
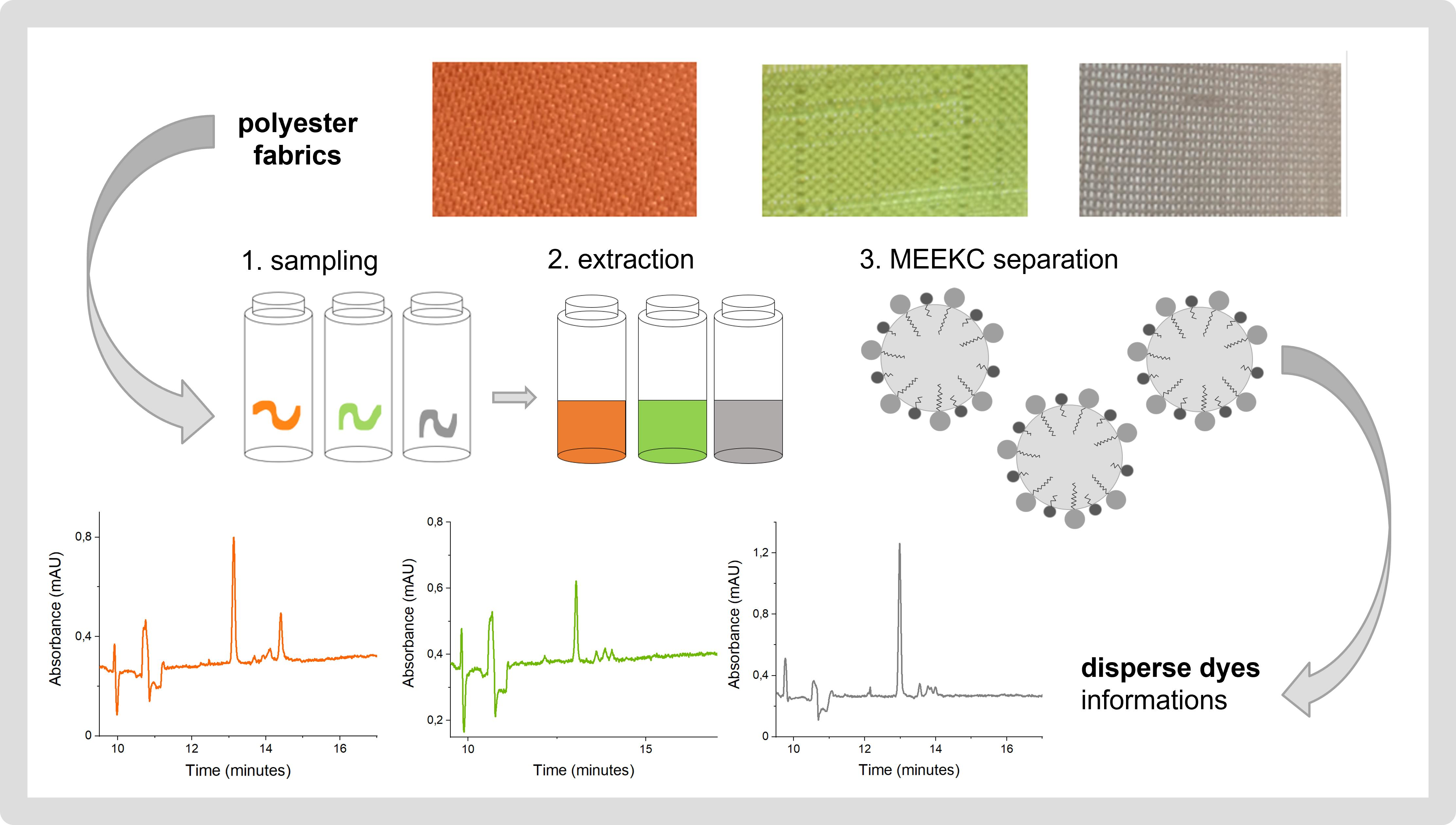
Development of the Microemulsion Electrokinetic Capillary Chromatography Method for the Analysis of Disperse Dyes Extracted from Polyester Fibers

 , , ,
, , ,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Preliminary Tests of the BGE Components
2.2. Optimization of Separation Conditions by Using Doehlert Experimental Design
2.3. Optimization of the Extraction Parameters
2.4. Method Validation
2.5. Analysis of Real Samples
- Total number of signals present at the electropherograms registered at 220 nm (signals with intensity above 0.5 mAU);
- Number of dye signals selected based on the UV-Vis spectra (including dye signals with intensity above 0.4 mAU at 220 nm);
- Bands in the Vis absorption spectrum recorded for dye signals—information corresponding to the color of analyzed dyes;
- Relative migration times of analytes, calculated in relation to the migration time of electroosmotic flow (EOF) signal, treated similarly to an internal standard.
