
Intramolecular Interactions in Derivatives of Uracil Tautomers


Abstract
:1. Introduction
2. Methodology
3. Results and Discussion
3.1. Electronic Properties of Substituents
3.2. Geometry
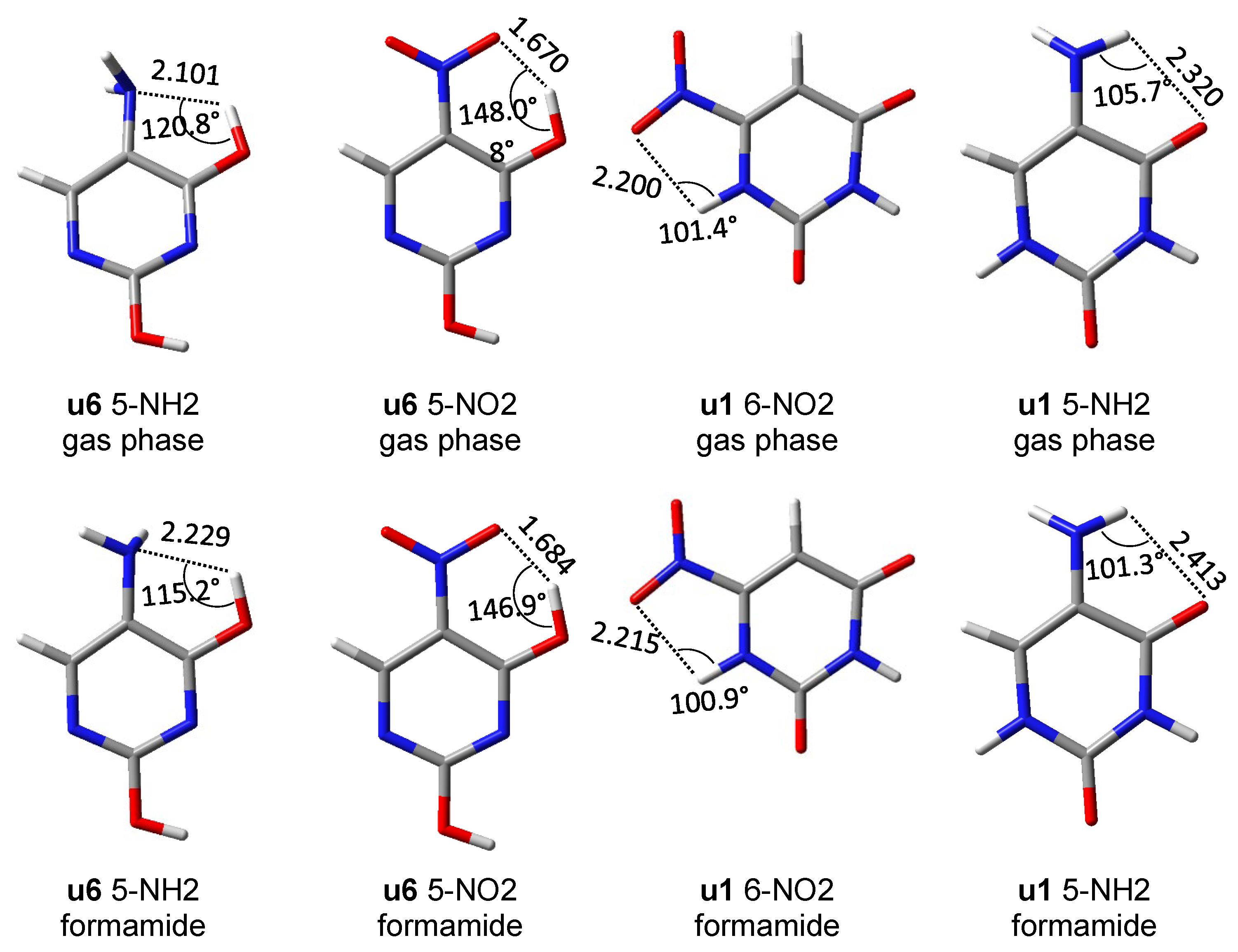
3.3. Intramolecular Interactions between Non-Covalently Bonded Atoms
3.4. Tautomer Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neidle, S.; Sanderson, M. Principles of Nucleic Acid Structure, 2nd ed.; Elsevier: San Diego, CA, USA, 2021. [Google Scholar]
- Chu, C.K.; Baker, D.C. (Eds.) Nucleosides and Nucleotides as Antitumor and Antiviral Agents; Springer US: Boston, MA, USA, 1993. [Google Scholar]
- Pozharskiĭ, A.F.; Katritzky, A.R.; Soldatenkov, A.T. Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry, and Applications, 2nd ed.; Wiley: Chichester, UK, 2011. [Google Scholar]
- Gong, L.; Zhang, Y.; Liu, C.; Zhang, M.; Han, S. Application of Radiosensitizers in Cancer Radiotherapy. Int. J. Nanomed. 2021, 16, 1083–1102. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, P.; Zhang, C.; Bu, Y. Mechanisms Responsible for High Energy Radiation Induced Damage to Single-Stranded DNA Modified by Radiosensitizing 5-Halogenated Deoxyuridines. J. Phys. Chem. B 2016, 120, 2649–2657. [Google Scholar] [CrossRef] [PubMed]
- Rak, J.; Chomicz, L.; Wiczk, J.; Westphal, K.; Zdrowowicz, M.; Wityk, P.; Żyndul, M.; Makurat, S.; Golon, Ł. Mechanisms of Damage to DNA Labeled with Electrophilic Nucleobases Induced by Ionizing or UV Radiation. J. Phys. Chem. B 2015, 119, 8227–8238. [Google Scholar] [CrossRef] [PubMed]
- Poštulka, J.; Slavíček, P.; Fedor, J.; Fárník, M.; Kočišek, J. Energy Transfer in Microhydrated Uracil, 5-Fluorouracil, and 5-Bromouracil. J. Phys. Chem. B 2017, 121, 8965–8974. [Google Scholar] [CrossRef] [PubMed]
- Chomicz, L.; Zdrowowicz, M.; Kasprzykowski, F.; Rak, J.; Buonaugurio, A.; Wang, Y.; Bowen, K.H. How to Find Out Whether a 5-Substituted Uracil Could Be a Potential DNA Radiosensitizer. J. Phys. Chem. Lett. 2013, 4, 2853–2857. [Google Scholar] [CrossRef]
- Spisz, P.; Kozak, W.; Chomicz-Mańka, L.; Makurat, S.; Falkiewicz, K.; Sikorski, A.; Czaja, A.; Rak, J.; Zdrowowicz, M. 5-(N-Trifluoromethylcarboxy)Aminouracil as a Potential DNA Radiosensitizer and Its Radiochemical Conversion into N-Uracil-5-Yloxamic Acid. Int. J. Mol. Sci. 2020, 21, 6352. [Google Scholar] [CrossRef]
- Zdrowowicz, M.; Chomicz, L.; Żyndul, M.; Wityk, P.; Rak, J.; Wiegand, T.J.; Hanson, C.G.; Adhikary, A.; Sevilla, M.D. 5-Thiocyanato-2′-Deoxyuridine as a Possible Radiosensitizer: Electron-Induced Formation of Uracil-C5-Thiyl Radical and Its Dimerization. Phys. Chem. Chem. Phys. 2015, 17, 16907–16916. [Google Scholar] [CrossRef]
- Sosnowska, M.; Makurat, S.; Zdrowowicz, M.; Rak, J. 5-Selenocyanatouracil: A Potential Hypoxic Radiosensitizer. Electron Attachment Induced Formation of Selenium Centered Radical. J. Phys. Chem. B 2017, 121, 6139–6147. [Google Scholar] [CrossRef]
