
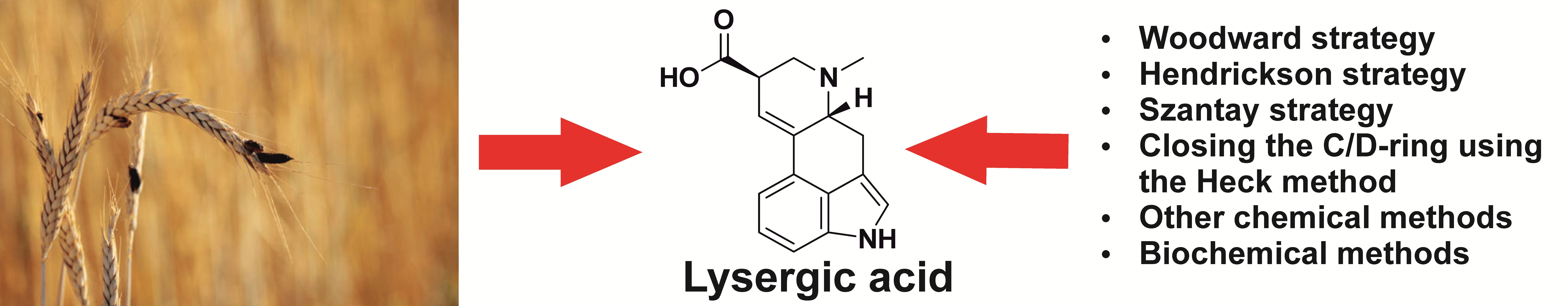
Methods of Lysergic Acid Synthesis—The Key Ergot Alkaloid



Abstract
:
1. Lysergic Acid as a Component of Ergot Alkaloids
1.1. Ergot
1.1.1. History of Ergot [2]
1.1.2. Properties of Ergot
1.2. Ergot Alkaloids
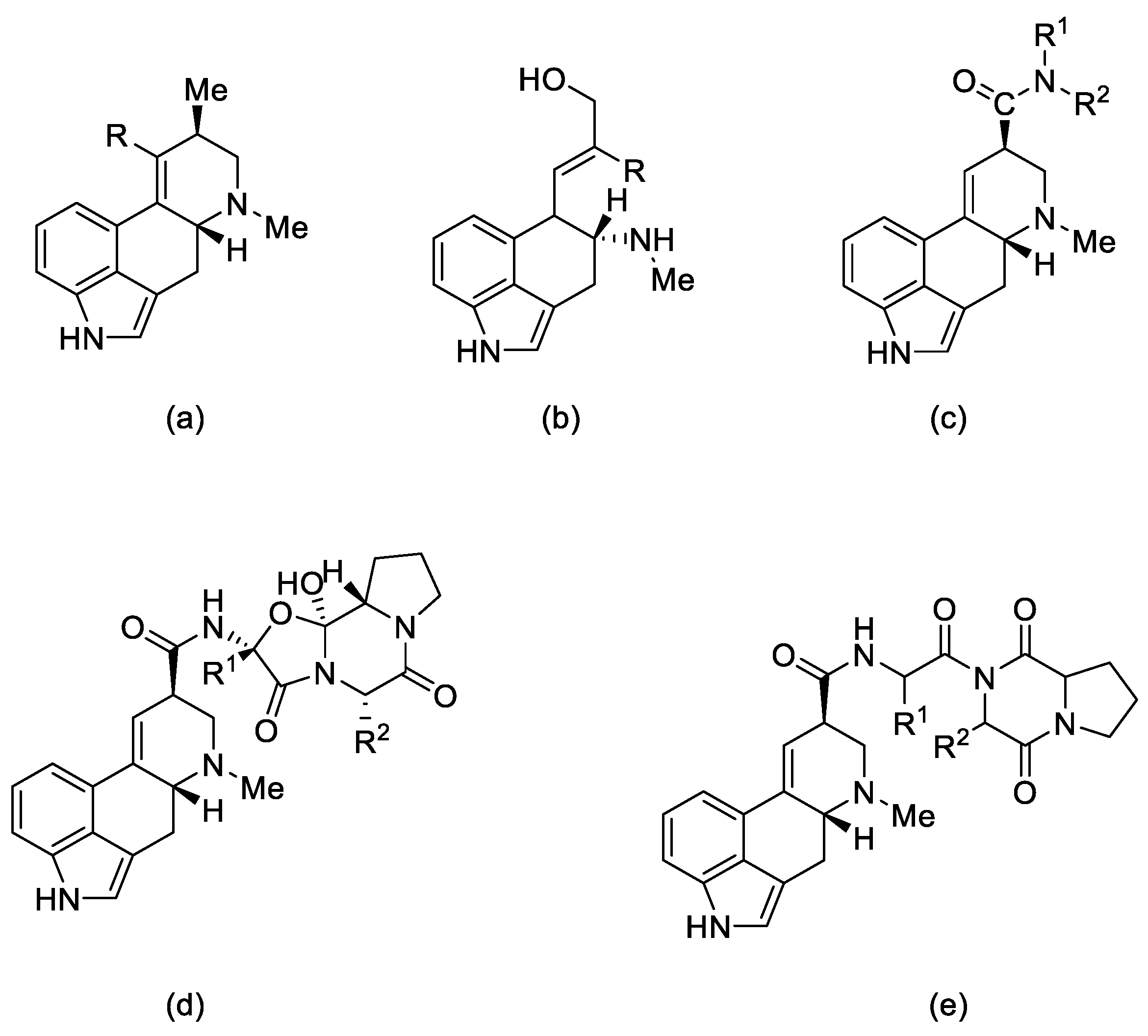
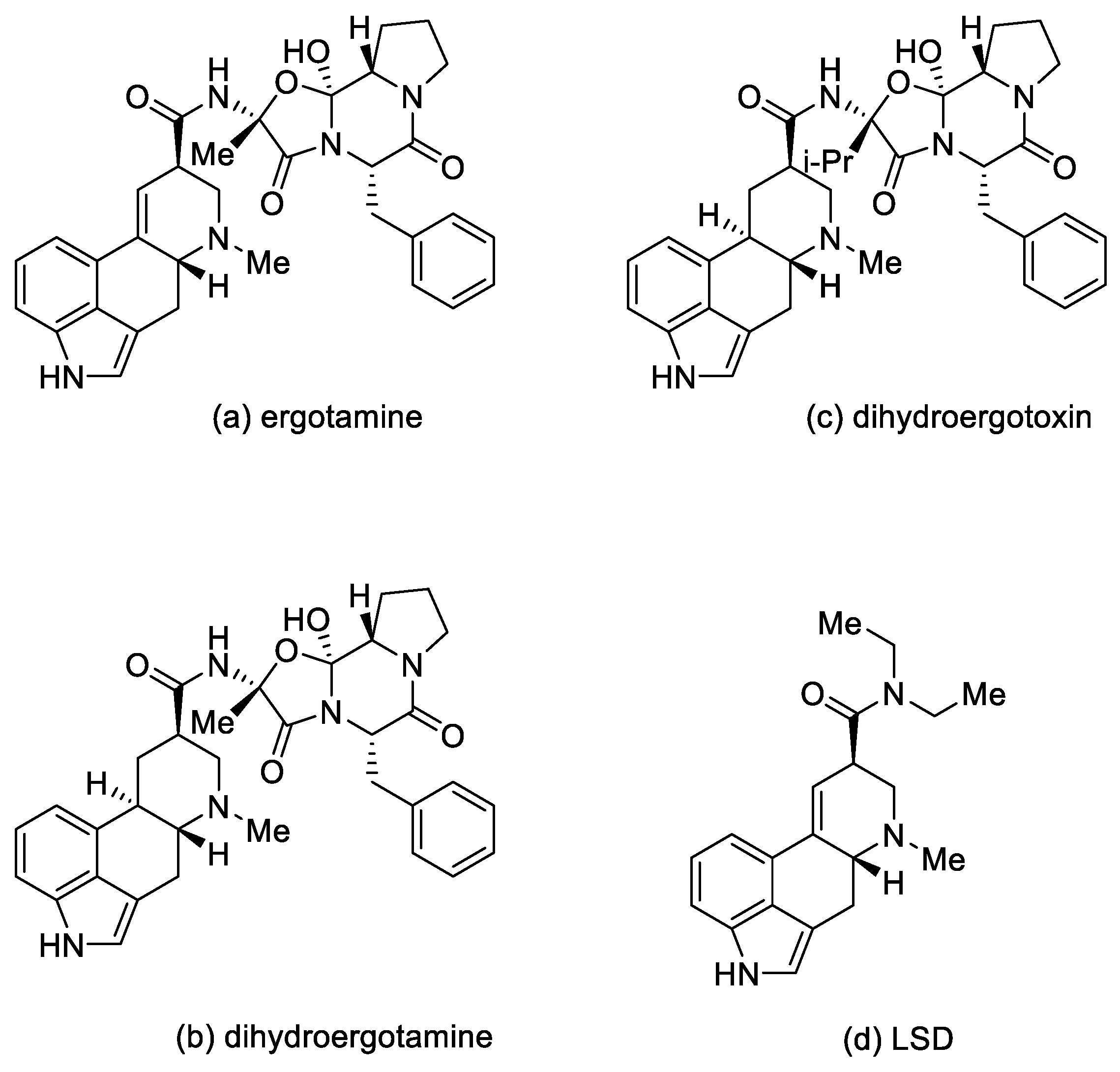
1.2.1. Types of Ergot Alkaloids
1.2.2. Biochemical and Pharmacological Properties
1.2.3. Other Application [20]
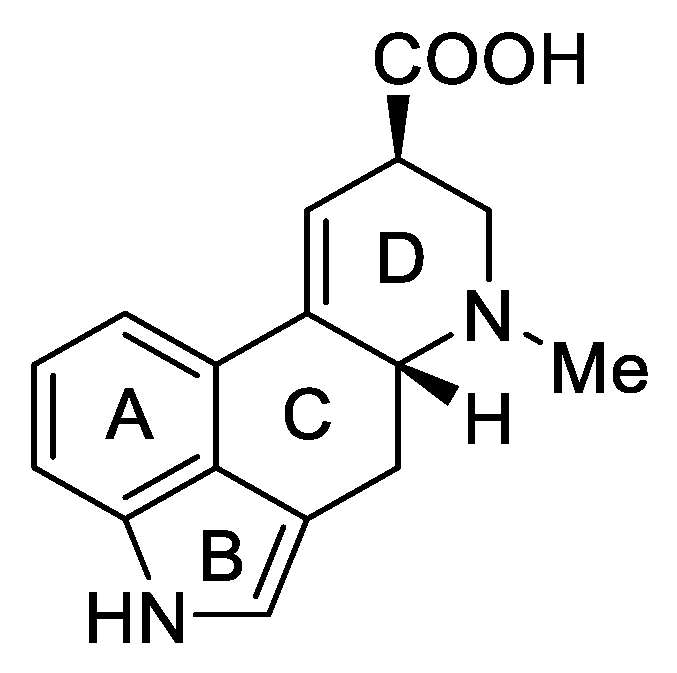
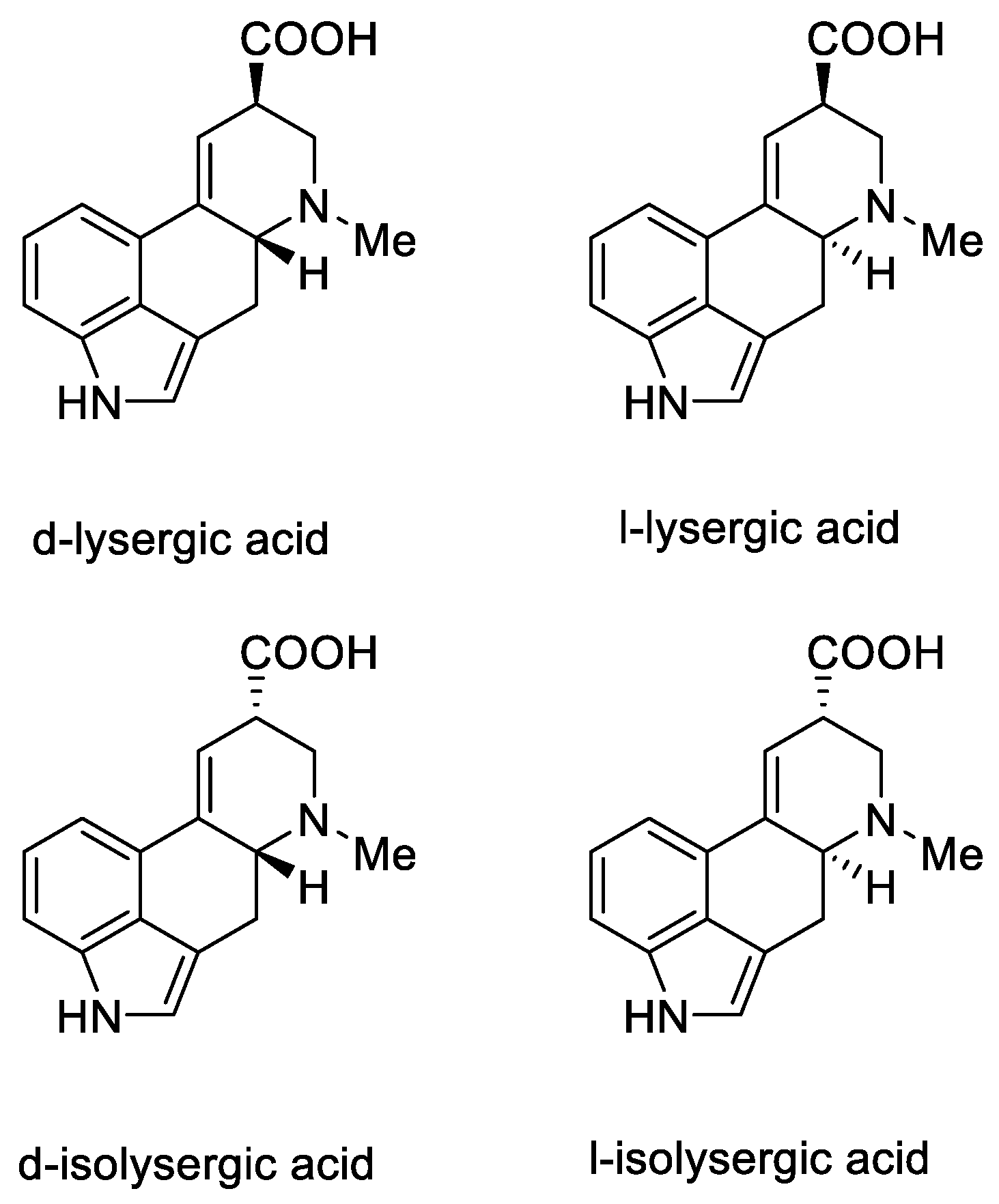
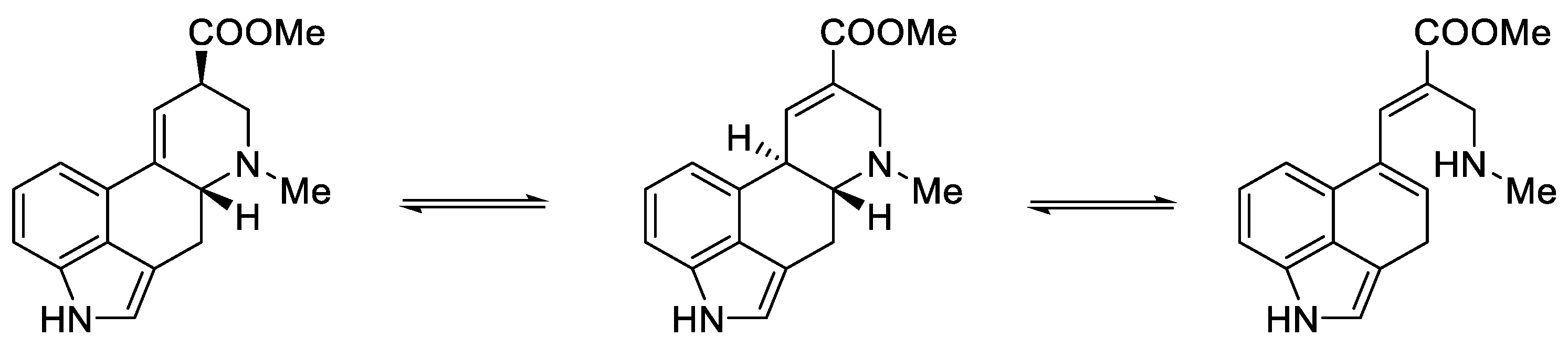
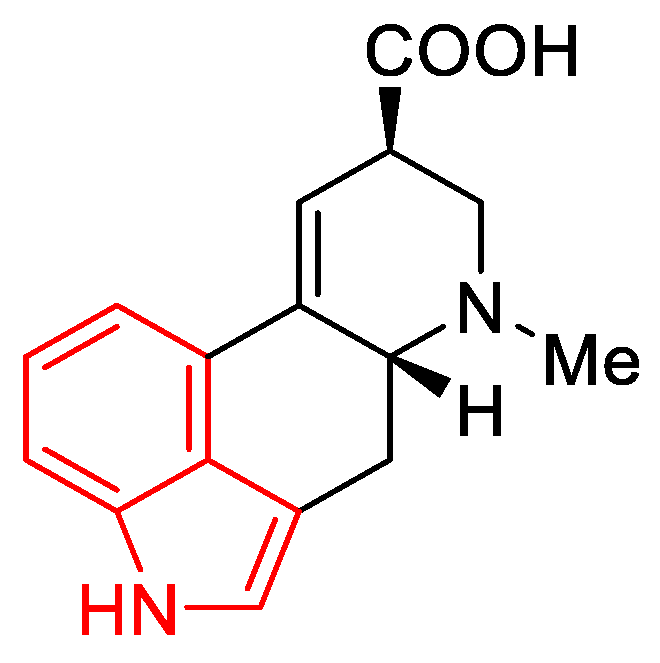
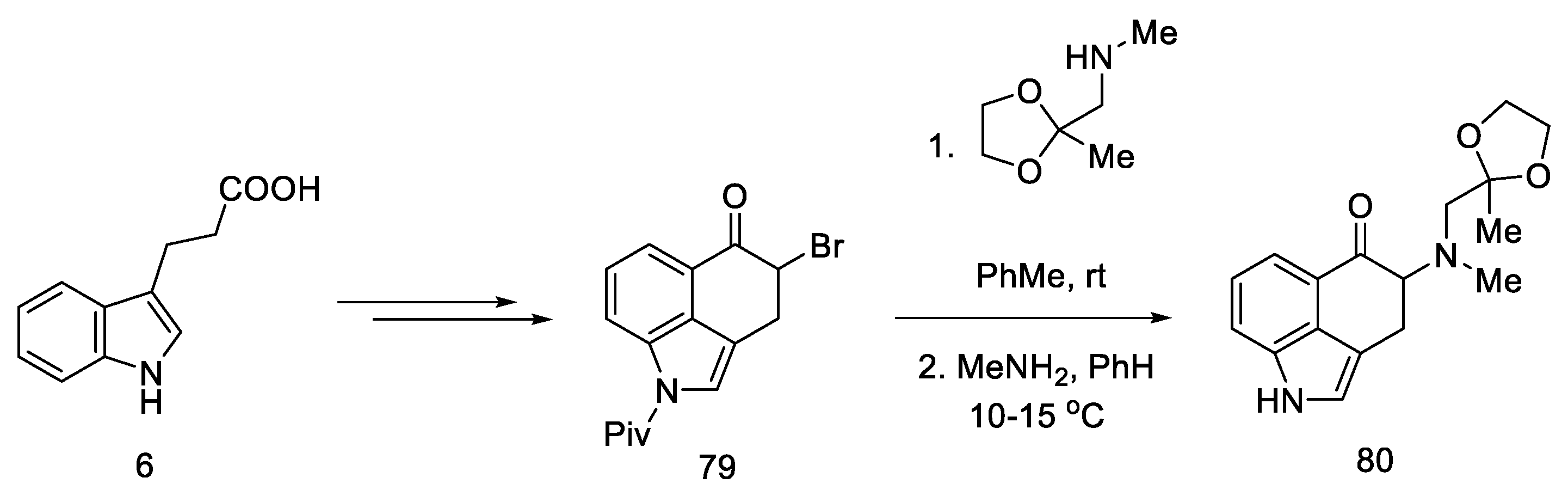
1.3. Lysergic Acid
History of Lysergic Acid [24]
2. Methods of Lysergic Acid Synthesis
2.1. Woodward’s Strategy
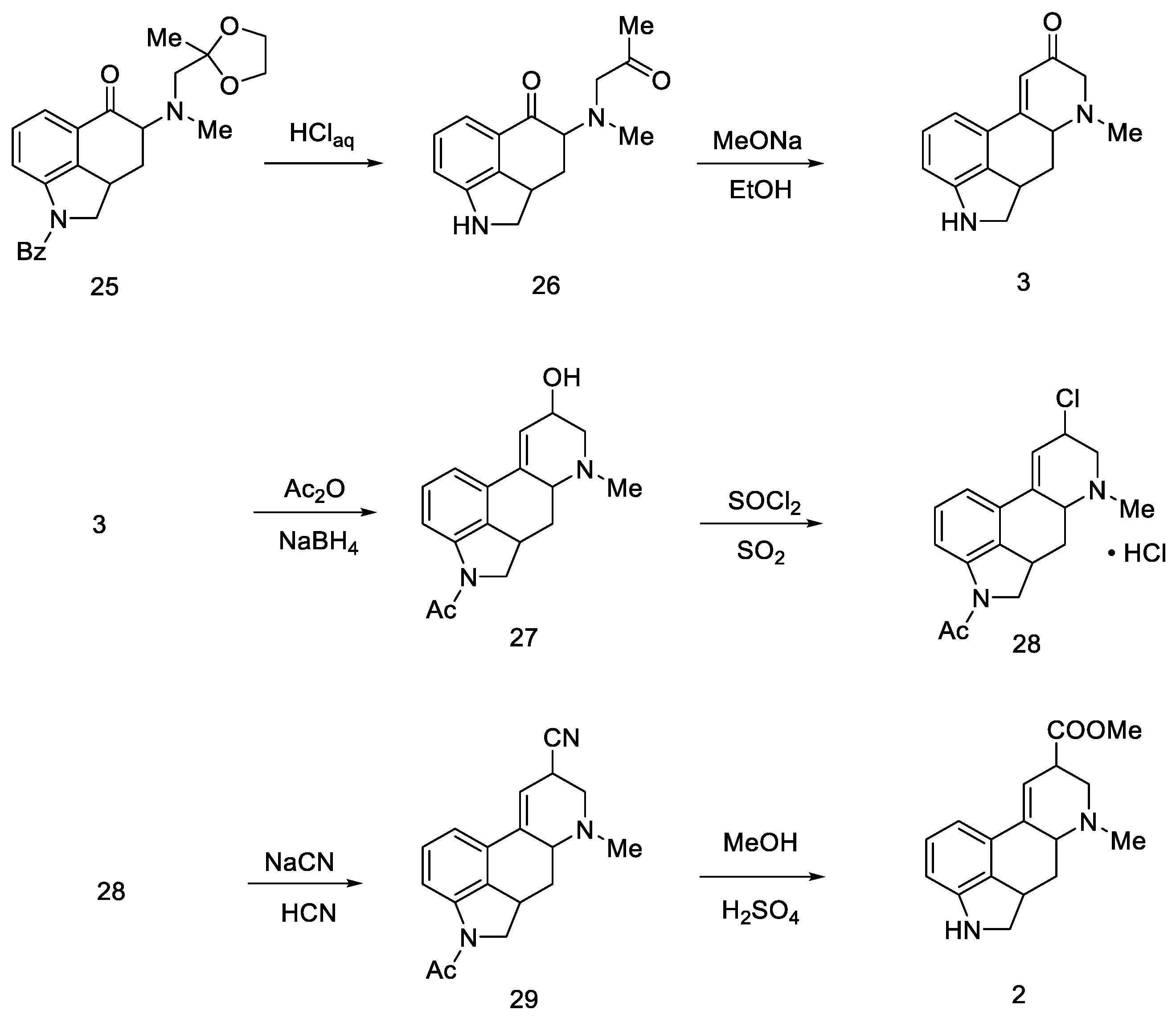
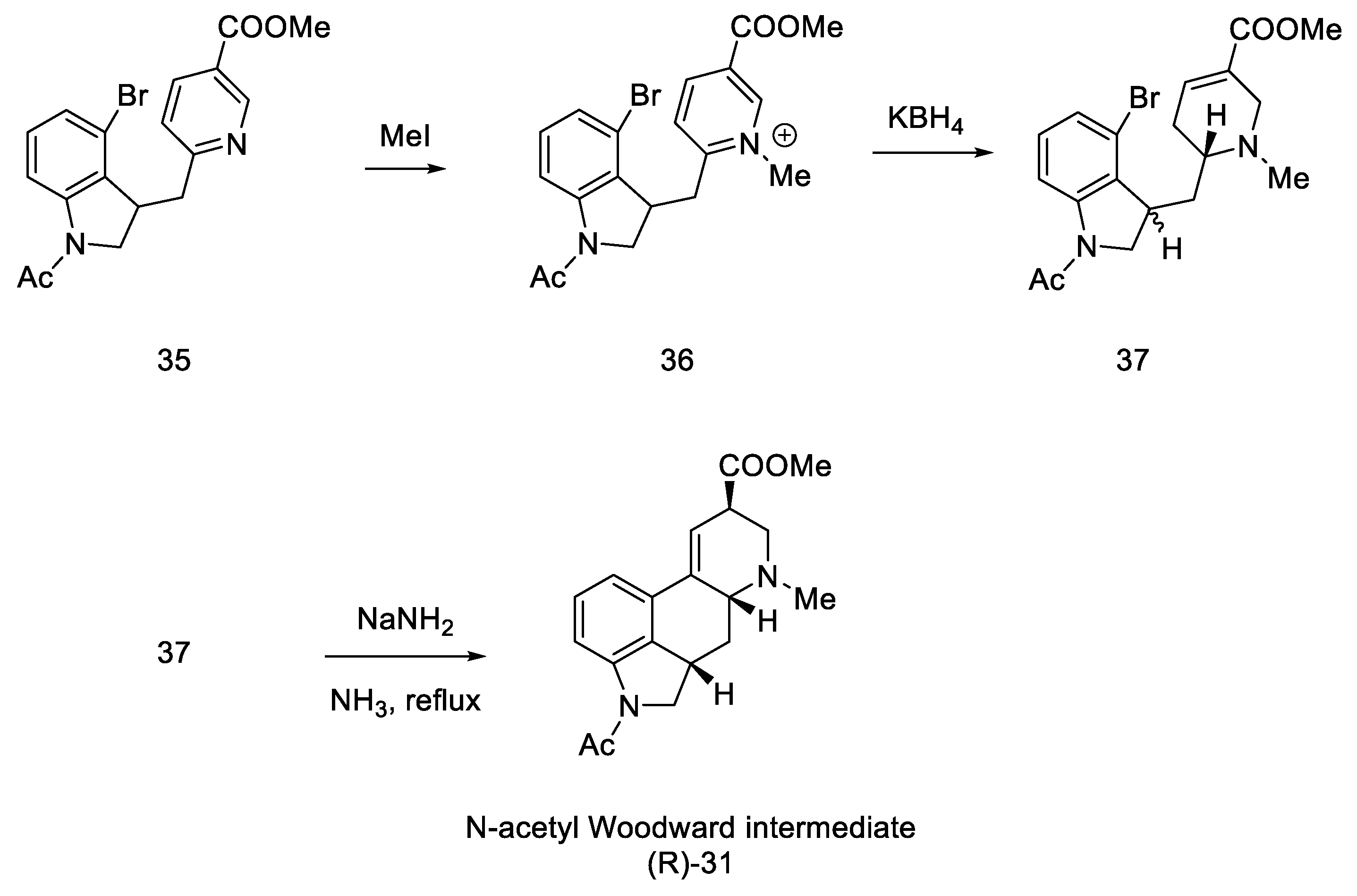
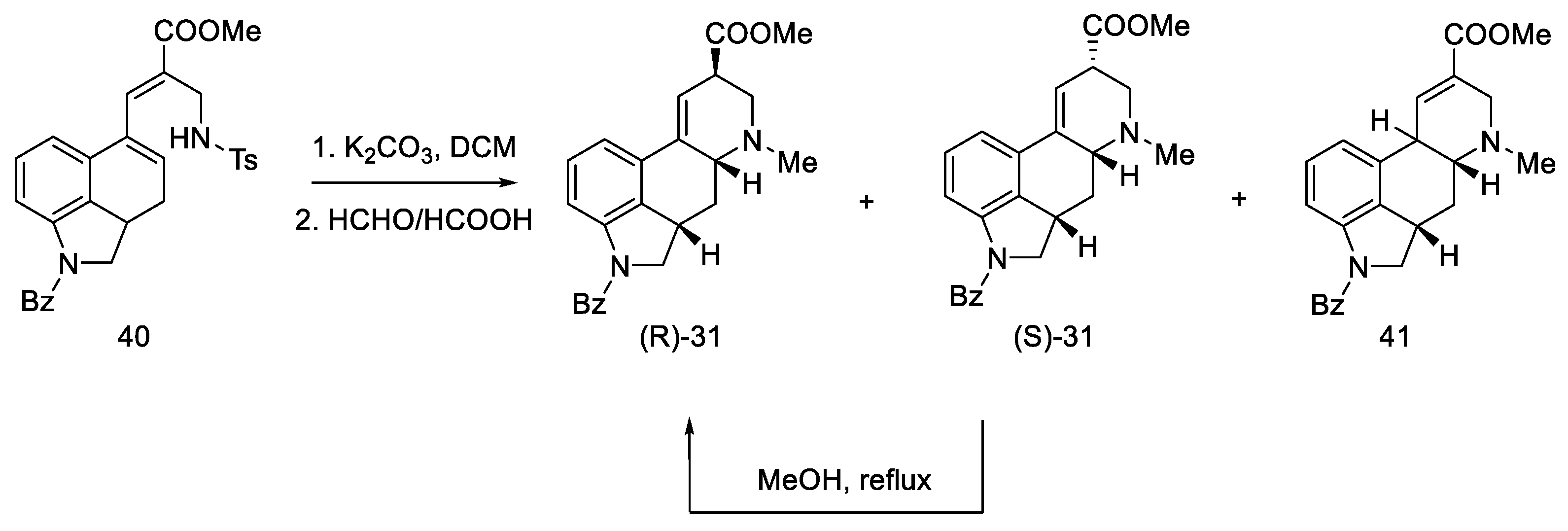
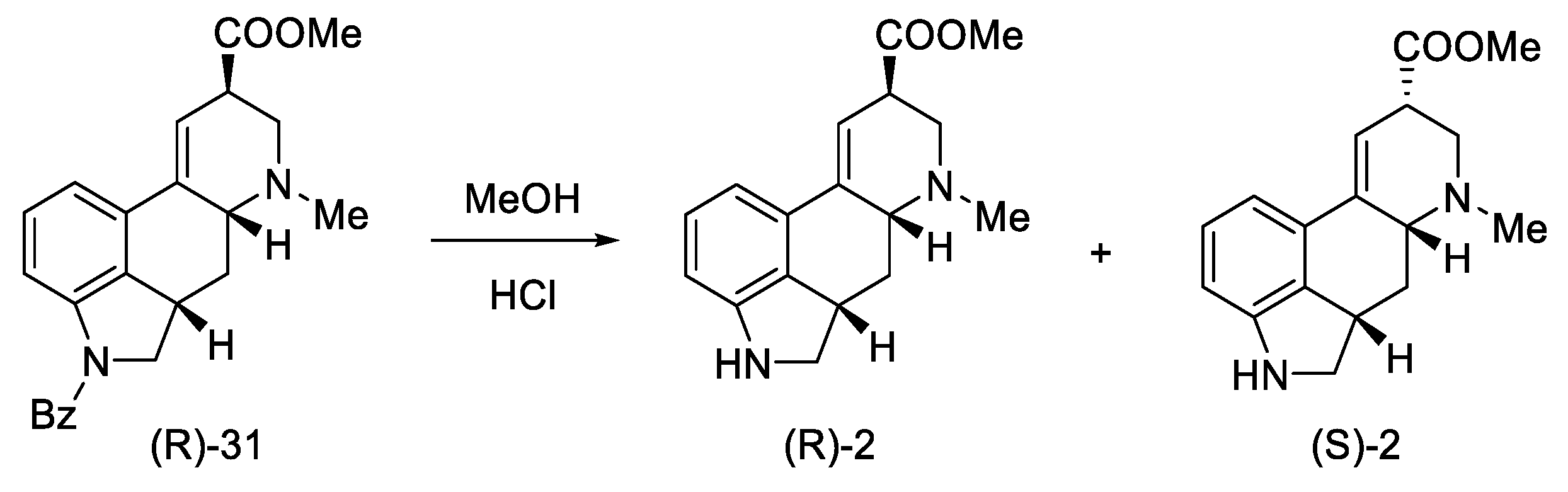
2.1.1. Woodward’s Research Group
2.1.2. Baillarge Group Research
2.1.3. Ramage’s Research Group
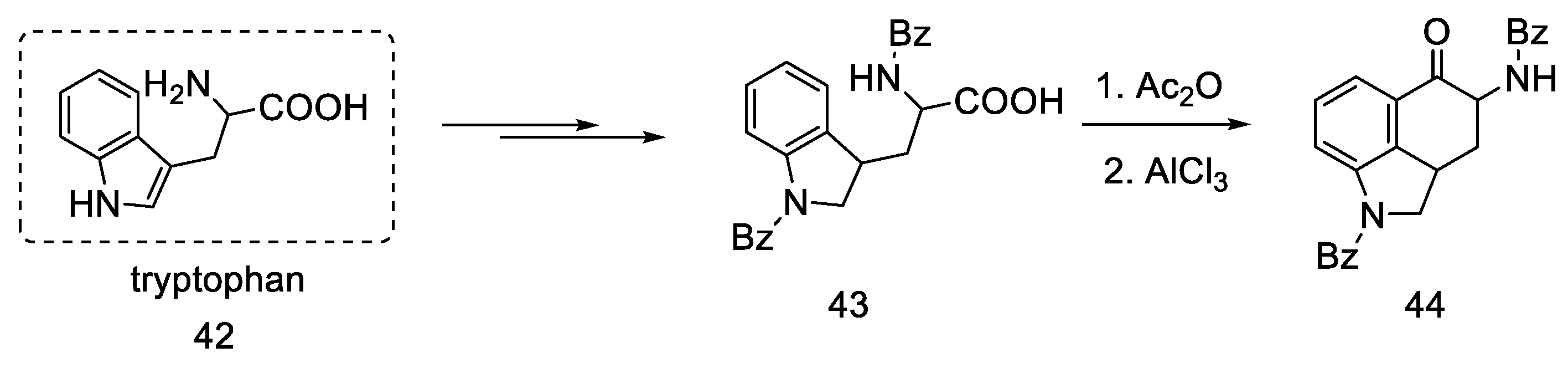
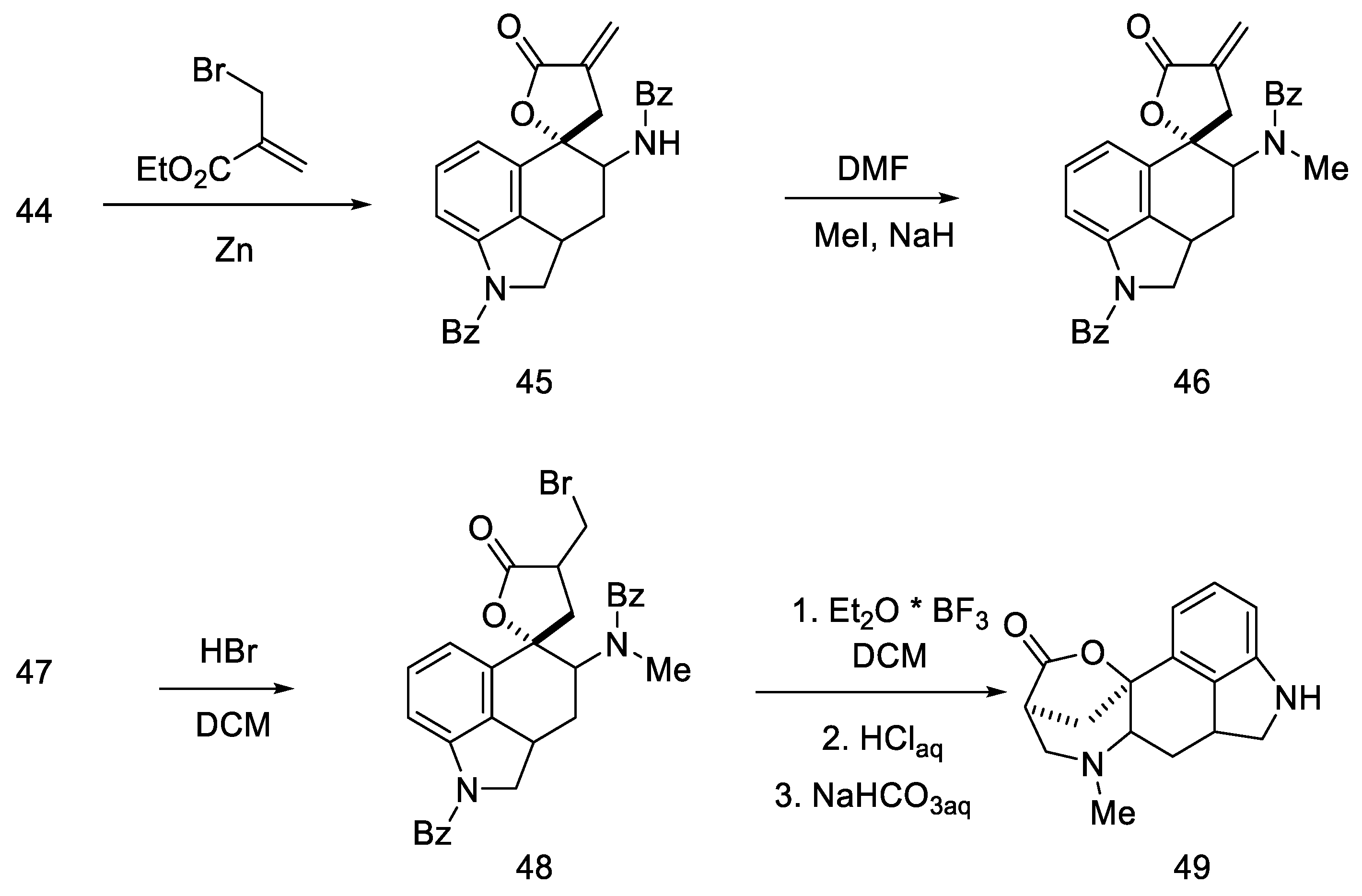
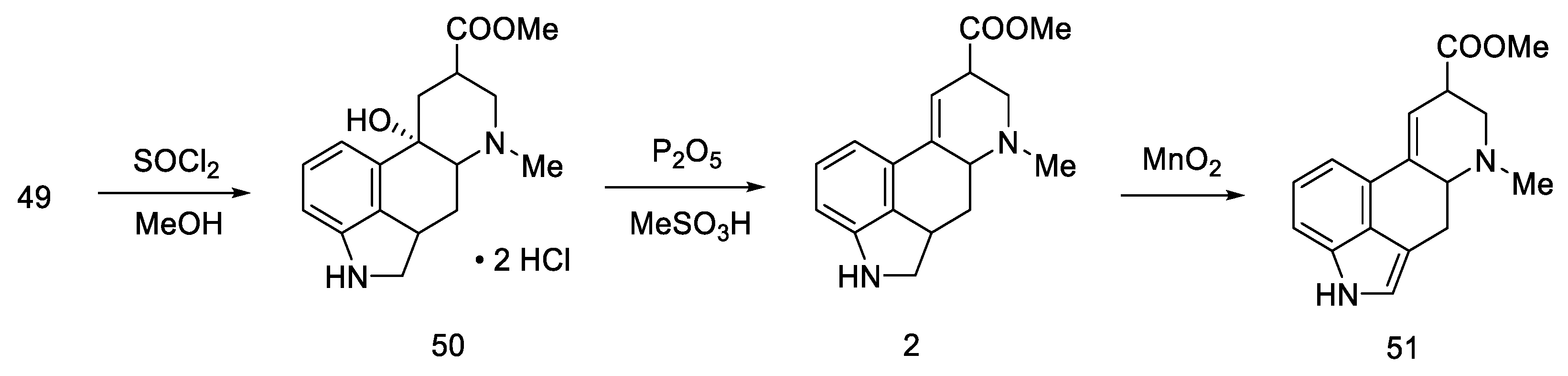
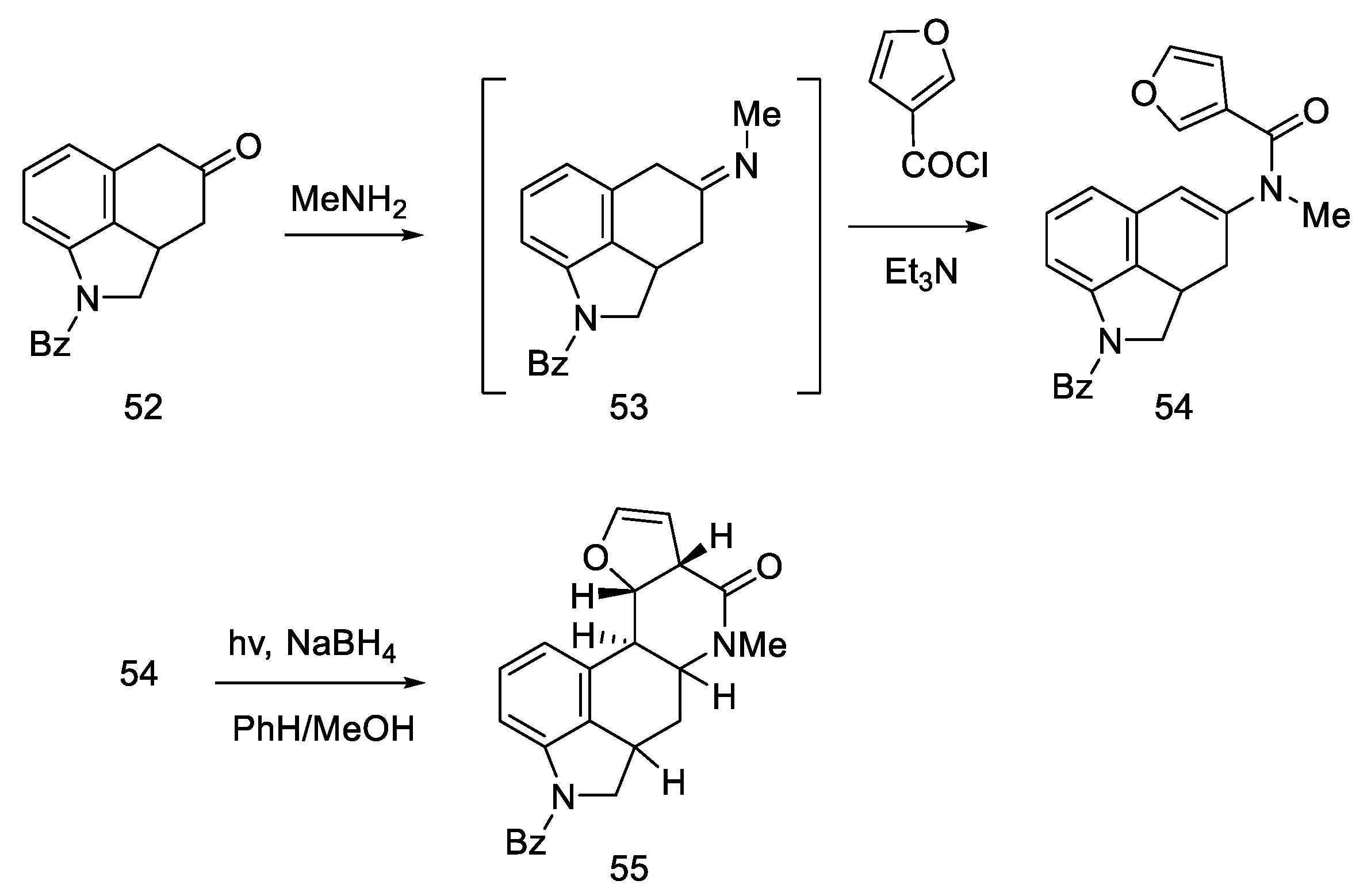
2.1.4. Rebek’s Research Group
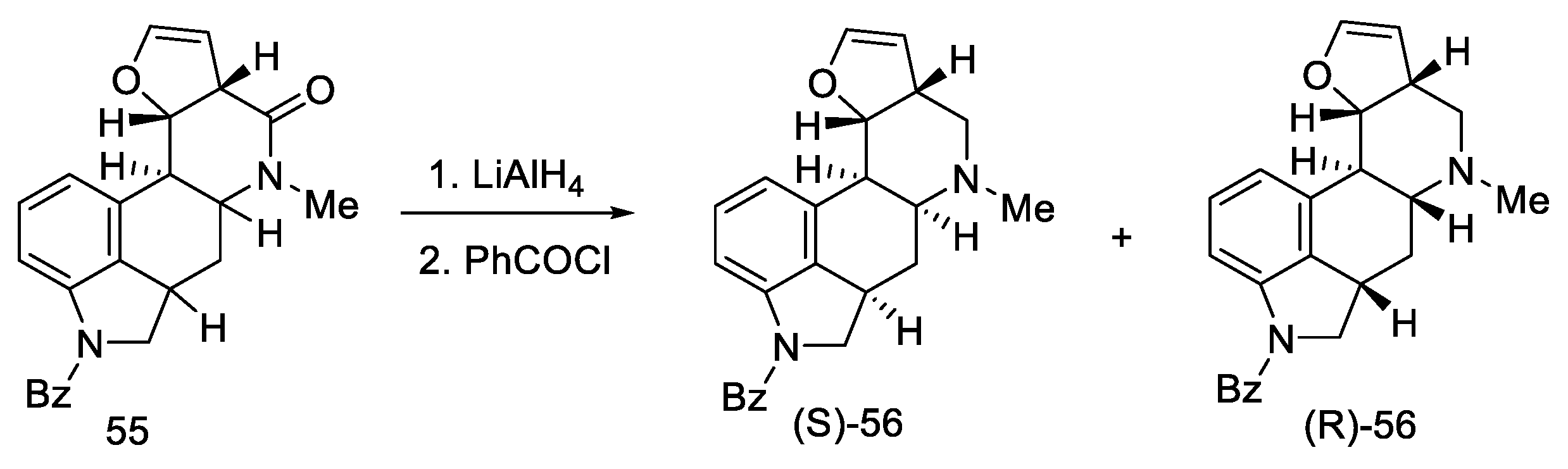
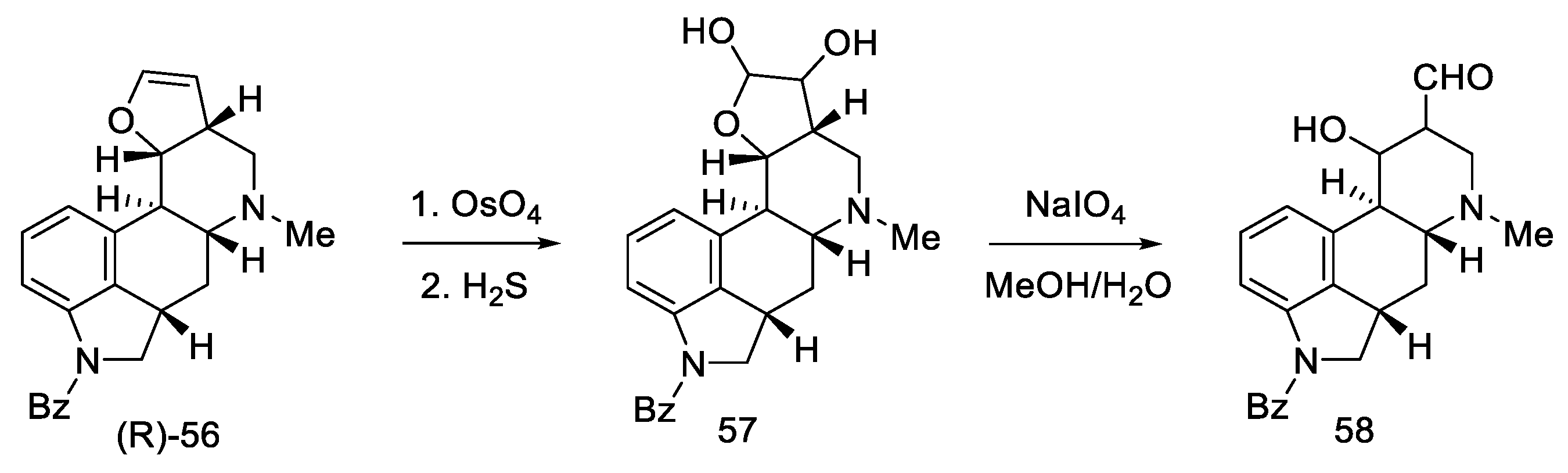
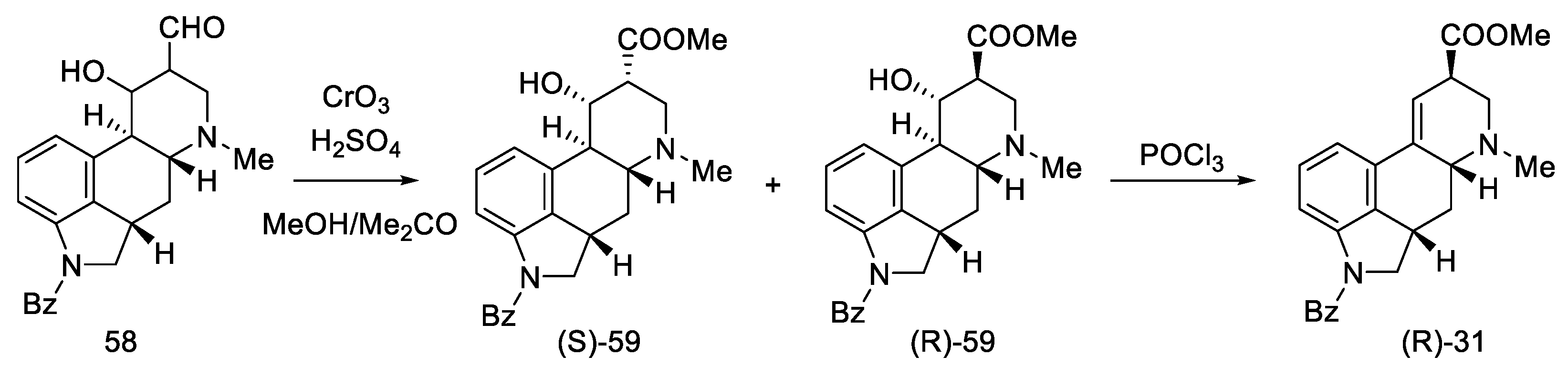
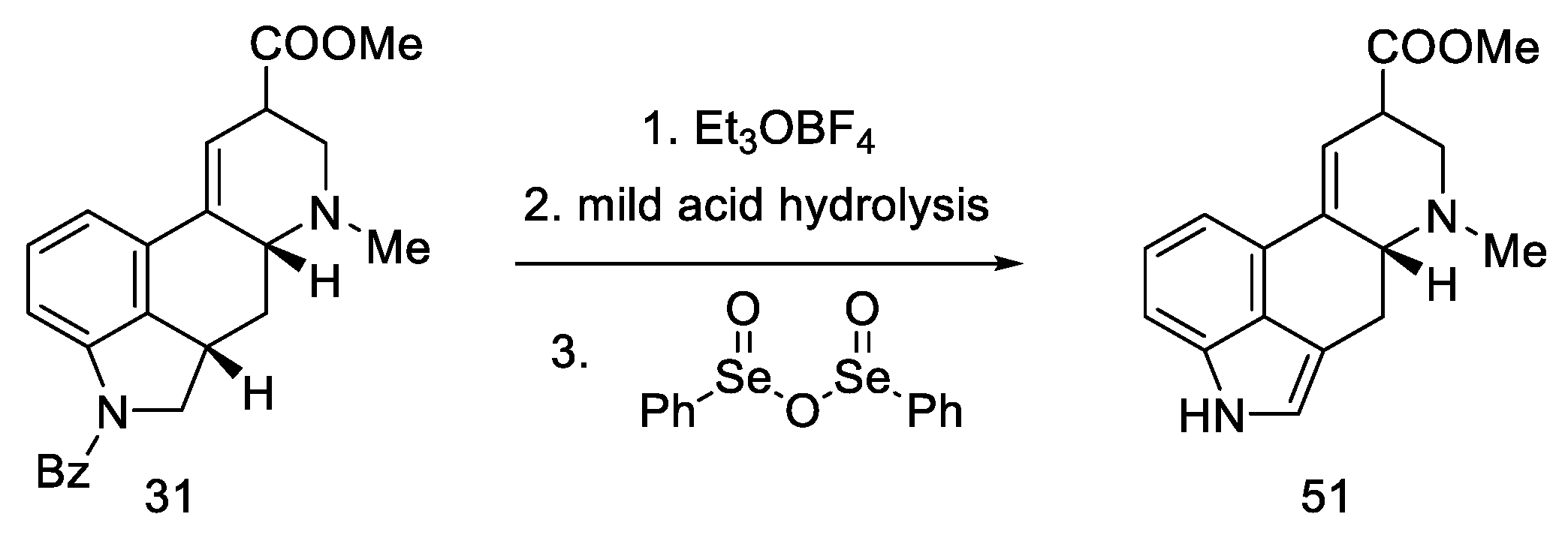
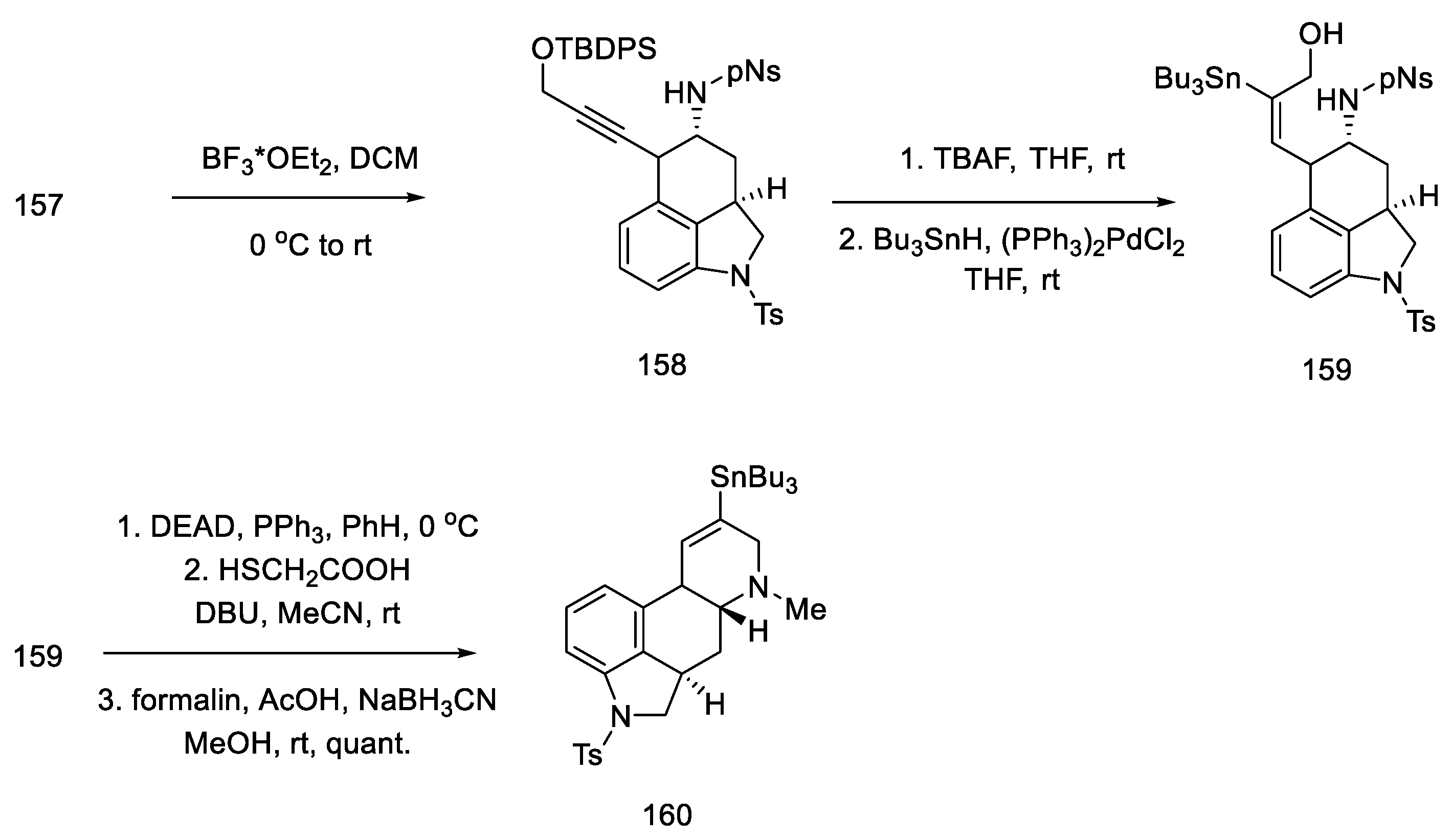
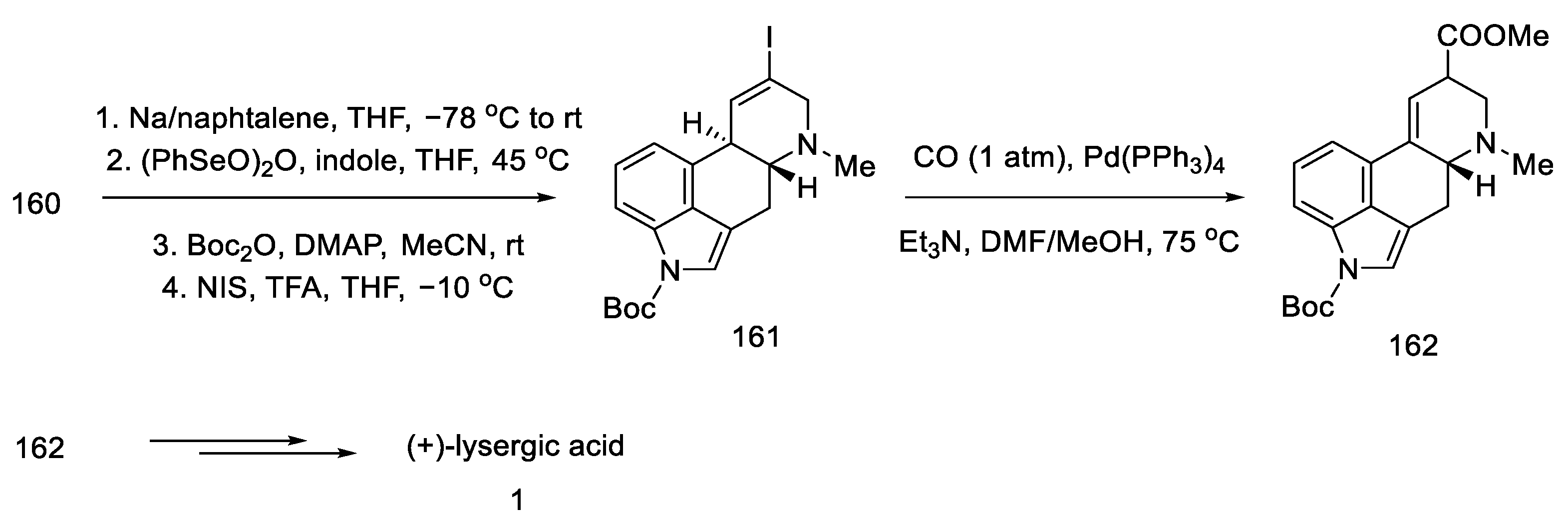
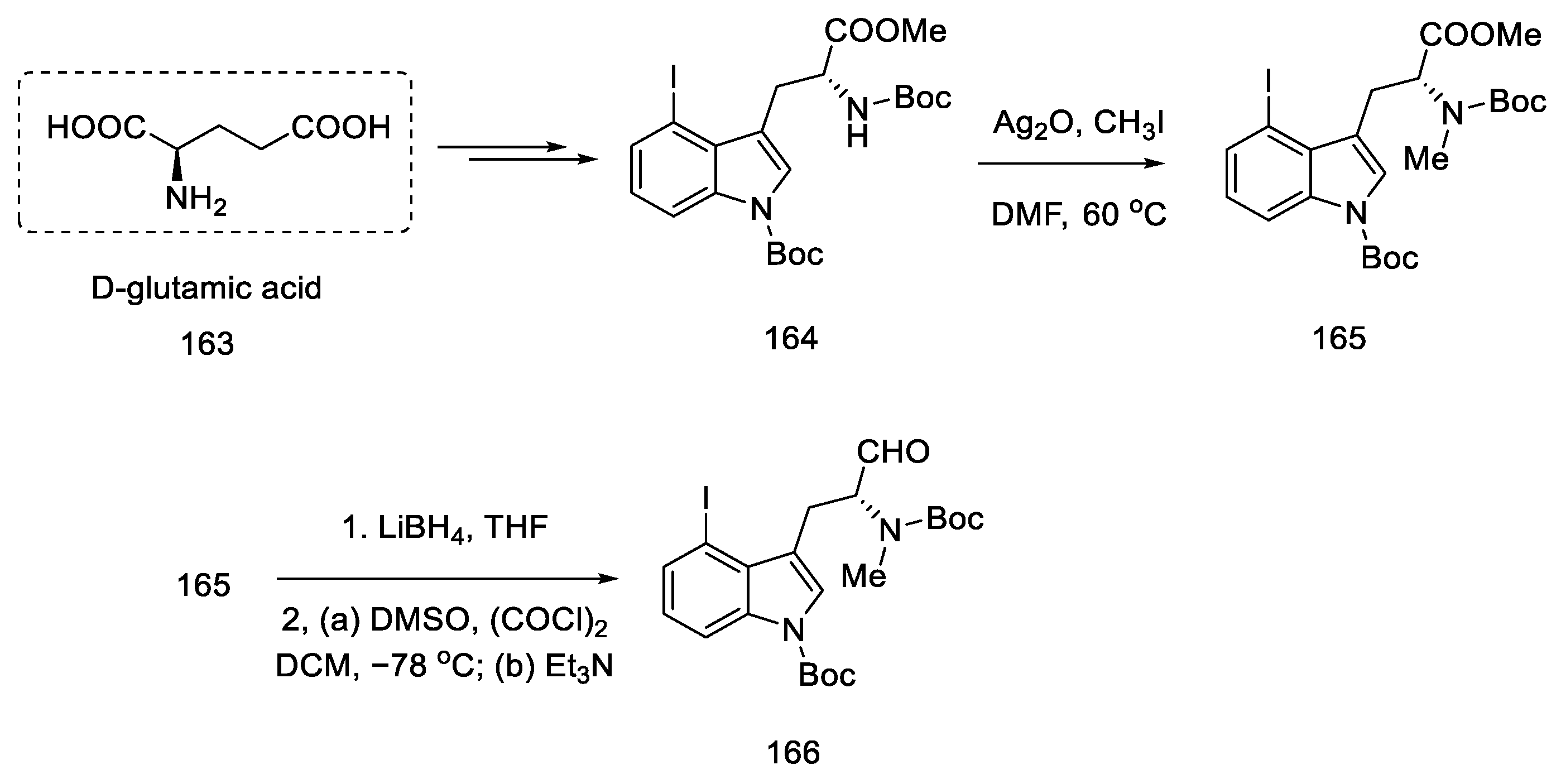
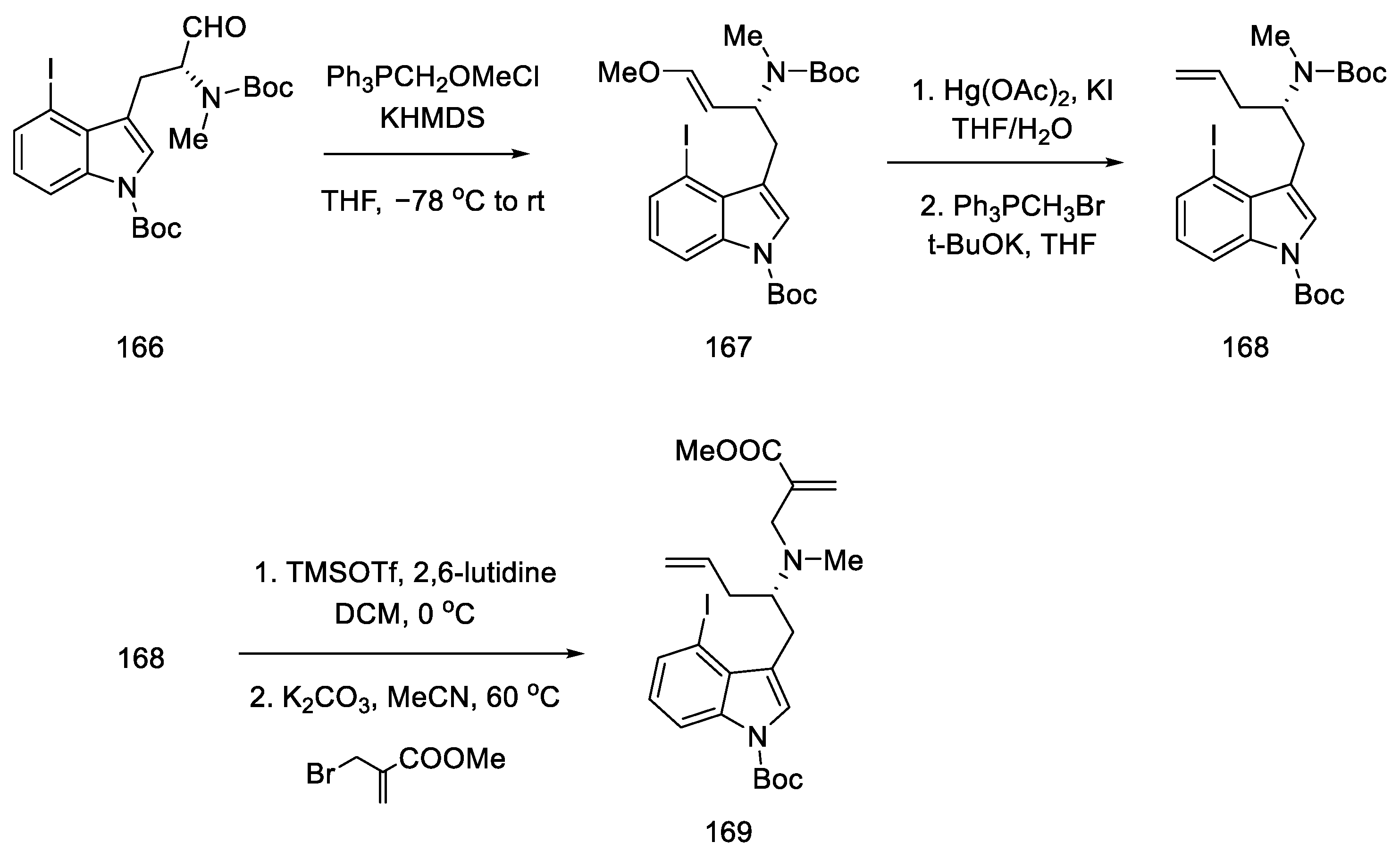
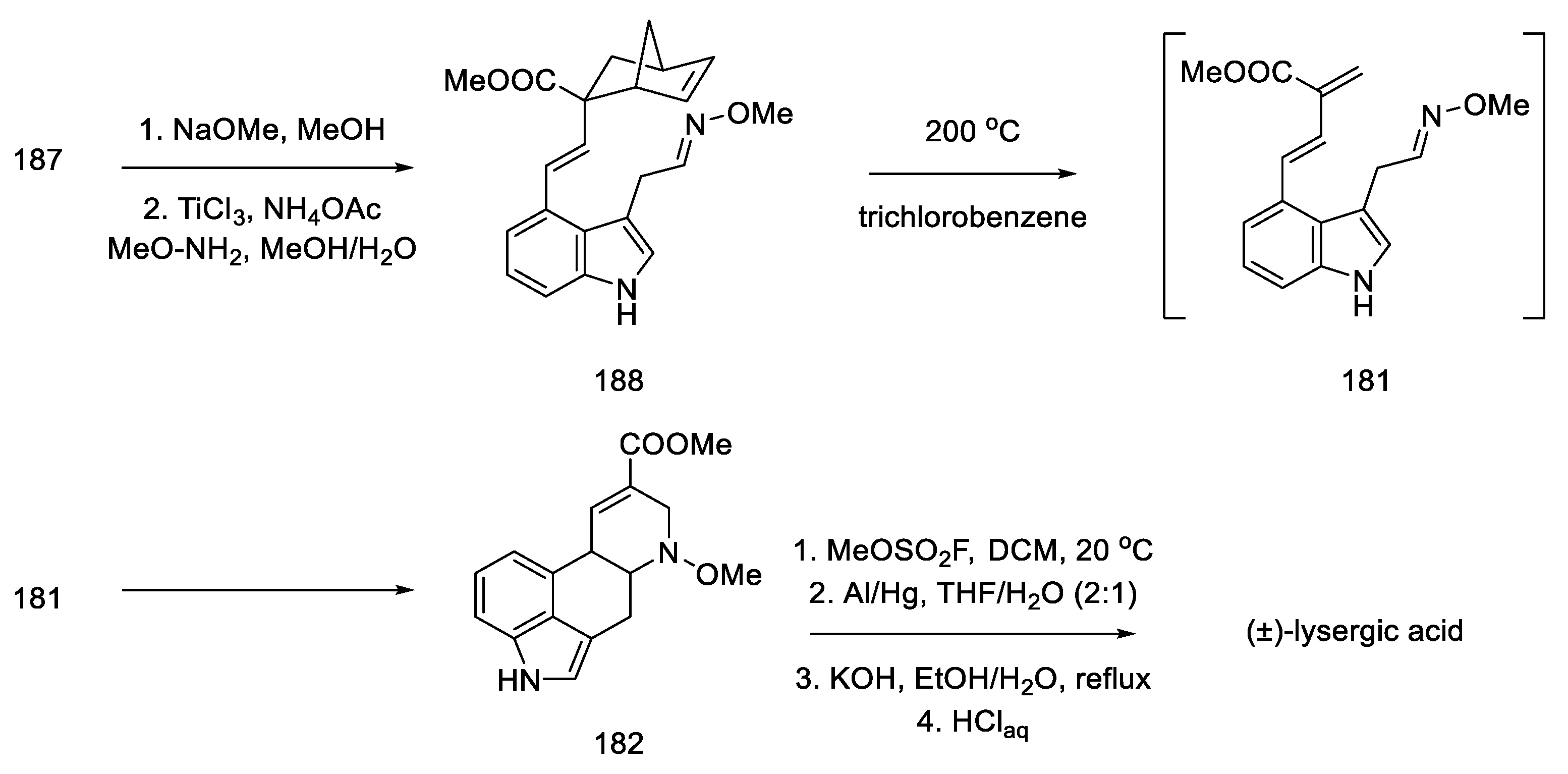
2.1.5. Kiguchi’s Research Group
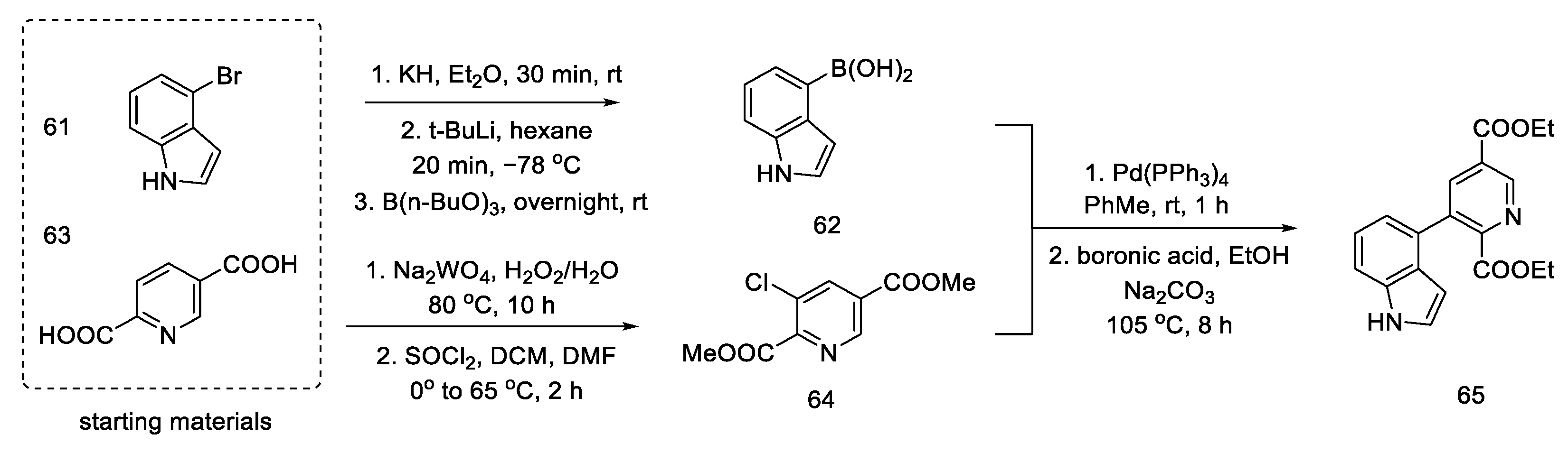
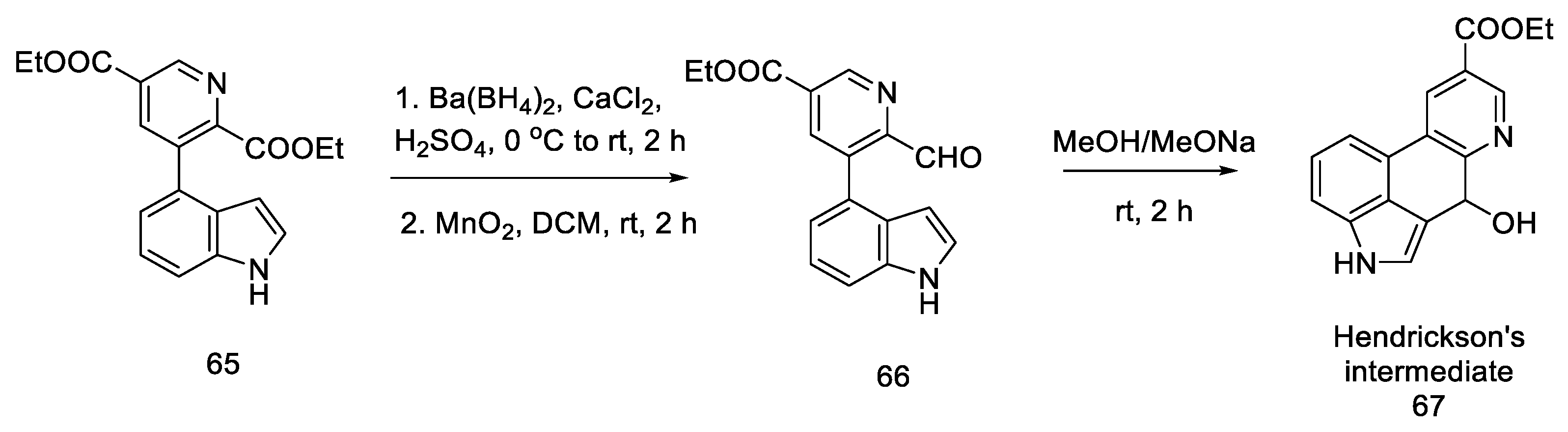
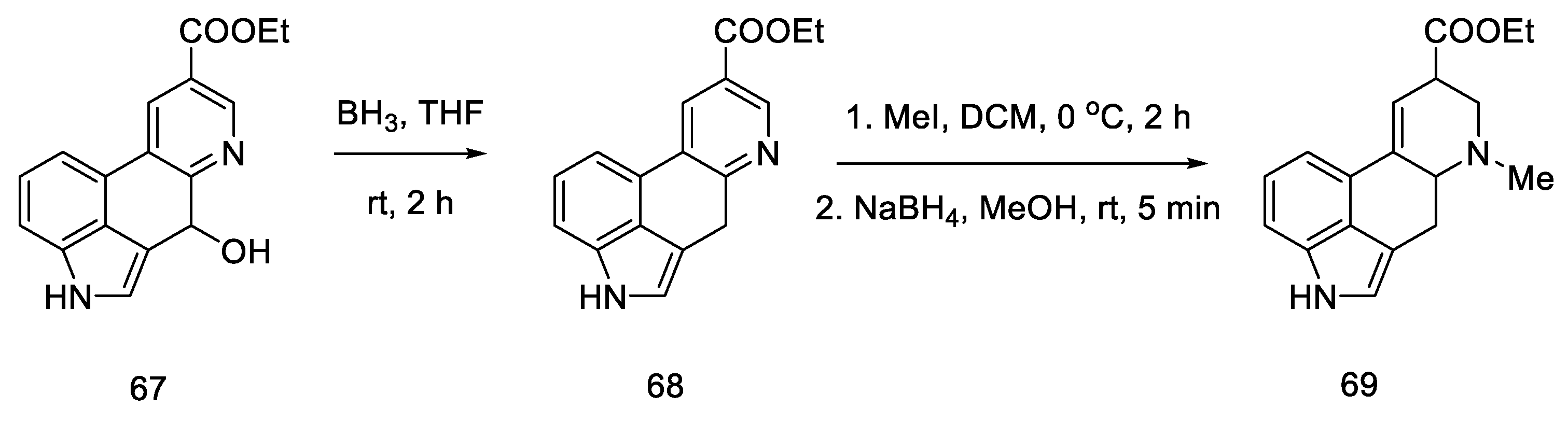
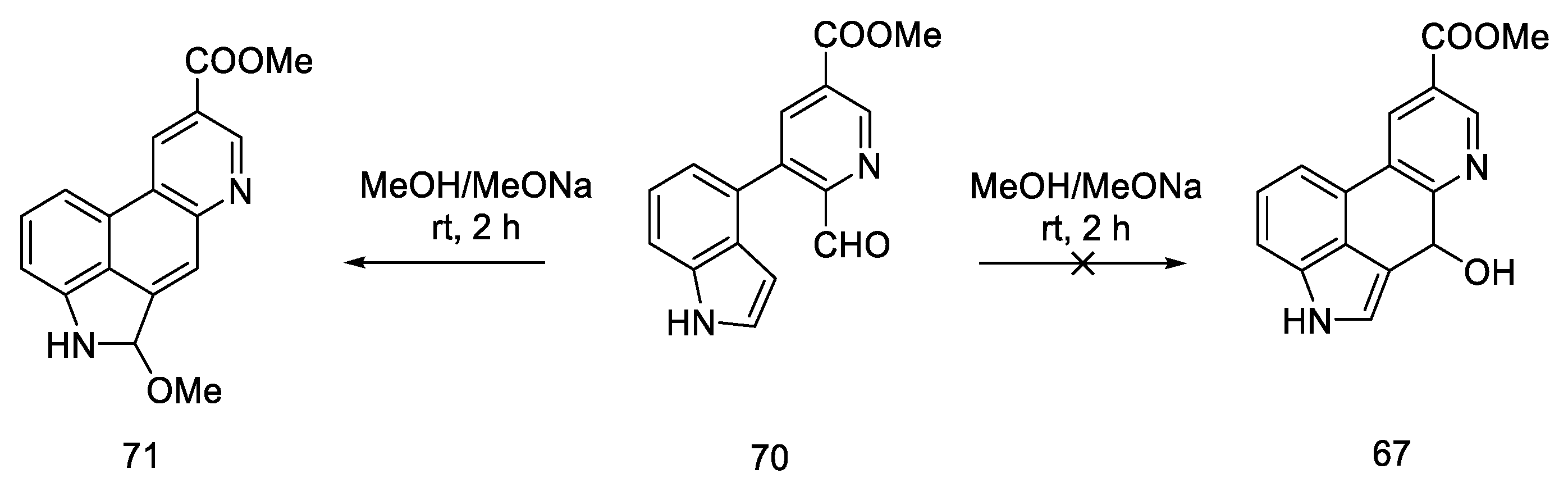
2.2. Hendrickson’s Strategy
2.2.1. Direct Hendrickson’s Research
2.2.2. Problems Noticed by Bekkam
2.2.3. Yigang Zhao Work
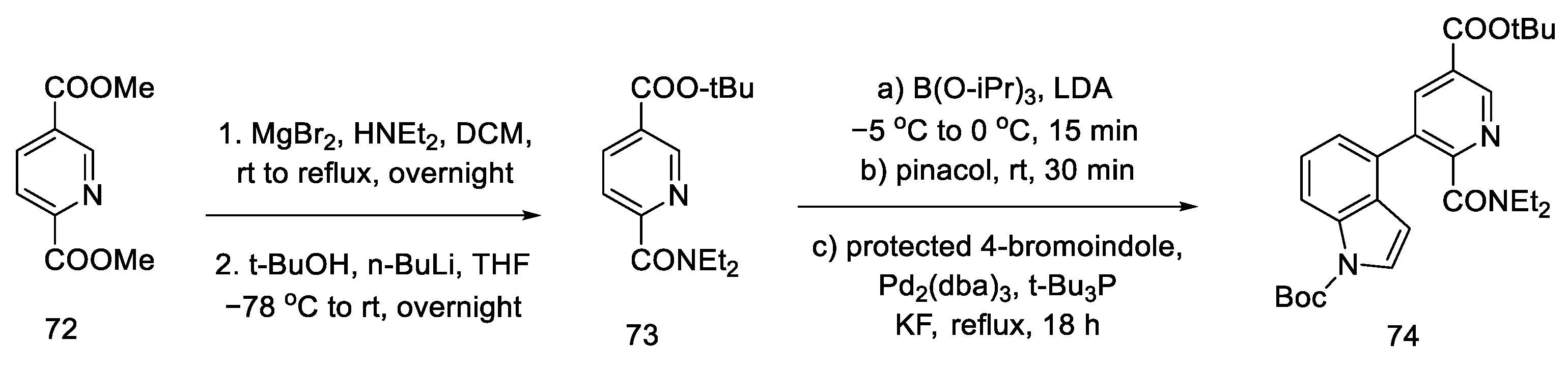
2.2.4. Beaundry’s Research
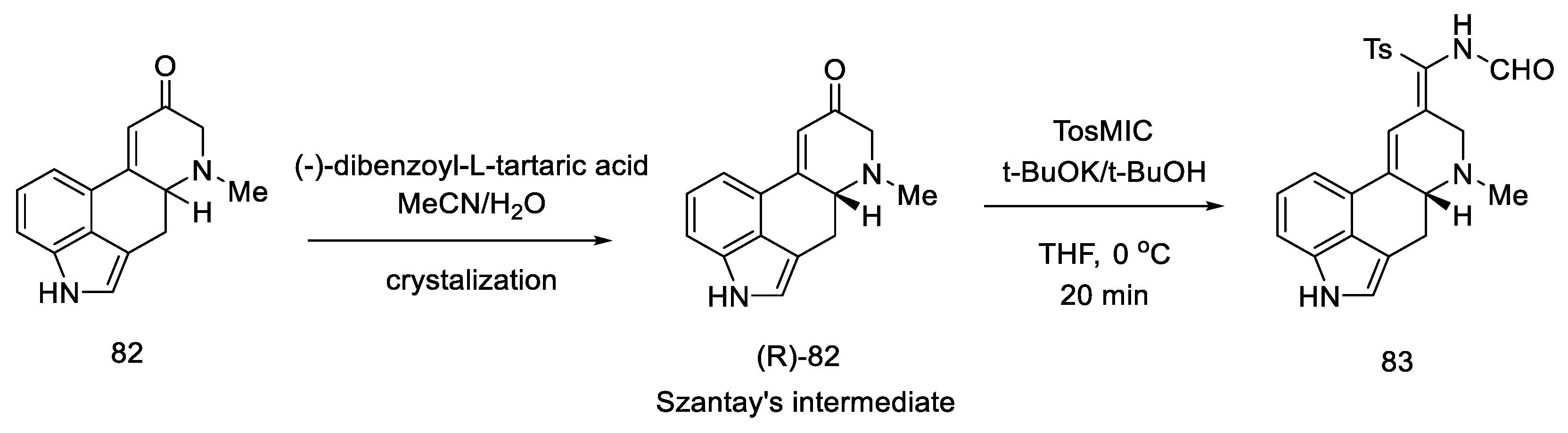
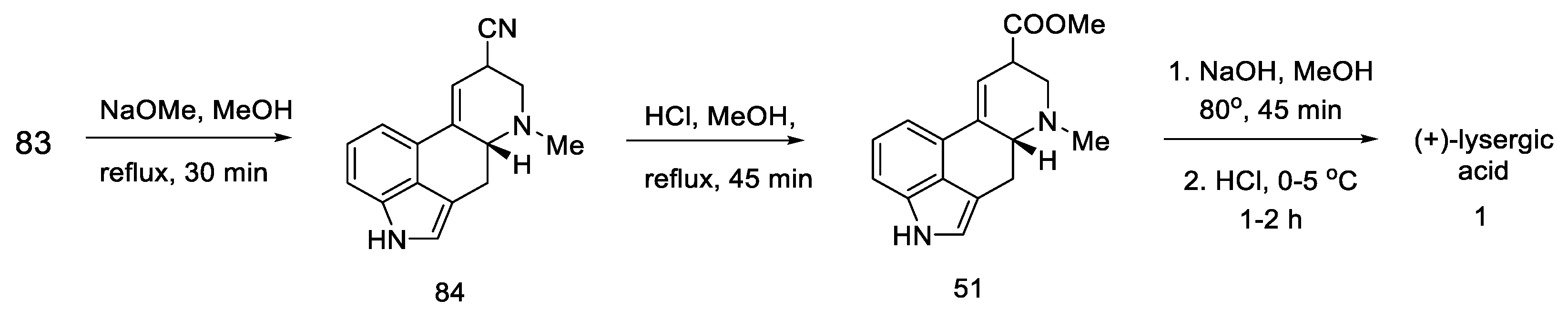
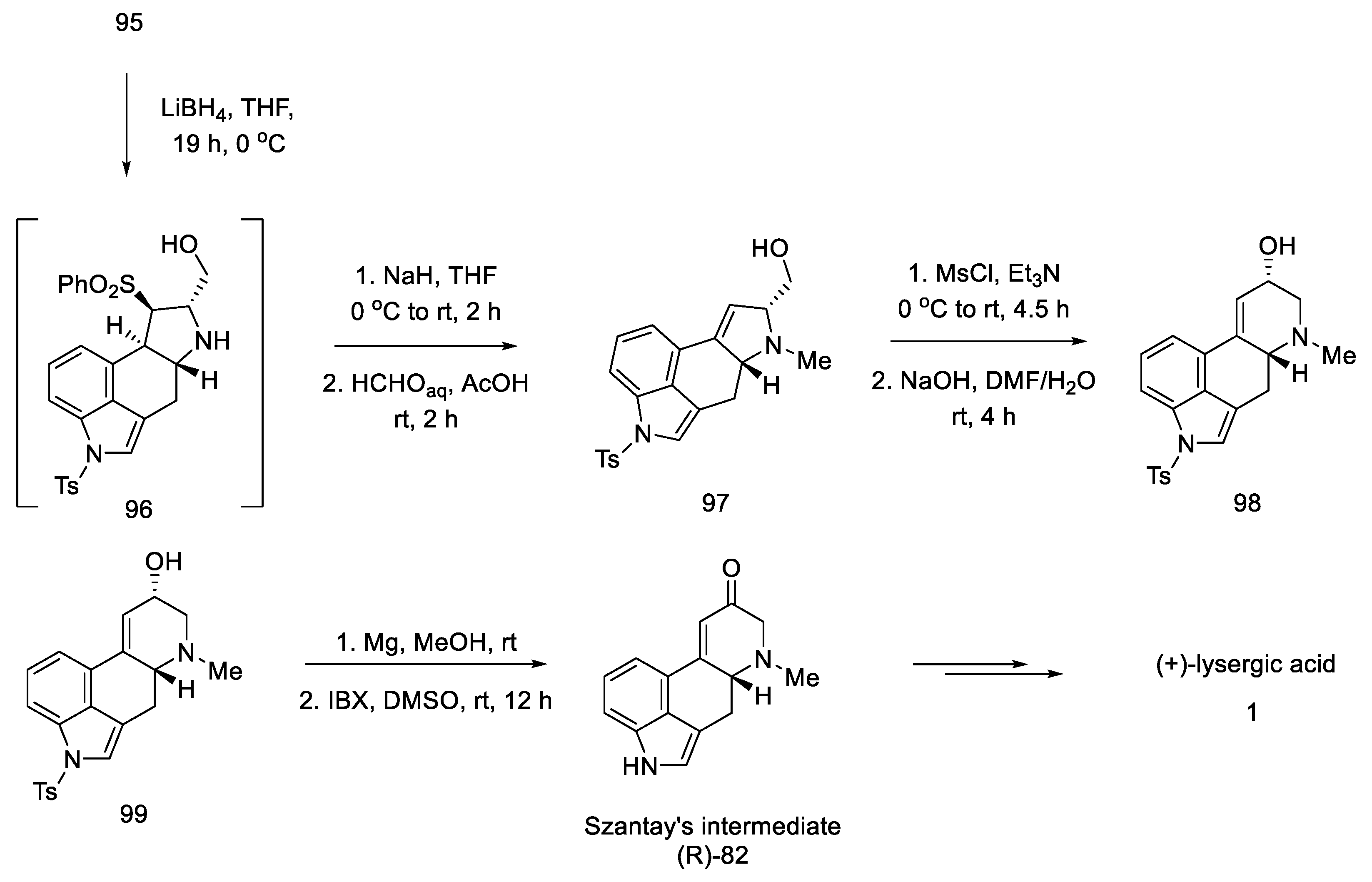
2.3. Szantay’s Strategy
2.3.1. Direct Szantay’s Research
2.3.2. Continuation of Szantay’s Research
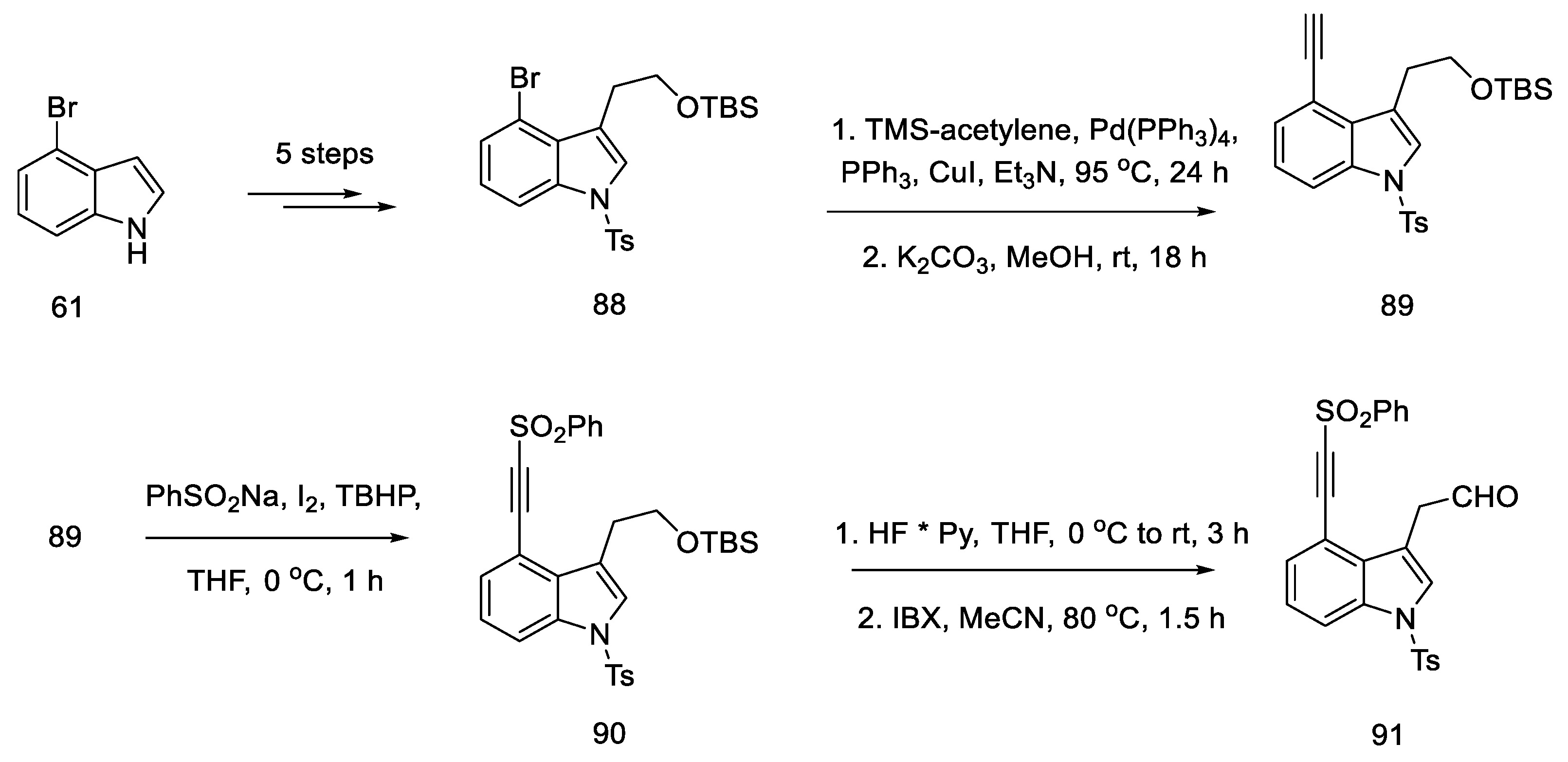
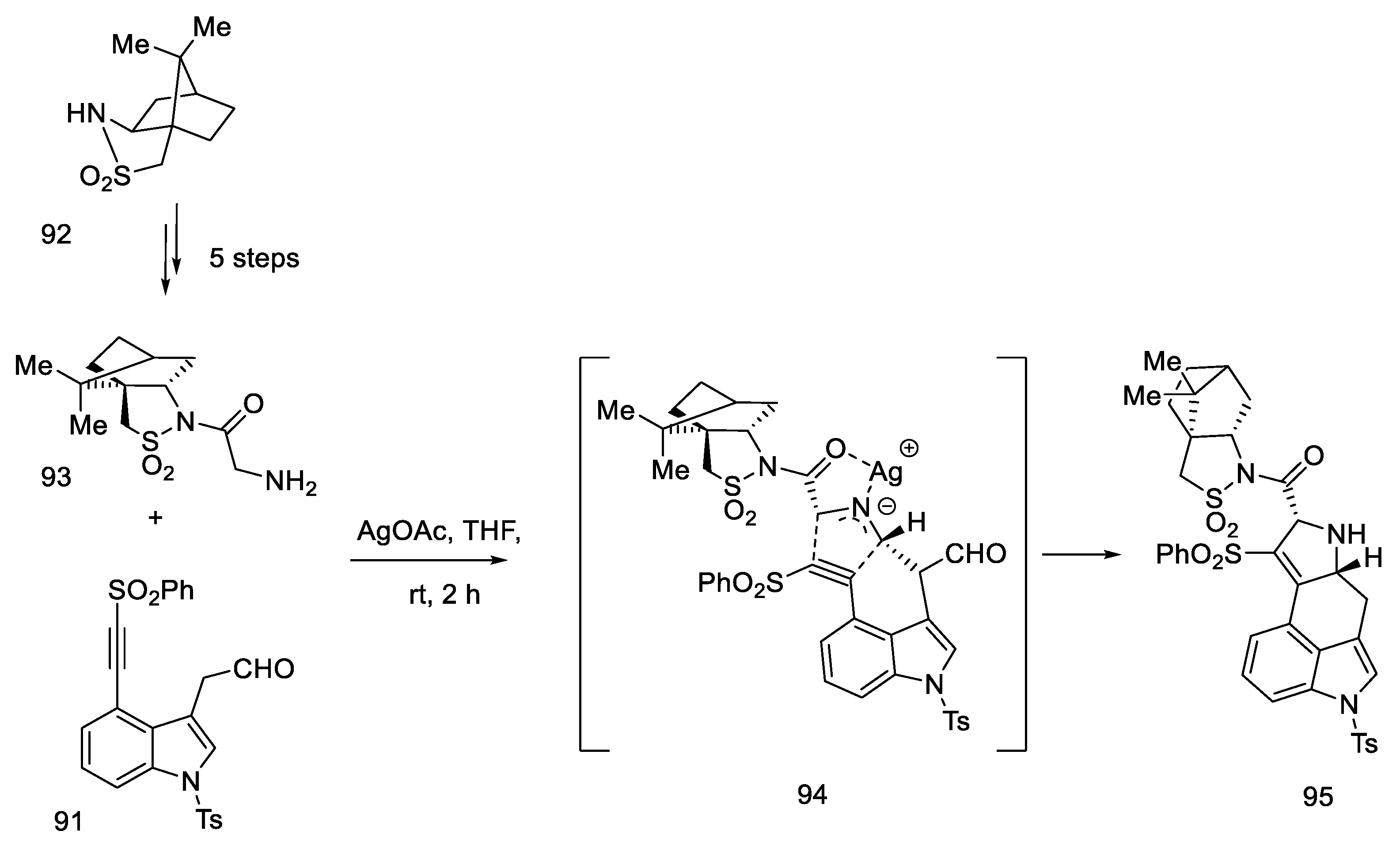
2.3.3. Garner’s Research
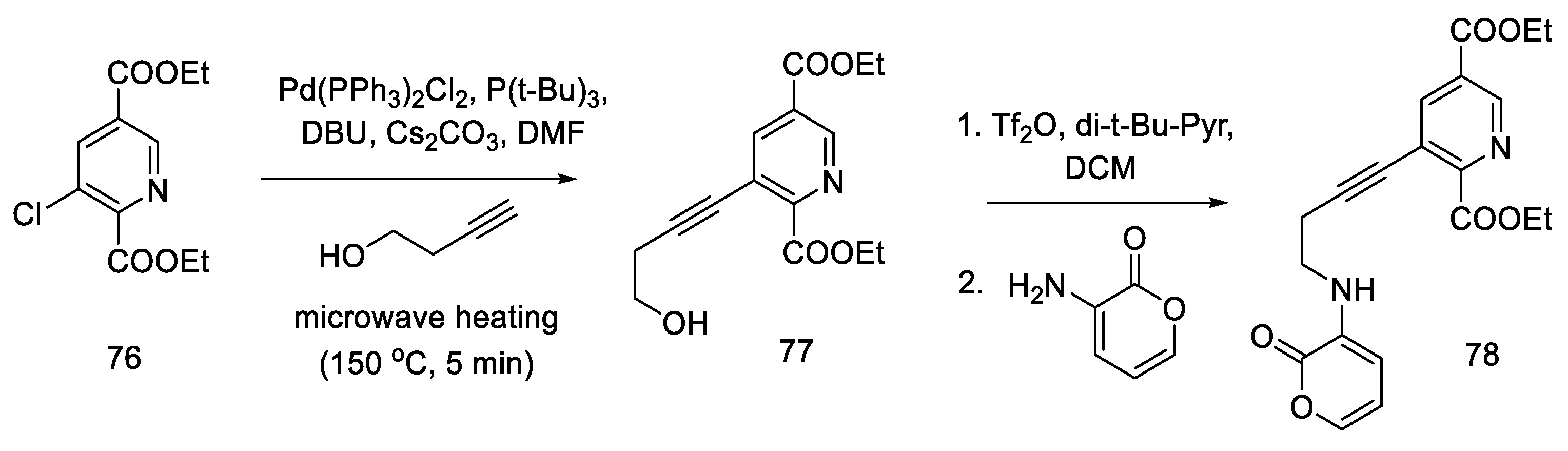
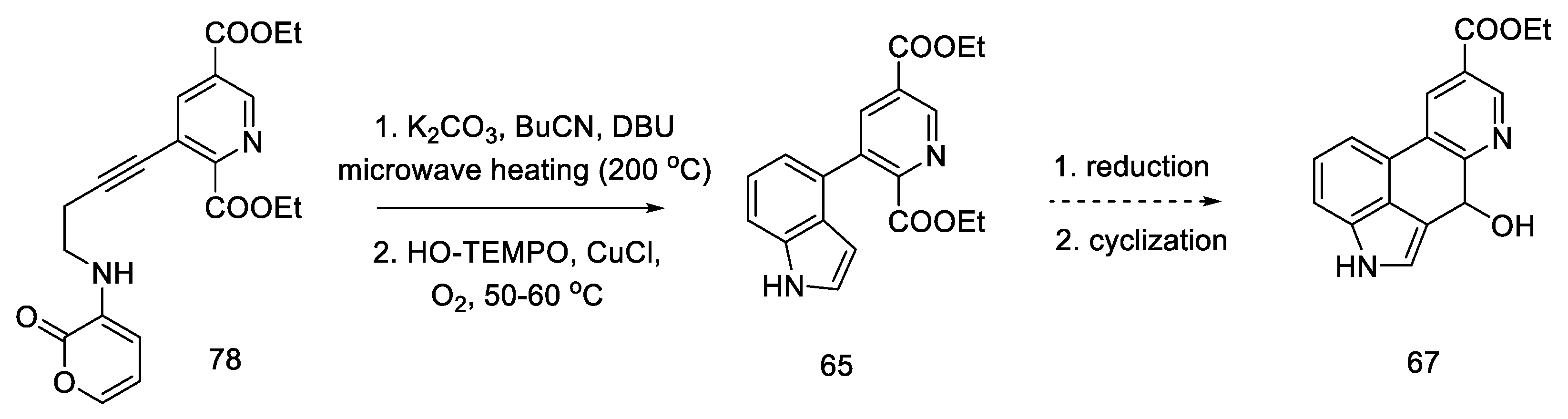
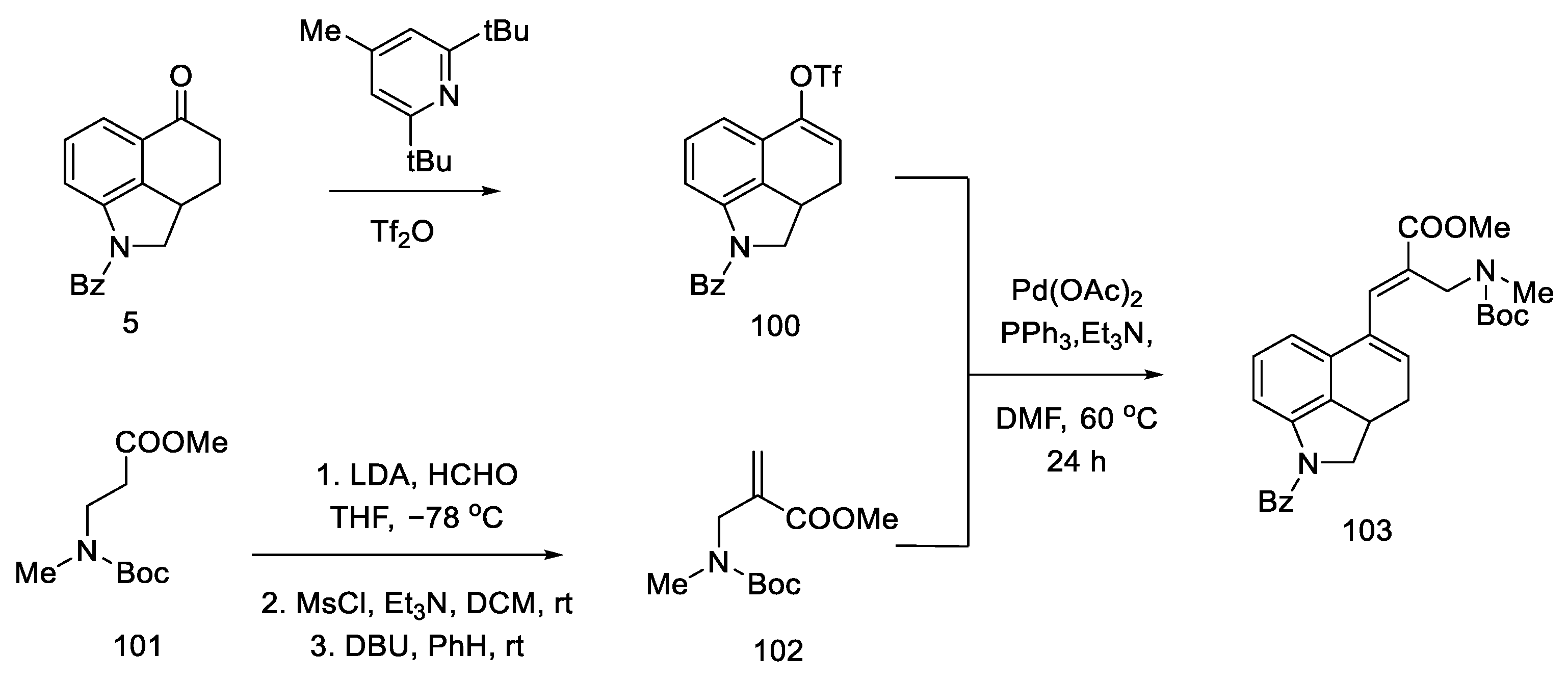
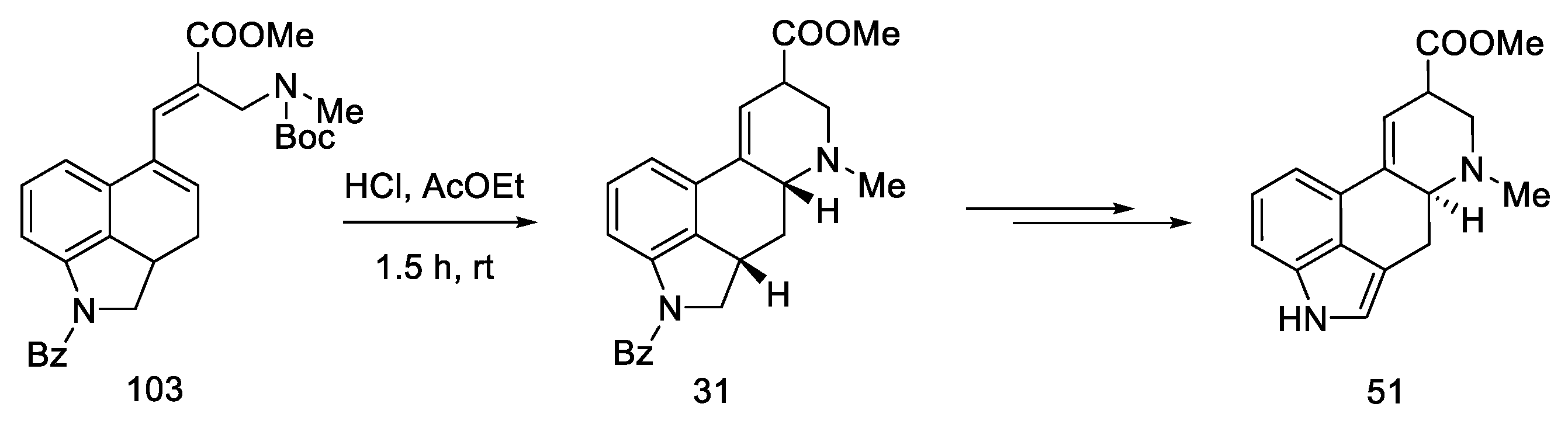
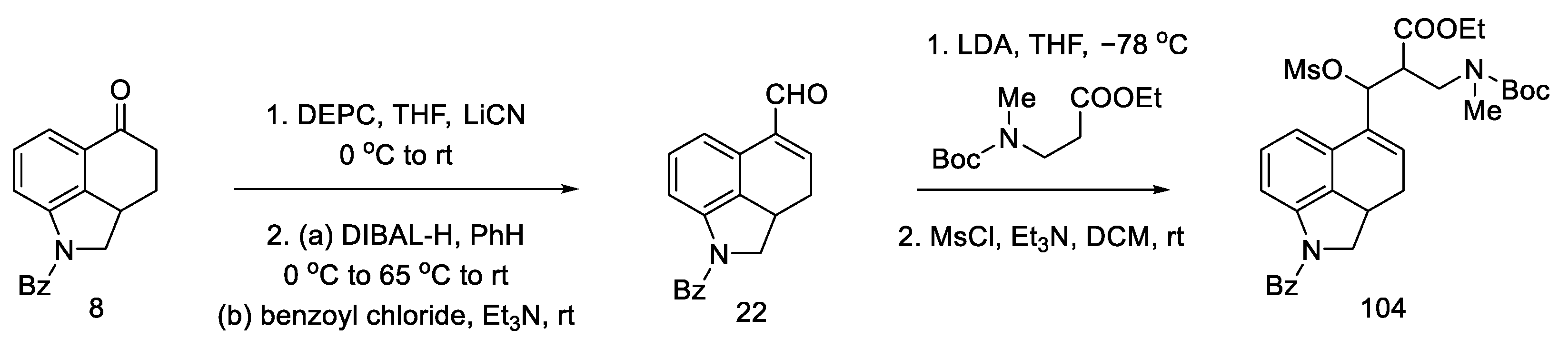
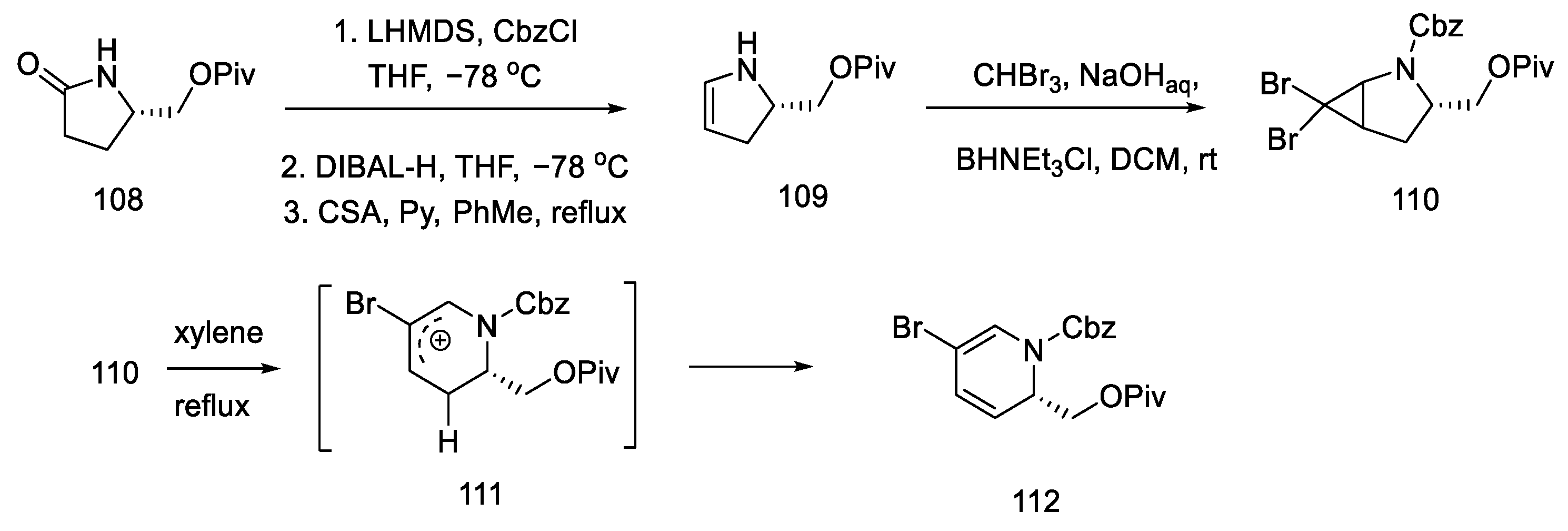
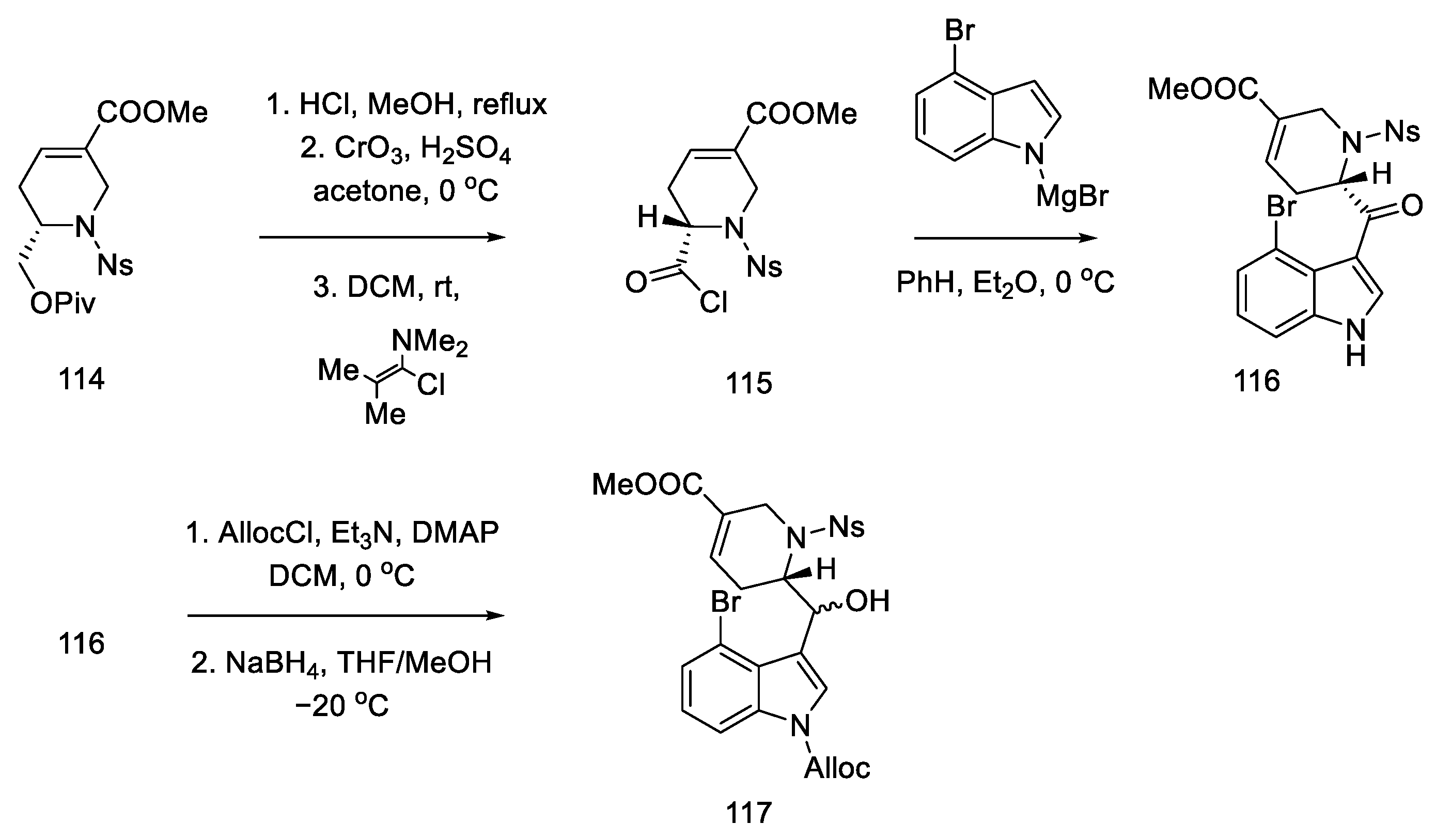
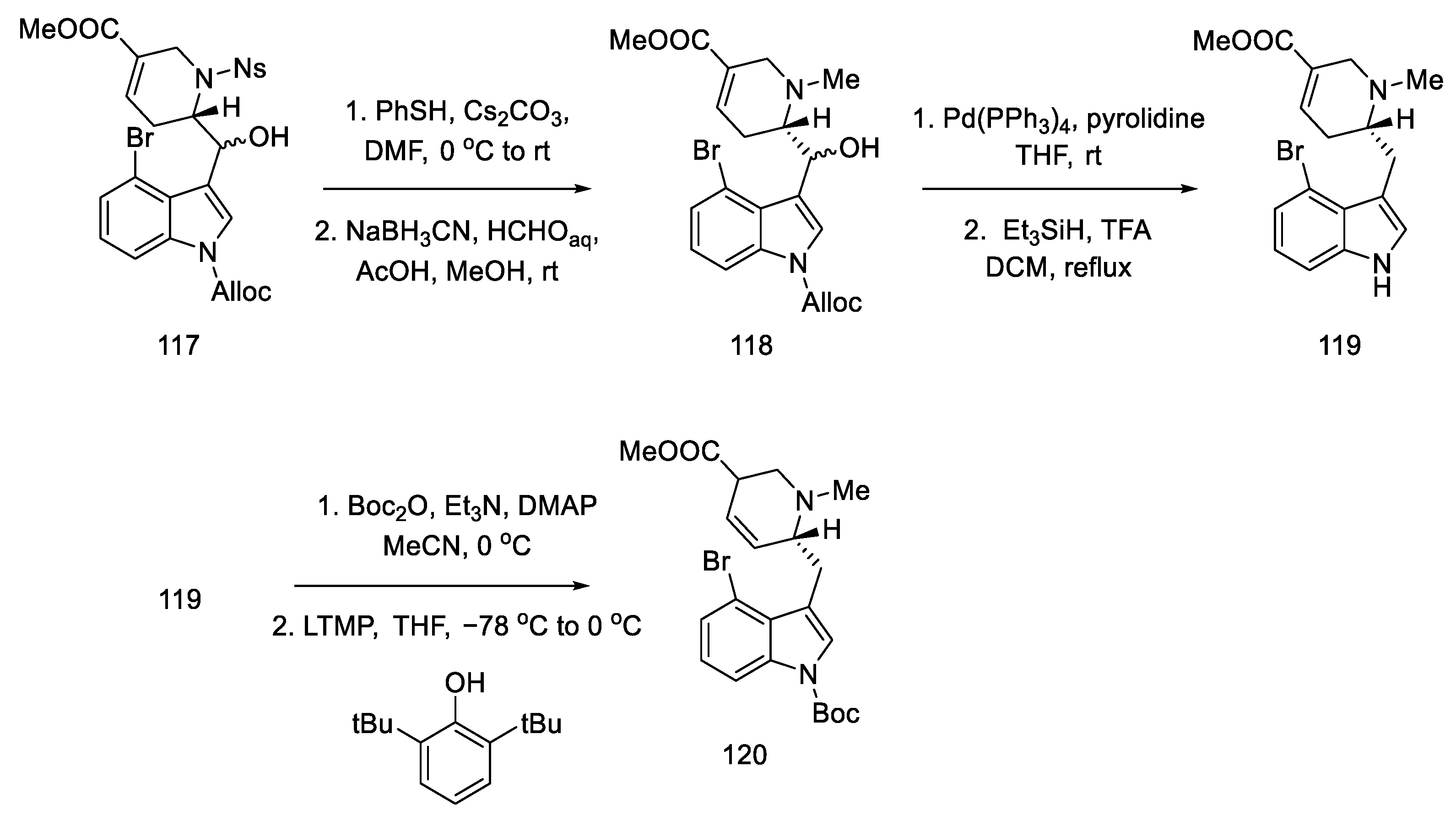
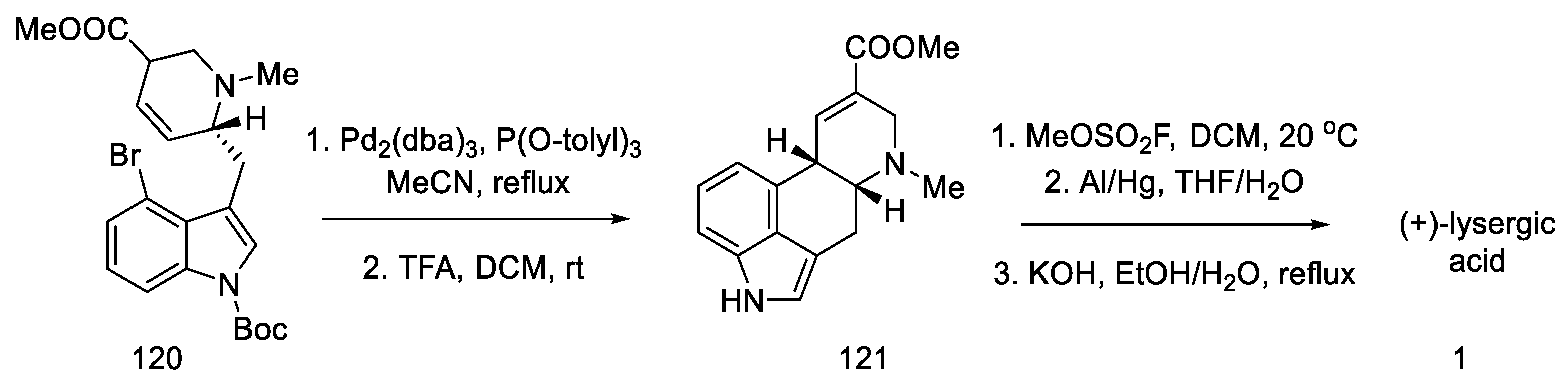
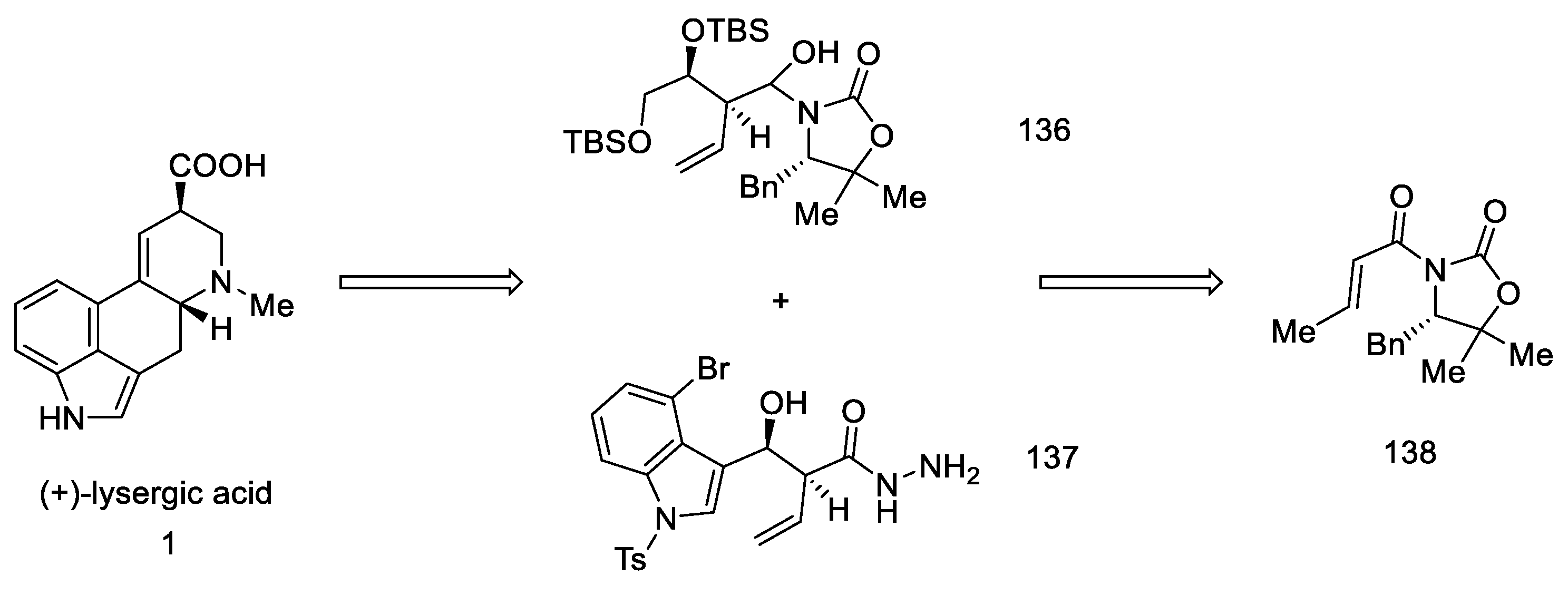
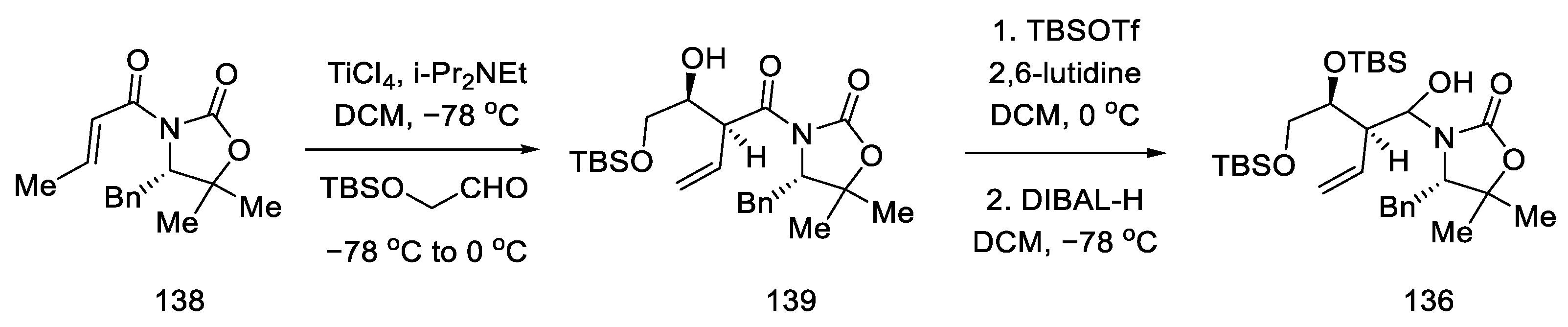
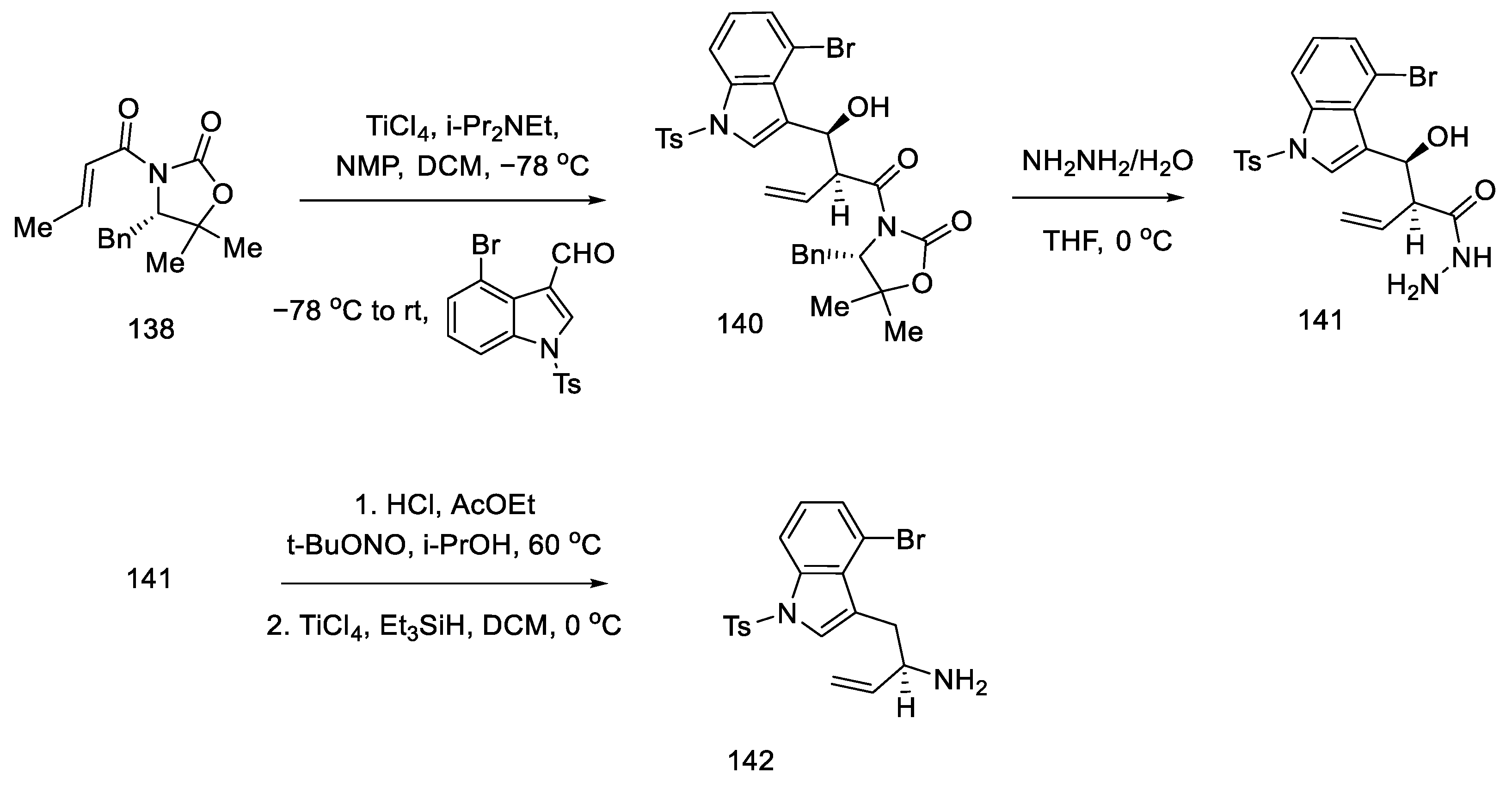
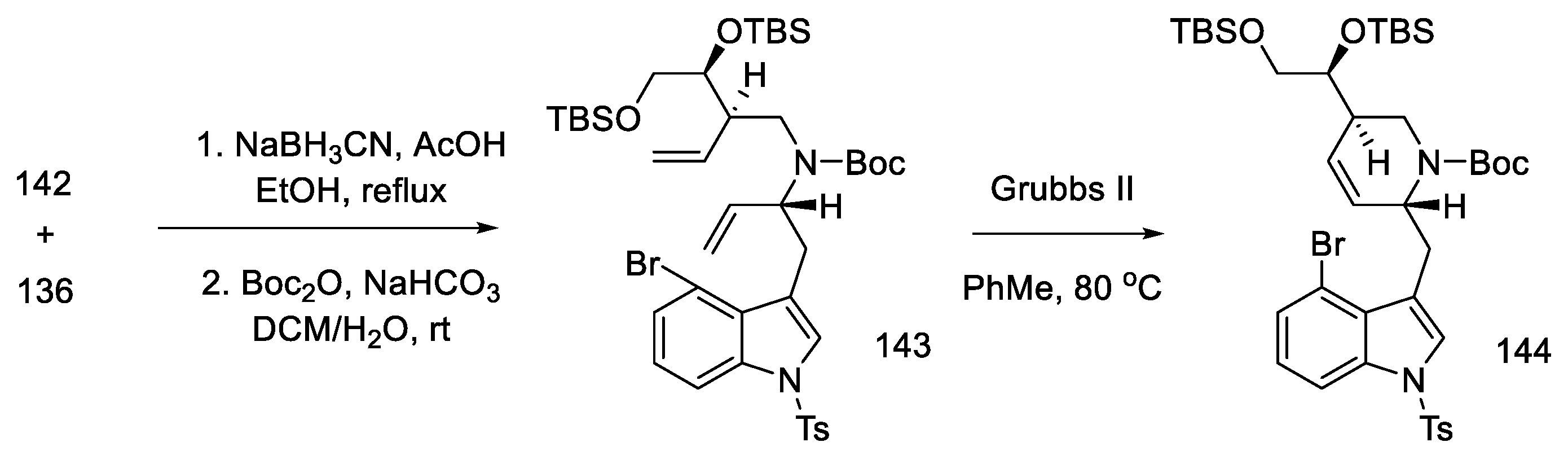
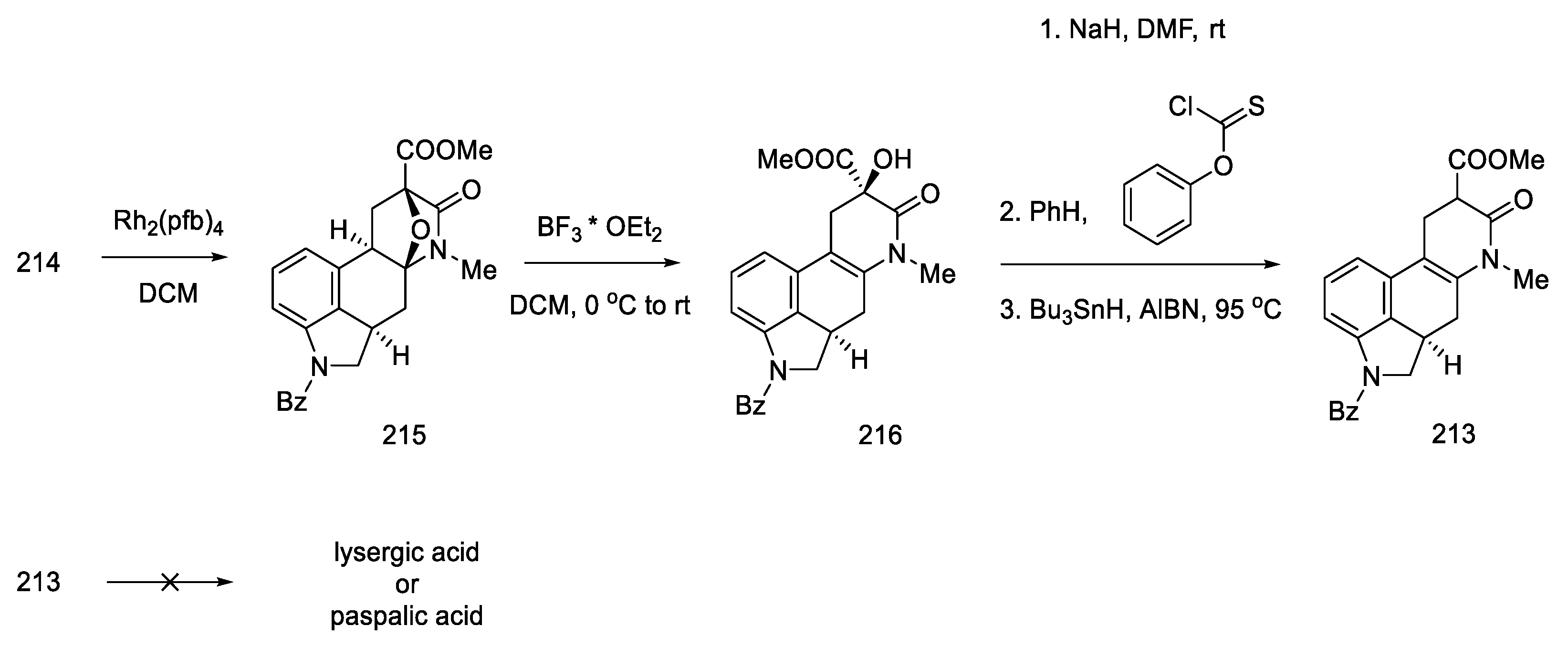
2.4. Closing the C/D-Ring Using the Heck Method
2.4.1. Ortar and Kurihara Groups
2.4.2. Fukuyama’s Research
2.4.3. Liu and Jia Research
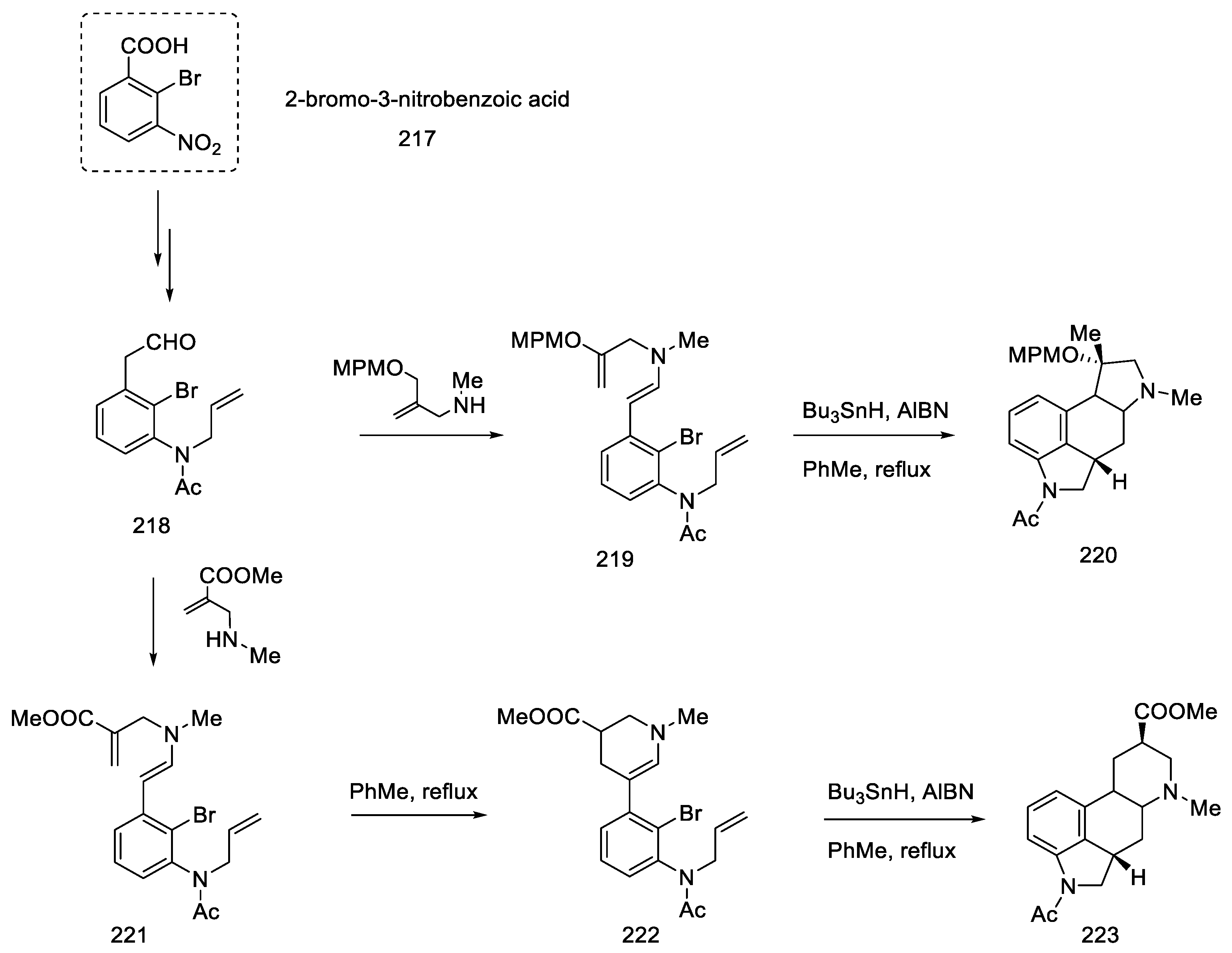
2.5. Other Chemical Methods of Obtaining Lysergic Acid
2.5.1. Oppolzer Group Research
2.5.2. Research Carried out by Inuki, Iwata. and Ohno
2.6. Attempts of Obtaining Lysergic Acid by Other Methods
2.6.1. Padwa Group Research
2.6.2. Parsons Group Research
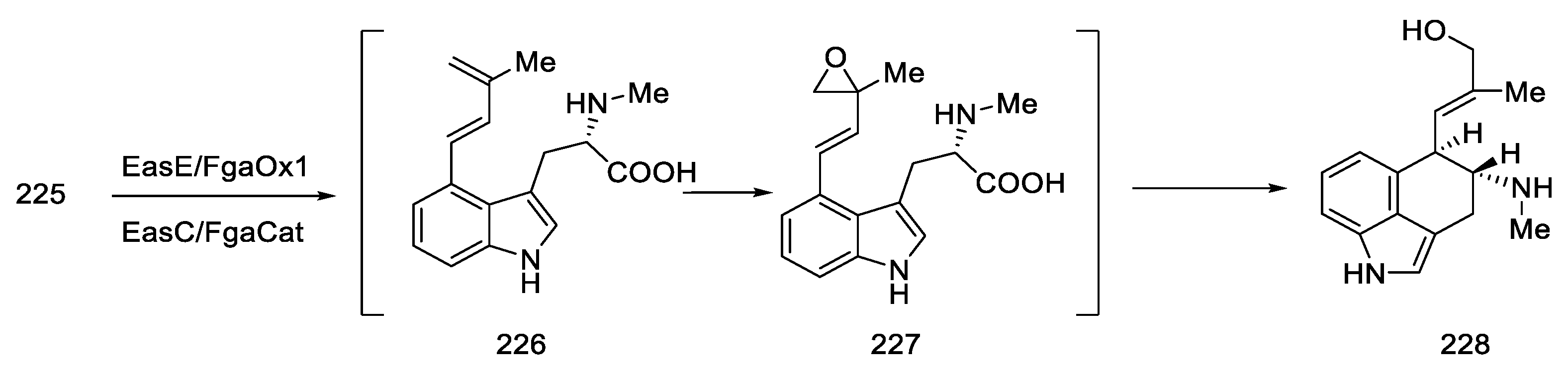
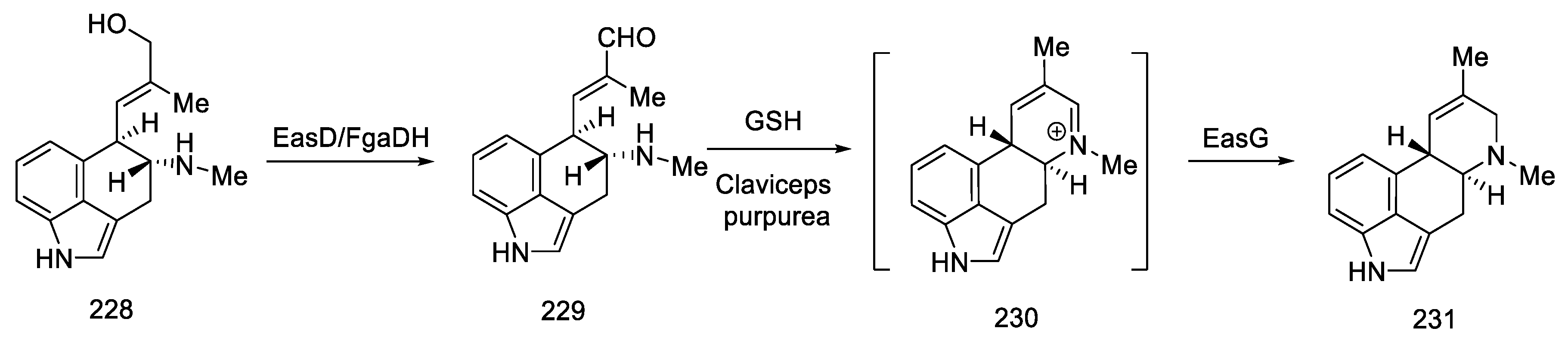
2.7. Biosynthetic Pathways of Lysergic Acid
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Ac | acetyl |
| AlBN | azobisisobutyronitrile |
| Alloc | allyloxycarbonyl |
| Aq | aqueous |
| BC | before Christ |
| Bn | benzyl |
| Boc | tert-Butyloxycarbonyl |
| Bz | benzoyl |
| °C | degree Celsius |
| Cat. | catalyst |
| Cbz | benzyloxycarbonyl |
| Cp | cyclopentadienyl |
| CSA | camphorsulfonic acid |
| Cy | cyclohexane |
| DAPCO | 1,4-diazabicyclo[2.2.2]octane |
| DBU | 1,8-diazabicyclo(5.4.0)undec-7-ene |
| DCE | 1,2-dichloroethane |
| DCM | dichloromethane or methylene chloride |
| DEAD | diethyl azodicarboxylate |
| DEPC | diethyl pyrocarbonate |
| DIAD | diisopropyl azodicarboxylate |
| DIBAL-H | diisobutylaluminium hydride |
| DMAP | 4-Dimethylaminopyridine (less frequently used for 4-Dimethylaminophenol) |