2.6. Application of the Developed Method for the Comparison Study of Polyester Fibers
2.7. Application of the Developed Method for the Identification Study of Polyester Fibers
3. Materials and Methods
3.1. Materials
3.2. Samples
3.3. Instrumentation
3.4. Stock ME and BGE Preparation Procedure
3.5. Sample Preparation Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Preferred Fiber and Materials Market Report. Available online: https://textileexchange.org/2020-preferred-fiber-and-materials-market-report-pfmr-released-2/ (accessed on 20 September 2022).
- Śmigiel-Kamińska, D.; Pośpiech, J.; Makowska, J.; Stepnowski, P.; Wąs-Gubała, J.; Kumirska, J. The Identification of Polyester Fibers Dyed with Disperse Dyes for Forensic Purposes. Molecules 2019, 24, 613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashed, H.S.; Mishra, P.; Nordon, A.; Palmer, D.S.; Baker, M.J. A Comparative Investigation of Two Handheld Near-Ir Spectrometers for Direct Forensic Examination of Fibres in-Situ. Vib. Spectrosc. 2021, 113, 103205. [Google Scholar] [CrossRef]
- Grieve, M.C.; Griffin, R.M.E.; Malone, R. Characteristic Dye Absorption Peaks Found in the FTIR Spectra of Coloured Acrylic Fibres. Sci. Justice J. Forensic Sci. Soc. 1998, 38, 27–37. [Google Scholar] [CrossRef]
- Ueland, M.; Howes, J.M.; Forbes, S.L.; Stuart, B.H. Degradation Patterns of Natural and Synthetic Textiles on a Soil Surface during Summer and Winter Seasons Studied Using ATR-FTIR Spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 185, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepot, L.; De Wael, K.; Gason, F.; Gilbert, B.; Eppe, G.; Malherbe, C. Discrimination of Textile Dyes in Binary Mixtures by Raman Spectroscopy. J. Raman Spectrosc. 2020, 51, 717–730. [Google Scholar] [CrossRef]
- Aljannahi, A.; Alblooshi, R.A.; Alremeithi, R.H.; Karamitsos, I.; Modak, S. Forensic Analysis of Textile Synthetic Fibers Using a FT-IR Spectroscopy Approach. Molecules 2022, 27, 4291. [Google Scholar] [CrossRef]
- Sharma, V.; Kumar, R.; Kaur, P. Forensic Examination of Textile Fibers Using UV-Vis Spectroscopy Combined with Multivariate Analysis. J. Appl. Spectrosc. 2019, 86, 96–100. [Google Scholar] [CrossRef]
- Almer, J.; McAnsh, E.; Doupe, B. Forensic Fibre Analysis by UV-Visible Microspectrophotometry. J. Can. Soc. Forensic Sci. 2010, 43, 16–30. [Google Scholar] [CrossRef]
- Komatsu, H.; Takahara, H.; Matsuda, W.; Nishiwaki, Y. Nondestructive Discrimination of Red Silk Single Fibers Using Total Reflection X-Ray Fluorescence Spectrometry and Synchrotron Radiation X-Ray Fluorescence Spectrometry. J. Forensic Sci. 2021, 66, 1658–1668. [Google Scholar] [CrossRef]
