A.S.; Hossain, M.U.; Khan, M.A.; Mahmood, T.B.; Mizan, S. Immunoinformatics aided-design of novel multi-epitope based peptide vaccine against Hendra henipavirus through proteome exploration. Inform. Med. Unlocked 2021, 25, 100678. [Google Scholar] [CrossRef]
- Freiberg, A.N.; Worthy, M.N.; Lee, B.; Holbrook, M.R. Combined chloroquine and ribavirin treatment does not prevent death in a hamster model of Nipah and Hendra virus infection. J. Gen. Virol. 2010, 91, 765. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Chemical Structure | AutoDock Vina Score | |
|---|---|---|---|
| G Attachement Glycoprotein | G Attachement Glycoprotein Complex with Ephrin-B2 Receptors | ||
| Top1 (C24H30FN3O3) |  | −9.7 | −9.5 |
| Top2 (C26H28N4O3) |  | −9.4 | −9.2 |
| Top3 (C22H24FN5O3) |  | −8.6 | −8.7 |
| Top4 (C22H21FN4O3) |  | −8.2 | −8.5 |
| Top5 (C21H26N4O3) |  | −7.9 | −7.4 |
| Compound ID | Physiochemical Properties | Drug-Likeliness (Violations for Different Rules) | Medicinal Chemistry | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular Weight (g/mol) | H-Bond Donor | H-Bond Acceptor | Topological Polar Surface Area (Å2) | Number of Rotatable Bonds | Lipinski Rule of 5 | Veber Rule | Egan Rule | Muegge Rule | PAINS Alert | Brenk Alert | Synthetic Accessibility | |
| Top 1 | 445.53 | 2 | 3 | 75.85 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 3.75 |
| Top 2 | 427.51 | 2 | 4 | 80.20 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 3.29 |
| Components | Top1-Combine | Top1 | Top2-Combine | Top2 |
|---|---|---|---|---|
| Energies (kcal/mol) | ||||
| ∆Evdw | −46.7359 | −19.2174 | −44.2806 | −35.5087 |
| ∆Eele | −157.7020 | −1.0961 | −60.6010 | −26.9743 |
| ∆EGB | 174.3174 | 13.9830 | 73.1834 | 44.9049 |
| ∆Esurf | −6.0934 | −2.4602 | −5.6009 | −4.8286 |
| ∆Ggas | −204.4379 | −20.3134 | −104.8816 | −62.4830 |
| ∆Gsolv/GB | 168.2240 | 11.5228 | 67.5825 | 40.0763 |
| ∆tot/GB | −36.2139 | −8.7906 | −37.2991 | −22.4067 |
| ∆EPB | 178.3334 | 12.0337 | 80.8110 | 53.6368 |
| ∆Gnpol | −4.5831 | −2.7127 | −4.6730 | −4.0441 |
| ∆Gsolv/PB | 173.7503 | 9.3210 | 76.1380 | 49.5926 |
| ∆tot/PB | −30.6876 | −10.9925 | −28.7436 | −12.8904 |
| ∆tot/PB | −30.6876 | −10.9925 | −28.7436 | −12.8904 |
| Algorithm | Waterswap Energy (kcal/mol) | |||
|---|---|---|---|---|
| Top1-Combine | Top1 | Top2-Combine | Top2 | |
| Bennetts | −27.69 | −25.89 | −35.53 | −30.80 |
| FEP | −24.75 | −20.15 | −33.23 | −26.60 |
| TI | −26.01 | −23.05 | −33.13 | −30.20 |
| Quadrature | −22.4 | −21.4 | −31.24 | −23.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, F.; Albutti, A.; Tariq, M.H.; Din, G.; Tahir ul Qamar, M.; Ahmad, S. Discovery of Potential Antiviral Compounds against Hendra Virus by Targeting Its Receptor-Binding Protein (G) Using Computational Approaches. Molecules 2022, 27, 554. https://doi.org/10.3390/molecules27020554
Ahmad F, Albutti A, Tariq MH, Din G, Tahir ul Qamar M, Ahmad S. Discovery of Potential Antiviral Compounds against Hendra Virus by Targeting Its Receptor-Binding Protein (G) Using Computational Approaches. Molecules. 2022; 27(2):554. https://doi.org/10.3390/molecules27020554
Chicago/Turabian StyleAhmad, Faisal, Aqel Albutti, Muhammad Hamza Tariq, Ghufranud Din, Muhammad Tahir ul Qamar, and Sajjad Ahmad. 2022. "Discovery of Potential Antiviral Compounds against Hendra Virus by Targeting Its Receptor-Binding Protein (G) Using Computational Approaches" Molecules 27, no. 2: 554. https://doi.org/10.3390/molecules27020554
APA StyleAhmad, F., Albutti, A., Tariq, M. H., Din, G., Tahir ul Qamar, M., & Ahmad, S. (2022). Discovery of Potential Antiviral Compounds against Hendra Virus by Targeting Its Receptor-Binding Protein (G) Using Computational Approaches. Molecules, 27(2), 554. https://doi.org/10.3390/molecules27020554