| DMAPP | dimethylallyl pyrophosphate |
| DMATS | dimethylallyl tryptophan synthase |
| DMF | dimethylformamide |
| DMSO | dimethyl sulfoxide |
| E. coli | lat. Escherichia coli |
| EAS | 5-epi-aristolochene synthase |
| e.g. | lat. exempli gratia, for example |
| Et | ethyl |
| Fmoc | fluorenylmethoxycarbonyl |
| Fr. | short name of discoverer (botanist) Elias Magnus Fries |
| Grubbs II | second-generation Grubbs catalyst |
| GSH | glutathione |
| HMPA | Hexamethylphosphoramide |
| hν | quantum of electromagnetic radiation |
| IBX | 2-Iodoxybenzoic acid |
| Im | imidazole |
| IPNBSH | N-Isopropylidene-N′-2-nitrobenzenesulfonyl hydrazine |
| iPr | isopropyl |
| HMDS | bis(trimethylsilyl)amide |
| Lat. | from Latin |
| LDA | lithium diisopropylamide |
| LSD | lysergic acid diethylamide |
| LTMP | lithium 2,2,6,6-tetramethylpiperidide |
| MDM | trisiloxane, octamethyl- |
| Me | methyl |
| MRSA | methicyllin-resistant Staphylococcus aureus |
| Ms | mesylate |
| NAD+/NADH | nicotinamide adenine dinucleotide (oxidized and reduced forms) |
| nBu | n-Buthyl |
| NCS | N-Chlorosuccinimide |
| NiRaney | Raney catalyst |
| NIS | N-Iodosuccinimide |
| NMO | N-Methylmorpholine N-oxide |
| NMP | N-Methyl-2-pyrrolidone |
| Ns (pNs) | nosyl, 4-Nitrobenzenesulfonyl |
| P-450 | cytochrome, superfamily of enzymes containing heme that functions as monooxygenases |
| Pfb | perfluorobutane |
| PG | protecting group |
| Ph | phenyl |
| Piv | pivalic |
| PNB | p-nitrobenzyl |
| PS (lipase) | Pseudomonas |
| PTSA | p-Toluenesulfonic acid, TsOH |
| Py/Pyr | pyridine |
| R | any functional group/further part of molecules |
| rt | room temperature |
| SAM | S-adenosyl methionine |
| SDR | short-chain dehydrogenases/reductases |
| Sia | 1,2-dimethylpropyl |
| TBAF | tetra-n-butylammonium fluoride |
| TBDMS | tert-Butyldimethylsilyl |
| TBHP | tert-Butyl hydroperoxide |
| tBu | tert-butyl |
| TEMPO | 2,2,6,6-Tetramethylpiperidine 1-oxyl |
| Tf | triflate |
| TFA | trifluoroacetic acid |
| TFE | trifluoroethyl alcohol |
| THF | tetrahydrofuran |
| TIPS | triisopropylsilane |
| TMS | tetramethylsilane |
| TosMIC | toluenesulfonylmethyl isocyanide |
| Ts | tosyl, toluenesulfonyl |
| Tul. | short name of discoverer (botanist) Louis Rene Edmond Tulasne |
| USA | United States of America |
| Var. | lat. Varietas, variety |
| Δ | gr. Delta, rise of temperature |
| μ | mikro (prefix) |
References
- Ochodzki, P.; National Research Institute. Zapobieganie Zagrożeniom Związanym z Występowaniem w Ziarnie Zbóż Alkaloidów Pasożytniczego Grzyba Buławinki Czerwonej (Sporysz)—Prevention of Risks Associated with the Presence of Alkaloids in the Cereal Grain Parasitic Fungus of Ergot; Expertise: Radzików, Poland, 2015. [Google Scholar]
- Schiff, P.L. Ergot and Its Alkaloids. Am. J. Pharm. Educ. 2006, 70, 98. [Google Scholar] [CrossRef]
- Smakosz, A.; Kurzyna, W.; Rudko, M.; Dąsal, M. The Usage of Ergot (Claviceps Purpurea (Fr.) Tul.) in Obstetrics and Gynecology: A Historical Perspective. Toxins 2021, 13, 492. [Google Scholar] [CrossRef] [PubMed]
- Krska, R.; Crews, C. Significance, Chemistry and Determination of Ergot Alkaloids: A Review. Food Addit. Contam. Part A 2008, 25, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Berde, B.; Schild, H.O. (Eds.) Ergot Alkaloids and Related Compounds; Springer: Berlin/Heidelberg, Germany, 1978. [Google Scholar]
- Stoll, A.; Petrzilka, T.; Rutschmann, J.; Hofmann, A.; Günthard, H.H. Über Die Stereochemie Der Lysergsäuren Und Der Dihydro-Lysergsäuren. 37. Mitteilung Über Mutterkornalkaloide. Helv. Chim. Acta 1954, 37, 2039–2057. [Google Scholar] [CrossRef]
- Hofmann, A.; Ott, H.; Griot, R.; Stadler, P.A.; Frey, A.J. Die Synthese Und Stereochemie Des Ergotamins. (58. Mitteilung Über Mutterkornalkaloide). Helv. Chim. Acta 1963, 46, 2306–2328. [Google Scholar] [CrossRef]
- Flieger, M.; Wurst, M.; Shelby, R. Ergot Alkaloids—Sources, Structures and Analytical Methods. Folia Microbiol. 1997, 42, 3–30. [Google Scholar] [CrossRef] [PubMed]
- Gerhards, N.; Neubauer, L.; Tudzynski, P.; Li, S.-M. Biosynthetic Pathways of Ergot Alkaloids. Toxins 2014, 6, 3281–3295. [Google Scholar] [CrossRef]
