
Association Complexes of Calix[6]arenes with Amino Acids Explained by Energy-Partitioning Methods


Abstract
:1. Introduction
2. Methodology
2.1. Geometry Optimization–Calixarenes
2.2. Geometry Optimization–Complexes
2.3. Energetics
2.4. Molecular Properties
2.5. Energy Partitioning
3. Results and Discussion
3.1. Pristine Calixarenes–Geometries
3.2. Pristine calixarenes–IR Spectra
3.3. Complexes with Amino Acids–Geometries
3.3.1. Dihedral Angles of Calixarenes
3.3.2. Binding Sites
3.3.3. Example: Complexes with Ala
3.4. Stability of Complexes with Amino Acids
3.5. Complexes with Amino Acids–Interaction Energies
3.6. Complexes with Amino Acids–IR Spectra
3.7. Pristine Calixarenes–Energy Partitioning
3.8. Pristine Calixarenes–Molecular Properties
3.9. Complexes with Amino Acids–Energy Partitioning with SSMF and F-SAPT
3.9.1. Special Case: SSMF3 and F-SAPT Partitioning Analyses for Complexes of CX⋯Gly
3.9.2. Special Case: SSMF3 and F-SAPT Partitioning Analyses for Complexes of BCX⋯Gly
3.9.3. Special Cases: Analysis of pc- CX Inclusion Complexes
3.9.4. Types of Noncovalent Bonding in Calixarene-Amino Acid Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| al | 1,2,3-alternate |
| BCX | hexa-p-tert-butylcalix[6]arene |
| CBS | Complete Basis Set |
| CCSD(T) | coupled cluster singles and doubles with perturbative triples correction |
| CSD | Cambridge Structural Database |
| CX | calix[6]arene |
| DF | density fitting |
| DFT | Density-Functional Theory |
| DFT+D | Density-Functional Theory with dispersion correction |
| FF | Finite-field |
| F-SAPT | Functional-group SAPT |
| I-SAPT | Intramolecular SAPT |
| HF | Hartree-Fock |
| MP2 | Møller-Plesset theory to the second order |
| pc | pinched-cone |
| SAPT | Symmetry-Adapted Perturbation Theory |
| SMFA | Systematic Molecular Fragmentation by Annihilation |
| SSMF | Symmetrized Systematic Molecular Fragmentation |
| SCS-MP2 | spin-component-scaled MP2 |
| wc | winged-cone |
| ZPVE | zero-point vibrational energy |
References
- Da Silva, E.; Lazar, A.; Coleman, A. Biopharmaceutical applications of calixarenes. J. Drug Deliv. Sci. Technol. 2004, 14, 3–20. [Google Scholar] [CrossRef]
- Guo, D.S.; Liu, Y. Supramolecular chemistry of p-sulfonatocalix[n]arenes and its biological applications. Accounts Chem. Res. 2014, 47, 1925–1934. [Google Scholar] [CrossRef]
- Yousaf, A.; Hamid, S.A.; Bunnori, N.M.; Ishola, A.A. Applications of calixarenes in cancer chemotherapy: Facts and perspectives. Drug Des. Dev. Ther. 2015, 9, 2831–2838. [Google Scholar] [CrossRef] [Green Version]
- Deraedt, C.; Astruc, D. Supramolecular nanoreactors for catalysis. Coord. Chem. Rev. 2016, 324, 106–122. [Google Scholar] [CrossRef]
- Mammino, L. Bowl-shaped structures from acylphloroglucinols: An ab initio and DFT study. Mol. Phys. 2017, 115, 2254–2266. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Olmedo-Romero, A.; Cruz-Flores, E.; Petrar, P.; Kunsagi-Mate, S.; Barroso-Flores, J. Calix[n]arene-based drug carriers: A DFT study of their electronic interactions with a chemotherapeutic agent used against leukemia. Comput. Theor. Chem. 2014, 1035, 84–91. [Google Scholar] [CrossRef]
- An, L.; Wang, J.W.; Liu, J.D.; Zhao, Z.M.; Song, Y.J. Design, Preparation, and Characterization of Novel Calix[4]arene Bioactive Carrier for Antitumor Drug Delivery. Front. Chem. 2019, 7, 732. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.T.; Li, Y.; Duan, X.; Wang, X.; Qi, C.; Lam, J.W.Y.; Ding, D.; Tang, B.Z. Substitution Activated Precise Phototheranostics through Supramolecular Assembly of AIEgen and Calixarene. J. Am. Chem. Soc. 2020, 142, 15966–15974. [Google Scholar] [CrossRef]
- Cacciapaglia, R.; Stefano, S.D.; Mandolini, L.; Salvio, R. Reactivity of carbonyl and phosphoryl groups at calixarenes. Supramol. Chem. 2013, 25, 537–554. [Google Scholar] [CrossRef] [Green Version]
- Rebilly, J.N.; Reinaud, O. Calixarenes and resorcinarenes as scaffolds for supramolecular metallo-enzyme mimicry. Supramol. Chem. 2014, 26, 454–479. [Google Scholar] [CrossRef]
- Hennrich, G.; Murillo, M.T.; Prados, P.; Song, K.; Asselberghs, I.; Clays, K.; Persoons, A.; Benet-Buchholz, J.; de Mendoza, J. Tetraalkynyl calix[4]arenes with advanced NLO properties. Chem. Commun. 2005, 2747–2749. [Google Scholar] [CrossRef]
- Pichierri, F. Cs+–π interactions and the design of macrocycles for the capture of environmental radiocesium (Cs-137): DFT, QTAIM, and CSD studies. Theor. Chem. Accounts 2018, 137, 118. [Google Scholar] [CrossRef]
- Chen, X.; Häkkinen, H. Protected but Accessible: Oxygen Activation by a Calixarene-Stabilized Undecagold Cluster. J. Am. Chem. Soc. 2013, 135, 12944–12947. [Google Scholar] [CrossRef]
- Ocak, Ü.; Ocak, M.; Bartsch, R.A. Calixarenes with dansyl groups as potential chemosensors. Inorganica Chim. Acta 2012, 381, 44–57. [Google Scholar] [CrossRef]
- Deska, M.; Dondela, B.; Sliwa, W. Selected applications of calixarene derivatives. Arkivoc 2015, 2015, 393–416. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Chawla, S.; Zou, M.C. Calixarenes based materials for gas sensing applications: A review. J. Incl. Phenom. Macrocycl. Chem. 2017, 88, 129–158. [Google Scholar] [CrossRef]
- Harris, S.J. Calixarene-Based Compounds Having Antibacterial, Antifungal, Anticancer-Hiv Activity. Patent WO 95/19974, 27 July 1995. [Google Scholar]
- Coleman, W.A.; Baggetto, L.G.; Lazar, A.N.; Michaud, M.H.; Magnard, S. Calixarene Derivatives as Anticancer Agent. U.S. Patent US2010/0056482A1, 4 March 2010. [Google Scholar]
- Naseer, M.M.; Ahmed, M.; Hameed, S. Functionalized calix[4]arenes as potential therapeutic agents. Chem. Biol. Drug Des. 2017, 89, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Pur, F.N.; Dilmaghani, K.A. Calixpenams: Synthesis, characterization, and biological evaluation of penicillins V and X clustered by calixarene scaffold. Turk. J. Chem. 2014, 38, 288–296. [Google Scholar] [CrossRef]
- Ben Salem, A.; Sautrey, G.; Fontanay, S.; Duval, R.E.; Regnouf-de Vains, J.B. Molecular drug-organiser: Synthesis, characterization and biological evaluation of penicillin V and/or nalidixic acid calixarene-based podands. Bioorg. Med. Chem. 2011, 19, 7534–7540. [Google Scholar] [CrossRef]
- Casnati, A.; Fabbi, M.; Pelizzi, N.; Pochini, A.; Sansone, F.; Unguro, R.; Di Modugno, E.; Tarzia, G. Synthesis, antimicrobial activity and binding properties of calix[4]arene based vancomycin mimics. Bioorg. Med. Chem. Lett. 1996, 6, 2699–2704. [Google Scholar] [CrossRef]
- Boukerb, A.M.; Rousset, A.; Galanos, N.; Méar, J.B.; Thépaut, M.; Grandjean, T.; Gillon, E.; Cecioni, S.; Abderrahmen, C.; Faure, K.; et al. Antiadhesive Properties of Glycoclusters against Pseudomonas aeruginosa Lung Infection. J. Med. Chem. 2014, 57, 10275–10289. [Google Scholar] [CrossRef] [PubMed]
- Mourer, M.; Dibama, H.M.; Fontanay, S.; Grare, M.; Duval, R.E.; Finance, C.; Regnouf-de Vains, J.B. p-Guanidinoethyl calixarene and parent phenol derivatives exhibiting antibacterial activities. Synthesis and biological evaluation. Bioorg. Med. Chem. 2009, 17, 5496–5509. [Google Scholar] [CrossRef]