- Was-Gubala, J.; Starczak, R. Nondestructive Identification of Dye Mixtures in Polyester and Cotton Fibers Using Raman Spectroscopy and Ultraviolet-Visible (UV-Vis) Microspectrophotometry. Appl. Spectrosc. 2015, 69, 296–303. [Google Scholar] [CrossRef]
- Was-Gubala, J.; Starczak, R. UV-Vis Microspectrophotometry as a Method of Differentiation between Cotton Fibre Evidence Coloured with Reactive Dyes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 142, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Groves, E.; Palenik, S.; Palenik, C.S. Reproducibility of High-Performance Thin-Layer Chromatography (HPTLC) in Textile Dye Analysis. Forensic Chem. 2018, 8, 104–110. [Google Scholar] [CrossRef]
- Hu, C.; Zhu, J.; Mei, H.; Shi, H.; Guo, H.; Zhang, G.; Wang, P.; Lu, L.; Zheng, X. A Sensitive HPLC-MS/MS Method for the Analysis of Fiber Dyes. Forensic Chem. 2018, 11, 1–6. [Google Scholar] [CrossRef]
- Carey, A.; Rodewijk, N.; Xu, X.; Van Der Weerd, J. Identification of Dyes on Single Textile Fibers by HPLC-DAD-MS. Anal. Chem. 2013, 85, 11335–11343. [Google Scholar] [CrossRef] [PubMed]
- Vasileiadou, A.; Karapanagiotis, I.; Zotou, A. Development and Validation of a Liquid Chromatographic Method with Diode Array Detection for the Determination of Anthraquinones, Flavonoids and Other Natural Dyes in Aged Silk. J. Chromatogr. A 2021, 1651, 462312. [Google Scholar] [CrossRef] [PubMed]
- Petrick, L.M.; Wilson, T.A.; Fawcett, W.R. High-Performance Liquid Chromatography-Ultraviolet-Visible Spectroscopy-Electrospray Ionization Mass Spectrometry Method for Acrylic and Polyester Forensic Fiber Dye Analysis. J. Forensic Sci. 2006, 51, 771–779. [Google Scholar] [CrossRef]
- Sabatini, F.; Giugliano, R.; Degano, I. Photo-Oxidation Processes of Rhodamine B: A Chromatographic and Mass Spectrometric Approach. Microchem. J. 2018, 140, 114–122. [Google Scholar] [CrossRef]
- Sebastiano, R.; Contiello, N.; Senatore, S.; Righetti, P.G.; Citterio, A. Analysis of Commercial Acid Black 194 and Related Dyes by Micellar Electrokinetic Chromatography. Dye. Pigment. 2012, 94, 258–265. [Google Scholar] [CrossRef]
- Burkinshaw, S.M.; Hinks, D.; Lewis, D.M. Capillary Zone Electrophoresis in the Analysis of Dyes and Other Compounds Employed in the Dye-Manufacturing and Dye-Using Industries. J. Chromatogr. A 1993, 640, 413–417. [Google Scholar] [CrossRef]
- Oxspring, D.A.; Franklin Smyth, W.; Marchant, R. Comparison of Reversed-Polarity Capillary Electrophoresis and Adsorptive Stripping Voltammetry for the Detection and Determination of Reactive Textile Dyes. Analyst 1995, 120, 1995–2000. [Google Scholar] [CrossRef]
- Sirén, H.; Sulkava, R. Determination of Black Dyes from Cotton and Wool Fibres by Capillary Zone Electrophoresis with UV Detection: Application of Marker Technique. J. Chromatogr. A 1995, 717, 149–155. [Google Scholar] [CrossRef]
