
Ultra-Large-Scale Screening of Natural Compounds and Free Energy Calculations Revealed Potential Inhibitors for the Receptor-Binding Domain (RBD) of SARS-CoV-2

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Molecular Screening of Natural Compounds Databases
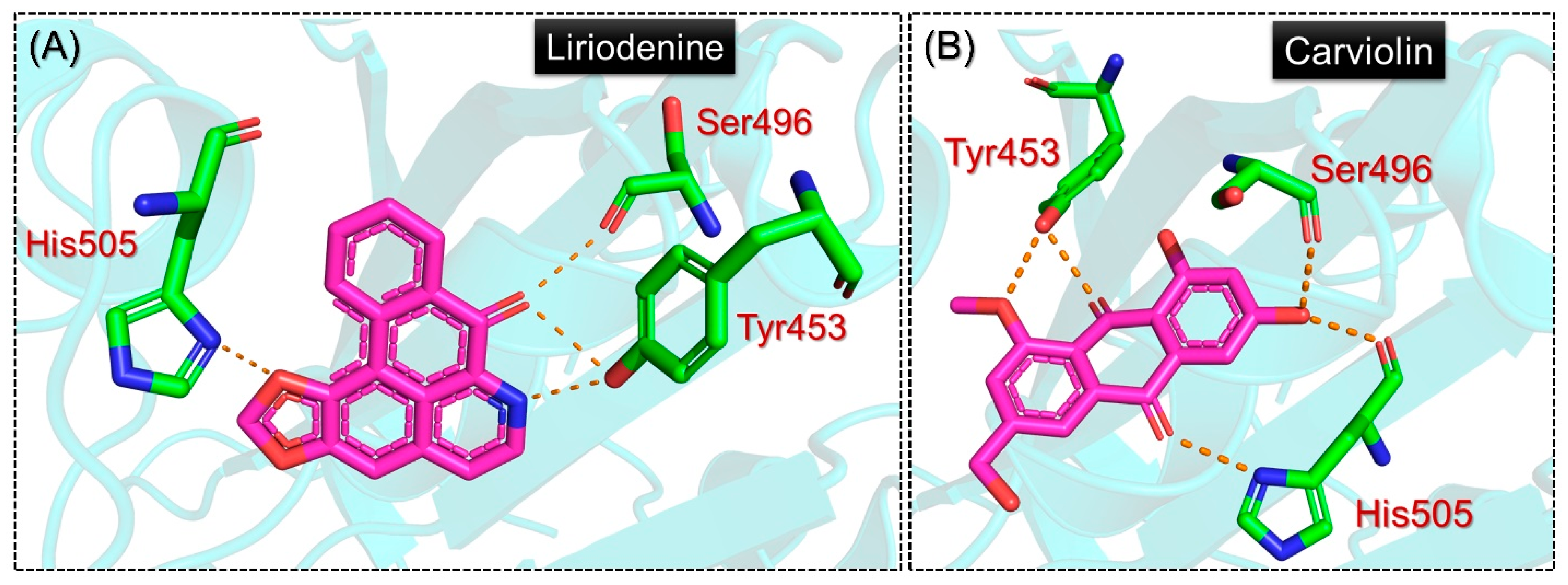
2.2. Structural and Dynamic Stability Assessment of the Top-Scoring Complexes
2.3. Assessing the Structural Packing in a Dynamic Environment
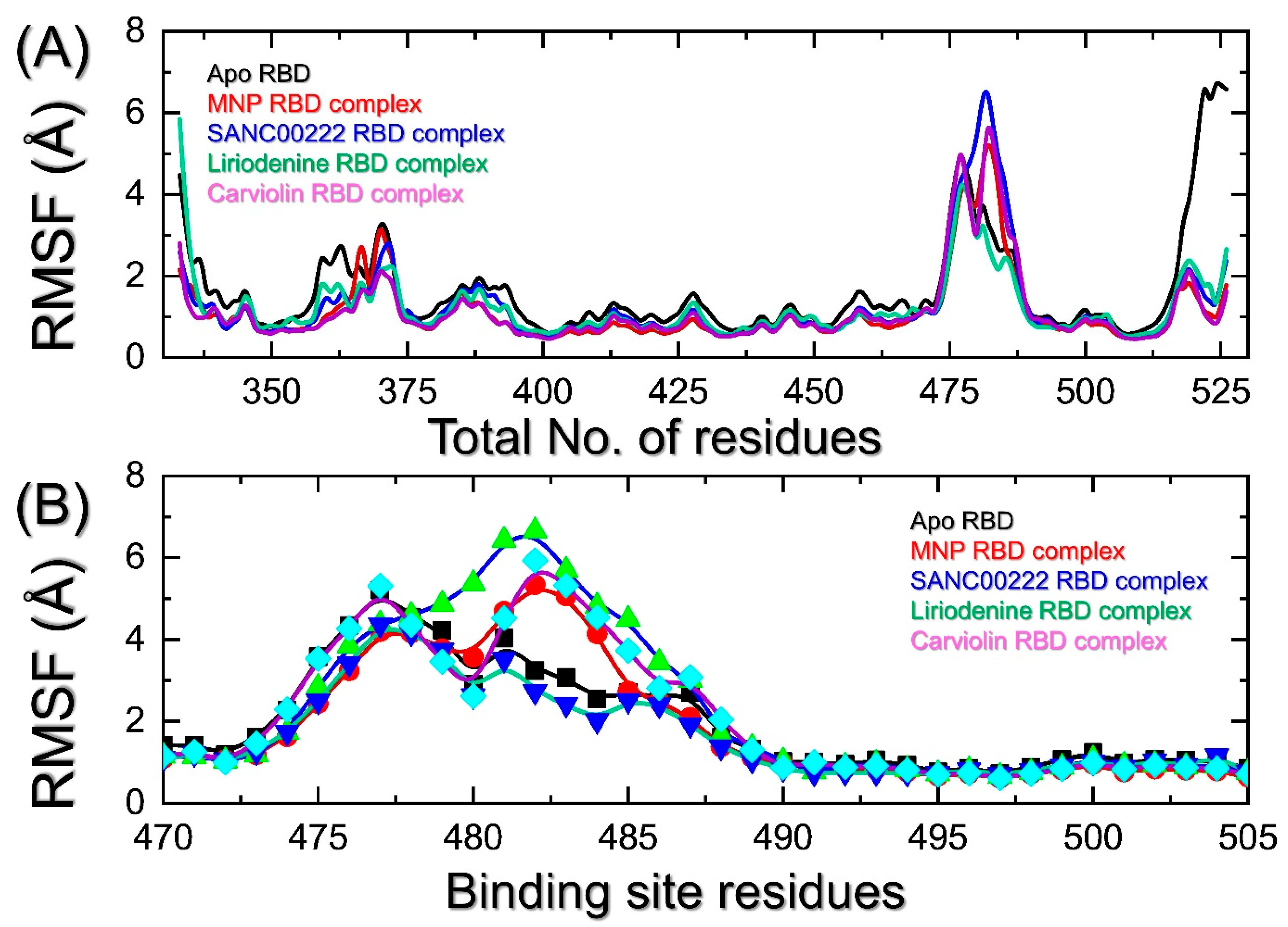
2.4. Indexing the Residual Flexibility
2.5. Analysis of the Hydrogen Bonding during Simulation
2.6. Gibbs Free Energy Calculation to Re-Evaluate the Best Conformations
2.7. Clustering of Protein Motion in Trajectories
2.8. Free Energy Landscape Analysis
3. Conclusions
4. Materials and Methods
4.1. Structures Retrieval and Molecular Screening
4.2. Molecular Dynamics Simulation of Protein-Ligand Complexes
4.3. The Binding Free Energy Calculations
4.4. Principal Component Analysis (PCA)
4.5. Free Energy Landscape (FEL)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Khan, T.; Ali, S.; Aftab, S.; Wang, Y.; Qiankun, W.; Khan, M.; Suleman, M.; Ali, S.; Heng, W. SARS-CoV-2 new variants: Characteristic features and impact on the efficacy of different vaccines. Biomed. Pharmacother. 2021, 143, 112176. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Waris, H.; Rafique, M.; Suleman, M.; Mohammad, A.; Ali, S.S.; Khan, T.; Waheed, Y.; Liao, C.; Wei, D.-Q. The Omicron (B. 1.1. 529) variant of SARS-CoV-2 binds to the hACE2 receptor more strongly and escapes the antibody response: Insights from structural and simulation data. Int. J. Biol. Macromol. 2022, 200, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell. Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V.J.N.R.M. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. Vol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Fontanet, A.; Autran, B.; Lina, B.; Kieny, M.P.; Karim, S.S.A.; Sridhar, D.J.T.L. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 2021, 397, 952–954. [Google Scholar] [CrossRef]