- Matławska, I. Farmakognozja; UM: Poznań, Poland, 2008. [Google Scholar]
- Seeman, P. Historical Overview: Introduction to the Dopamine Receptors. In The Dopamine Receptors; Humana Press: Totowa, NJ, USA, 2010; pp. 1–21. [Google Scholar]
- Pucadyil, T.J.; Kalipatnapu, S.; Chattopadhyay, A. The Serotonin1A A Receptor: A Representative Member of the Serotonin Receptor Family. Cell. Mol. Neurobiol. 2005, 25, 553–580. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L.; Geyer, M.A. Multiple Receptors Contribute to the Behavioral Effects of Indoleamine Hallucinogens. Neuropharmacology 2011, 61, 364–381. [Google Scholar] [CrossRef] [Green Version]
- Nichols, D.E. Psychedelics. Pharmacol. Rev. 2016, 68, 264–355. [Google Scholar] [CrossRef] [PubMed]
- Watts, V.J.; Mailman, R.B.; Lawler, C.P.; Neve, K.A.; Nichols, D.E. LSD and Structural Analogs: Pharmacological Evaluation at D1 Dopamine Receptors. Psychopharmacology 1995, 118, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Bijoch, Ł.; Pękała, M.; Beroun, A. Molekularne Podstawy Działania Wybranych Substancji Psychoaktywnych. Postepy Biochem. 2021, 67, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, J.J.; Fonseca, F.; Elices, M.; Farré, M.; Torrens, M. Therapeutic Use of LSD in Psychiatry: A Systematic Review of Randomized-Controlled Clinical Trials. Front. Psychiatry 2020, 10, 943. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.E.; Raswyck, G.E.; Dickerson Davidson, L. From Hofmann to the Haight Ashbury, and into the Future: The Past and Potential of Lysergic Acid Diethlyamide. J. Psychoact. Drugs 2014, 46, 3–10. [Google Scholar] [CrossRef]
- de Groot, A.N.; van Dongen, P.W.J.; Vree, T.B.; Hekster, Y.A.; van Roosmalen, J. Ergot Alkaloids. Drugs 1998, 56, 523–535. [Google Scholar] [CrossRef]
- Johnson, J.W.; Ellis, M.J.; Piquette, Z.A.; MacNair, C.; Carfrae, L.; Bhando, T.; Ritchie, N.E.; Saliba, P.; Brown, E.D.; Magolan, J. Antibacterial Activity of Metergoline Analogues: Revisiting the Ergot Alkaloid Scaffold for Antibiotic Discovery. ACS Med. Chem. Lett. 2022, 13, 284–291. [Google Scholar] [CrossRef]
- Webster, J. Dopamine Agonist Therapy in Hyperprolactinemia. J. Reprod. Med. 1999, 44, 1105–1110. [Google Scholar]
- Hamon, M.; Mallat, M.; Herbet, A.; Nelson, D.L.; Audinot, M.; Pichat, L.; Glowinski, J. [3 H]Metergoline: A New Ligand of Serotonin Receptors in the Rat Brain. J. Neurochem. 1981, 36, 613–626. [Google Scholar] [CrossRef]
- Miller, K.J.; King, A.; Demchyshyn, L.; Niznik, H.; Teitler, M. Agonist Activity of Sumatriptan and Metergoline at the Human 5-HT1Dβ Receptor: Further Evidence for a Role of the 5-HT1D Receptor in the Action of Sumatriptan. Eur. J. Pharmacol. Mol. Pharmacol. 1992, 227, 99–102. [Google Scholar] [CrossRef]
- Hofmann, A. Historical View on Ergot Alkaloids. Pharmacology 1978, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Barger, G.; Carr, F.H. The Alkaloids of Ergot. J. Chem. Soc. Trans. 1907, 91, 337–353. [Google Scholar] [CrossRef]
- Barger, G.; Dale, H.H. Ergotoxine and Some Other Constituents of Ergot. Biochem. J. 1907, 2, 240–299. [Google Scholar] [CrossRef] [Green Version]
- Stoll, A. Zur Kenntnis Der Mutterkornalkaloide. Verh. Schweiz. Nat. Ges. 1918, 101, 190–191. [Google Scholar]
- Rothlin, E.; Konzett, H.; Cerletti, A. The antagonism of ergot alkaloids towards the inhibitory response of the isolated rabbit intestine to epinephrine and norepinephrine. J. Pharmacol. Exp. Ther. 1954, 112, 185–190. [Google Scholar]
- Jacobs, W.A.; Craig, L.C. The structure of the ergot alkaloids. J. Am. Chem. Soc. 1935, 57, 383–384. [Google Scholar] [CrossRef]
- Stoll, A.; Hofmann, A.; Troxler, F. Über Die Isomerie von Lysergsäure Und Isolysergsäure. 14. Mitteilung Über Mutterkornalkaloide. Helv. Chim. Acta 1949, 32, 506–521. [Google Scholar] [CrossRef]
- Kornfeld, E.C.; Fornefeld, E.J.; Kline, G.B.; Mann, M.J.; Morrison, D.E.; Jones, R.G.; Woodward, R.B. The Total Synthesis of Lysergic Acid. J. Am. Chem. Soc. 1956, 78, 3087–3114. [Google Scholar] [CrossRef]
- Ramage, R.; Armstrong, V.W.; Coulton, S. A New Synthetic Route to (±) Lysergic Acid. Tetrahedron 1981, 37, 157–164. [Google Scholar] [CrossRef]