- Zdrowowicz, M.; Datta, M.; Rychłowski, M.; Rak, J. Radiosensitization of PC3 Prostate Cancer Cells by 5-Thiocyanato-2′-Deoxyuridine. Cancers 2022, 14, 2035. [Google Scholar] [CrossRef]
- Pałasz, A.; Cież, D. In Search of Uracil Derivatives as Bioactive Agents. Uracils and Fused Uracils: Synthesis, Biological Activity and Applications. Eur. J. Med. Chem. 2015, 97, 582–611. [Google Scholar] [CrossRef]
- Tsuda, H.; Ohshima, Y.; Nomoto, H.; Fujita, K.; Matsuda, E.; Iigo, M.; Takasuka, N.; Moore, M.A. Cancer Prevention by Natural Compounds. Drug Metab. Pharmacokinet. 2004, 19, 245–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, K. Ferrocenyl-Nucleobase Complexes: Synthesis, Chemistry and Applications. Coord. Chem. Rev. 2016, 317, 132–156. [Google Scholar] [CrossRef]
- Raczyńska, E.D. Quantum-Chemical Studies on the Favored and Rare Isomers of Isocytosine. Comput. Theor. Chem. 2017, 1121, 58–67. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Kamińska, B. Variations of the Tautomeric Preferences and π-Electron Delocalization for the Neutral and Redox Forms of Purine When Proceeding from the Gas Phase (DFT) to Water (PCM). J. Mol. Model. 2013, 19, 3947–3960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raczyńska, E.D.; Makowski, M.; Hallmann, M.; Kamińska, B. Geometric and Energetic Consequences of Prototropy for Adenine and Its Structural Models—A Review. RSC Adv. 2015, 5, 36587–36604. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Wu, D.-Y.; Tian, Z.-Q. Theoretical Investigation on the Substituent Effect of Halogen Atoms at the C 8 Position of Adenine: Relative Stability, Vibrational Frequencies, and Raman Spectra of Tautomers. J. Phys. Chem. A 2016, 120, 4049–4058. [Google Scholar] [CrossRef]
- Laxer, A.; Major, D.T.; Gottlieb, H.E.; Fischer, B. (15 N5)-Labeled Adenine Derivatives: Synthesis and Studies of Tautomerism by 15N NMR Spectroscopy and Theoretical Calculations. J. Org. Chem. 2001, 66, 5463–5481. [Google Scholar] [CrossRef]
- Lippert, B.; Gupta, D. Promotion of Rare Nucleobase Tautomers by Metal Binding. Dalton Trans. 2009, 24, 4619–4634. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C.; Kamińska, B.; Igielska, M.; Kurpiewski, J.; Juras, W. Purine Tautomeric Preferences and Bond-Length Alternation in Relation with Protonation-Deprotonation and Alkali Metal Cationization. J. Mol. Model. 2020, 26, 93. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Fedeles, B.I.; Essigmann, J.M. Role of Tautomerism in RNA Biochemistry. RNA 2015, 21, 1–13. [Google Scholar] [CrossRef]