- Li, Y.; Xie, Z.; Lin, W.; Cai, W.; Wen, C.; Guan, Y.; Mo, X.; Wang, J.; Wang, Y.; Peng, P. Efficacy and safety of lopinavir/ritonavir or arbidol in adult patients with mild/moderate COVID-19: An exploratory randomized controlled trial. Med 2020, 1, 105–113. [Google Scholar] [CrossRef]
- Lian, N.; Xie, H.; Lin, S.; Huang, J.; Zhao, J.; Lin, Q. Umifenovir treatment is not associated with improved outcomes in patients with coronavirus disease 2019: A retrospective study. Clin. Microbiol. Infect. 2020, 26, 917–921. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19 - Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Wu, X.; Yu, K.; Wang, Y.; Xu, W.; Ma, H.; Hou, Y.; Li, Y.; Cai, B.; Zhu, L.; Zhang, M.; et al. Efficacy and Safety of Triazavirin Therapy for Coronavirus Disease 2019: A Pilot Randomized Controlled Trial. Engineering 2020, 6, 1185–1191. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.-X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Randhawa, A.W.; Balouch, A.R.; Mukhtar, N.; Sayaf, A.M.; Suleman, M.; Khan, T.; Ali, S.; Ali, S.S.; Wang, Y. Blocking key mutated hotspot residues in the RBD of the omicron variant (B. 1.1. 529) with medicinal compounds to disrupt the RBD-hACE2 complex using molecular screening and simulation approaches. RSC Adv. 2022, 12, 7318–7327. [Google Scholar] [CrossRef] [PubMed]
- Baildya, N.; Khan, A.A.; Ghosh, N.N.; Dutta, T.; Chattopadhyay, A.P. Screening of potential drug from Azadirachta Indica (Neem) extracts for SARS-CoV-2: An insight from molecular docking and MD-simulation studies. J. Mol. Struct. 2021, 1227, 129390. [Google Scholar] [CrossRef]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 4659–4670. [Google Scholar] [CrossRef]
- Khan, A.; Heng, W.; Wang, Y.; Qiu, J.; Wei, X.; Peng, S.; Saleem, S.; Khan, M.; Ali, S.S.; Wei, D.-Q. In silico and in vitro evaluation of kaempferol as a potential inhibitor of the SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 4659–4670. [Google Scholar] [CrossRef]
- Khan, A.; Khan, M.; Saleem, S.; Babar, Z.; Ali, A.; Khan, A.A.; Sardar, Z.; Hamayun, F.; Ali, S.S.; Wei, D.Q. Phylogenetic Analysis and Structural Perspectives of RNA-Dependent RNA-Polymerase Inhibition from SARs-CoV-2 with Natural Products. Interdiscip. Sci. Comput. Life Sci. 2020, 12, 335–348. [Google Scholar] [CrossRef]