- Stadler, P.A.; Frey, A.J.; Troxler, F.; Hofmann, A. Selektive Reduktions- Und Oxydationsreaktionen an Lysergsäure- Derivaten. 2.3-Dihydro- Und 12-Hydroxy-Lysergsäureamide. 59. Mitteilung Über Mutterkornalkaloide. Helv. Chim. Acta 1964, 47, 756–769. [Google Scholar] [CrossRef]
- Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry. Angew. Chem. Int. Ed. Engl. 1969, 8, 781–853. [Google Scholar] [CrossRef]
- Stoll, A.; Hofmann, A. Die Optisch Aktiven Hydrazide Der Lysergsäure Und Der Isolysergsäure. (4. Mitteilung Über Mutterkornalkaloide). Helv. Chim. Acta 1943, 26, 922–928. [Google Scholar] [CrossRef]
- Woodward, R.B. Private Communication between Woodward and Ramage. Ph.D. Thesis, Harvard, Cambridge, MA, USA, 1952. [Google Scholar]
- Křen, V.; Cvak, V. Ergot The Genus Claviceps (Medicinal and Aromatic Plants—Industrial Profiles); CRC Press: Boca Raton, FL, USA; Informa UK Limited: London, England, 1999; Volume 6. [Google Scholar]
- Martínková, L.; Křen, V.; Cvak, L.; Ovesná, M.; Přepechalová, I. Hydrolysis of Lysergamide to Lysergic Acid by Rhodococcus Equi A4. J. Biotechnol. 2000, 84, 63–66. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1942. [Google Scholar]
- Blount, B.K.; Robinson, R. CCCCXXXVI.—Strychnine and Brucine. Part XIII. Note on Dihydroindolylpropionic and Dihydroindolylbutyric Acids. J. Chem. Soc. 1931, 3158–3160. [Google Scholar] [CrossRef]
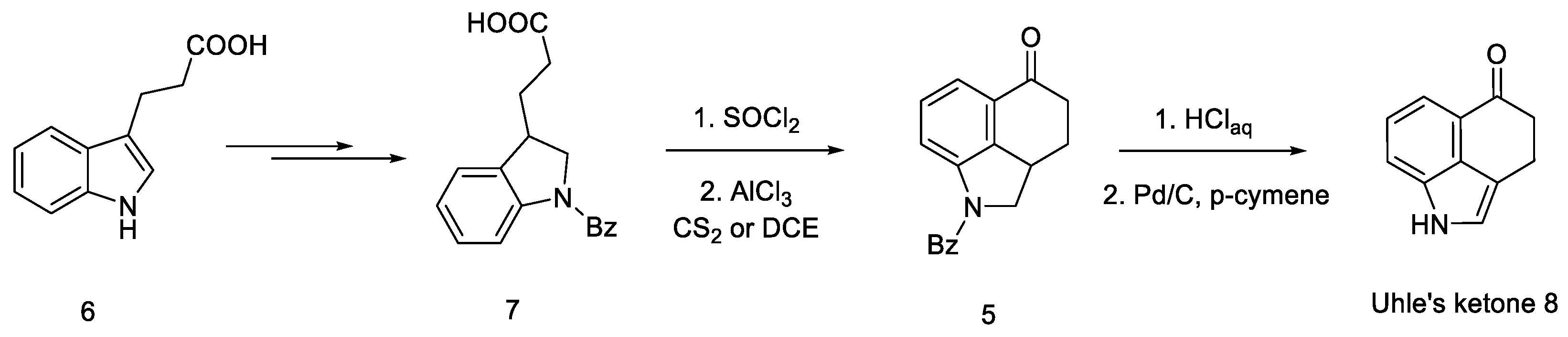
- Uhle, F.C. The Synthesis of 5-Keto-1,3,4,5-Tetrahydrobenz[Cd]Indole. A Synthesis of 4-Substituted Indoles. J. Am. Chem. Soc. 1949, 71, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.S.; Belew, J.S.; Chinn, L.J.; Hunt, R.H. Studies Relating to the Formation and Reactions of Glycidic Esters. J. Am. Chem. Soc. 1953, 75, 4995–5001. [Google Scholar] [CrossRef]
- Price, C.C.; Krishnamurti, I.V. The Acid-Catalyzed Reaction of Formaldehyde with Allyl Cyanide. J. Am. Chem. Soc. 1950, 72, 5334–5335. [Google Scholar] [CrossRef]
- Kleiderer, E.C.; Kornfeld, E.C. Raney nickel as an organic oxidation-reduction catalyst. J. Org. Chem. 1948, 13, 455–458. [Google Scholar] [CrossRef]
- Stoll, A.; Hofmann, A. Racemische Lysergsäure Und Ihre Auflösung in Die Optischen Antipoden.-Zweite, Vorläufige Mitteilung Über Mutterkornalkaloide. Hoppe Seyler’s Z. Für Physiol. Chem. 1937, 250, 7–10. [Google Scholar] [CrossRef]
- Smith, S.; Timmis, G.M. A New Alkaloid of Ergot. Nature 1936, 137, 111. [Google Scholar] [CrossRef]
- Julia, M.; Le Goffic, F.; Igolen, J.; Baillarge, M. Une Nouvelle Synthese de l’acide Lysergique. Tetrahedron Lett. 1969, 10, 1569–1571. [Google Scholar] [CrossRef]
- House, H.O.; Rasmussen, G.H. Stereoselective Synthesis of α-Substituted α,β-Unsaturated Esters. J. Org. Chem. 1961, 26, 4278. [Google Scholar] [CrossRef]
- Jackson, A.G.; Kenner, G.W.; Moore, G.A.; Ramage, R.; Thorpe, W.D. Activation of Carboxylic Acids as Diphenylphosphinic Mixed Anhydrides: Application to Peptide Chemistry. Tetrahedron Lett. 1976, 17, 3627–3630. [Google Scholar] [CrossRef]
- Bu’Lock, J.D.; Harley-Mason, J. 497. Melanin and Its Precursors. Part III. New Syntheses of 5: 6-Dihydroxyindole and Its Derivatives. J. Chem. Soc. 1951, 2248–2252. [Google Scholar] [CrossRef]
- Troxler, F. Beiträge Zur Chemie Der 6-Methy1-8-Ergolen-8-Carbonsäure. 69. Mitteilung Über Mutterkornalkaloide. Helv. Chim. Acta 1968, 51, 1372–1381. [Google Scholar] [CrossRef]
- Eschweiler, W. Ersatz von an Stickstoff Gebundenen Wasserstoffatomen Durch Die Methylgruppe Mit Hülfe von Formaldehyd. Ber. Dtsch. Chem. Ges. 1905, 38, 880–882. [Google Scholar] [CrossRef] [Green Version]
- Clarke, H.T.; Gillespie, H.B.; Weisshaus, S.Z. The Action of Formaldehyde on Amines and Amino Acids 1. J. Am. Chem. Soc. 1933, 55, 4571–4587. [Google Scholar] [CrossRef]
- Fehr, T.; Stadler, P.A.; Hofmann, A. Demethylierung Des Lysergsäuregerüstes. 73. Mitteilung Über Mutterkornalkaloide. Helv. Chim. Acta 1970, 53, 2197–2201. [Google Scholar] [CrossRef]
- Rebek, J.; Tai, D.F. A New Synthesis of Lysergic Acid. Tetrahedron Lett. 1983, 24, 859–860. [Google Scholar] [CrossRef]
- Rebek, J.; Tai, D.F.; Shue, Y.K. Synthesis of Ergot Alkaloids from Tryptophan. J. Am. Chem. Soc. 1984, 106, 1813–1819. [Google Scholar] [CrossRef]