- Shurpik, D.N.; Padnya, P.L.; Stoikov, I.I.; Cragg, P.J. Antimicrobial Activity of Calixarenes and Related Macrocycles. Molecules 2020, 25, 5145. [Google Scholar] [CrossRef]
- Neagu, M.; Ion, R.M.; Manda, G.; Constantin, C.; Radu, E.; Cristu, Z. Antitumoral Effect of Calixarenes in Experimental Photodynamic Therapy with K562 Tumor Cell Line. Rom. J. Biochem. 2010, 47, 17–35. [Google Scholar]
- Ugozzoli, F.; Andreetti, G.D. Symbolic Representation of the Molecular Conformation of Calixarenes. J. Incl. Phenom. Mol. Recognit. Chem. 1992, 13, 337–348. [Google Scholar] [CrossRef]
- Matthews, S.E.; Cecioni, S.; O’Brien, J.E.; MacDonald, C.J.; Hughes, D.L.; Jones, G.A.; Ashworth, S.H.; Vidal, S. Fixing the Conformation of Calix[4]arenes: When Are Three Carbons Not Enough? Chem. A Eur. J. 2018, 24, 4436–4444. [Google Scholar] [CrossRef] [Green Version]
- Gassoumi, B.; Chaabene, M.; Ghalla, H.; Chaabane, R.B. Physicochemical properties of the three cavity form of calix[n = 4, 6, 8] aren molecules: DFT investigation. Theor. Chem. Accounts 2019, 138, 58. [Google Scholar] [CrossRef]
- Kumar, S.; Kaur, J.; Verma, A.; Mukesh; Kumar, A.; Dominic, S. Influence of polyether chain on the non-covalent interactions and stability of the conformers of calix[4]crown ethers. J. Incl. Phenom. Macrocycl. Chem. 2018, 91, 81–93. [Google Scholar] [CrossRef]
- Özkinali, S.; Karayel, A. Synthesis, characterization, conformational equilibrium and intramolecular hydrogen bond analysis of Novel Azocalix[4]arenes including acryloyl moiety using DFT studies. J. Mol. Struct. 2019, 1176, 303–313. [Google Scholar] [CrossRef]
- Furer, V.L.; Potapova, L.I.; Vatsouro, I.M.; Kovalev, V.V.; Shokova, E.A.; Kovalenko, V.I. Investigation of the conformation and hydrogen bonds in adamantylthiacalix[4]arene by IR spectroscopy and DFT. J. Mol. Struct. 2018, 1171, 207–213. [Google Scholar] [CrossRef]
- Malinska, M. Insights into molecular recognition from the crystal structures of p-tert-butylcalix[6]arene complexed with different solvents. IUCrJ 2022, 9, 55–64. [Google Scholar] [CrossRef]
- Furer, V.L.; Potapova, L.I.; Vatsouro, I.M.; Kovalev, V.V.; Shokova, E.A.; Kovalenko, V.I. Study of conformation and hydrogen bonds in the p-1-adamantylcalix[8]arene by IR spectroscopy and DFT. J. Incl. Phenom. Macrocycl. Chem. 2019, 95, 63–71. [Google Scholar] [CrossRef]
- Kieliszek, A.; Malinska, M. Conformations of p-tert-Butylcalix[8]arene in Solvated Crystal Structures. Cryst. Growth Des. 2021, 21, 6862–6871. [Google Scholar] [CrossRef]
- Puchta, R.; Clark, T.; Bauer, W. The formation of endo-complexes between calixarenes and amines–a reinvestigation. J. Mol. Model. 2006, 12, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.M.; Yu, C.J.; Kim, J.S.; Kim, S.U. Ab initio design of drug carriers for zoledronate guest molecule using phosphonated and sulfonated calix[4]arene and calix[4]resorcinarene host molecules. J. Mater. Sci. 2018, 53, 5125–5139. [Google Scholar] [CrossRef] [Green Version]
- Kryuchkova, N.A.; Kostin, G.A.; Korotaev, E.V.; Kalinkin, A.V. XPS and quantum chemical investigation of electronic structure of Co complexes with calix[4]arenes modified by R2PO groups in upper or lower rim. J. Electron Spectrosc. Relat. Phenom. 2018, 229, 114–123. [Google Scholar] [CrossRef]
- Murphy, P.; Dalgarno, S.J.; Paterson, M.J. Systematic Study of the Effect of Lower-Rim Methylation on Small Guest Binding within the Host Cavity of Calix[4]arene. J. Phys. Chem. A 2017, 121, 7986–7992. [Google Scholar] [CrossRef]
- Sharafdini, R.; Mosaddeghi, H. Inhibition of Insulin Amyloid Fibrillation by Salvianolic Acids and Calix[n]arenes: Molecular Docking Insight. J. Comput. Biophys. Chem. 2021, 20, 539–555. [Google Scholar] [CrossRef]
- Shinde, M.N.; Barooah, N.; Bhasikuttan, A.C.; Mohanty, J. Inhibition and disintegration of insulin amyloid fibrils: A facile supramolecular strategy with p-sulfonatocalixarenes. Chem. Commun. 2016, 52, 2992–2995. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, X.H.; Pan, Y.C.; Tian, H.W.; Hu, X.Y.; Guo, D.S. Inhibition of insulin fibrillation by amphiphilic sulfonatocalixarene. Chin. Chem. Lett. 2020, 31, 1873–1876. [Google Scholar] [CrossRef]
- Böhmer, V. Calixarenes, Macrocycles with (Almost) Unlimited Possibilities. Angew. Chem. Int. Ed. Engl. 1995, 34, 713–745. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Szalewicz, K. Symmetry-Adapted Perturbation Theory of Intermolecular Forces. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 254–272. [Google Scholar] [CrossRef]
- Parrish, R.M.; Sherrill, C.D. Spatial assignment of symmetry adapted perturbation theory interaction energy components: The atomic SAPT partition. J. Chem. Phys. 2014, 141, 044115. [Google Scholar] [CrossRef] [PubMed]
- Parrish, R.M.; Parker, T.M.; Sherrill, C.D. Chemical Assignment of Symmetry-Adapted Perturbation Theory Interaction Energy Components: The Functional-Group SAPT Partition. J. Chem. Theory Comput. 2014, 10, 4417–4431. [Google Scholar] [CrossRef]
- Collins, M.A.; Bettens, R.P. Energy-Based Molecular Fragmentation Methods. Chem. Rev. 2015, 115, 5607–5642. [Google Scholar] [CrossRef]
- Masoumifeshani, E.; Korona, T. Symmetrized Systematic Molecular Fragmentation Model and its Application for Molecular Properties. Comput. Theor. Chem. 2021, 1202, 113303. [Google Scholar] [CrossRef]
- Masoumifeshani, E.; Chojecki, M.; Korona, T. Electronic Correlation Contribution to the Intermolecular Interaction Energy from Symmetrized Systematic Molecular Fragmentation Model. Comput. Theor. Chem. 2022, 1211, 113684. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Izgorodina, E.I.; MacFarlane, D.R. Nature of Hydrogen Bonding in Charged Hydrogen-Bonded Complexes and Imidazolium-Based Ionic Liquids. J. Phys. Chem. B 2011, 115, 14659–14667. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Ilnicka, A.; Sadlej, J. Inverse hydrogen bond: Theoretical investigation on the nature of interaction and spectroscopic properties. Struct. Chem. 2012, 23, 1323–1332. [Google Scholar] [CrossRef] [Green Version]
- Smaga, A.; Sadlej, J. Computational study on interaction energy changes during double proton transfer process. Comput. Theor. Chem. 2012, 998, 120–128. [Google Scholar] [CrossRef]
- Jabłoński, M. Theoretical insight into the nature of the intermolecular charge-inverted hydrogen bond. Comput. Theor. Chem. 2012, 998, 39–45. [Google Scholar] [CrossRef]
- Gallardo, A.; Fanfrlík, J.; Hobza, P.; Jelinek, P. Nature of Binding in Planar Halogen-Benzene Assemblies and Their Possible Visualization in Scanning Probe Microscopy. J. Phys. Chem. C 2019, 123, 8379–8386. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Significance of hydrogen bonding and other noncovalent interactions in determining octahedral tilting in the CH3NH3PbI3 hybrid organic-inorganic halide perovskite solar cell semiconductor. Sci. Rep. 2019, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Jabłoński, M. Ten years of charge-inverted hydrogen bonds. Struct. Chem. 2020, 31, 61–80. [Google Scholar] [CrossRef]