- Lord, G.A.; Gordon, D.B.; Tetler, L.W.; Carr, C.M. Electrochromatography-Electrospray Mass Spectrometry of Textile Dyes. J. Chromatogr. A 1995, 700, 27–33. [Google Scholar] [CrossRef]
- Stefan, A.R.; Dockery, C.R.; Nieuwland, A.A.; Roberson, S.N.; Baguley, B.M.; Hendrix, J.E.; Morgan, S.L. Forensic Analysis of Anthraquinone, Azo, and Metal Complex Acid Dyes from Nylon Fibers by Micro-Extraction and Capillary Electrophoresis. Anal. Bioanal. Chem. 2009, 394, 2077–2085. [Google Scholar] [CrossRef] [PubMed]
- Tetler, L.W.; Cooper, P.A.; Carr, C.M. The Application of Capillary Electrophoresis/Mass Spectrometry Using Negative-ion Electrospray Ionization to Areas of Importance in the Textile Industry. Rapid Commun. Mass Spectrom. 1994, 8, 179–182. [Google Scholar] [CrossRef]
- Xu, X.; Leijenhorst, H.A.L.; Van Den Hoven, P.; De Koeijer, J.A.; Logtenberg, H. Analysis of Single Textile Fibres by Sample-Induced Isotachophoresis—Micellar Electrokinetic Capillary Chromatography. Sci. Justice J. Forensic Sci. Soc. 2001, 41, 93–105. [Google Scholar] [CrossRef]
- Morgan, S.L.; Vann, B.C.; Baguley, B.M.; Stefan, A.R. Advances in Discrimination of Dyed Textile Fibers Using Capillary Electrophoresis/Mass Spectrometry. In Proceedings of the FBI Trace Evidence Symposium, Clearwater, FL, USA, 16 August 2007; pp. 1–41. [Google Scholar]
- Peláez-Cid, A.A.; Blasco-Sancho, S.; Matysik, F.M. Determination of Textile Dyes by Means of Non-Aqueous Capillary Electrophoresis with Electrochemical Detection. Talanta 2008, 75, 1362–1368. [Google Scholar] [CrossRef]
- Dockery, C.R.; Stefan, A.R.; Nieuwland, A.A.; Roberson, S.N.; Baguley, B.M.; Hendrix, J.E.; Morgan, S.L. Automated Extraction of Direct, Reactive, and Vat Dyes from Cellulosic Fibers for Forensic Analysis by Capillary Electrophoresis. Anal. Bioanal. Chem. 2009, 394, 2095–2103. [Google Scholar] [CrossRef]
- Croft, S.N.; Hinks, D. Analysis of Dyes by Capillary Electrophoresis. J. Soc. Dye. Colour. 1992, 108, 546–551. [Google Scholar] [CrossRef]
- Stefan, A.R.; Dockery, C.R.; Baguley, B.M.; Vann, B.C.; Nieuwland, A.A.; Hendrix, J.E.; Morgan, S.L. Microextraction, Capillary Electrophoresis, and Mass Spectrometry for Forensic Analysis of Azo and Methine Basic Dyes from Acrylic Fibers. Anal. Bioanal. Chem. 2009, 394, 2087–2094. [Google Scholar] [CrossRef]
- Hansa, A.; Pillay, V.L.; Buckley, C.A. Analysis of Reactive Dyes Using High Performence Capillary Electrophoresis. Water Sci. Technol. 1999, 39, 169–172. [Google Scholar] [CrossRef]