- Hanessian, S. Selective Hydrolysis of Amide Bonds in Acetamido Deoxy Sugars.-Ethyl Acetamidium Fluoroborates. Tetrahedron Lett. 1967, 8, 1549–1552. [Google Scholar] [CrossRef]
- Smith, S.; Timmis, G.M. 311. The Alkaloids of Ergot. Part VII. IsoErgine and Isolysergic Acids. J. Chem. Soc. 1936, 1440–1444. [Google Scholar] [CrossRef]
- Ninomiya, I.; Kiguchi, T.; Hashimoto, C.; Naito, T. A New Synthesis of (±)-Lysergic Acid. Heterocycles 1982, 19, 2279. [Google Scholar] [CrossRef]
- Ninomiya, I.; Naito, T. Enamide Cyclization and Its Application to the Synthesis of Heterocycles. J. Synth. Org. Chem. Japan 1984, 42, 225–246. [Google Scholar] [CrossRef]
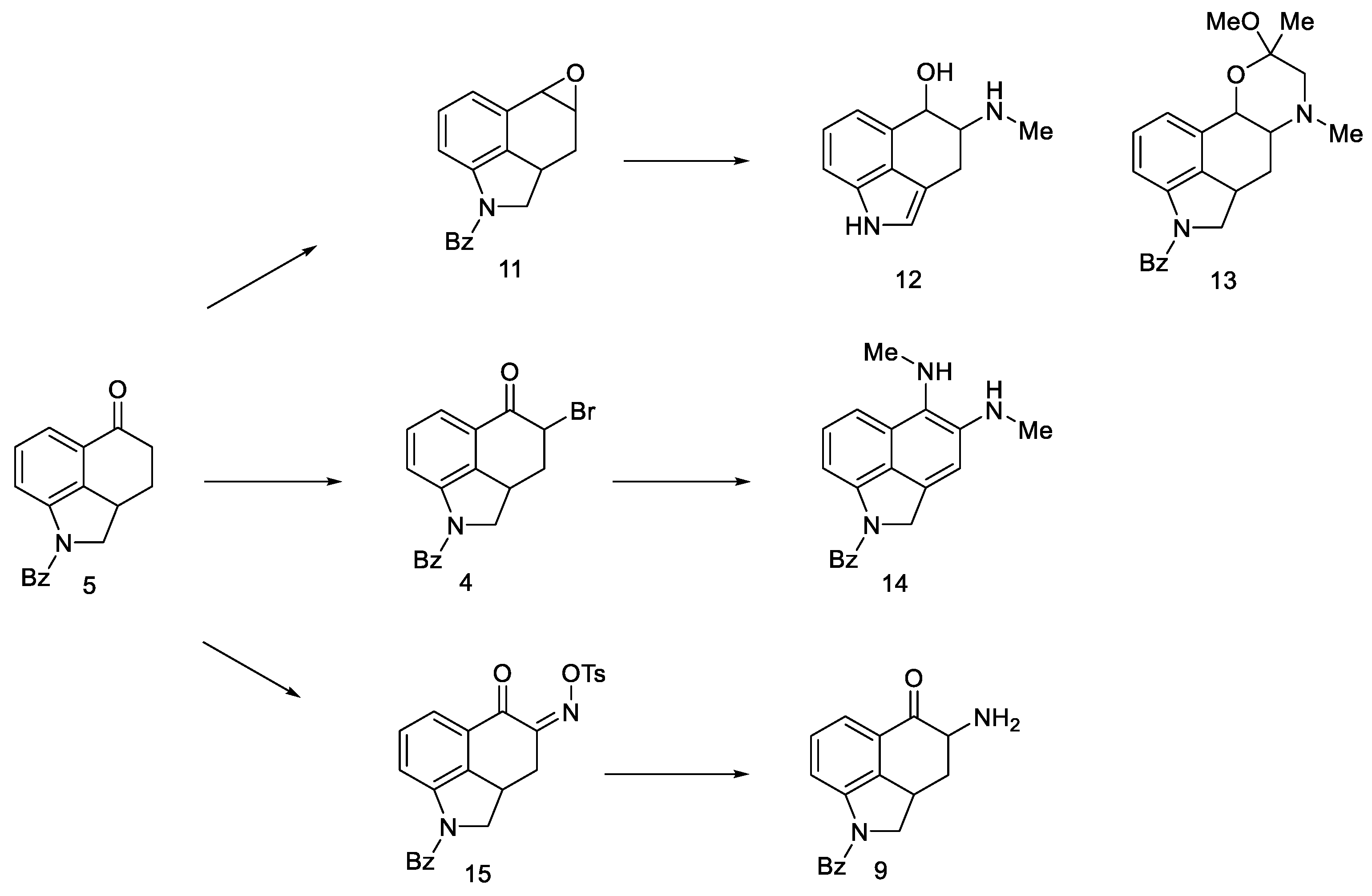
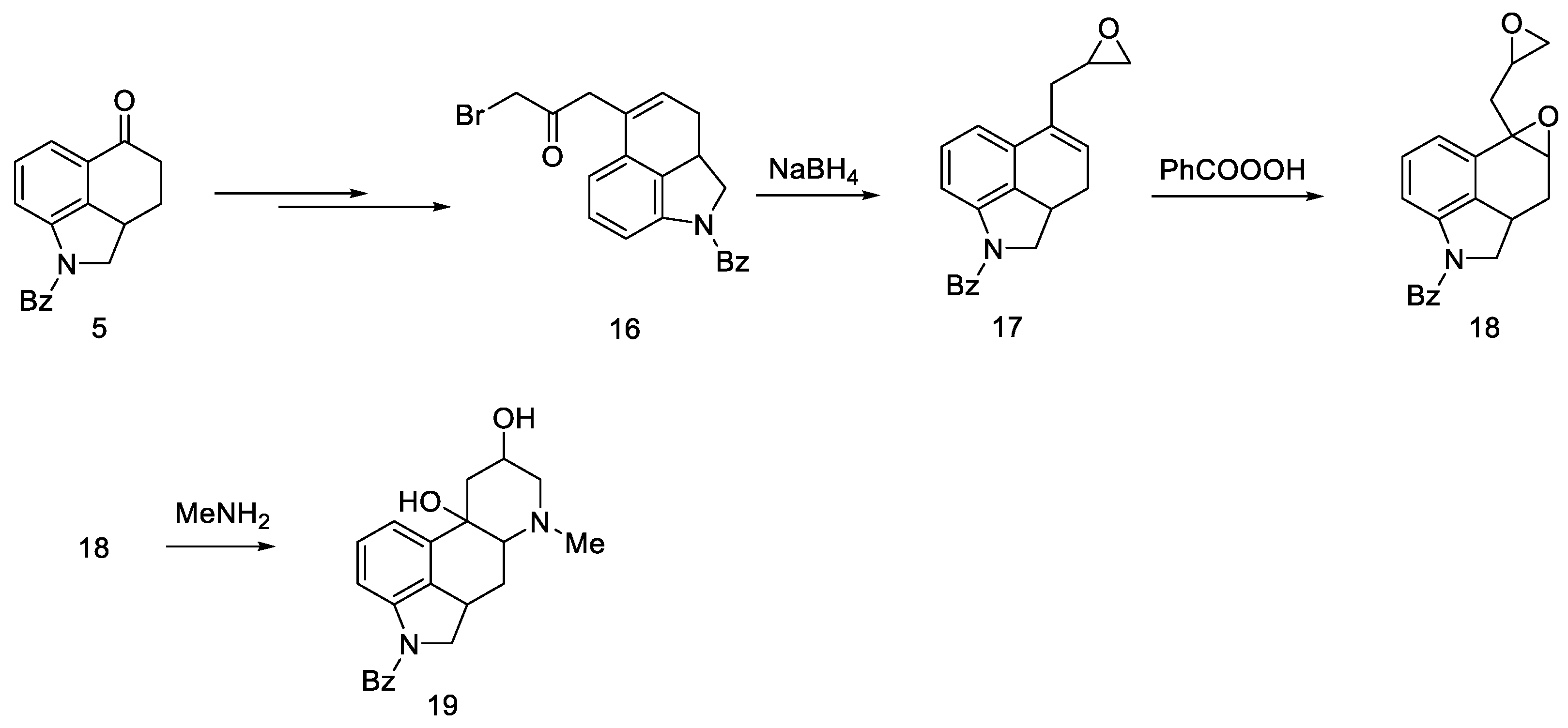
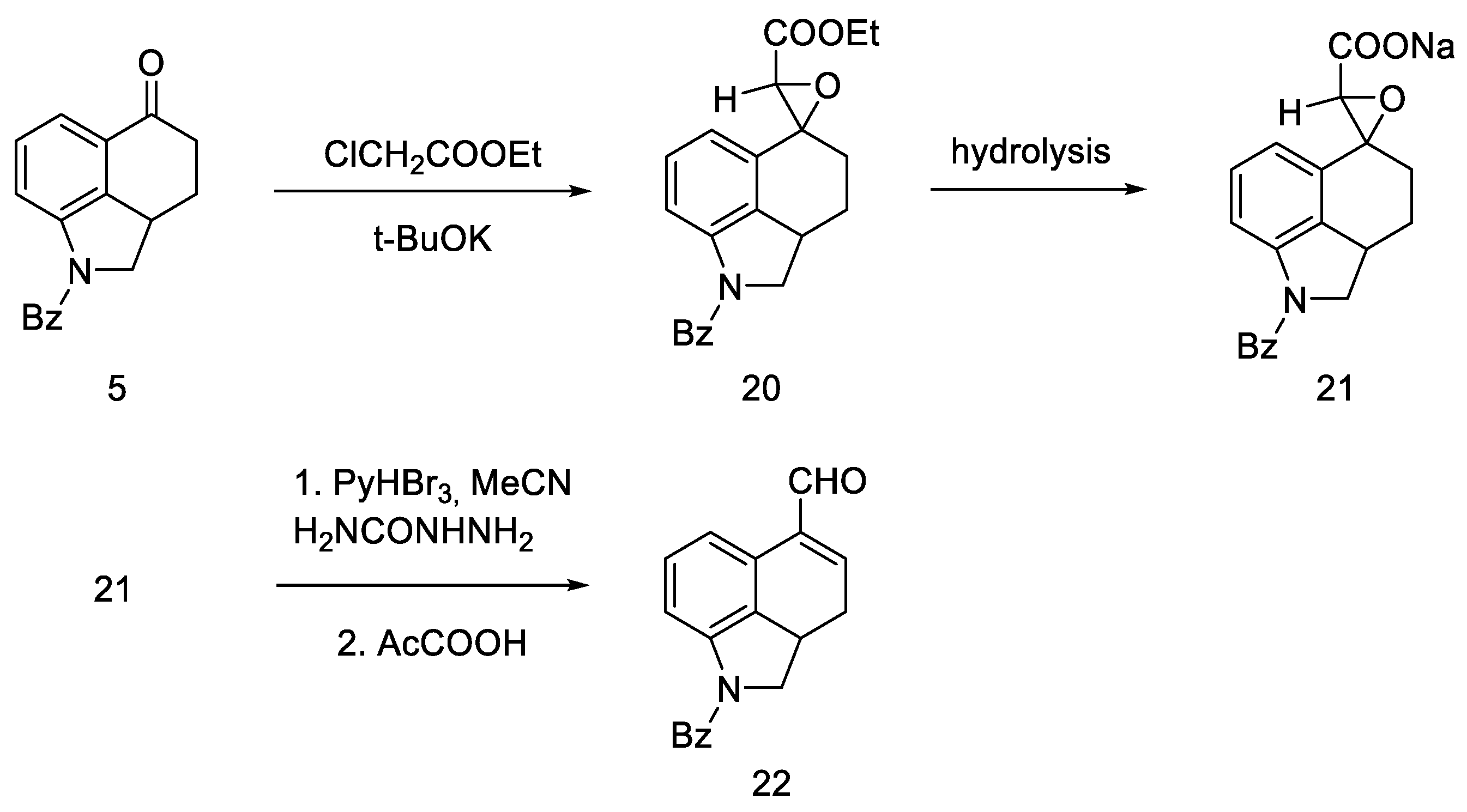
- Nichols, D.E.; Robinson, J.M.; Li, G.S.; Cassady, J.M.; Floss, H.G. An improved synthesis of 1-benzoyl-4-keto-1,2,2a,3,4,5-hexahydrobenz [cd]indole. Org. Prep. Proced. Int. 1977, 9, 277–280. [Google Scholar] [CrossRef]
- Ninomiya, I.; Hashimoto, C.; Kiguchi, T.; Naito, T. Photocyclisation of Enamides. Part 24. Total Synthesis of (±)-Isofumigaclavine B and (±)-Lysergic Acid. J. Chem. Soc. Perkin Trans. 1 1985, 941–948. [Google Scholar] [CrossRef]
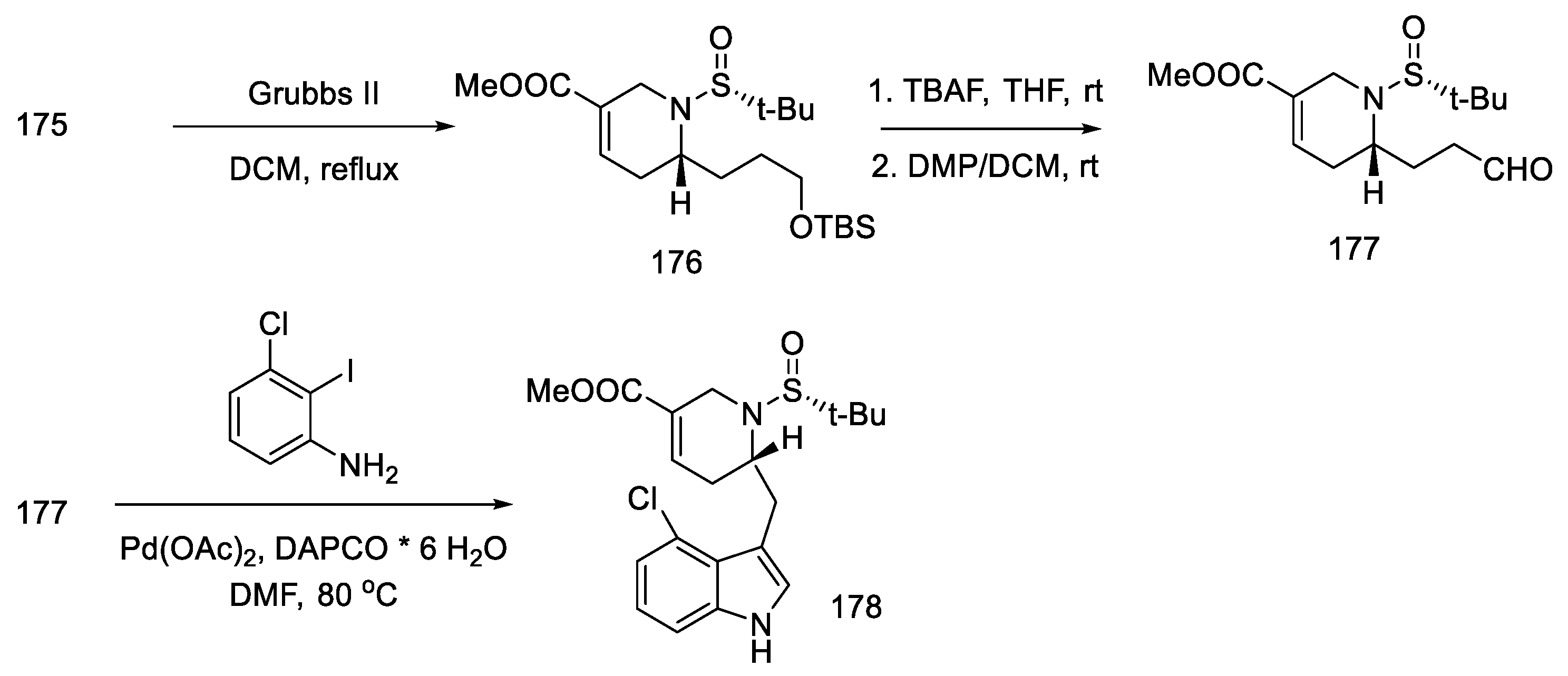
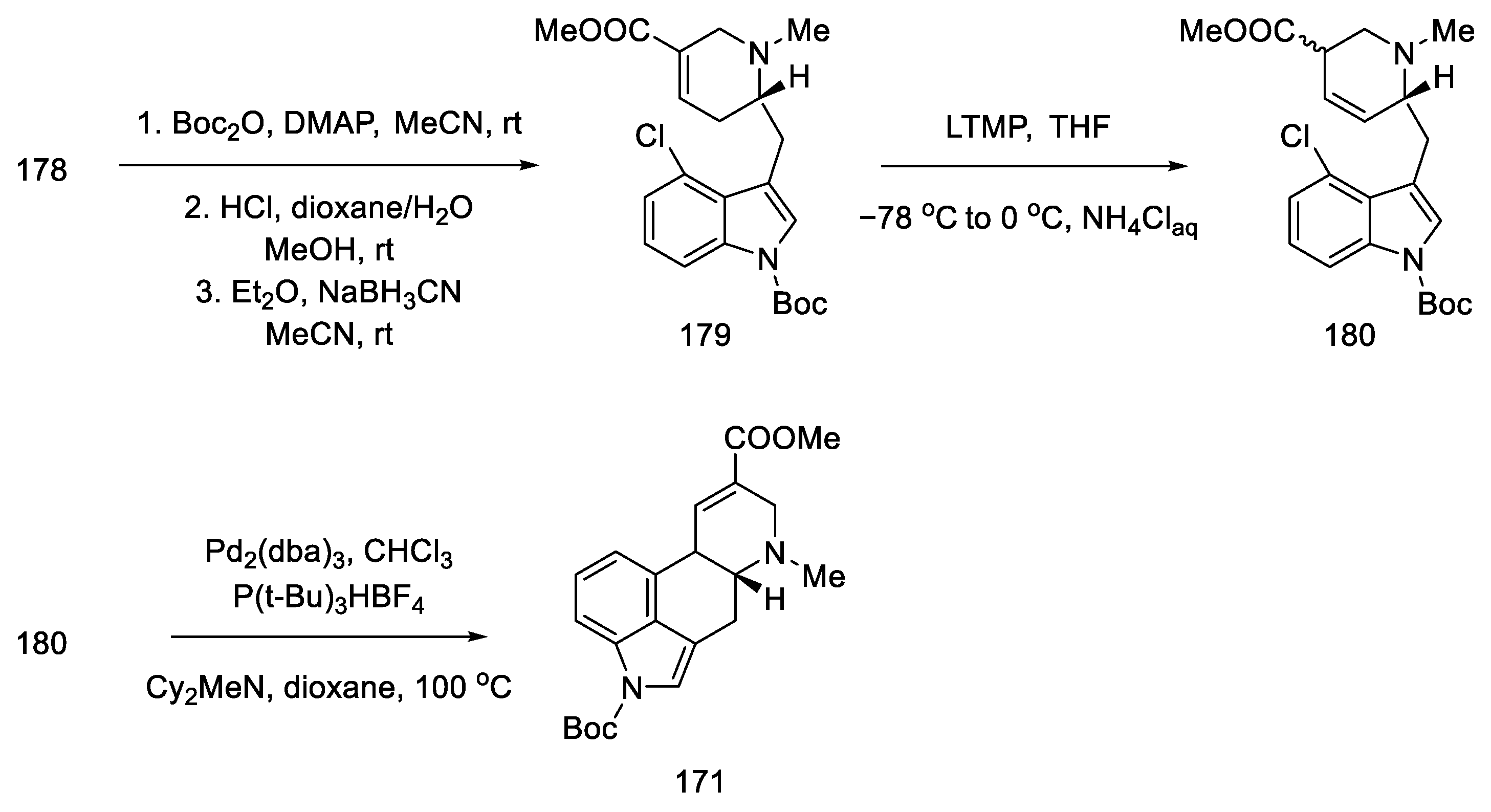
- Tasker, N.R.; Wipf, P. A Short Synthesis of Ergot Alkaloids and Evaluation of the 5-HT 1/2 Receptor Selectivity of Lysergols and Isolysergols. Org. Lett. 2022, 24, 7255–7259. [Google Scholar] [CrossRef]
- Hendrickson, J.B.; Wang, J. A New Synthesis of Lysergic Acid. Org. Lett. 2004, 6, 3–5. [Google Scholar] [CrossRef]
- Doll, M.K.-H. A Short Synthesis of the 8-Azaergoline Ring System by Intramolecular Tandem Decarboxylation−Cyclization of the Minisci-Type Reaction. J. Org. Chem. 1999, 64, 1372–1374. [Google Scholar]
- Bekkam, M.; Mo, H.; Nichols, D.E. A Reported “New Synthesis of Lysergic Acid” Yields Only The Derailment Product: Methyl 5-Methoxy-4,5-Dihydroindolo[4,3-f,g ]Quinoline-9-Carboxylate. Org. Lett. 2012, 14, 296–298. [Google Scholar]
- Zhao, Y. Reduction of Tertiary Benzamides to Benzaldehydes by an in Situ–Generated Schwartz Reagent (Cp2Zr(H)Cl); Formal Synthesis of Lysergic Acid; Queen’s University Kingston: Kingston, ON, Canada, 2010. [Google Scholar]
- Points, G.L.; Stout, K.T.; Beaudry, C.M. Regioselective Formation of Substituted Indoles: Formal Synthesis of Lysergic Acid. Chem. Eur. J. 2020, 26, 16655–16658. [Google Scholar] [CrossRef]
- Zhao, Y.; Snieckus, V. A Practical in Situ Generation of the Schwartz Reagent. Reduction of Tertiary Amides to Aldehydes and Hydrozirconation. Org. Lett. 2014, 16, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Moldvai, I.; Temesvári-Major, E.; Incze, M.; Szentirmay, É.; Gács-Baitz, E.; Szántay, C. Enantioefficient Synthesis of α-Ergocryptine: First Direct Synthesis of (+)-Lysergic Acid. J. Org. Chem. 2004, 69, 5993–6000. [Google Scholar] [CrossRef] [PubMed]
- Teranishi, K.; Hayashi, S.; Nakatsuka, S.; Goto, T. A Facile Biomimetic Synthesis of Uhle’s Ketone by the Regioselective Friedel-Crafts Cyclization of Indole-3-Ylpropionyl Chloride. Tetrahedron Lett. 1994, 35, 8173–8176. [Google Scholar] [CrossRef]
- Roth, M.; Dubs, P.; Götschi, E.; Eschenmoser, A. Sulfidkontraktion via Alkylative Kupplung: Eine Methode Zur Darstellung von β-Dicarbonylderivaten. Über Synthetische Methoden, 1. Mitteilung. Helv. Chim. Acta 1971, 54, 710–734. [Google Scholar] [CrossRef]
- Moldvai, I.; Temesvári-Major, E.; Incze, M.; Dörnyei, G.; Szentirmay, É.; Szántay, C. Synthetic Route to Ergot Alkaloids. Helv. Chim. Acta 2005, 88, 1344–1356. [Google Scholar] [CrossRef]
- Rathnayake, U.; Garner, P. Asymmetric Synthesis of Lysergic Acid via an Intramolecular (3+2) Dipolar Cycloaddition/Ring-Expansion Sequence. Org. Lett. 2021, 23, 6756–6759. [Google Scholar] [CrossRef]
- Zuo, Z.; Ma, D. Enantioselective Total Syntheses of Communesins A and B. Angew. Chem. Int. Ed. Engl. 2011, 50, 12008–12011. [Google Scholar] [CrossRef]
- Métro, T.-X.; Appenzeller, J.; Gomez Pardo, D.; Cossy, J. Highly Enantioselective Synthesis of β-Amino Alcohols. Org. Lett. 2006, 8, 3509–3512. [Google Scholar] [CrossRef]