- Korona, T.; Dodziuk, H. Small Molecules in C60 and C70: Which Complexes Could Be Stabilized? J. Chem. Theory Comput. 2011, 7, 1476–1483. [Google Scholar] [CrossRef]
- Pan, Y.C.; Hu, X.Y.; Guo, D.S. Biomedical Applications of Calixarenes: State of the Art and Perspectives. Angew. Chem. Int. Ed. 2021, 60, 2768–2794. [Google Scholar] [CrossRef]
- Arena, G.; Contino, A.; Gulino, F.G.; Magrì, A.; Sansone, F.; Sciotto, D.; Ungaro, R. Complexation of native L-α-aminoacids by water soluble calix [4] arenes. Tetrahedron Lett. 1999, 40, 1597–1600. [Google Scholar] [CrossRef]
- Douteau-Guével, N.; Coleman, A.W.; Morel, J.P.; Morel-Desrosiers, N. Complexation of basic amino acids by water-soluble calixarene sulphonates as a study of the possible mechanisms of recognition of calixarene sulphonates by proteins. J. Phys. Org. Chem. 1998, 11, 693–696. [Google Scholar] [CrossRef]
- Giuliani, M.; Morbioli, I.; Sansone, F.; Casnati, A. Moulding calixarenes for biomacromolecule targeting. Chem. Commun. 2015, 51, 14140–14159. [Google Scholar] [CrossRef] [PubMed]
- Antipin, I.S.; Stoikov, I.I.; Pinkhassik, E.M.; Fitseva, N.A.; Stibor, I.; Konovalov, A.I. Calix[4]arene based α-aminophosphonates: Novel carriers for zwitterionic amino acids transport. Tetrahedron Lett. 1997, 38, 5865–5868. [Google Scholar] [CrossRef]
- Selkti, M.; Coleman, A.W.; Nicolis, I.; Douteau-Guével, N.; Villain, F.; Tomas, A.; de Rango, C. The first example of a substrate spanning the calix[4]arene bilayer: The solid state complex of sulfonatocalix[4]arene with lysine. Chem. Commun. 2000, 161–162. [Google Scholar] [CrossRef]
- Buschmann, H.J.; Mutihac, L.; Jansen, K. Complexation of some amine compounds by macrocyclic receptors. J. Incl. Phenom. Macrocycl. Chem. 2001, 39, 1–11. [Google Scholar] [CrossRef]
- Atwood, J.L.; Ness, T.; Nichols, P.J.; Raston, C.L. Confinement of Amino Acids in Tetra-p-Sulfonated Calix[4]arene Bilayers. Cryst. Growth Des. 2002, 2, 171–176. [Google Scholar] [CrossRef]
- Hassen, W.M.; Martelet, C.; Davis, F.; Higson, S.P.; Abdelghani, A.; Helali, S.; Jaffrezic-Renault, N. Calix[4]arene based molecules for amino-acid detection. Sens. Actuators B Chem. 2007, 124, 38–45. [Google Scholar] [CrossRef]
- Mutihac, L.; Lee, J.H.; Kim, J.S.; Vicens, J. Recognition of amino acids by functionalized calixarenes. Chem. Soc. Rev. 2011, 40, 2777–2796. [Google Scholar] [CrossRef]
- Español, E.S.; Villamil, M.M. Calixarenes: Generalities and Their Role in Improving the Solubility, Biocompatibility, Stability, Bioavailability, Detection, and Transport of Biomolecules. Biomolecules 2019, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Parikh, J.; Bhatt, K.; Modi, K.; Patel, N.; Desai, A.; Kumar, S.; Mohan, B. A versatile enrichment of functionalized calixarene as a facile sensor for amino acids. Luminescence 2022, 37, 370–390. [Google Scholar] [CrossRef]
- Stone, M.M.; Franz, A.H.; Lebrilla, C.B. Non-covalent calixarene–amino acid complexes formed by MALDI-MS. J. Am. Soc. Mass Spectrom. 2002, 13, 964–974. [Google Scholar] [CrossRef]
- Oshima, T.; Inoue, K.; Furusaki, S.; Goto, M. Liquid membrane transport of amino acids by a calix[6]arene carboxylic acid derivative. J. Membr. Sci. 2003, 217, 87–97. [Google Scholar] [CrossRef]
- Douteau-Guével, N.; Coleman, A.W.; Morel, J.P.; Morel-Desrosiers, N. Complexation of the basic amino acids lysine and arginine by three sulfonatocalix[n]arenes (n = 4, 6 and 8) in water: Microcalorimetric determination of the Gibbs energies, enthalpies and entropies of complexation. J. Chem. Soc. Perkin Trans. 1999, 2, 629–634. [Google Scholar] [CrossRef]
- Oshima, T.; Goto, M.; Furusaki, S. Extraction Behavior of Amino Acids by Calix[6]arene Carboxylic Acid Derivatives. J. Incl. Phenom. Macrocycl. Chem. 2002, 43, 77–86. [Google Scholar] [CrossRef]
- Martins, J.N.; Lima, J.C.; Basílio, N. Selective Recognition of Amino Acids and Peptides by Small Supramolecular Receptors. Molecules 2021, 26, 106. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Dale, S.H.; Elsegood, M.R.; Redshaw, C. Polymorphism and Pseudopolymorphism in Calixarenes: Acetonitrile Clathrates of p-But-Calix[n]arenes (n= 6 and 8). CrystEngComm 2003, 5, 368–373. [Google Scholar] [CrossRef]
- Wolfgong, W.J.; Talafuse, L.K.; Smith, J.M.; Adams, M.J.; Adeogba, F.; Valenzuela, M.; Rodriguez, E.; Contreras, K.; Carter, D.M.; Bacchus, A.; et al. The Influence of Solvent of Crystallization upon the Solid-State Conformation of Calix[6]Arenes. Supramol. Chem. 1996, 7, 67–78. [Google Scholar] [CrossRef]
- Martins, F.T.; De Freitas Oliveira, B.G.; Sarotti, A.M.; De Fátima, Â. Winged-Cone Conformation in Hexa-p-tert-Butylcalix[6]Arene Driven by the Unusually Strong Guest Encapsulation. ACS Omega 2017, 2, 5315–5323. [Google Scholar] [CrossRef]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.P.; Ren, P.; Ponder, J.W. Tinker 8: Software Tools for Molecular Design. J. Chem. Theory Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary Basis Sets to Approximate Coulomb Potentials. Chem. Phys. Lett. 1995, 240, 283–290. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Garcia, J.; Podeszwa, R.; Szalewicz, K. SAPT codes for calculations of intermolecular interaction energies. J. Chem. Phys. 2020, 152, 184109. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Grimme, S. Improved second-order Møller–Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies. J. Chem. Phys. 2003, 118, 9095–9102. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef]
- Weigend, F.; Köhn, A.; Hättig, C. Efficient Use of the Correlation Consistent Basis Sets in Resolution of the Identity MP2 Calculations. J. Chem. Phys. 2002, 116, 3175–3183. [Google Scholar] [CrossRef]
- Weigend, F. A fully direct RI-HF algorithm: Implementation, optimised auxiliary basis sets, demonstration of accuracy and efficiency. Phys. Chem. Chem. Phys. 2002, 4, 4285–4291. [Google Scholar] [CrossRef]
- Available online: http://xxx.lanl.gov/abs/https://github.com/psi4/psi4/blob/master/psi4/share/psi4/basis/aug-cc-pvdz-jkfit.gbs (accessed on 22 December 2021).
- Parrish, R.M.; Burns, L.A.; Smith, D.G.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helgaker, T.; Klopper, W.; Koch, H.; Noga, J. Basis-Set Convergence of Correlated Calculations on Water. J. Chem. Phys. 1997, 106, 9639–9646. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Collins, M.A. Systematic fragmentation of large molecules by annihilation. Phys. Chem. Chem. Phys. 2012, 14, 7744–7751. [Google Scholar] [CrossRef]
- Collins, M.A.; Deev, V.A. Accuracy and efficiency of electronic energies from systematic molecular fragmentation. J. Chem. Phys. 2006, 125, 104104. [Google Scholar] [CrossRef]
- Addicoat, M.A.; Collins, M.A. Accurate treatment of nonbonded interactions within systematic molecular fragmentation. J. Chem. Phys. 2009, 131, 104103. [Google Scholar] [CrossRef] [Green Version]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; Lindh, R.; et al. MOLPRO, version 2020.1, a Package of ab Initio Programs. Available online: https://www.molpro.net (accessed on 15 September 2022).