- Khuu, P.; Ho, P.S. A Rare Nucleotide Base Tautomer in the Structure of an Asymmetric DNA Junction. Biochemistry 2009, 48, 7824–7832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brovarets’, O.O.; Hovorun, D.M. Renaissance of the Tautomeric Hypothesis of the Spontaneous Point Mutations in DNA: New Ideas and Computational Approaches. In Mitochondrial DNA-New Insights; Seligmann, H., Ed.; InTech: London, UK, 2018. [Google Scholar]
- Srivastava, R. The Role of Proton Transfer on Mutations. Front. Chem. 2019, 7, 536. [Google Scholar] [CrossRef] [PubMed]
- Slocombe, L.; Al-Khalili, J.S.; Sacchi, M. Quantum and Classical Effects in DNA Point Mutations: Watson–Crick Tautomerism in AT and GC Base Pairs. Phys. Chem. Chem. Phys. 2021, 23, 4141–4150. [Google Scholar] [CrossRef] [PubMed]
- Jalbout, A.F.; Trzaskowski, B.; Xia, Y.; Li, Y.; Hu, X.; Li, H.; El-Nahas, A.; Adamowicz, L. Structures, Stabilities and Tautomerizations of Uracil and Diphosphouracil Tautomers. Chem. Phys. 2007, 332, 152–161. [Google Scholar] [CrossRef]
- Beak, P.; White, J.M. Relative Enthalpies of 1,3-Dimethyl-2,4-Pyrimidinedione, 2,4-Dimethoxypyrimidine, and 4-Methoxy-1-Methyl-1-2-Pyrimidinone: Estimation of the Relative Stabilities of Two Protomers of Uracil. J. Am. Chem. Soc. 1982, 104, 7073–7077. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Tamura, T.; Fujii, M.; Ito, M. Keto-Enol Tautomer of Uracil and Thymine. J. Phys. Chem. 1988, 92, 1760–1765. [Google Scholar] [CrossRef]
- Jezuita, A.; Wieczorkiewicz, P.A.; Szatylowicz, H.; Krygowski, T.M. Effect of the Solvent and Substituent on Tautomeric Preferences of Amine-Adenine Tautomers. ACS Omega 2021, 6, 18890–18903. [Google Scholar] [CrossRef]
- Jezuita, A.; Wieczorkiewicz, P.A.; Szatylowicz, H.; Krygowski, T.M. Solvent Effect on the Stability and Reverse Substituent Effect in Nitropurine Tautomers. Symmetry 2021, 13, 1223. [Google Scholar] [CrossRef]
- Wieczorkiewicz, P.A.; Szatylowicz, H.; Krygowski, T.M. Energetic and Geometric Characteristics of the Substituents: Part 2: The Case of NO2, Cl, and NH2 Groups in Their Mono-Substituted Derivatives of Simple Nitrogen Heterocycles. Molecules 2021, 26, 6543. [Google Scholar] [CrossRef]
- Becke, A.D. Perspective: Fifty Years of Density-Functional Theory in Chemical Physics. J. Chem. Phys. 2014, 140, 18A301. [Google Scholar] [CrossRef]
- Jones, R.O. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87, 897–923. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; J. Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Marek, P.H.; Szatylowicz, H.; Krygowski, T.M. Stacking of Nucleic Acid Bases: Optimization of the Computational Approach—The Case of Adenine Dimers. Struct. Chem. 2019, 30, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Sadlej-Sosnowska, N. On the Way to Physical Interpretation of Hammett Constants: How Substituent Active Space Impacts on Acidity and Electron Distribution in p-Substituted Benzoic Acid Molecules. Polish J. Chem. 2007, 81, 1123–1134. [Google Scholar]