- Jarvis, S.B.D.; Charette, A.B. Synthesis of Enantiopure Substituted Piperidines via an Aziridinium Ring Expansion. Org. Lett. 2011, 13, 3830–3833. [Google Scholar] [CrossRef]
- Oestreich, M. (Ed.) The Mizoroki–Heck Reaction; John Wiley & Sons, Ltd.: Chichester, UK, 2009. [Google Scholar]
- Cacchi, S.; Giuseppe Ciattini, P.; Morera, E.; Ortar, G. A Concise, Palladium-Catalyzed Approach to (±)-Lysergic Acid. Tetrahedron Lett. 1988, 29, 3117–3120. [Google Scholar] [CrossRef]
- Kurihara, T.; Terada, T.; Yoneda, R. A New Synthesis of (±)-Lysergic Acid. Chem. Pharm. Bull. 1986, 34, 442–443. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, T.; Terada, T.; Harusawa, S.; Yoneda, R. Synthetic Studies of (±)-Lysergic Acid and Related Compounds. Chem. Pharm. Bull. 1987, 35, 4793–4802. [Google Scholar] [CrossRef] [Green Version]
- Busqué, F.; de March, P.; Figueredo, M.; Font, J.; Gallagher, T.; Milán, S. Efficient Synthesis of (S)-3,4-Dihydro-2-Pivaloyloxymethyl-2H-Pyrrole 1-Oxide. Tetrahedron Asymmetry 2002, 13, 437–445. [Google Scholar] [CrossRef]
- Birkofer, L.; Bierwirth, E.; Ritter, A. Decarbobenzoxylierungen Mit Triäthylsilan. Chem. Ber. 1961, 94, 821–824. [Google Scholar] [CrossRef]
- Ghosez, L.; Haveaux, B.; Viehe, H.G. Alkyl and Arylα-Chloro Enamines. Angew. Chem. Int. Ed. Engl. 1969, 8, 454–455. [Google Scholar] [CrossRef]
- Oppolzer, W.; Francotte, E.; Bättig, K. Total Synthesis of (±)-Lysergic Acid by an Intramolecular Imino- Diels—Alder Reaction. Preliminary Communication. Helv. Chim. Acta 1981, 64, 478–481. [Google Scholar] [CrossRef]
- Kurokawa, T.; Isomura, M.; Tokuyama, H.; Fukuyama, T. Synthesis of Lysergic Acid Methyl Ester via the Double Cyclization Strategy. Synlett 2009, 2009, 775–778. [Google Scholar]
- Ninomiya, I.; Hashimoto, C.; Kiguchi, T.; Naito, T.; Barton, D.H.R.; Lusinchi, X.; Milliet, P. Dehydrogenation with Benzeneseleninic Anhydride in the Total Synthesis of Ergot Alkaloids. J. Chem. Soc. Perkin Trans. 1 1990, 707–713. [Google Scholar] [CrossRef]
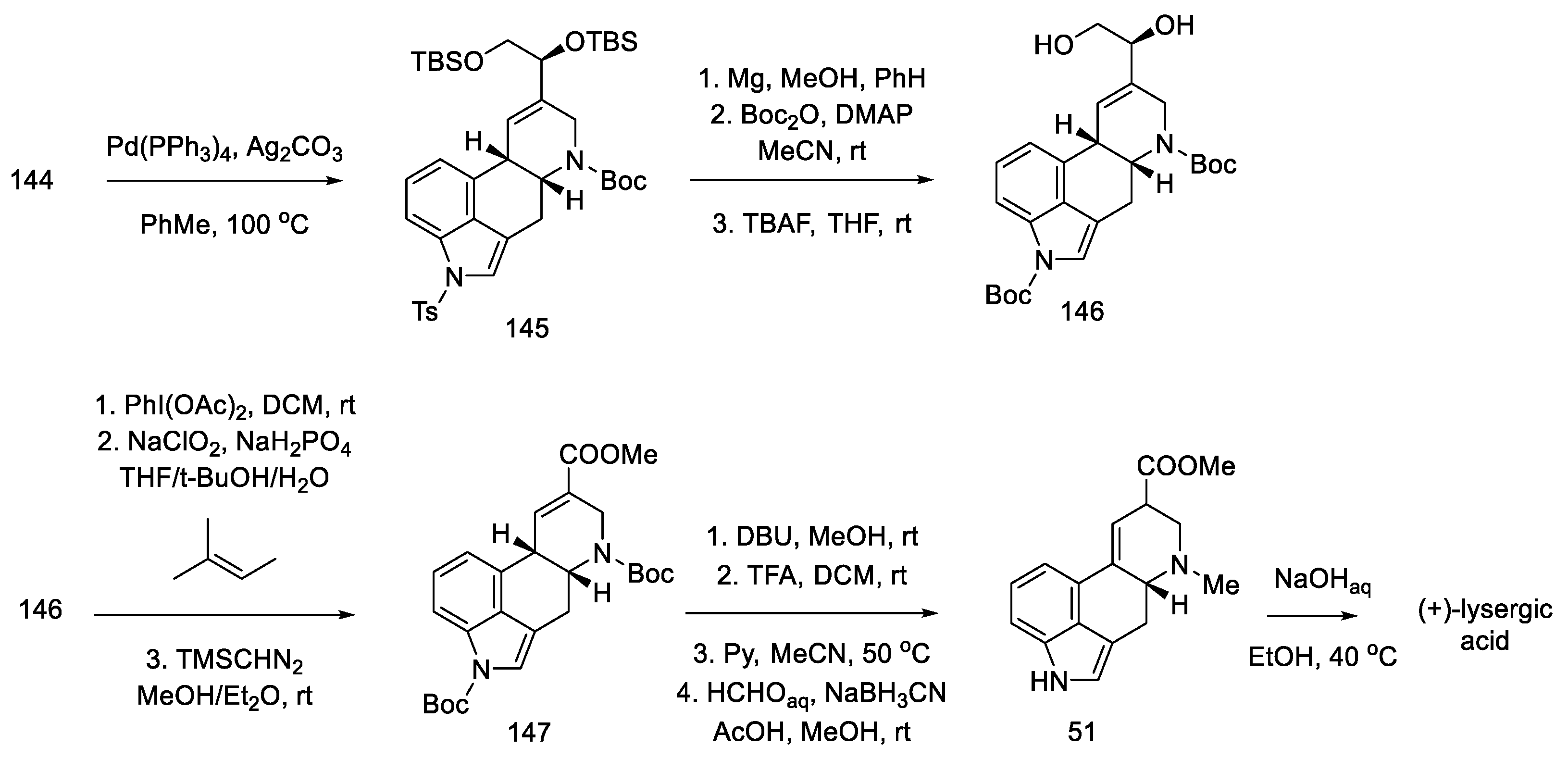
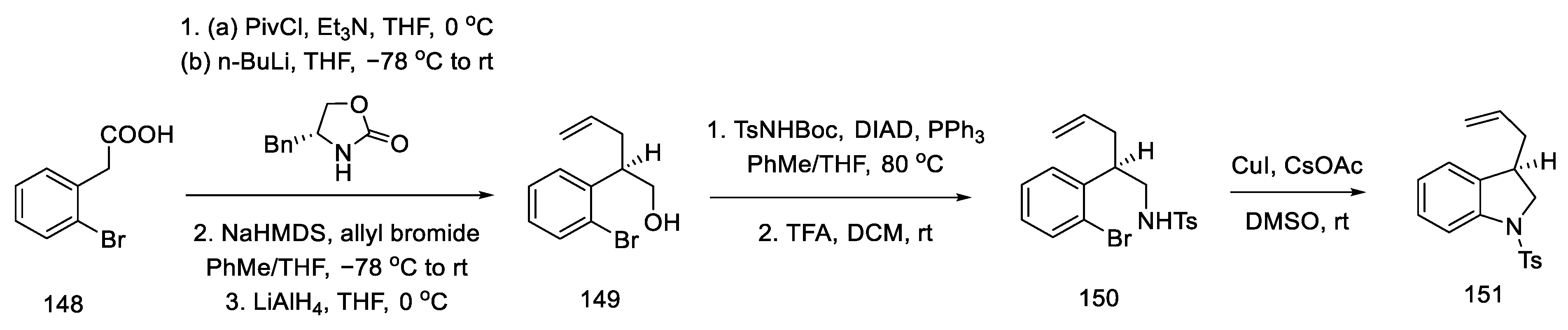
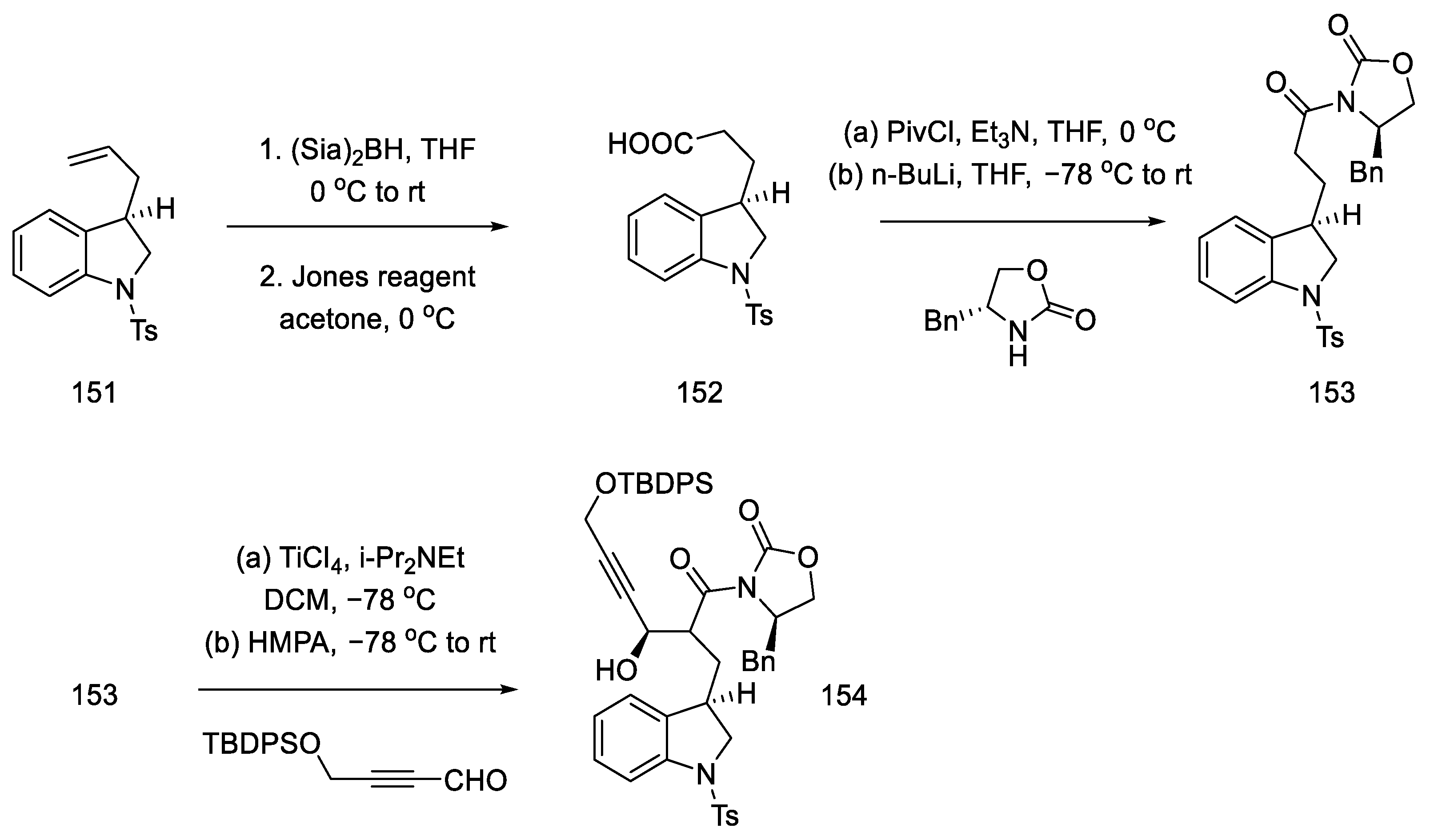
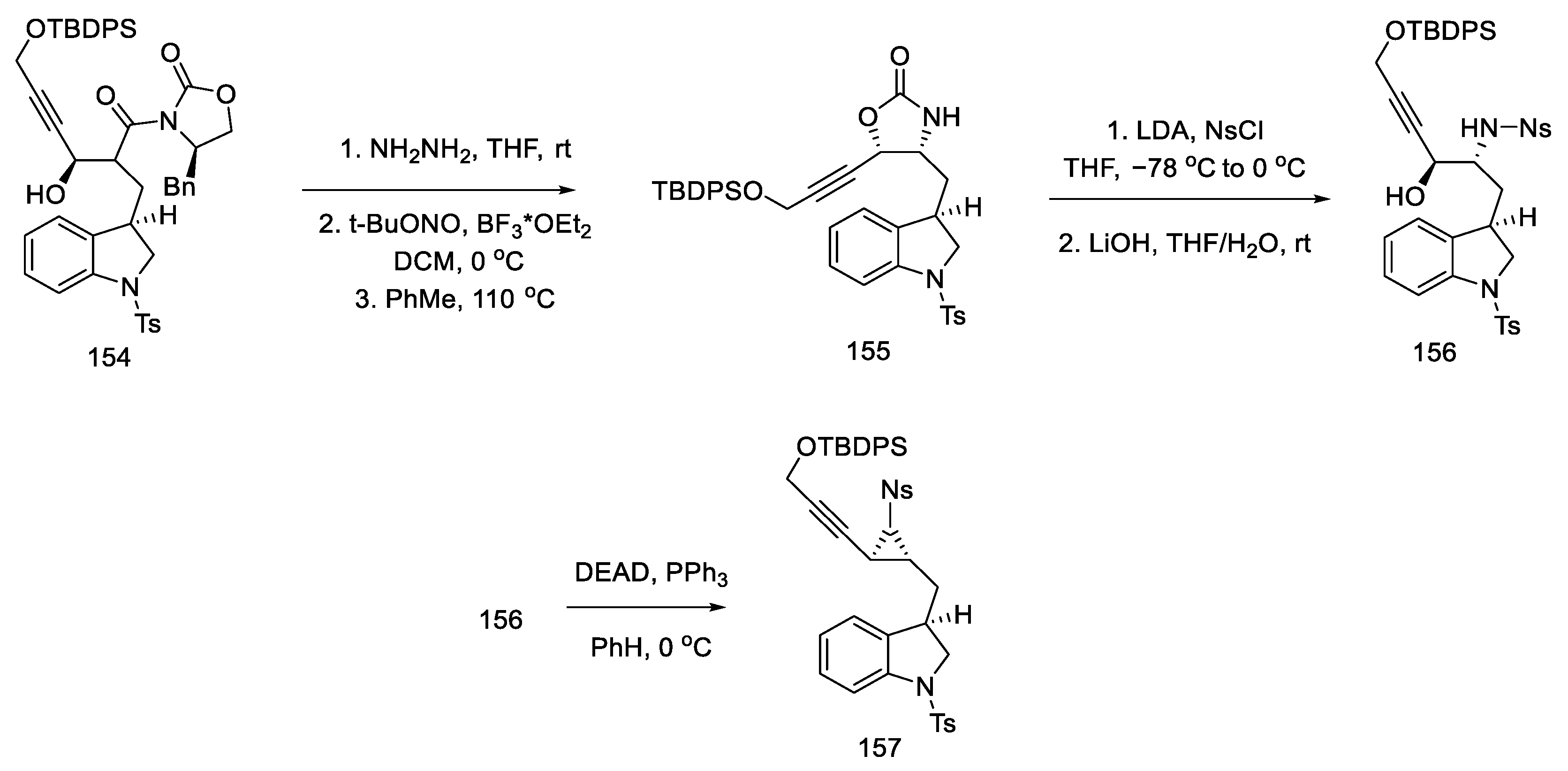
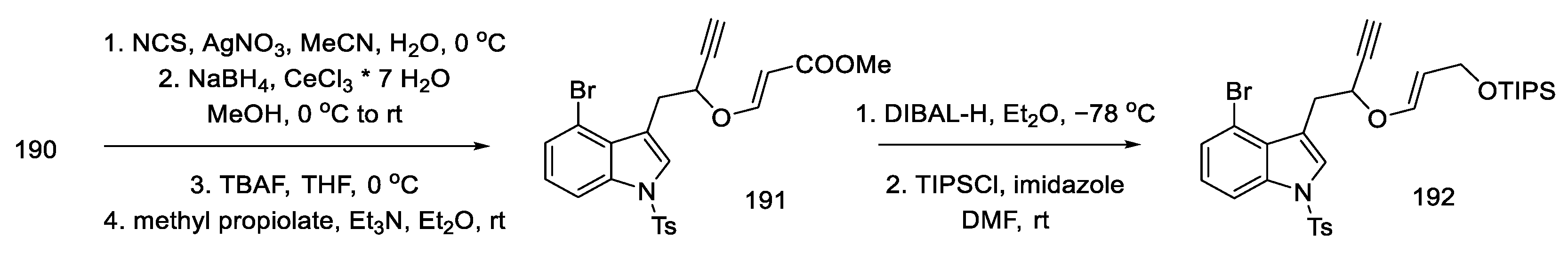
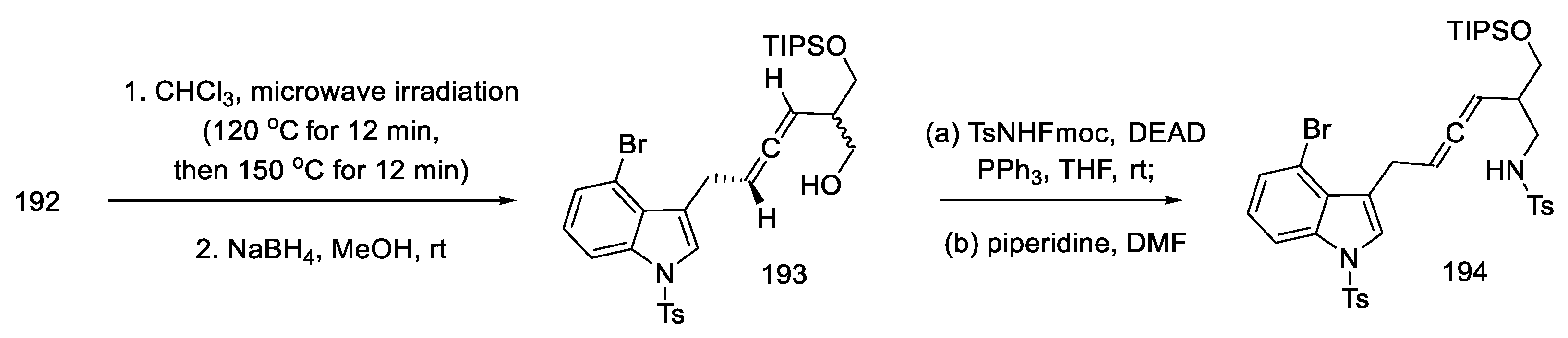
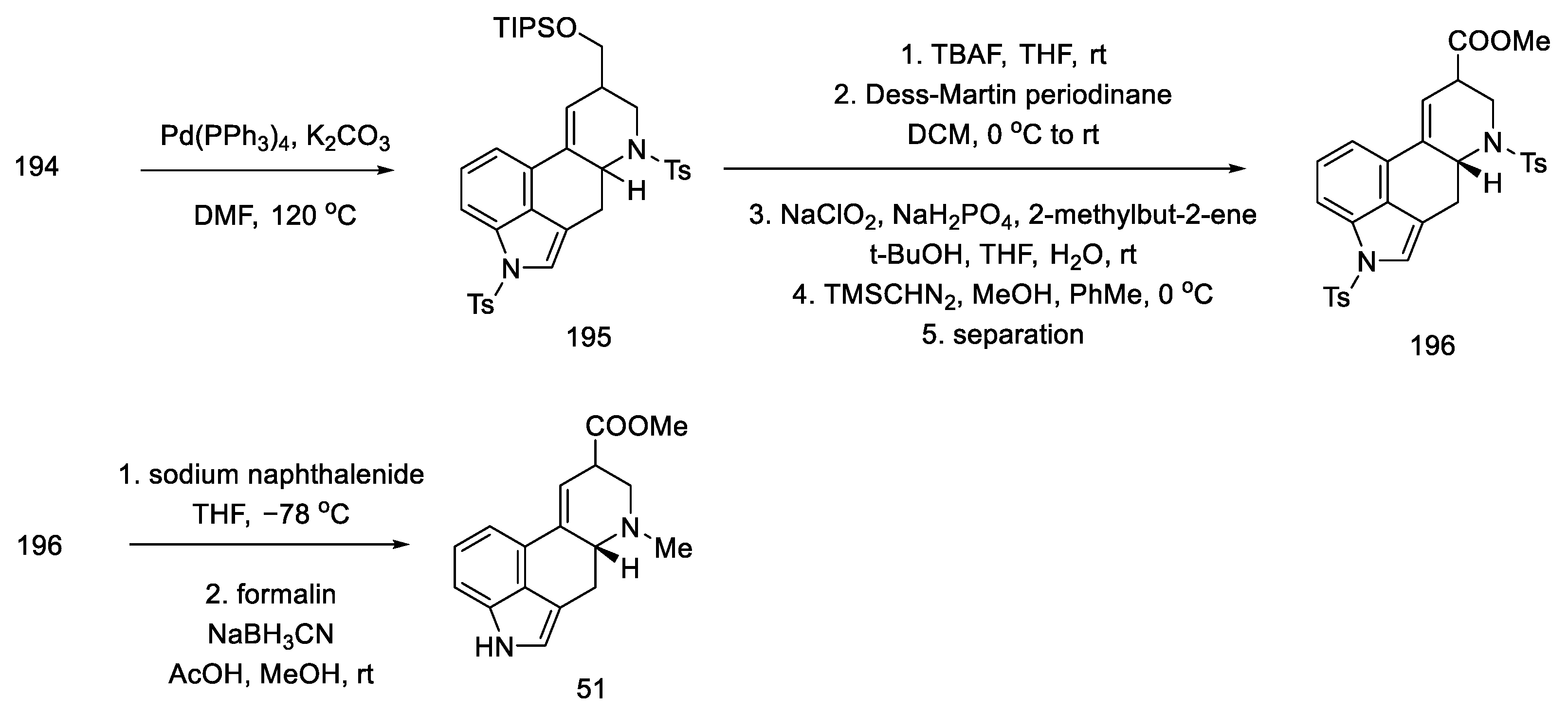
- Umezaki, S.; Yokoshima, S.; Fukuyama, T. Total Synthesis of Lysergic Acid. Org. Lett. 2013, 15, 4230–4233. [Google Scholar] [CrossRef]
- Crimmins, M.T.; King, B.W.; Tabet, E.A.; Chaudhary, K. Asymmetric Aldol Additions: Use of Titanium Tetrachloride and (−)-Sparteine for the Soft Enolization of N-Acyl Oxazolidinones, Oxazolidinethiones, and Thiazolidinethiones. J. Org. Chem. 2001, 66, 894–902. [Google Scholar] [CrossRef]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-Dihydroimidazol-2-Ylidene Ligands. Org. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef]
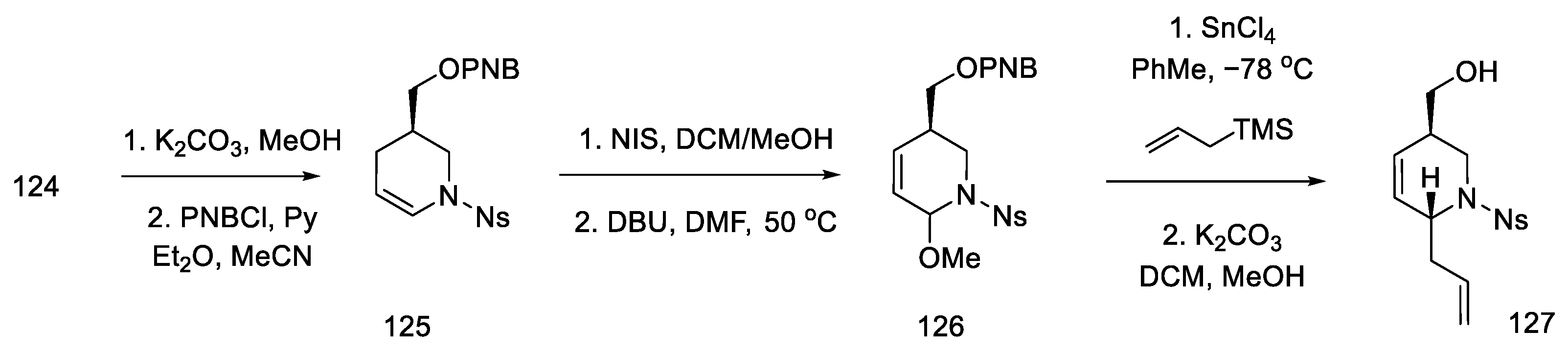
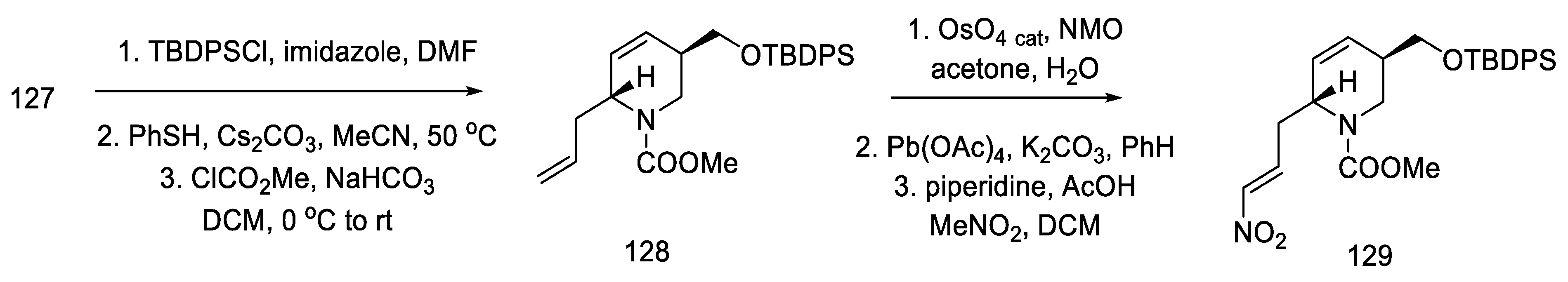
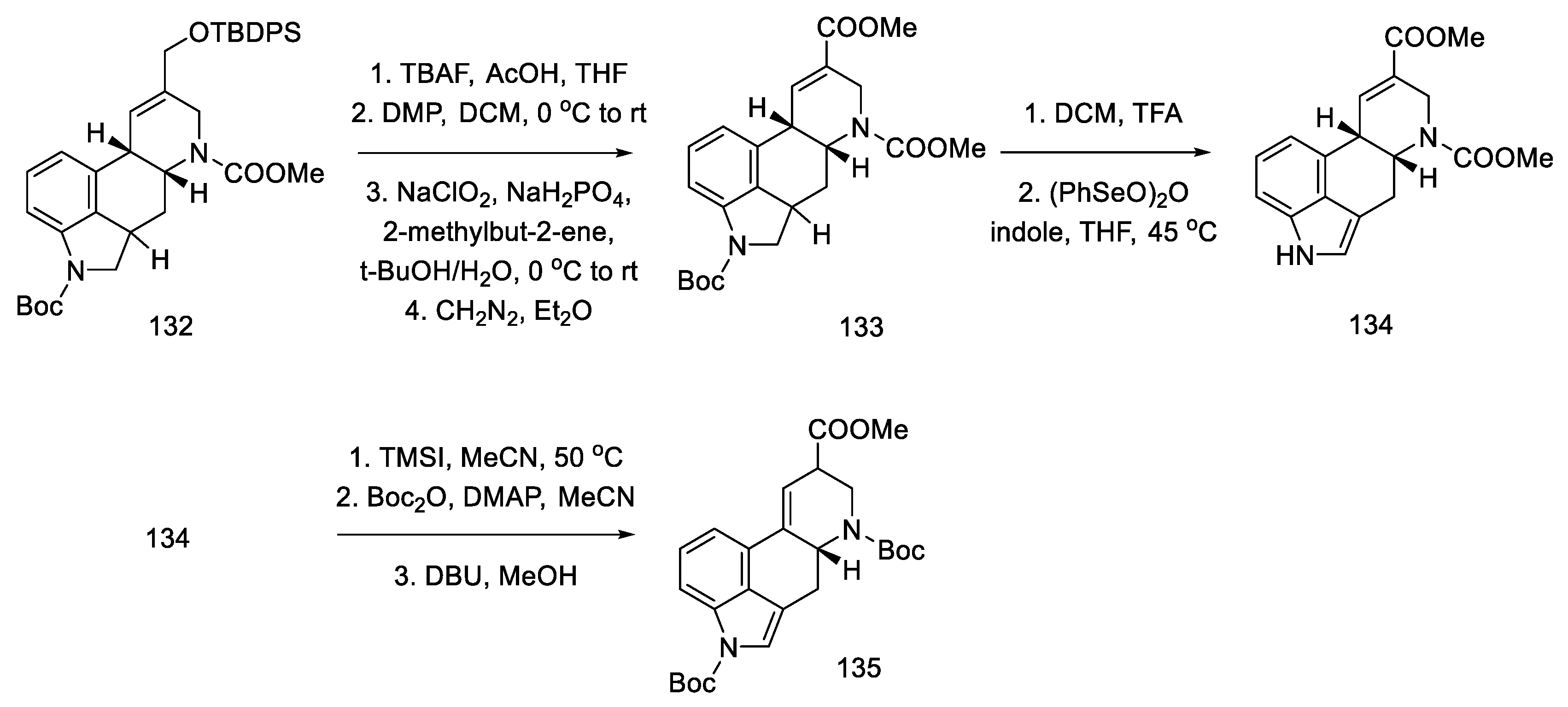
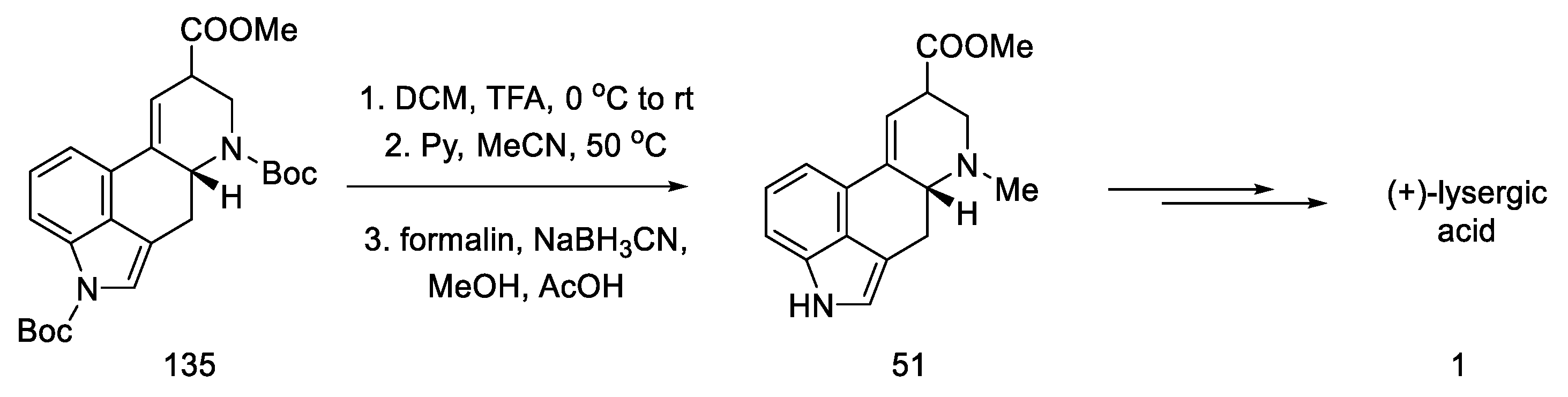
- Kanno, R.; Yokoshima, S.; Kanai, M.; Fukuyama, T. Total Synthesis of (+)-Lysergic Acid. J. Antibiot. 2018, 71, 240–247. [Google Scholar] [CrossRef]
- Evans, D.A.; Ennis, M.D.; Mathre, D.J. Asymmetric Alkylation Reactions of Chiral Imide Enolates. A Practical Approach to the Enantioselective Synthesis of Alpha.-Substituted Carboxylic Acid Derivatives. J. Am. Chem. Soc. 1982, 104, 1737–1739. [Google Scholar] [CrossRef]
- Liu, Q.; Jia, Y. Total Synthesis of (+)-Lysergic Acid. Org. Lett. 2011, 13, 4810–4813. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, Y.-A.; Xu, P.; Jia, Y. Total Synthesis of (+)-Lysergic Acid. J. Org. Chem. 2013, 78, 10885–10893. [Google Scholar] [CrossRef]
- González-Gómez, J.C.; Medjahdi, M.; Foubelo, F.; Yus, M. Stereoselective α-Aminoallylation of Aldehydes with Chiral Tert-Butanesulfinamides and Allyl Bromides. J. Org. Chem. 2010, 75, 6308–6311. [Google Scholar] [CrossRef]
- Oppolzer, W.; Grayson, J.I. Stereoselective Total Syntheses of (±)-Chanoclavine I and (±)-Isochanoclavine I by an Intramolecular Nitrone-Olefin/Cycloaddition. Preliminary Communication. Helv. Chim. Acta 1980, 63, 1706–1710. [Google Scholar] [CrossRef]