- Korona, T.; Pflüger, K.; Werner, H.J. The effect of local approximations in coupled-cluster wave functions on dipole moments and static dipole polarisabilities. Phys. Chem. Chem. Phys. 2004, 6, 2059–2065. [Google Scholar] [CrossRef]
- Parrish, R.M.; Gonthier, J.F.; Corminbœuf, C.; Sherrill, C.D. Communication: Practical intramolecular symmetry adapted perturbation theory via Hartree-Fock embedding. J. Chem. Phys. 2015, 143, 051103. [Google Scholar] [CrossRef] [Green Version]
- Chojecki, M.; Rutkowska-Zbik, D.; Korona, T. On the applicability of functional-group symmetry-adapted perturbation theory and other partitioning models for chiral recognition - the case of popular drug molecules interacting with chiral phases. Phys. Chem. Chem. Phys. 2019, 21, 22491–22510. [Google Scholar] [CrossRef] [PubMed]
- Sirianni, D.A.; Zhu, X.; Sitkoff, D.F.; Cheney, D.L.; Sherrill, C.D. The influence of a solvent environment on direct non-covalent interactions between two molecules: A symmetry-adapted perturbation theory study of polarization tuning of π-π interactions by water. J. Chem. Phys. 2022, 156, 194306. [Google Scholar] [CrossRef] [PubMed]
- Masumian, E.; Daniel Boese, A. Intramolecular resonance-assisted hydrogen bonds: Insights from symmetry adapted perturbation theory. Chem. Phys. 2022, 557, 111474. [Google Scholar] [CrossRef]
- Muchowska, K.B.; Pascoe, D.J.; Borsley, S.; Smolyar, I.V.; Mati, I.K.; Adam, C.; Nichol, G.S.; Ling, K.B.; Cockroft, S.L. Reconciling Electrostatic and n →π* Orbital Contributions in Carbonyl Interactions. Angew. Chem. Int. Ed. 2020, 59, 14602–14608. [Google Scholar] [CrossRef]
- Li, C.; Liu, X.; Han, Y.; Guo, Q.; Yang, W.; Liu, Q.; Song, B.; Zheng, X.; Tao, S. Ultra-stable anti-counterfeiting materials inspired by water stains. Cell Rep. Phys. Sci. 2021, 2, 100571. [Google Scholar] [CrossRef]
- Cukras, J.; Sadlej, J. Towards quantum-chemical modeling of the activity of anesthetic compounds. Int. J. Mol. Sci. 2021, 22, 9272. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; et al. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef]
- Petković, M. O-H Stretch in Phenol and Its Hydrogen-Bonded Complexes: Band Position and Relaxation Pathways. J. Phys. Chem. A 2012, 116, 364–371. [Google Scholar] [CrossRef]
- Kalescky, R.; Zou, W.; Kraka, E.; Cremer, D. Local vibrational modes of the water dimer-Comparison of theory and experiment. Chem. Phys. Lett. 2012, 554, 243–247. [Google Scholar] [CrossRef]
- Malenov, D.P.; Janjić, G.V.; Veljković, D.Z.; Zarić, S.D. Mutual influence of parallel, CH/O, OH/π and lone pair/π interactions in water/benzene/water system. Comput. Theor. Chem. 2013, 1018, 59–65. [Google Scholar] [CrossRef]
- Podeszwa, R.; Bukowski, R.; Szalewicz, K. Potential Energy Surface for the Benzene Dimer and Perturbational Analysis of π-π Interactions. J. Phys. Chem. A 2006, 110, 10345–10354. [Google Scholar] [CrossRef] [PubMed]
- Fink, R.F. Why does MP2 work? J. Chem. Phys. 2016, 145, 184101. [Google Scholar] [CrossRef] [PubMed]
- Szabados, A. Theoretical interpretation of Grimme’s spin-component-scaled second order Møller-Plesset theory. J. Chem. Phys. 2006, 125, 214105. [Google Scholar] [CrossRef] [PubMed]
- Nekoei, A.R.; Vatanparast, M. π-Hydrogen bonding and aromaticity: A systematic interplay study. Phys. Chem. Chem. Phys. 2019, 21, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, I.; Lee, H.M.; Kim, K.S. Phenol vs Water Molecule Interacting with Various Molecules: σ-type, π-type, and χ-type Hydrogen Bonds, Interaction Energies, and Their Energy Components. J. Phys. Chem. A 2005, 109, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Defining the hydrogen bond: An account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef]
- Forbes, C.R.; Sinha, S.K.; Ganguly, H.K.; Bai, S.; Yap, G.P.A.; Patel, S.; Zondlo, N.J. Insights into Thiol-Aromatic Interactions: A Stereoelectronic Basis for S-H/π Interactions. J. Am. Chem. Soc. 2017, 139, 1842–1855. [Google Scholar] [CrossRef] [Green Version]
- Tafipolsky, M. Challenging dogmas: Hydrogen bond revisited. J. Phys. Chem. A 2016, 120, 4550–4559. [Google Scholar] [CrossRef]