- Celik, I.; Yadav, R.; Duzgun, Z.; Albogami, S.; El-Shehawi, A.M.; Fatimawali; Idroes, R.; Tallei, T.E.; Emran, T.B. Interactions of the Receptor Binding Domain of SARS-CoV-2 Variants with hACE2: Insights from Molecular Docking Analysis and Molecular Dynamic Simulation. Biology 2021, 10, 880. [Google Scholar] [CrossRef]
- Stepto, R.; Chang, T.; Kratochvíl, P.; Hess, M.; Horie, K.; Sato, T.; Vohlídal, J. Definitions of terms relating to individual macromolecules, macromolecular assemblies, polymer solutions, and amorphous bulk polymers (IUPAC Recommendations 2014). Pure Appl. Chem. 2015, 87, 71–120. [Google Scholar] [CrossRef]
- Bornot, A.; Etchebest, C.; de Brevern, A.G. Predicting protein flexibility through the prediction of local structures. Proteins 2011, 79, 839–852. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [PubMed]
- Diallo, B.N.t.; Glenister, M.; Musyoka, T.M.; Lobb, K.; Tastan Bishop, Ö. SANCDB: An update on South African natural compounds and their readily available analogs. J. Cheminform. 2021, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Modeling 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [Green Version]
- Masters, L.; Eagon, S.; Heying, M. Evaluation of consensus scoring methods for AutoDock Vina, smina and idock. J. Mol. Graph. Model. 2020, 96, 107532. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Hoang, T.X.; Trovato, A.; Seno, F.; Banavar, J.R.; Maritan, A. Geometry and symmetry presculpt the free-energy landscape of proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 7960–7964. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | Salt/Hydrophobic Interactions | Hydrogen Bonding | Docking Scores (kcal/mol) |
|---|---|---|---|
| 2-(2,3-dihydroxyphenyl)-N-[2-(1H-imidazol-5-yl)ethyl]-1,3-thiazole-4-carboxamide | Arg403, Tyr453, Ty495, Ty501, His505 | Arg403, Gln409x2, Asn417x2, Tyr453, Ser496 | −6.6 kcal/mol |
| SANC00222 | Tyr501, His505 | Asn417x2, Tyr453, Tyr501x2, His505x2 | −6.3 kcal/mol |
| Liriodenine | Phe497 | Tyr453, Ser496, Tyr501, His505 | −5.9 kcal/mol |