- Sałdan, A.; Król, M.; Woźniakiewicz, M.; Kościelniak, P. Application of Capillary Electromigration Methods in the Analysis of Textile Dyes—Review. Molecules 2022, 27, 2767. [Google Scholar] [CrossRef] [PubMed]
- Glenz, O.; Beckmann, W.; Wulder, W. The Mechanism of the Dyeing of Polyester Fibres with Disperse Dyes. J. Soc. Dye. Colour. 2008, 75, 141–147. [Google Scholar] [CrossRef]
- Benkhaya, S.; M’rabet, S.; El Harfi, A. A Review on Classifications, Recent Synthesis and Applications of Textile Dyes. Inorg. Chem. Commun. 2020, 115, 107891. [Google Scholar]
- Tehrani-Bagha, A.R.; Holmberg, K. Solubilization of Hydrophobic Dyes in Surfactant Solutions. Materials 2013, 6, 580–608. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chu, K.; Ye, H.; Liu, X.; Yu, L.; Xu, X.; Chen, G. Recent Advances in Microemulsion Electrokinetic Chromatography. TrAC Trends Anal. Chem. 2012, 34, 140–151. [Google Scholar] [CrossRef]
- Hitoshi, W. Microemulsion capillary electrophoresis. Chem. Lett. 1991, 20, 391–394. [Google Scholar] [CrossRef]
- Ryan, R.; Altria, K.; Mcevoy, E.; Donegan, S.; Power, J. A Review of Developments in the Methodology and Application of Microemulsion Electrokinetic Chromatography. Electrophoresis 2013, 34, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.; Donegan, S.; Power, J.; Altria, K. Advances in the Theory and Application of MEEKC. Electrophoresis 2010, 31, 755–767. [Google Scholar] [CrossRef]
- Altria, K.D. Highly Efficient and Selective Separations of a Wide Range of Analytes Obtained by an Optimised Microemulsion Electrokinetic Chromatography Method. Chromatographia 1999, 49, 457–464. [Google Scholar] [CrossRef]
- Mahuzier, P.; Clark, B.J.; Bryant, S.M.; Altria, K.D. High-Speed Microemulsion Electrokinetic Chromatography. Electrophoresis 2001, 22, 3819–3823. [Google Scholar] [CrossRef]
- Ryan, R.; McEvoy, E.; Donegan, S.; Power, J.; Altria, K. Recent Developments in the Methodology and Application of MEEKC. Electrophoresis 2011, 32, 184–201. [Google Scholar] [CrossRef] [PubMed]
- Beldean-Galea, M.S.; Copaciu, F.M.; Coman, M.V. Chromatographic Analysis of Textile Dyes. J. AOAC Int. 2018, 101, 1353–1370. [Google Scholar] [CrossRef] [PubMed]
- Groves, E.; Palenik, C.S.; Palenik, S. A Survey of Extraction Solvents in the Forensic Analysis of Textile Dyes. Forensic Sci. Int. 2016, 268, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Śmigiel-Kamińska, D.; Pośpiech, J.; Stepnowski, P.; Wąs-Gubała, J.; Kumirska, J. Development of HPLC-DAD and UPLC-QTOF-MS Chromatographic Systems for the Identification for Forensic Purposes of Disperse Dyes of Polyester. Measurment 2021, 174, 108994. [Google Scholar] [CrossRef]
- Society of Dyers and Colourists & AATCC. Colour-Index. Available online: https://colour-index.com/ (accessed on 7 August 2022).