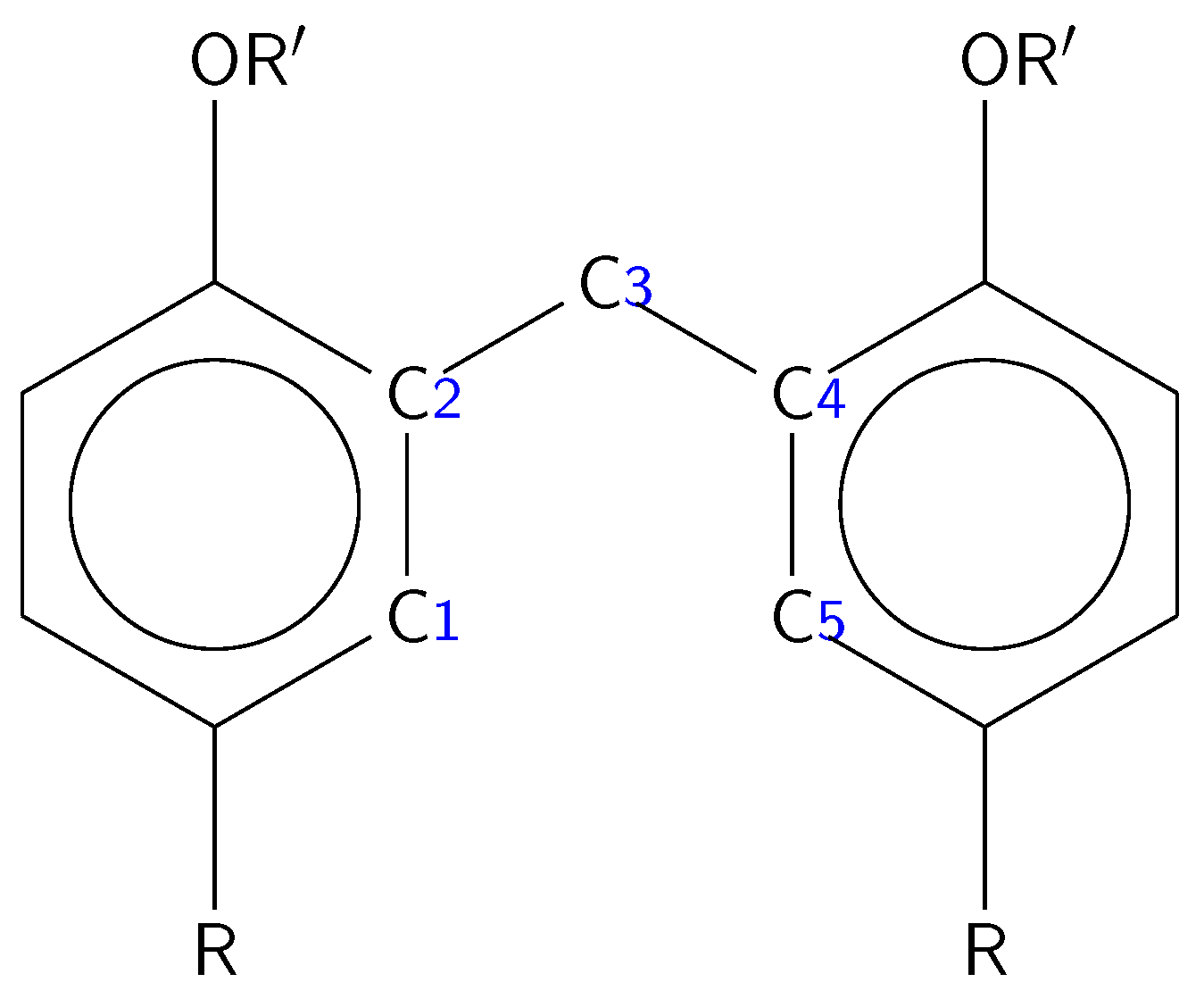{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
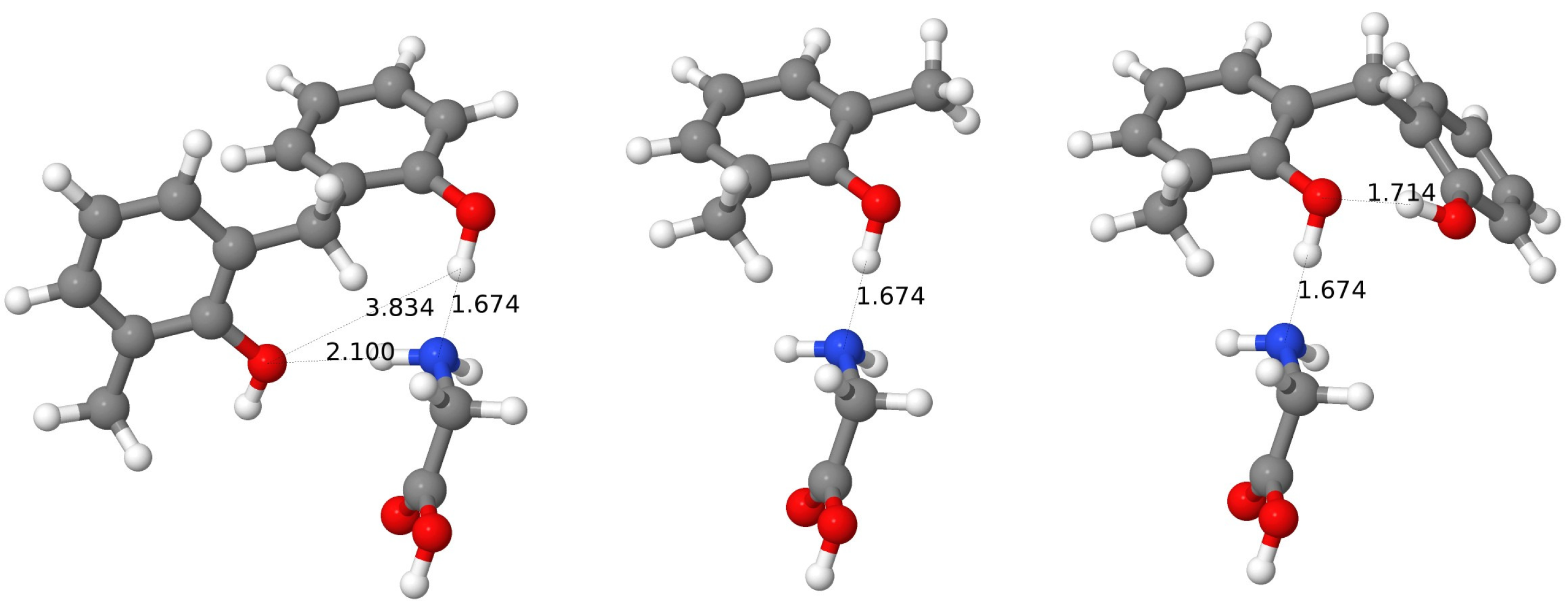{kind=link}
| CX Electronic Energy | CX Total Energy | BCX Electronic Energy | BCX Total Energy | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino Acid | al-CX | pc-CX | wc-CX | al-CX | pc-CX | wc-CX | al-BCX | pc-BCX | wc-BCX | al-BCX | pc-BCX | wc-BCX |
| - | 13.2 | 0.0 | 15.8 | 12.0 | 0.0 | 14.9 | 10.7 | 0.0 | 1.7 | 9.7 | 0.0 | 2.4 |
| Ala | 2.7 | 2.4 | 0.0 | 2.0 | 1.5 | 0.0 | 3.5 | 3.6 | 0.0 | 1.6 | 1.5 | 0.0 |
| Asn | 0.0 | 16.1 | 15.7 | 0.0 | 16.3 | 17.5 | 1.8 | 14.4 | 0.0 | 0.0 | 12.0 | 0.0 |
| AspH | 0.0 | 4.1 | 8.7 | 0.0 | 4.2 | 9.4 | 3.9 | 11.1 | 0.0 | 3.5 | 11.0 | 0.0 |
| Cys | 0.0 | 4.7 | 6.0 | 0.0 | 4.3 | 6.3 | 0.0 | 11.2 | 0.2 | 0.0 | 10.0 | 1.8 |
| Gln | 0.0 | 1.6 | 1.8 | 0.0 | 0.1 | 1.9 | 4.2 | 6.9 | 0.0 | 2.9 | 6.4 | 0.0 |
| GluH | 0.0 | 1.1 | 1.1 | 0.0 | 1.5 | 1.7 | 9.9 | 10.2 | 0.0 | 9.1 | 9.2 | 0.0 |
| Gly | 0.0 | 8.7 | 7.0 | 0.0 | 8.3 | 8.0 | 7.6 | 17.0 | 0.0 | 6.0 | 14.7 | 0.0 |
| HisD | 2.0 | 2.4 | 0.0 | 0.9 | 1.6 | 0.0 | 5.1 | 11.9 | 0.0 | 4.5 | 9.9 | 0.0 |
| HisE | 0.0 | 3.5 | 4.8 | 0.0 | 4.1 | 5.6 | 5.2 | 4.8 | 0.0 | 2.5 | 2.9 | 0.0 |
| Ile | 2.9 | 0.0 | 1.7 | 3.6 | 0.0 | 2.5 | 8.8 | 7.9 | 0.0 | 7.2 | 6.5 | 0.0 |
| Leu | 0.8 | 0.0 | 13.7 | 0.3 | 0.0 | 13.5 | 9.8 | 6.6 | 0.0 | 7.9 | 6.1 | 0.0 |
| Lys | 0.0 | 1.0 | 1.6 | 0.0 | 2.5 | 2.2 | 1.6 | 12.6 | 0.0 | 0.0 | 13.3 | 0.0 |
| Met | 0.0 | 5.9 | 4.8 | 0.0 | 5.1 | 4.6 | 2.4 | 1.1 | 0.0 | 0.4 | 0.0 | 0.2 |
| Phe | 1.9 | 0.0 | 8.6 | 2.5 | 0.0 | 8.8 | 1.6 | 0.1 | 0.0 | 1.1 | 0.0 | 0.3 |
| Pro | 2.1 | 0.0 | 9.5 | 0.7 | 0.0 | 9.4 | 4.5 | 0.0 | 3.8 | 4.2 | 0.0 | 4.8 |
| Ser | 0.0 | 11.8 | 9.4 | 0.0 | 9.8 | 9.8 | 2.1 | 13.5 | 0.0 | 0.0 | 10.4 | 0.0 |
| Thr | 0.0 | 7.6 | 11.2 | 0.0 | 5.9 | 11.2 | 4.4 | 11.0 | 0.0 | 3.1 | 8.1 | 0.0 |
| Trp | 0.1 | 0.0 | 3.7 | 0.8 | 0.0 | 4.5 | 13.7 | 8.2 | 0.0 | 10.8 | 6.7 | 0.0 |
| Tyr | 2.7 | 2.2 | 0.0 | 1.9 | 1.2 | 0.0 | 8.6 | 0.0 | 5.7 | 6.9 | 0.0 | 5.3 |
| Val | 7.9 | 0.0 | 8.4 | 7.3 | 0.0 | 8.8 | 7.4 | 5.4 | 0.0 | 5.6 | 3.9 | 0.0 |
| al−CX-Ala | −66.5 (−0.3) | 97.9 (0.6) | −46.2 (1.4) | 29.4 (0.7) | −56.6 (−1.4) | 8.3 (−2.9) | −0.1 (−423.9) | −48.4 (−1.9) | −40.7 (−1.0) | −29.6 (−1.1) |
| al−CX-Asn | −86.9 (0.4) | 110.4 (0.1) | −58.9 (−1.1) | 35.0 (−1.4) | −57.0 (−0.9) | 8.5 (−1.3) | −20.3 (−3.8) | −68.8 (−1.7) | −37.9 (−1.4) | −26.8 (−1.7) |
| al−CX-AspH | −67.5 (−0.3) | 94.5 (0.3) | −46.7 (−1.0) | 29.9 (−1.9) | −55.3 (−1.1) | 7.6 (−1.4) | −3.7 (−24.7) | −51.3 (−2.8) | −40.1 (−1.4) | −29.2 (−1.5) |
| al−CX-Cys | −84.5 (0.4) | 104.7 (0.1) | −51.6 (−0.4) | 31.4 (−1.1) | −58.8 (−1.5) | 8.6 (−2.7) | −15.8 (−0.8) | −66.0 (−1.1) | −38.4 (−1.8) | −26.9 (−2.2) |
| al−CX-Gln | −102.6 (0.0) | 128.1 (0.6) | −59.0 (−0.3) | 36.0 (−0.5) | −68.9 (−0.9) | 10.1 (−1.4) | −16.5 (−4.8) | −75.3 (−1.7) | −44.4 (−1.3) | −30.8 (−1.6) |
| al−CX-GluH | −59.4 (1.2) | 66.9 (0.1) | −30.4 (0.5) | 18.7 (0.0) | −54.0 (−1.8) | 6.5 (−3.5) | −11.8 (7.1) | −59.2 (0.2) | −34.9 (−2.0) | −24.3 (−2.7) |
| al−CX-Gly | −74.7 (0.5) | 91.3 (0.2) | −46.8 (−0.4) | 28.2 (−1.2) | −50.6 (−1.6) | 7.5 (−2.7) | −16.6 (−1.2) | −59.7 (−1.3) | −32.6 (−1.4) | −22.7 (−1.6) |
| al−CX-HisD | −57.1 (1.7) | 67.4 (0.2) | −30.6 (2.7) | 18.3 (0.3) | −54.6 (−1.4) | 6.4 (−3.2) | −10.2 (18.7) | −58.4 (2.3) | −38.4 (−1.1) | −27.4 (−1.4) |
| al−CX-HisE | −67.6 (−0.1) | 96.5 (0.4) | −49.0 (−1.4) | 30.6 (−0.8) | −63.7 (−0.8) | 8.7 (−0.8) | −5.2 (−26.7) | −60.1 (−3.1) | −45.9 (−1.3) | −33.8 (−1.4) |
| al−CX-Ile | −56.1 (−0.3) | 76.0 (0.1) | −32.6 (0.9) | 21.5 (−0.6) | −61.6 (−1.5) | 7.6 (−3.3) | 0.4 (−73.8) | −53.6 (−0.7) | −43.6 (−1.5) | −31.4 (−1.9) |
| al−CX-Leu | −58.3 (−0.1) | 83.7 (0.3) | −39.0 (−1.1) | 25.0 (−1.7) | −57.6 (−1.2) | 7.5 (−2.1) | −0.3 (−267.8) | −50.4 (−2.7) | −43.1 (−1.5) | −31.9 (−1.6) |
| al−CX-Lys | −75.7 (0.2) | 102.7 (0.4) | −48.3 (−0.6) | 29.8 (−1.5) | −63.0 (−0.9) | 8.7 (−2.0) | −7.9 (−11.3) | −62.2 (−2.1) | −44.7 (−1.1) | −32.5 (−1.3) |