- Plieninger, H.; Wagner, C.; Immel, H. Synthese von 4-[E-4-Hydroxy-3-Methyl-Δ2-Butenyl]-Tryptophan-[4-14C] Sowie -Tryptamin-[4-14C] Und Einbau in Clavin-Alkaloide. Justus Liebigs Ann. Chem. 1971, 743, 95–111. [Google Scholar] [CrossRef]
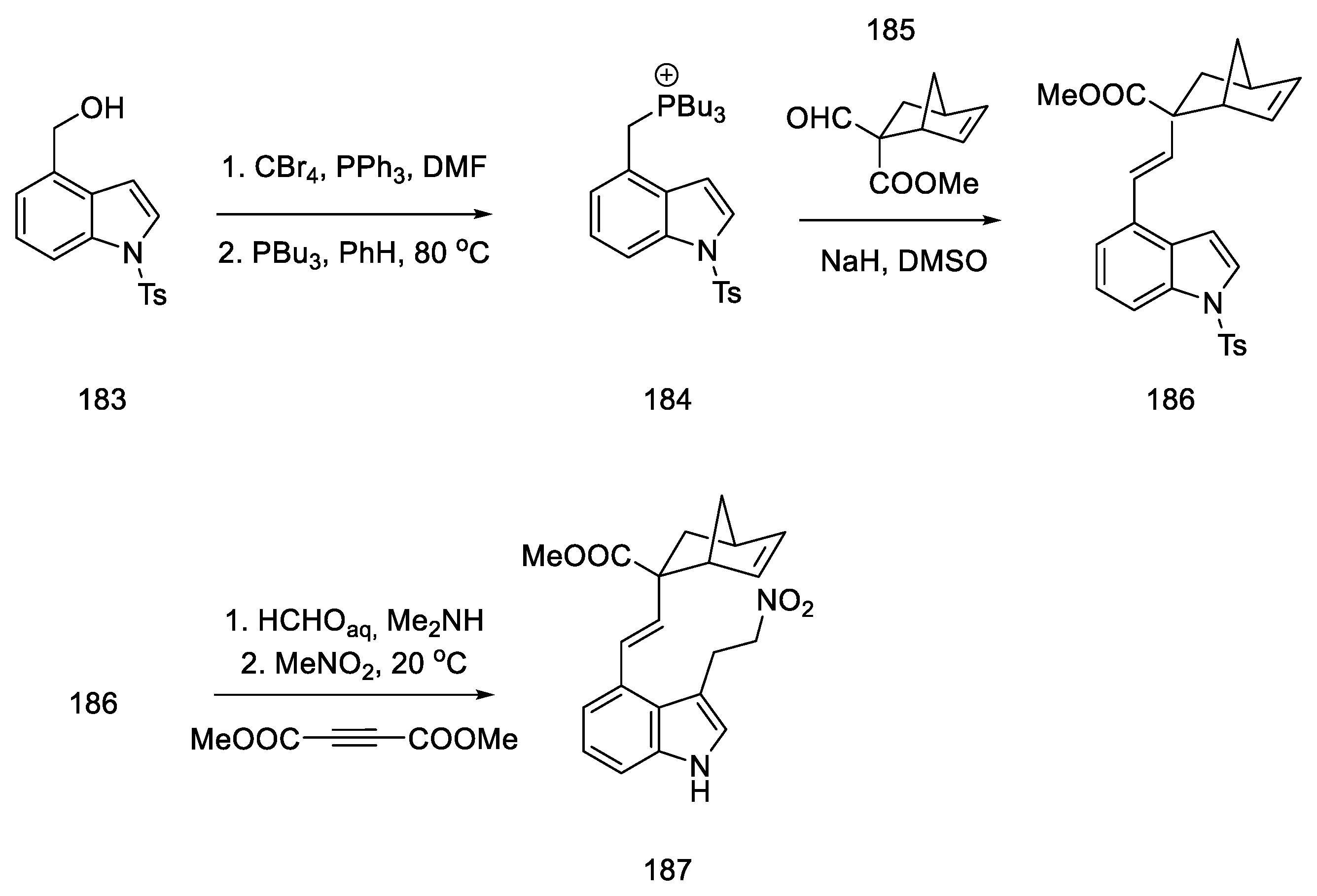
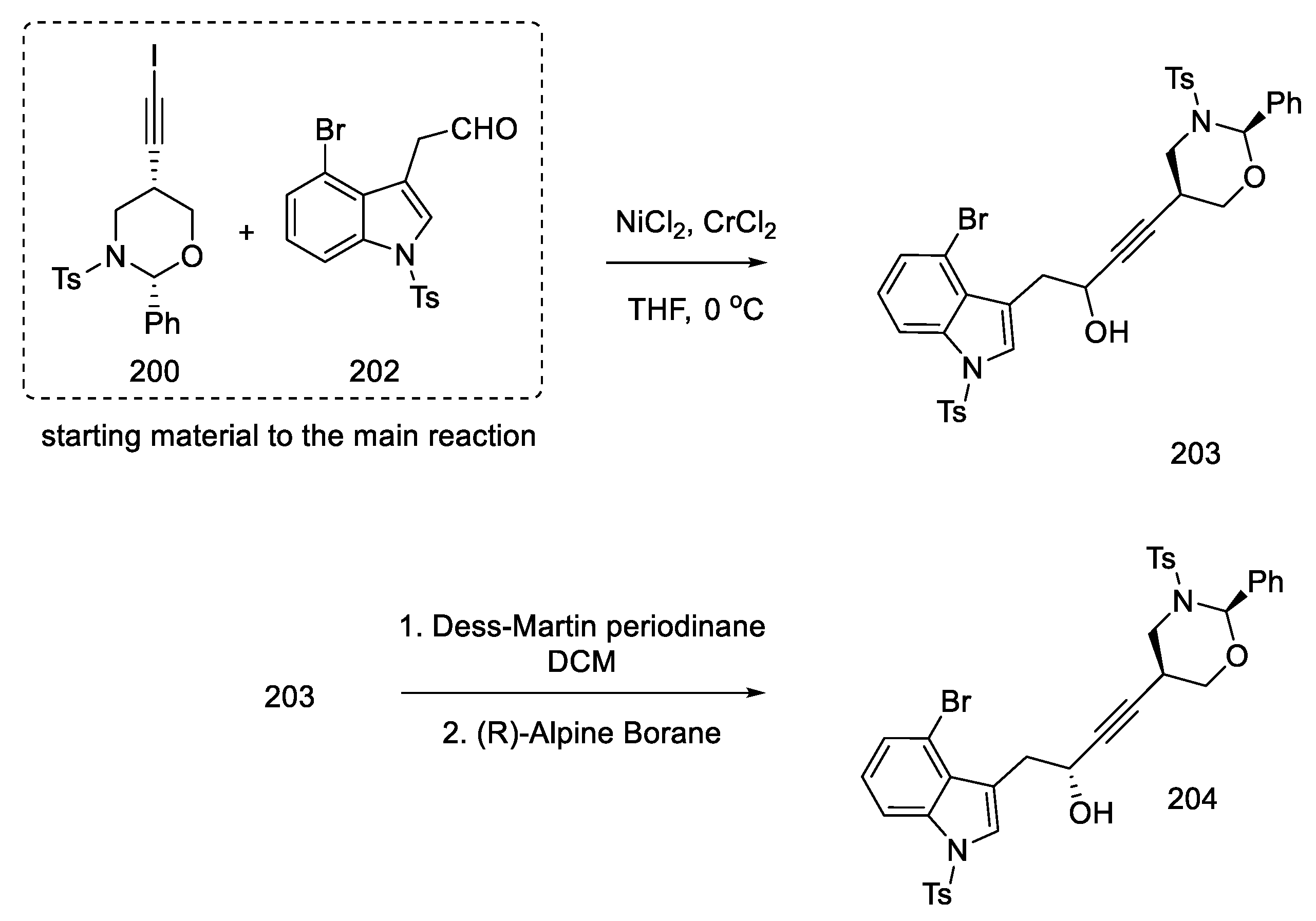
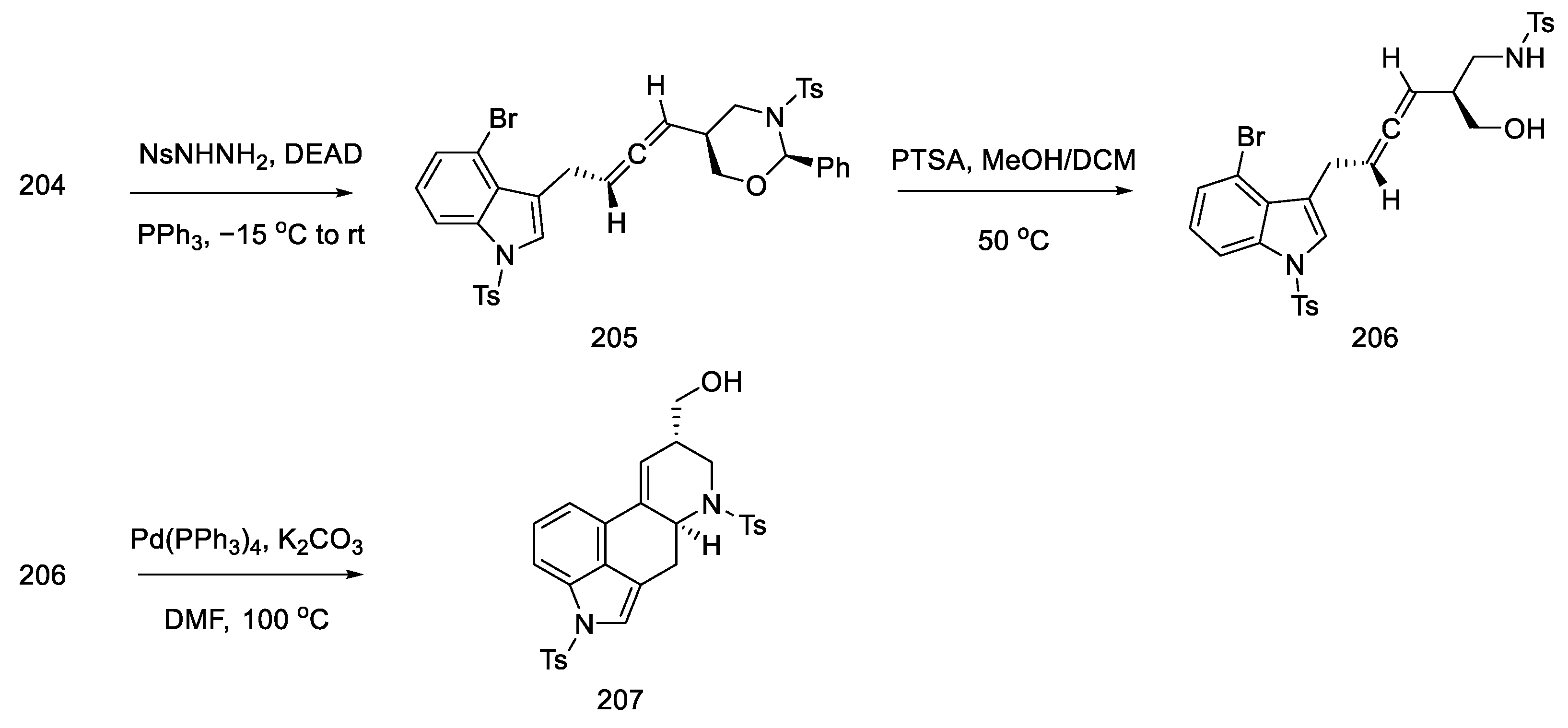
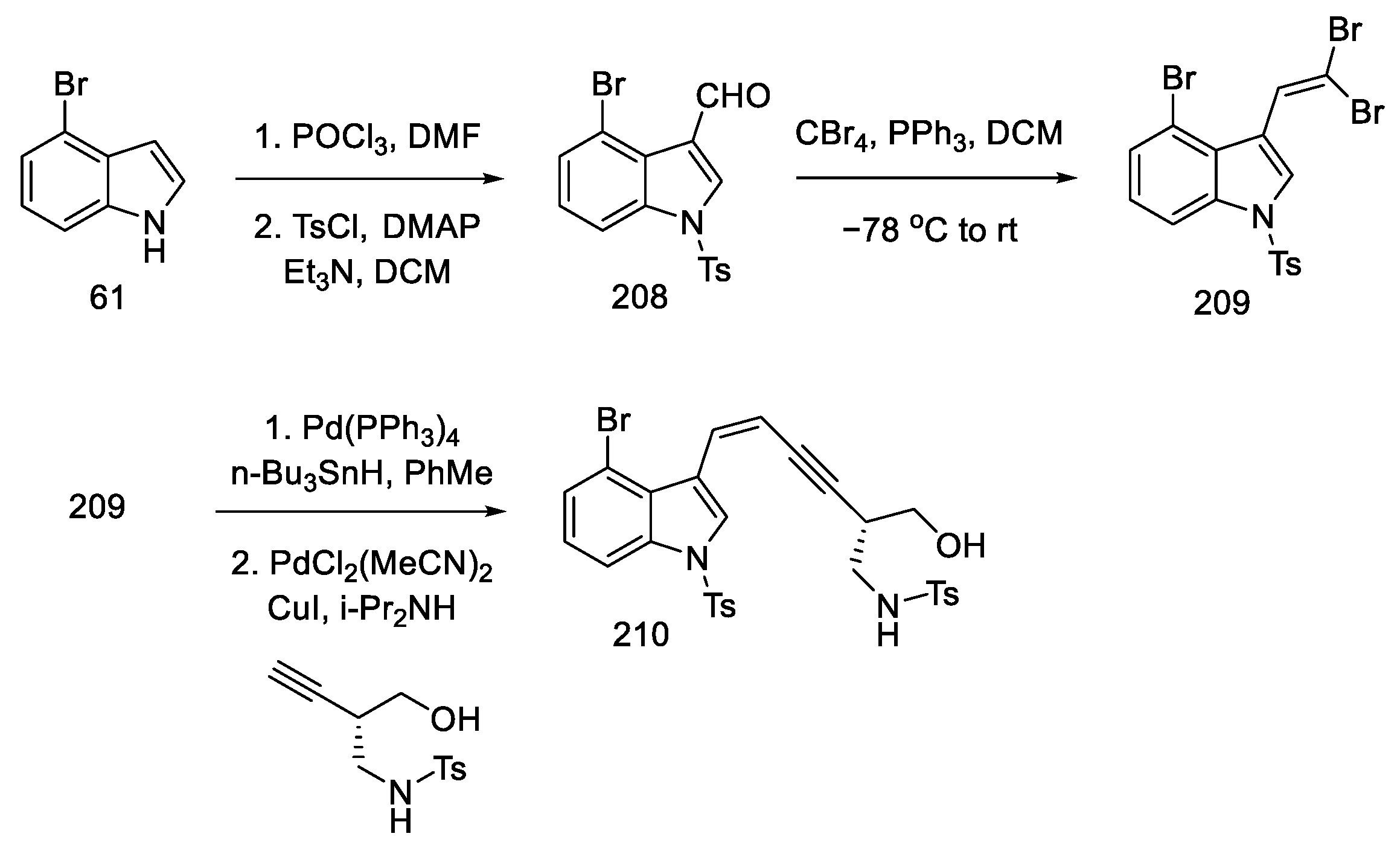
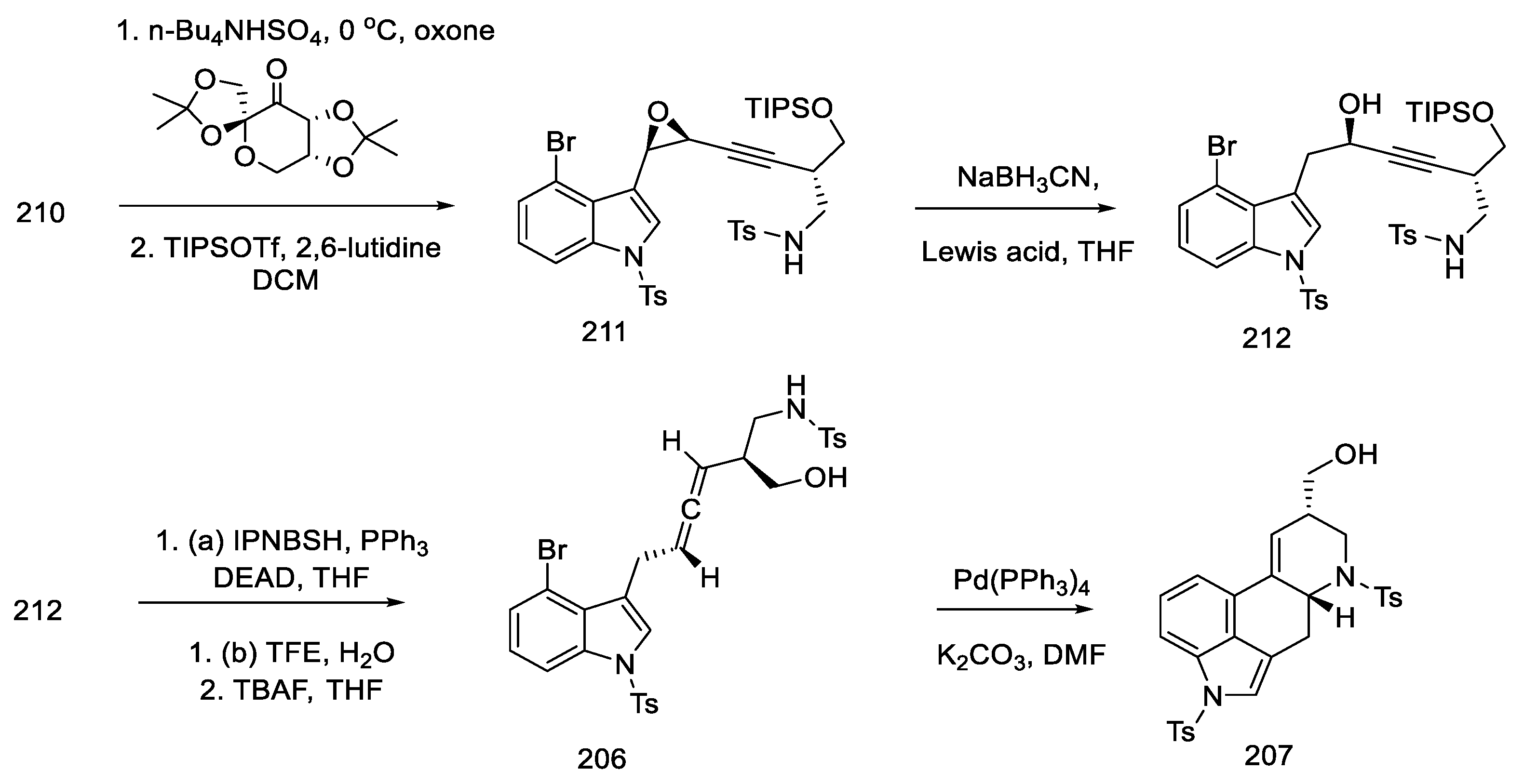
- Inuki, S.; Oishi, S.; Fujii, N.; Ohno, H. Total Synthesis of (±)-Lysergic Acid, Lysergol, and Isolysergol by Palladium-Catalyzed Domino Cyclization of Amino Allenes Bearing a Bromoindolyl Group. Org. Lett. 2008, 10, 5239–5242. [Google Scholar] [CrossRef]
- Iwata, A.; Inuki, S.; Oishi, S.; Fujii, N.; Ohno, H. Formal Total Synthesis of (+)-Lysergic Acid via Zinc(II)-Mediated Regioselective Ring-Opening Reduction of 2-Alkynyl-3-Indolyloxirane. J. Org. Chem. 2011, 76, 5506–5512. [Google Scholar] [CrossRef]
- Inuki, S.; Iwata, A.; Oishi, S.; Fujii, N.; Ohno, H. Enantioselective Total Synthesis of (+)-Lysergic Acid, (+)-Lysergol, and (+)-Isolysergol by Palladium-Catalyzed Domino Cyclization of Allenes Bearing Amino and Bromoindolyl Groups. J. Org. Chem. 2011, 76, 2072–2083. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Futamata, M.; Mukai, R.; Tamaru, Y. Pd-Catalyzed C3-Selective Allylation of Indoles with Allyl Alcohols Promoted by Triethylborane. J. Am. Chem. Soc. 2005, 127, 4592–4593. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, H.; Annen, K.; Laurent, H.; Wiechert, R. A Novel Entry to 17α-Bromo-and 17α-Iodoethynyl Steroids. Angew. Chem. Int. Ed. Engl. 1984, 23, 727–729. [Google Scholar] [CrossRef]
- Myers, A.G.; Zheng, B. New and Stereospecific Synthesis of Allenes in a Single Step from Propargylic Alcohols. J. Am. Chem. Soc. 1996, 118, 4492–4493. [Google Scholar] [CrossRef]
- Lauchli, R.; Shea, K.J. A Synthesis of the Welwistatin Core. Org. Lett. 2006, 8, 5287–5289. [Google Scholar] [CrossRef] [PubMed]
- Uenishi, J.; Kawahama, R.; Shiga, Y.; Yonemitsu, O.; Tsuji, J. A General and Convenient Synthetic Method of Geometrically Pure (Z)-1-Bromo-1-Alkenes. Tetrahedron Lett. 1996, 37, 6759–6762. [Google Scholar] [CrossRef]
- Movassaghi, M.; Ahmad, O.K. N -Isopropylidene- N’-2-Nitrobenzenesulfonyl Hydrazine, a Reagent for Reduction of Alcohols via the Corresponding Monoalkyl Diazenes. J. Org. Chem. 2007, 72, 1838–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, J.P.; Osterhout, M.H.; Padwa, A. An Approach to Lysergic Acid Utilizing an Intramolecular Isomuenchnone Cycloaddition Pathway. J. Org. Chem. 1995, 60, 2704–2713. [Google Scholar] [CrossRef]
- Padwa, A.; Austin, D.J. Ligand Effects on the Chemoselectivity of Transition Metal Catalyzed Reactions Ofα-Diazo Carbonyl Compounds. Angew. Chem. Int. Ed. Engl. 1994, 33, 1797–1815. [Google Scholar] [CrossRef]
- Barton, D.H.R.; McCombie, S.W. A New Method for the Deoxygenation of Secondary Alcohols. J. Chem. Soc. Perkin Trans. 1 1975, 1574–1585. [Google Scholar] [CrossRef]
- Parsons, P.J.; Penkett, C.S.; Shell, A.J. Tandem Reactions in Organic Synthesis: Novel Strategies for Natural Product Elaboratoin and the Development of New Synthetic Methodology. Chem. Rev. 1996, 96, 195–206. [Google Scholar] [CrossRef]
- Cladingboel, D.E.; Parsons, P.J. Synthesis of Lysergic Acid Derivatives by Triple Radical Cyclisation. J. Chem. Soc. Chem. Commun. 1990, 1543–1544. [Google Scholar] [CrossRef]
- Özlü, Y.; Cladingboel, D.E.; Parsons, P.J. Tandem Radical Cyclisations: Synthesis of Lysergic Acid Derivatives. Tetrahedron 1994, 50, 2183–2206. [Google Scholar] [CrossRef]
- Floss, H.G. Biosynthesis of Ergot Alkaloids and Related Compounds. Tetrahedron 1976, 32, 873–912. [Google Scholar] [CrossRef]
- Mukherjee, J.; Menge, M. Progress and Prospects of Ergot Alkaloid Research. In New Products and New Areas of Bioprocess Engineering. Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2000; Volume 68, pp. 1–20. [Google Scholar]
- Tsai, H.F.; Wang, H.; Gebler, J.C.; Poulter, C.D.; Schardl, C.L. The Claviceps Purpurea Gene Encoding Dimethylallyltryptophan Synthase, the Committed Step for Ergot Alkaloid Biosynthesis. Biochem. Biophys. Res. Commun. 1995, 216, 119–125. [Google Scholar] [CrossRef]