- Mohorič, U.; Beutner, A.; Krickl, S.; Touraud, D.; Kunz, W.; Matysik, F.M. Surfactant-Free Microemulsion Electrokinetic Chromatography (SF-MEEKC) with UV and MS Detection—A Novel Approach for the Separation and ESI-MS Detection of Neutral Compounds. Anal. Bioanal. Chem. 2016, 408, 8681–8689. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Coded Variables | Real Variables | Measured Response | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| x1 | x2 | x3 | ciPr [% v/v] | cCD [mM] | T [°C] | n | tD1 | a | F | |
| 1 | 0 | 0 | 0 | 15 | 40 | 30 | 7 | 11.74 | 1 | 0.60 |
| 2 | 0 | 1 | 0 | 15 | 60 | 30 | 6 | 11.98 | 1 | 0.50 |
| 3 | 0.866 | 0.5 | 0 | 30 | 50 | 30 | 8 | 18.42 | 1 | 0.43 |
| 4 | 0.289 | 0.5 | 0.817 | 20 | 50 | 35 | 9 | 10.43 | 1 | 0.86 |
| 5 | 0 | −1 | 0 | 15 | 20 | 30 | 6 | 11.79 | 1 | 0.51 |
| 6 | −0.866 | −0.5 | 0 | 0 | 30 | 30 | 7 | 9.67 | 1 | 0.72 |
| 7 | −0.289 | −0.5 | −0.817 | 10 | 30 | 25 | 7 | 12.10 | 1 | 0.58 |
| 8 | −0.866 | 0.5 | 0 | 0 | 50 | 30 | 7 | 7.85 | 0 | 0.00 |
| 9 | −0.289 | 0.5 | −0.817 | 10 | 50 | 25 | 8 | 12.55 | 1 | 0.64 |
| 10 | 0.866 | −0.5 | 0 | 30 | 30 | 30 | 8 | 16.78 | 1 | 0.48 |
| 11 | 0.577 | 0 | −0.817 | 25 | 40 | 25 | 9 | 18.90 | 1 | 0.48 |
| 12 | 0.289 | −0.5 | 0.817 | 20 | 30 | 35 | 8 | 10.33 | 0 | 0.00 |
| 13 | −0.577 | 0 | 0.817 | 5 | 40 | 35 | 7 | 7.14 | 0 | 0.00 |
| Peak | 1 (D8) | 2 (D3) | 3 (D1) | 4 (D4) | 5 (D2) | 6 (D7) | 7 (D5) | 8 (D9) | 9 (D6) |
|---|---|---|---|---|---|---|---|---|---|
| Injection repeatability (1 sample × 4 repetitions × 1 day = 4 runs), 3-day average | |||||||||
| rel tm, av [min] | 1.37 | 1.39 | 1.41 | 1.43 | 1.46 | 2.05 | 2.11 | 2.13 | 2.37 |
| SD | 0.01 | 0.01 | 0.01 | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
| CV [%] | 0.72 | 0.91 | 0.82 | 1.24 | 0.86 | 0.42 | 0.43 | 0.39 | 0.32 |
| Sampling repeatability (4 samples × 1 repetition × 1 day = 4 runs), 3-day average | |||||||||
| rel tm, av [min] | 1.39 | 1.40 | 1.43 | 1.44 | 1.47 | 2.01 | 2.09 | 2.10 | 2.34 |
| SD | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.003 | 0.002 | 0.004 |
| CV [%] | 0.60 | 0.63 | 0.71 | 0.52 | 0.65 | 0.56 | 0.16 | 0.11 | 0.19 |
| The intermediate precision of injection (1 sample × 4 repetitions × 3 days = 12 runs) | |||||||||
| rel tm, av [min] | 1.37 | 1.39 | 1.41 | 1.43 | 1.46 | 2.04 | 2.11 | 2.12 | 2.37 |
| SD | 0.01 | 0.02 | 0.01 | 0.02 | 0.01 | 0.07 | 0.05 | 0.05 | 0.06 |
| CV [%] | 0.58 | 0.72 | 0.62 | 1.03 | 0.78 | 3.21 | 1.67 | 1.61 | 2.13 |
| The intermediate precision of sampling (4 samples × 1 repetition × 3 days = 12 runs) | |||||||||
| rel tm, av [min] | 1.39 | 1.40 | 1.42 | 1.44 | 1.47 | 2.01 | 2.09 | 2.11 | 2.34 |
| SD | 0.01 | 0.02 | 0.01 | 0.01 | 0.01 | 0.05 | 0.02 | 0.02 | 0.03 |
| CV [%] | 0.88 | 1.08 | 0.99 | 0.93 | 1.01 | 2.67 | 0.87 | 0.88 | 1.28 |
| The intermediate precision in relation to the capillary (4 samples × 1 repetition × 2 capillaries × 1 day = 8 runs) | |||||||||
| rel tm, av [min] | 1.39 | 1.41 | 1.43 | 1.45 | 1.47 | 2.03 | 2.09 | 2.11 | 2.35 |