- Sadlej-Sosnowska, N. Substituent Active Region—A Gate for Communication of Substituent Charge with the Rest of a Molecule: Monosubstituted Benzenes. Chem. Phys. Lett. 2007, 447, 192–196. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theoret. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. Theochem 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Romero, E.E.; Hernandez, F.E. Solvent Effect on the Intermolecular Proton Transfer of the Watson and Crick Guanine–Cytosine and Adenine–Thymine Base Pairs: A Polarizable Continuum Model Study. Phys. Chem. Chem. Phys. 2018, 20, 1198–1209. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Todd, A.; Keith, T.K. Gristmill Software, Overland Park KS, AIMAll (Version 19.10.12), USA. 2019. Available online: Aim.tkgristmill.com (accessed on 23 September 2022).
- Afonin, A.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the Energy of Intramolecular Hydrogen Bonds from 1H NMR and QTAIM Calculations. Org. Biomol. Chem. 2016, 14, 11199–11211. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Jabłoński, M. A Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions. Molecules 2020, 25, 5512. [Google Scholar] [CrossRef]
- Desiraju, G.R. A Bond by Any Other Name. Angew. Chem. Int. Ed. 2011, 50, 52–59. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formamide | Water | DMSO | Ethanol | Pyridine | THF | o-Cresol | Chloroform | Toluene | Gas Phase |
|---|---|---|---|---|---|---|---|---|---|
| 108.94 | 78.36 | 46.83 | 24.85 | 12.98 | 7.43 | 6.76 | 4.71 | 2.37 | 1.00 |
| Taut. | 5-NH2 | Δ | 5-NO2 | Δ | 5-NO2 (90°) | Δ | 6-NH2 | Δ | 6-NO2 | Δ | 6-NO2 (90°) | Δ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| u1 | 0.067 | 0.011 | −0.163 | −0.070 | −0.121 | −0.045 | 0.215 | 0.102 | −0.001 | 0.002 | 0.001 | 0.003 |
| u2 | 0.078 | 0.003 | −0.166 | −0.082 | −0.127 | −0.053 | 0.201 | 0.052 | −0.052 | −0.035 | −0.030 | −0.034 |
| u3 | 0.057 | 0.032 | −0.180 | −0.047 | −0.136 | −0.026 | 0.234 | 0.103 | 0.004 | 0.003 | 0.013 | 0.005 |
| u4 | 0.068 | 0.025 | −0.173 | −0.055 | −0.138 | −0.037 | 0.206 | 0.048 | −0.042 | −0.036 | −0.021 | −0.034 |
| u5 | 0.068 | 0.025 | −0.172 | −0.055 | −0.139 | −0.037 | 0.214 | 0.038 | −0.035 | −0.044 | −0.015 | −0.041 |
| u6 | 0.042 | 0.055 | −0.136 | −0.018 | −0.157 | −0.009 | 0.208 | 0.045 | −0.036 | −0.040 | −0.018 | −0.037 |
| range | 0.037 | 0.052 | 0.045 | 0.064 | 0.036 | 0.044 | 0.032 | 0.065 | 0.056 | 0.047 | 0.043 | 0.046 |
| Tautomer | 5-NH2 | 5-NO2 | 5-NO2 (90°) | 6-NO2 | 6-NO2 (90°) | 6-NH2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a | R2 | a | R2 | a | R2 | a | R2 | a | R2 | a | R2 | |
| u1 | −0.011 | 0.993 | 0.071 | 0.957 | 0.044 | 0.996 | −0.002 | 0.772 | −0.002 | 0.683 | −0.103 | 0.952 |