| al−CX-Met | −58.3 (1.9) | 74.6 (0.1) | −33.7 (1.5) | 21.2 (−0.4) | −62.2 (−1.7) | 7.6 (−3.5) | −4.9 (38.4) | −59.5 (1.8) | −43.7 (−1.1) | −31.5 (−1.4) |
| al−CX-Phe | −52.8 (−0.2) | 68.5 (−0.1) | −29.5 (−0.4) | 18.2 (−0.4) | −53.5 (−1.0) | 6.4 (−2.0) | −4.5 (−11.2) | −51.6 (−1.8) | −36.4 (−1.5) | −26.0 (−1.7) |
| al−CX-Pro | −85.3 (−0.1) | 115.5 (0.3) | −59.8 (−1.5) | 36.2 (−1.1) | −58.3 (−1.1) | 8.8 (−1.7) | −13.8 (−11.9) | −63.3 (−3.4) | −39.5 (−1.2) | −28.2 (−1.4) |
| al−CX-Ser | −61.7 (−0.4) | 74.4 (0.4) | −31.1 (1.6) | 19.8 (0.8) | −53.1 (−1.8) | 6.8 (−4.3) | −6.2 (3.1) | −52.5 (−0.9) | −37.6 (−1.4) | −27.0 (−1.7) |
| al−CX-Thr | −62.4 (−0.2) | 72.3 (0.2) | −29.8 (1.2) | 18.9 (0.2) | −57.1 (−1.7) | 7.1 (−3.6) | −8.3 (3.8) | −58.3 (−0.7) | −41.0 (−1.4) | −29.5 (−1.7) |
| al−CX-Trp | −53.4 (−0.3) | 79.1 (0.3) | −34.7 (−0.4) | 24.3 (−0.7) | −67.7 (−0.7) | 8.3 (−1.4) | 5.7 (5.4) | −53.6 (−1.2) | −53.0 (−0.9) | −39.6 (−1.0) |
| al−CX-Tyr | −60.8 (−0.1) | 73.3 (0.4) | −31.7 (1.9) | 20.1 (0.8) | −60.1 (−1.6) | 7.4 (−3.5) | −6.9 (7.7) | −59.6 (−0.3) | −43.4 (−1.5) | −31.7 (−1.8) |
| al−CX-Val | −60.1 (−0.8) | 78.8 (0.2) | −36.6 (−0.2) | 22.9 (−1.7) | −57.7 (−1.2) | 7.3 (−2.9) | −5.4 (−11.2) | −55.9 (−2.0) | −42.3 (−1.7) | −31.0 (−2.0) |
| pc−CX-Ala | −55.1 (−0.2) | 62.7 (0.1) | −29.5 (2.9) | 18.2 (0.0) | −30.1 (−1.4) | 4.6 (−1.9) | −12.0 (2.8) | −37.6 (0.0) | −18.0 (−1.4) | −12.3 (−1.8) |
| pc−CX-Asn | −34.1 (1.2) | 37.7 (0.0) | −14.4 (13.3) | 10.1 (2.1) | −31.2 (−2.0) | 3.4 (−2.6) | −3.3 (65.5) | −31.1 (5.3) | −20.3 (−2.0) | −14.4 (−2.6) |
| pc−CX-AspH | −26.6 (2.1) | 32.8 (0.4) | −13.5 (13.9) | 8.7 (2.7) | −26.9 (−1.4) | 2.8 (−1.8) | −1.4 (147.3) | −25.6 (7.0) | −18.3 (−1.6) | −13.2 (−2.1) |
| pc−CX-Cys | −14.6 (2.7) | 19.6 (0.3) | −7.0 (14.8) | 5.0 (1.9) | −19.7 (−1.1) | 1.8 (−1.0) | 1.4 (−95.5) | −16.6 (6.7) | −14.4 (−1.1) | −10.5 (−1.3) |
| pc−CX-Gln | −50.1 (0.2) | 57.3 (0.3) | −26.1 (6.5) | 16.2 (1.3) | −35.4 (−1.5) | 4.5 (−1.8) | −9.7 (15.8) | −40.6 (2.7) | −21.1 (−1.2) | −14.4 (−1.7) |
| pc−CX-GluH | −41.8 (0.6) | 45.6 (0.2) | −20.7 (9.7) | 13.3 (2.0) | −29.8 (−1.6) | 3.6 (−2.1) | −8.5 (25.2) | −34.7 (5.0) | −17.8 (−2.0) | −12.3 (−2.6) |
| pc−CX-Gly | −33.4 (0.8) | 36.0 (0.0) | −15.9 (6.8) | 9.7 (0.9) | −20.1 (−1.4) | 2.6 (−1.9) | −8.0 (15.3) | −25.5 (3.9) | −11.4 (−1.3) | −7.7 (−1.9) |
| pc−CX-HisD | −39.8 (0.6) | 52.5 (0.1) | −22.6 (7.0) | 16.3 (1.2) | −47.0 (−1.1) | 5.4 (−1.1) | 2.0 (−84.7) | −39.6 (3.2) | −34.1 (−0.9) | −25.1 (−1.1) |
| pc−CX-HisE | −24.9 (0.2) | 36.2 (0.3) | −15.2 (7.0) | 11.8 (1.6) | −39.4 (−0.6) | 4.3 (−0.1) | 5.0 (−18.9) | −30.1 (2.3) | −30.3 (−0.7) | −22.6 (−0.7) |
| pc−CX-Ile | −22.2 (2.4) | 39.8 (0.8) | −14.7 (4.1) | 11.4 (1.2) | −41.2 (−1.3) | 4.4 (−1.8) | 11.1 (−6.8) | −25.7 (1.1) | −32.1 (−1.6) | −24.1 (−1.8) |
| pc−CX-Leu | −31.0 (1.3) | 43.8 (0.3) | −16.1 (10.4) | 11.8 (1.7) | −38.5 (−1.0) | 4.1 (−1.4) | 4.9 (−40.4) | −29.5 (5.5) | −28.1 (−0.8) | −20.7 (−1.0) |
| pc−CX-Lys | −19.3 (0.3) | 38.9 (0.2) | −12.4 (3.2) | 9.4 (0.5) | −43.2 (−1.0) | 4.5 (−1.4) | 13.4 (−3.5) | −25.2 (0.4) | −35.4 (−0.9) | −26.7 (−1.0) |
| pc−CX-Met | −17.4 (2.1) | 25.6 (0.5) | −8.6 (17.7) | 6.0 (2.2) | −26.9 (−0.9) | 2.3 (−0.2) | 3.5 (−49.2) | −21.0 (7.1) | −20.1 (−1.6) | −14.8 (−1.9) |
| pc−CX-Phe | −39.9 (0.9) | 56.2 (0.0) | −24.9 (5.8) | 18.4 (0.9) | −49.1 (−1.0) | 6.0 (−1.7) | 4.6 (−36.2) | −38.5 (3.3) | −36.4 (−0.8) | −27.1 (−1.0) |
| pc−CX-Pro | −20.1 (0.0) | 34.2 (0.2) | −12.8 (3.4) | 9.6 (0.7) | −39.0 (−0.9) | 4.1 (−0.9) | 7.7 (−4.9) | −27.2 (0.3) | −30.8 (−1.1) | −22.9 (−1.2) |
| pc−CX-Ser | −30.0 (1.4) | 34.7 (0.3) | −14.4 (11.5) | 9.6 (2.0) | −24.8 (−1.5) | 2.8 (−1.8) | −3.5 (57.0) | −25.6 (6.6) | −16.2 (−1.3) | −11.5 (−1.7) |
| pc−CX-Thr | −37.9 (0.7) | 39.6 (−0.3) | −17.7 (4.3) | 10.5 (0.2) | −22.5 (−1.3) | 2.8 (−1.7) | −10.4 (11.0) | −30.1 (3.0) | −12.1 (−1.4) | −8.0 (−2.0) |
| pc−CX-Trp | −35.8 (1.0) | 54.5 (0.0) | −22.2 (7.7) | 16.9 (1.1) | −55.7 (−0.9) | 6.2 (−1.1) | 8.9 (−22.9) | −40.6 (3.9) | −42.7 (−0.7) | −32.1 (−0.8) |
| pc−CX-Tyr | −17.3 (1.8) | 27.4 (0.1) | −10.8 (4.6) | 7.0 (1.2) | −31.4 (−0.8) | 3.1 (−0.7) | 4.0 (−18.4) | −24.3 (2.1) | −24.3 (−0.5) | −17.9 (−0.5) |