| SD | 0.01 | 0.01 | 0.02 | 0.01 | 0.02 | 0.06 | 0.02 | 0.02 | 0.04 |
| CV [%] | 1.02 | 1.04 | 1.15 | 0.90 | 1.21 | 2.93 | 1.05 | 1.03 | 1.50 |
| Similarity | Similar | Different |
|---|---|---|
| expanded uncertainty (U, k = 2) | ≤0.11 | >0.11 |
| annotations | U values lower than parameters obtained in method validation | U values higher than parameters obtained in method validation |
| Signal Number | Relative Migration Times of the Analytes | U (k = 2) for Samples B2 and X | |||
|---|---|---|---|---|---|
| B1 | B2 | B3 | X | ||
| 1 | 1.32 | 1.32 | 1.33 | 1.33 | 0.02 |
| 2 | 1.39 | 1.40 | 1.41 | 1.41 | 0.02 |
| 3 | 1.44 | 1.44 | 1.48 | 1.47 | 0.03 |
| 4 | 1.47 | 1.47 | 1.56 | 1.49 | 0.03 |
| 5 | 1.54 | 1.54 | 1.59 | 1.57 | 0.04 |
| 6 | 1.57 | 1.57 | - | 1.60 | 0.05 |
| total | 6 | 6 | 5 | 6 | |
| dye | 4 | 5 | 3 | 5 | |
| Signal Number | Relative Migration Times of the Analytes | U (k = 2) for Samples P16a and A2 | |||||
|---|---|---|---|---|---|---|---|
| P2 | P8 | P16a | P21 | P25 | A2 | ||
| 1 | 1.46 | 1.48 | 1.33 | 1.32 | 1.24 | 1.32 | 0.01 |
| 2 | 1.49 | 1.79 | 1.43 | 1.38 | 1.33 | 1.42 | 0.02 |
| 3 | - | - | 1.46 | 1.40 | 1.44 | 1.45 | 0.02 |
| 4 | 2.26 | 1.42 | 1.47 | 2.25 | 0.01 | ||
| 5 | - | - | 1.49 | - | - | ||
| 6 | 2.39 | ||||||
| total | 2 | 2 | 4 | 4 | 6 | 4 | - |
| dye peaks | 2 | 2 | 2 | 2 | 4 | 2 | |
| Mixture Symbol | Commercial Name | Dyeing Component CIGN, CICN | Color of Methanolic Solution | Fabric Symbol (Color) |
|---|---|---|---|---|
| D1 | Setapers scarlet SE-3GFL | Disperse Red 54 (11131) | red | FA (orange) |
| D2 | Serilene blau HBGL 200% | Disperse Blue 73 (63265) | blue | |
| D3 | Serilene pink A-2GN | Disperse Red 86 (62175) | rose | |
| D4 | Serilene blau RG 200% | Disperse Blue 165 (11077) Disperse Blue 60 (61104) | blue | FB (light green) |
| D5 | Serilene yellow 6 GLS 200% | Disperse Yellow 114 (128455) | yellow | |
| D6 | Serilene golden yellow K-2 G | Disperse Orange 25 (11227) Disperse Yellow 86 (10353) Disperse Yellow 211 (12755) | yellow-orange | |
| D7 | Serilene yellow A-GL | Disperse Yellow 42 (10338) | yellow | FC (light grey) |
| D8 | Serilene red A-TB | Disperse Red 92 (60752) | red | |
| D9 | Serilene blau A-BL | Disperse Blue 77 (60766) | blue |
| Sample | Fabric Composition | Color | Distributor (Origin Country) | Clothing Type |
|---|---|---|---|---|
| P1 | 100% polyester | navy | OTCF S.A. (Poland) | sweatshirt |
| P2 | - | green | MiLady (Italy) | dress |
| P3 | 50% polyester 50% viscose | black | unknown (unknown) | sweater |
| P4 | 50% cotton 50% polyester | grey | Medicine (China) | sweatshirt |
| P5 | 100% polyester | red | Asos (China) | dress lining |
| P6 | 100% polyester | navy | Reserved (China) | jacket |
| P7 | 100% polyester | blue | Mohito (China) | sweater |
| P8 | 100% polyester | green | Asos (China) | dress |
| P9 | 100% polyester | blue | Janina (China) | dress |
| P10 | 100% polyester | gold | MANGO (Spain) | skirt lining |
| P11 | 100% polyester | black | MISSGUIDED (UK) | top |
| P12 | 100% polyester | red | Pepco (unknown) | pillowcase |