| Carviolin | Tyr495, Tyr501 | Tyr453x2, Ser496, His505x2 | −5.9 kcal/mol |
| Parameters | MNP-RBD Complex | SANC00222-RBD Complex | Liriodenine-RBD Complex | Carviolin-RBD Complex |
|---|---|---|---|---|
| VDWAALS | −41.11 ± 0.11 | −29.96 ± 0.10 | −31.73 ± 0.08 | −28.98 ± 0.13 |
| EEL | −10.34 ± 0.10 | −13.31 ± 0.13 | −8.12 ± 0.08 | −6.54 ± 0.09 |
| EGB | 23.29 ± 0.10 | 22.96 ± 0.13 | 7.74 ± 0.10 | 10.45 ± 0.08 |
| ESURF | −4.69 ± 0.14 | −3.10 ±0.08 | −2.18 ± 0.90 | −2.61 ± 0.01 |
| DELTA G gas | −51.46 ± 0.17 | −43.27 ± 0.18 | −25.86 ± 0.13 | −28.52 ± 0.116 |
| DELTA G solv | 18.59 ± 0.09 | 10.82 ± 0.11 | 5.56 ± 0.07 | 9.84 ± 0.08 |
| DELTA TOTAL | −32.86 ± 0.10 | −23.41 ± 0.15 | −34.29 ± 0.07 | −27.67 ± 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, L.; Zafar, F.; Moeen, N.; Alshabrmi, F.M.; Lin, J.; Ali, S.S.; Munir, M.; Khan, A.; Wei, D. Ultra-Large-Scale Screening of Natural Compounds and Free Energy Calculations Revealed Potential Inhibitors for the Receptor-Binding Domain (RBD) of SARS-CoV-2. Molecules 2022, 27, 7317. https://doi.org/10.3390/molecules27217317
Guo L, Zafar F, Moeen N, Alshabrmi FM, Lin J, Ali SS, Munir M, Khan A, Wei D. Ultra-Large-Scale Screening of Natural Compounds and Free Energy Calculations Revealed Potential Inhibitors for the Receptor-Binding Domain (RBD) of SARS-CoV-2. Molecules. 2022; 27(21):7317. https://doi.org/10.3390/molecules27217317
Chicago/Turabian StyleGuo, Lisha, Faryar Zafar, Nawal Moeen, Fahad M. Alshabrmi, Junqi Lin, Syed Shujait Ali, Muhammad Munir, Abbas Khan, and Dongqing Wei. 2022. "Ultra-Large-Scale Screening of Natural Compounds and Free Energy Calculations Revealed Potential Inhibitors for the Receptor-Binding Domain (RBD) of SARS-CoV-2" Molecules 27, no. 21: 7317. https://doi.org/10.3390/molecules27217317
APA StyleGuo, L., Zafar, F., Moeen, N., Alshabrmi, F. M., Lin, J., Ali, S. S., Munir, M., Khan, A., & Wei, D. (2022). Ultra-Large-Scale Screening of Natural Compounds and Free Energy Calculations Revealed Potential Inhibitors for the Receptor-Binding Domain (RBD) of SARS-CoV-2. Molecules, 27(21), 7317. https://doi.org/10.3390/molecules27217317