| pc−CX-Val | −23.1 (0.7) | 37.5 (0.5) | −13.0 (4.1) | 9.6 (1.4) | −39.5 (−1.1) | 4.2 (−1.3) | 8.0 (−5.8) | −27.3 (0.3) | −30.4 (−1.1) | −22.6 (−1.3) |
| wc−CX-Ala | −72.9 (−0.1) | 80.5 (0.1) | −39.3 (−1.3) | 23.0 (−1.9) | −36.4 (−1.2) | 5.7 (−3.0) | −20.9 (−0.4) | −51.7 (−0.7) | −21.3 (−1.3) | −14.2 (−1.6) |
| wc−CX-Asn | −92.1 (−0.1) | 109.0 (0.2) | −55.3 (−0.6) | 31.5 (−1.4) | −45.4 (−1.0) | 7.1 (−2.5) | −23.8 (−1.0) | −62.1 (−0.8) | −25.6 (−1.3) | −16.9 (−1.8) |
| wc−CX-AspH | −49.7 (0.6) | 48.1 (−0.3) | −22.4 (−0.8) | 12.5 (−1.2) | −30.0 (−1.2) | 3.6 (−2.3) | −17.6 (1.6) | −44.1 (0.0) | −16.7 (−2.0) | −10.8 (−2.8) |
| wc−CX-Cys | −54.9 (−0.3) | 58.0 (−0.3) | −27.8 (−0.2) | 16.0 (−0.7) | −30.9 (−1.3) | 4.1 (−3.2) | −17.7 (−1.4) | −44.4 (−1.2) | −16.7 (−2.0) | −10.8 (−2.6) |
| wc−CX-Gln | −83.6 (−0.2) | 85.2 (−0.4) | −41.1 (0.1) | 22.2 (−1.0) | −39.3 (−1.3) | 5.7 (−2.9) | −31.4 (0.7) | −65.0 (−0.2) | −18.1 (−2.0) | −10.8 (−2.9) |
| wc−CX-GluH | −71.5 (0.2) | 73.2 (−0.4) | −37.1 (−0.9) | 19.9 (−0.9) | −34.0 (−1.3) | 4.8 (−3.1) | −28.0 (−0.7) | −57.2 (−0.9) | −16.0 (−2.6) | −9.6 (−3.7) |
| wc−CX-Gly | −74.2 (0.0) | 80.0 (0.2) | −39.5 (−1.2) | 23.0 (−1.8) | −34.7 (−1.3) | 5.6 (−3.3) | −22.9 (−0.4) | −52.0 (−0.7) | −19.8 (−1.5) | −13.0 (−1.9) |
| wc−CX-HisD | −89.0 (0.2) | 104.4 (0.1) | −52.6 (−0.5) | 31.5 (−1.2) | −52.9 (−1.0) | 7.9 (−2.4) | −22.0 (0.0) | −67.1 (−0.5) | −34.9 (−1.1) | −24.6 (−1.3) |
| wc−CX-HisE | −71.0 (0.1) | 83.3 (0.1) | −40.8 (−0.6) | 25.9 (−1.2) | −51.2 (−1.0) | 7.1 (−2.0) | −13.7 (1.4) | −57.7 (−0.3) | −36.3 (−0.7) | −26.4 (−0.8) |
| wc−CX-Ile | −68.1 (0.0) | 75.6 (0.0) | −35.0 (−1.0) | 20.5 (−1.8) | −39.4 (−1.1) | 5.5 (−2.7) | −17.7 (0.7) | −51.6 (−0.3) | −24.6 (−1.2) | −16.8 (−1.5) |
| wc−CX-Leu | −73.8 (0.1) | 86.4 (−0.1) | −42.5 (−0.2) | 25.2 (−1.1) | −43.6 (−1.2) | 6.4 (−2.2) | −17.0 (2.1) | −54.2 (0.0) | −26.3 (−1.4) | −17.7 (−1.7) |
| wc−CX-Lys | −92.1 (0.2) | 105.1 (−0.3) | −55.9 (−0.6) | 32.2 (−0.9) | −44.8 (−1.4) | 7.1 (−2.9) | −29.3 (0.1) | −66.9 (−0.5) | −24.7 (−2.1) | −16.1 (−2.8) |
| wc−CX-Met | −77.5 (−0.1) | 90.1 (0.1) | −43.9 (−0.8) | 25.6 (−1.9) | −42.4 (−1.1) | 6.3 (−2.8) | −19.7 (−0.1) | −55.8 (−0.6) | −25.7 (−1.1) | −17.4 (−1.4) |
| wc−CX-Phe | −78.8 (0.1) | 88.2 (0.1) | −43.6 (−1.2) | 25.3 (−2.1) | −41.3 (−1.2) | 6.2 (−3.1) | −22.4 (0.5) | −57.4 (−0.4) | −24.4 (−1.3) | −16.4 (−1.7) |
| wc−CX-Pro | −70.7 (0.9) | 81.1 (−0.1) | −39.8 (−0.1) | 24.7 (−0.6) | −46.7 (−1.6) | 6.6 (−3.1) | −15.4 (6.1) | −55.5 (0.7) | −29.0 (−1.6) | −20.0 (−2.0) |
| wc−CX-Ser | −81.1 (0.0) | 84.1 (−0.1) | −41.5 (−0.4) | 23.3 (−1.9) | −39.4 (−1.0) | 5.9 (−2.8) | −27.3 (1.4) | −60.8 (0.3) | −22.0 (−1.3) | −14.2 (−1.7) |
| wc−CX-Thr | −77.1 (0.3) | 85.9 (0.0) | −40.0 (−0.2) | 23.7 (−1.9) | −43.5 (−1.3) | 6.3 (−3.0) | −19.3 (3.8) | −56.5 (0.6) | −25.8 (−1.7) | −17.1 (−2.2) |
| wc−CX-Trp | −79.9 (0.2) | 93.8 (−0.1) | −43.3 (0.1) | 27.4 (−1.6) | −60.4 (−1.1) | 8.0 (−2.6) | −13.9 (5.8) | −66.3 (0.5) | −40.8 (−1.3) | −28.9 (−1.6) |
| wc−CX-Tyr | −72.2 (0.1) | 83.8 (−0.1) | −40.2 (−0.7) | 26.7 (−1.3) | −53.7 (−0.7) | 7.5 (−1.7) | −12.7 (1.5) | −58.9 (−0.1) | −37.1 (−0.7) | −26.6 (−0.8) |
| wc−CX-Val | −71.5 (0.2) | 78.8 (−0.1) | −37.3 (−1.1) | 21.8 (−2.0) | −40.3 (−1.1) | 5.7 (−2.8) | −19.4 (1.2) | −54.0 (−0.1) | −24.6 (−1.3) | −16.7 (−1.6) |
| OH-1 | OH-2 | OH-3 | OH-4 | OH-5 | OH-6 | |
|---|---|---|---|---|---|---|
| OH-1 | - | −10.9/−12.3/−9.3 | −0.5/−1.6/0.0 | −0.4/−1.0/0.0 | −0.7/−3.2/−0.3 | −11.4/−15.1/−9.7 |
| OH-2 | −11.8/−12.6/−9.6 | - | −0.1/−14.3/−1.2 | −0.2/−3.2/−0.3 | 0.4 /2.1/0.0 | −4.0 /−5.7/−2.7 |
| OH-3 | −0.3/−1.5/−0.3 | −0.1/−13.9/−1.1 | - | −11.6/−15.1/−9.6 | −2.9/−5.7/−2.7 | −0.3 /−1.5 /1.2 |
| OH-4 | −0.4/−0.9/0.0 | −0.6/−3.2/0.1 | −11.2/−15.2/−9.2 | - | −10.5/−12.3/−9.3 | −0.3/−1.6 /0.0 |
| OH-5 | −0.3/−3.2/0.1 | −0.4/ 2.2/1.4 | −4.0/−5.9/−2.7 | −11.7/−12.6/−9.6 | - | −1.8/−14.3/−1.2 |
| OH-6 | −11.2/−15.3/−9.2 | −3.0/−5.9/−2.7 | 0.4 /−1.2/0.0 | −0.8/−1.5/−0.3 | −1.8/−13.8/−1.1 | - |
| Weight | al-CX | pc-CX | wc-CX | |
|---|---|---|---|---|