| P13a * | 95% polyester 5% spandex | rose | Mohito (Poland) | dress |
| P13b * | unknown | black | Mohito (Poland) | dress |
| P14 | 94% polyester 6% spandex | black | AMISU (China) | skirt |
| P15 | 90% polyester 10% spandex | black | F&F (India) | top |
| P16a | 85% polyester 13%viscose 2% spandex | green | House (China) | skirt |
| P16b | 85% polyester 13%viscose 2% spandex | black | House (China) | skirt |
| P17 | 64% polyamide 28% polyester 8% spandex | blue | Primark (China) | swimsuit |
| P18 | 73% cotton 23% polyester 4% spandex | black | H&M (China) | shirt |
| P19 | 100% polyester | navy | Alena firany (Poland) | curtains |
| P20 | 100% polyester | black | Sinsay (China) | top |
| P21 | 100% polyester | green | Reserved (China) | dress |
| P22 | 95% polyester 5% spandex | black | Mohito (Cambodia) | jumpsuit |
| P23 | unknown | red | Labres LINGERIE (Poland) | nightshirt |
| P24 | 100% polyester | blue | Eva & Lola China | dress lining |
| P25 | 94% polyester 5% spandex | green | Sinsay (Burma) | dress |
| P26 | unknown | red | unknown (unknown) | hairband |
| Sample Symbol | Comment | Adjustment |
|---|---|---|
| P10 P17 | Poor extraction yield: only a pale beige color was observed after reducing the solvent volume | The amount of sample increased to a sheet of fabric ca. 0.5 × 0.5 cm |
| P13a P13b | Poor extraction yield: colorless extract. The fibers were not entirely colored, only the top layer of the fabric was colored, | The amount of sample increased to a sheet of fabric ca. 0.5 × 0.5 cm |
| P18 | Poor extraction yield: colorless extract. Probably due to the high content of cotton and low content of polyester in the fabrics which indicate dying with other classes of dyes than disperse ones | Excluded from the study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sałdan, A.; Król, M.; Śmigiel-Kamińska, D.; Woźniakiewicz, M.; Kościelniak, P. Development of the Microemulsion Electrokinetic Capillary Chromatography Method for the Analysis of Disperse Dyes Extracted from Polyester Fibers. Molecules 2022, 27, 6974. https://doi.org/10.3390/molecules27206974
Sałdan A, Król M, Śmigiel-Kamińska D, Woźniakiewicz M, Kościelniak P. Development of the Microemulsion Electrokinetic Capillary Chromatography Method for the Analysis of Disperse Dyes Extracted from Polyester Fibers. Molecules. 2022; 27(20):6974. https://doi.org/10.3390/molecules27206974
Chicago/Turabian StyleSałdan, Anna, Małgorzata Król, Daria Śmigiel-Kamińska, Michał Woźniakiewicz, and Paweł Kościelniak. 2022. "Development of the Microemulsion Electrokinetic Capillary Chromatography Method for the Analysis of Disperse Dyes Extracted from Polyester Fibers" Molecules 27, no. 20: 6974. https://doi.org/10.3390/molecules27206974
APA StyleSałdan, A., Król, M., Śmigiel-Kamińska, D., Woźniakiewicz, M., & Kościelniak, P. (2022). Development of the Microemulsion Electrokinetic Capillary Chromatography Method for the Analysis of Disperse Dyes Extracted from Polyester Fibers. Molecules, 27(20), 6974. https://doi.org/10.3390/molecules27206974