| u2 | −0.003 | 0.831 | 0.084 | 0.961 | 0.052 | 0.996 | 0.036 | 0.981 | 0.034 | 0.998 | −0.053 | 0.962 |
| u3 | −0.032 | 0.974 | 0.047 | 0.969 | 0.026 | 1.000 | −0.003 | 0.654 | −0.004 | 0.838 | −0.104 | 0.950 |
| u4 | −0.025 | 0.976 | 0.055 | 0.967 | 0.037 | 0.998 | 0.036 | 0.981 | 0.034 | 0.999 | −0.049 | 0.965 |
| u5 | −0.025 | 0.974 | 0.056 | 0.968 | 0.037 | 0.998 | 0.045 | 0.972 | 0.041 | 0.997 | −0.039 | 0.979 |
| u6 | −0.056 | 0.968 | 0.018 | 0.983 | 0.010 | 0.989 | 0.040 | 0.974 | 0.037 | 0.998 | −0.046 | 0.963 |
| Rotational | Afonin | |
|---|---|---|
| u6 5-NO2 | −7.49 | −7.55 |
| u6 5-NH2 | −3.36 * | −2.61 |
| Taut. | H | 5-NH2 | Δ | 5-NO2 | Δ | 5-NO2 (90°) | Δ | 6-NH2 | Δ | 6-NO2 | Δ | 6-NO2 (90°) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| u1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| u2 | 11.1 | 9.5 | 2.7 | 10.1 | 2.1 | 9.1 | 2.5 | 6.6 | 3.8 | 9.9 | 1.1 | 5.4 |
| u3 | 11.4 | 14.6 | −2.2 | 10.2 | 1.1 | 11.8 | 0.2 | 10.3 | −0.6 | 11.4 | 0.4 | 11.1 |
| u4 | 13.2 | 14.4 | 3.6 | 11.8 | 5.9 | 11.6 | 5.6 | 8.1 | 6.5 | 11.7 | 1.6 | 7.2 |
| u5 | 14.2 | 15.5 | 2.9 | 12.9 | 5.0 | 12.6 | 4.8 | 8.5 | 6.4 | 12.9 | 0.5 | 8.1 |
| u6 | 17.9 | 18.1 | 2.1 | 5.4 | 7.3 | 15.8 | 4.8 | 12.4 | 3.9 | 16.2 | 1.3 | 11.7 |
| Tautomer | 5-NH2 | 5-NO2 | 5-NO2 (90°) | 6-NO2 | 6-NO2 (90°) | 6-NH2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a | μ | a | μ | a | μ | a | μ | a | μ | a | μ | |
| u1 | 0.0180 | 4.5 | 0.0234 | 4.9 | 0.0207 | 4.7 | 0.0152 | 0.5 | 0.0164 | 1.1 | 0.0231 | 6.2 |
| u2 | 0.0135 | 2.3 | 0.0200 | 7.0 | 0.0168 | 6.3 | 0.0170 | 4.5 | 0.0155 | 4.2 | 0.0168 | 4.8 |
| u3 | 0.0216 | 5.9 | 0.0217 | 2.2 | 0.0203 | 2.3 | 0.0159 | 2.8 | 0.0167 | 2.6 | 0.0242 | 6.6 |
| u4 | 0.0120 | 2.6 | 0.0137 | 3.8 | 0.0120 | 2.8 | 0.0126 | 3.3 | 0.0112 | 2.8 | 0.0125 | 3.2 |
| u5 | 0.0133 | 3.3 | 0.0152 | 4.7 | 0.0132 | 3.7 | 0.0144 | 5.8 | 0.0125 | 5.4 | 0.0127 | 1.9 |
| u6 | 0.0145 | 3.8 | 0.0115 | 1.8 | 0.0130 | 1.3 | 0.0173 | 4.7 | 0.0157 | 4.5 | 0.0168 | 4.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieczorkiewicz, P.A.; Krygowski, T.M.; Szatylowicz, H. Intramolecular Interactions in Derivatives of Uracil Tautomers. Molecules 2022, 27, 7240. https://doi.org/10.3390/molecules27217240
Wieczorkiewicz PA, Krygowski TM, Szatylowicz H. Intramolecular Interactions in Derivatives of Uracil Tautomers. Molecules. 2022; 27(21):7240. https://doi.org/10.3390/molecules27217240
Chicago/Turabian StyleWieczorkiewicz, Paweł A., Tadeusz M. Krygowski, and Halina Szatylowicz. 2022. "Intramolecular Interactions in Derivatives of Uracil Tautomers" Molecules 27, no. 21: 7240. https://doi.org/10.3390/molecules27217240
APA StyleWieczorkiewicz, P. A., Krygowski, T. M., & Szatylowicz, H. (2022). Intramolecular Interactions in Derivatives of Uracil Tautomers. Molecules, 27(21), 7240. https://doi.org/10.3390/molecules27217240