| No Fragmentation | −59.7 | −25.5 | −52.0 | |
| Fragment #1 | 1 | −25.3 | −8.7 | −9.5 |
| Fragment #2 | −1 | −18.1 | −1.7 | −6.1 |
| Fragment #3 | 1 | −22.6 | −4.8 | −24.3 |
| Fragment #4 | −1 | −22.6 | −4.7 | −24.1 |
| Fragment #5 | 1 | −22.7 | −4.7 | −24.2 |
| Fragment #6 | −1 | −4.3 | −2.7 | −16.3 |
| Fragment #7 | 1 | −9.6 | −18.7 | −22.5 |
| Fragment #8 | −1 | −6.9 | −18.5 | −23.1 |
| Fragment #9 | 1 | −7.3 | −18.6 | −23.0 |
| Fragment #10 | −1 | −6.5 | −13.3 | −3.5 |
| Fragment #11 | 1 | −25.5 | −9.7 | −10.0 |
| Fragment #12 | −1 | −25.0 | −9.4 | −9.8 |
| Fragment #13 | 1 | −26.4 | −9.7 | −9.5 |
| Fragment #14 | −1 | −19.4 | 2.5 | −5.4 |
| Fragment #15 | 1 | −21.8 | 2.7 | −16.1 |
| Fragment #16 | −1 | −21.1 | 3.1 | −16.0 |
| Fragment #17 | 1 | −21.2 | 1.9 | −16.2 |
| Fragment #18 | −1 | −3.6 | −0.9 | −10.8 |
| Fragment #19 | 1 | −7.8 | −8.6 | −14.4 |
| Fragment #20 | −1 | −7.2 | −8.7 | −14.5 |
| Fragment #21 | 1 | −7.4 | −8.7 | −14.5 |
| Fragment #22 | −1 | −4.0 | −7.8 | −2.9 |
| Fragment #23 | 1 | −24.5 | −9.4 | −10.9 |
| Fragment #24 | −1 | −24.6 | −8.6 | −10.8 |
| Sum | −58.9 | −26.5 | −51.6 |
| Complex | Group A | Group B | (Ratio) | (Ratio) | (Ratio) | (Ratio) | Bond Type | |
|---|---|---|---|---|---|---|---|---|
| al-CX-Asn | OH-2 | NH2-1 | −53.9 (1.8) | 60.7 (2.0) | −29.0 (1.0) | −7.5 (0.3) | −29.7 | typical H-bond |
| al-BCX-Lys | OH-2 | NH2-1 | −48.3 (1.6) | 47.9 (1.6) | −23.1 (0.8) | −6.5 (0.2) | −29.9 | typical H-bond |
| al-CX-Pro | OH-2 | Ring | −44.7 (1.7) | 54.0 (2.1) | −26.2 (1.0) | −8.7 (0.3) | −25.5 | typical H-bond+disp |
| wc-CX-Pro | OH-3 | Ring | −26.8 (1.8) | 29.1 (2.0) | −11.2 (0.8) | −5.8 (0.4) | −14.6 | typical H-bond+disp |
| wc-BCX-Pro | OH-6 | Ring | −25.7 (1.8) | 28.4 (2.0) | −11.0 (0.8) | −5.6 (0.4) | −13.9 | typical H-bond+disp |
| wc-CX-Leu | Ph-6 | COOH | −19.7 (0.9) | 7.5 (0.3) | −4.7 (0.2) | −4.8 (0.2) | −21.7 | H⋯ |
| wc-CX-Pro | Ph-6 | COOH | −16.6 (0.9) | 6.2 (0.3) | −3.8 (0.2) | −4.5 (0.2) | −18.6 | H⋯ |
| al-BCX-Ser | Ph-5 | COOH | −19.4 (0.9) | 10.2 (0.5) | −5.8 (0.3) | −5.8 (0.3) | −20.8 | H⋯ |
| pc-BCX-Tyr | Ph-4 | OH | −10.2 (1.0) | 5.5 (0.6) | −2.5 (0.3) | −2.8 (0.3) | −9.9 | H⋯ |
| pc-CX-HisE | Ph-6 | Ring | −11.9 (0.9) | 10.1 (0.8) | −2.7 (0.2) | −8.5 (0.7) | −13.0 | disp |
| wc-CX-HisE | Ph-2 | Ring | −11.8 (0.9) | 7.5 (0.6) | −1.7 (0.1) | −7.2 (0.5) | −13.2 | disp |
| pc-BCX-HisE | Ph-5 | Ring | −13.4 (0.9) | 10.6 (0.7) | −3.5 (0.2) | −8.8 (0.6) | −15.1 | disp |
| wc-BCX-HisE | Ph-5 | Ring | −13.9 (0.9) | 10.0 (0.7) | −2.3 (0.2) | −9.0 (0.6) | −15.1 | disp |
| wc-BCX-Trp | Ph-5 | Ring | −12.1 (0.8) | 10.7 (0.7) | −2.6 (0.2) | −10.8 (0.7) | −14.8 | disp |
| pc-CX-Tyr | Ph-3 | COOH | −15.7 (0.9) | 1.2 (0.1) | −1.4 (0.1) | −2.2 (0.1) | −18.1 | elst |
| al-CX-HisD | Ph-4 | COOH | −11.7 (0.8) | 0.3 (0.0) | −2.3 (0.2) | −1.5 (0.1) | −15.1 | elst |
| al-BCX-Gly | Ph-5 | COOH | −7.7 (0.7) | 0.8 (0.1) | −1.4 (0.1) | −2.1 (0.2) | −10.5 | elst |
| pc-BCX-GluH | Ph-3 | COOH-1 | −15.4 (0.8) | 2.3 (0.1) | −2.2 (0.1) | −3.2 (0.2) | −18.5 | elst |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masoumifeshani, E.; Chojecki, M.; Rutkowska-Zbik, D.; Korona, T. Association Complexes of Calix[6]arenes with Amino Acids Explained by Energy-Partitioning Methods. Molecules 2022, 27, 7938. https://doi.org/10.3390/molecules27227938
Masoumifeshani E, Chojecki M, Rutkowska-Zbik D, Korona T. Association Complexes of Calix[6]arenes with Amino Acids Explained by Energy-Partitioning Methods. Molecules. 2022; 27(22):7938. https://doi.org/10.3390/molecules27227938
Chicago/Turabian StyleMasoumifeshani, Emran, Michał Chojecki, Dorota Rutkowska-Zbik, and Tatiana Korona. 2022. "Association Complexes of Calix[6]arenes with Amino Acids Explained by Energy-Partitioning Methods" Molecules 27, no. 22: 7938. https://doi.org/10.3390/molecules27227938
APA StyleMasoumifeshani, E., Chojecki, M., Rutkowska-Zbik, D., & Korona, T. (2022). Association Complexes of Calix[6]arenes with Amino Acids Explained by Energy-Partitioning Methods. Molecules, 27(22), 7938. https://doi.org/10.3390/molecules27227938