- Lee, S.-L.; Floss, H.G.; Heinstein, P. Purification and Properties of Dimethylallylpyrophosphate: Tryptophan Dimethylallyl Transferase, the First Enzyme of Ergot Alkaloid Biosynthesis in Claviceps Sp. SD 58. Arch. Biochem. Biophys. 1976, 177, 84–94. [Google Scholar] [CrossRef]
- Metzger, U.; Schall, C.; Zocher, G.; Unsöld, I.; Stec, E.; Li, S.-M.; Heide, L.; Stehle, T. The Structure of Dimethylallyl Tryptophan Synthase Reveals a Common Architecture of Aromatic Prenyltransferases in Fungi and Bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 14309–14314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
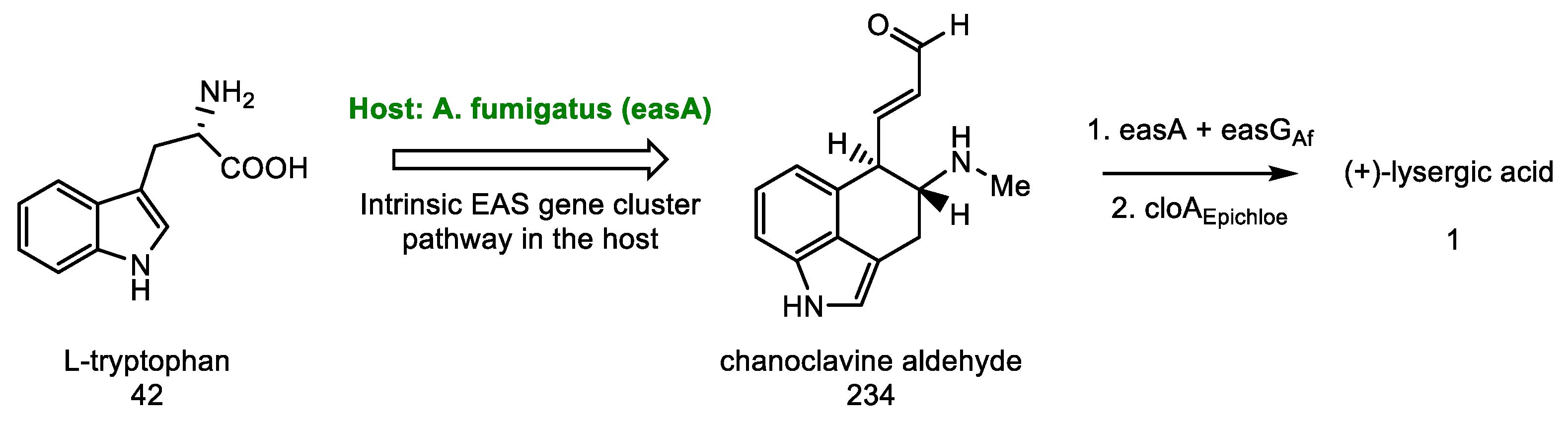
- Rigbers, O.; Li, S.-M. Ergot Alkaloid Biosynthesis in Aspergillus Fumigatus. J. Biol. Chem. 2008, 283, 26859–26868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tudzynski, P.; Correia, T.; Keller, U. Biotechnology and Genetics of Ergot Alkaloids. Appl. Microbiol. Biotechnol. 2001, 57, 593–605. [Google Scholar] [CrossRef]
- Lorenz, N.; OlŠovská, J.; šulc, M.; Tudzynski, P. Alkaloid Cluster Gene CcsA of the Ergot Fungus Claviceps Purpurea Encodes Chanoclavine I Synthase, a Flavin Adenine Dinucleotide-Containing Oxidoreductase Mediating the Transformation of N-Methyl-Dimethylallyltryptophan to Chanoclavine I. Appl. Environ. Microbiol. 2010, 76, 1822–1830. [Google Scholar] [CrossRef] [Green Version]
- Goetz, K.E.; Coyle, C.M.; Cheng, J.Z.; O’Connor, S.E.; Panaccione, D.G. Ergot Cluster-Encoded Catalase Is Required for Synthesis of Chanoclavine-I in Aspergillus Fumigatus. Curr. Genet. 2011, 57, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.; Moore, C.; Panaccione, D. Partial Reconstruction of the Ergot Alkaloid Pathway by Heterologous Gene Expression in Aspergillus Nidulans. Toxins 2013, 5, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Wallwey, C.; Matuschek, M.; Li, S.-M. Ergot Alkaloid Biosynthesis in Aspergillus Fumigatus: Conversion of Chanoclavine-I to Chanoclavine-I Aldehyde Catalyzed by a Short-Chain Alcohol Dehydrogenase FgaDH. Arch. Microbiol. 2010, 192, 127–134. [Google Scholar] [CrossRef]
- Cheng, J.Z.; Coyle, C.M.; Panaccione, D.G.; O’Connor, S.E. Controlling a Structural Branch Point in Ergot Alkaloid Biosynthesis. J. Am. Chem. Soc. 2010, 132, 12835–12837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matuschek, M.; Wallwey, C.; Xie, X.; Li, S.-M. New Insights into Ergot Alkaloid Biosynthesis in Claviceps Purpurea: An Agroclavine Synthase EasG Catalyses, via a Non-Enzymatic Adduct with Reduced Glutathione, the Conversion of Chanoclavine-I Aldehyde to Agroclavine. Org. Biomol. Chem. 2011, 9, 4328–4335. [Google Scholar] [CrossRef] [PubMed]
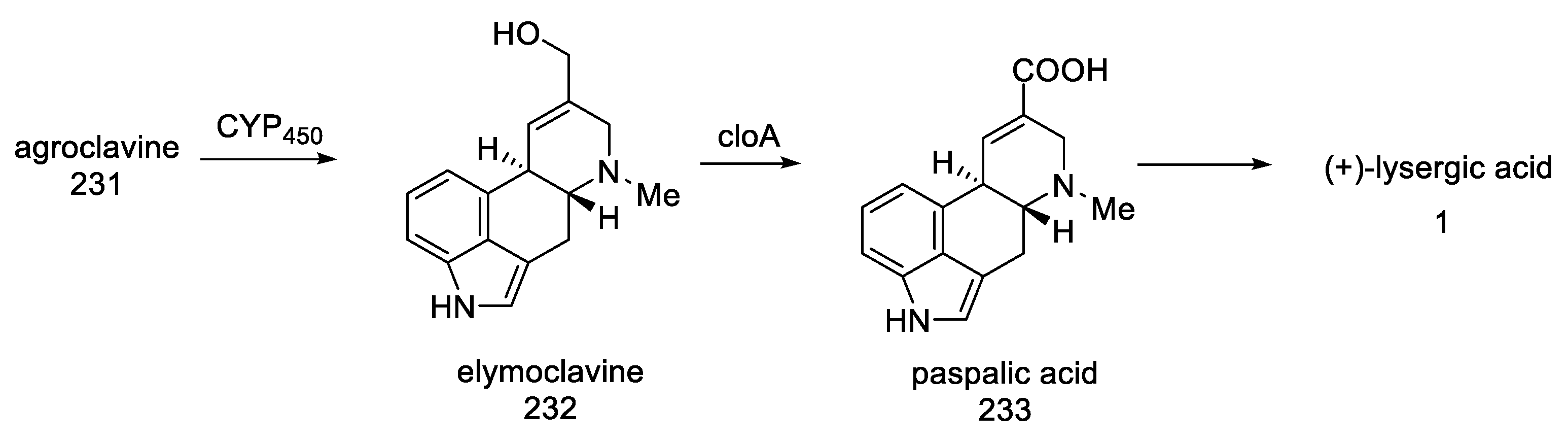
- Kim, S.-U.; Cho, Y.-J.; Floss, H.; Anderson, J. Conversion of Elymoclavine to Paspalic Acid by a Particulate Fraction from an Ergotamine-Producing Strain of Claviceps sp. Planta Med. 1983, 48, 145–148. [Google Scholar] [CrossRef]
- Maier, W.; Schumann, B.; Gröger, D. Microsomal Oxygenases Involved in Ergoline Alkaloid Biosynthesis of VariousClaviceps Strains. J. Basic Microbiol. 1988, 28, 83–93. [Google Scholar] [CrossRef]
- Britton, K.N.; Steen, C.R.; Davis, K.A.; Sampson, J.K.; Panaccione, D.G. Contribution of a Novel Gene to Lysergic Acid Amide Synthesis in Metarhizium Brunneum. BMC Res. Notes 2022, 15, 183. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-J.; Han, M.-Y.; Gong, T.; Yang, J.-L.; Zhu, P. Recent Progress in Ergot Alkaloid Research. RSC Adv. 2017, 7, 27384–27396. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.L.; Panaccione, D.G. Heterologous Expression of Lysergic Acid and Novel Ergot Alkaloids in Aspergillus Fumigatus. Appl. Environ. Microbiol. 2014, 80, 6465–6472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, G.; Lim, L.R.; Tan, Y.Q.; Go, M.K.; Bell, D.J.; Freemont, P.S.; Yew, W.S. Reconstituting the Complete Biosynthesis of D-Lysergic Acid in Yeast. Nat. Commun. 2022, 13, 712. [Google Scholar] [CrossRef] [PubMed]








































































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
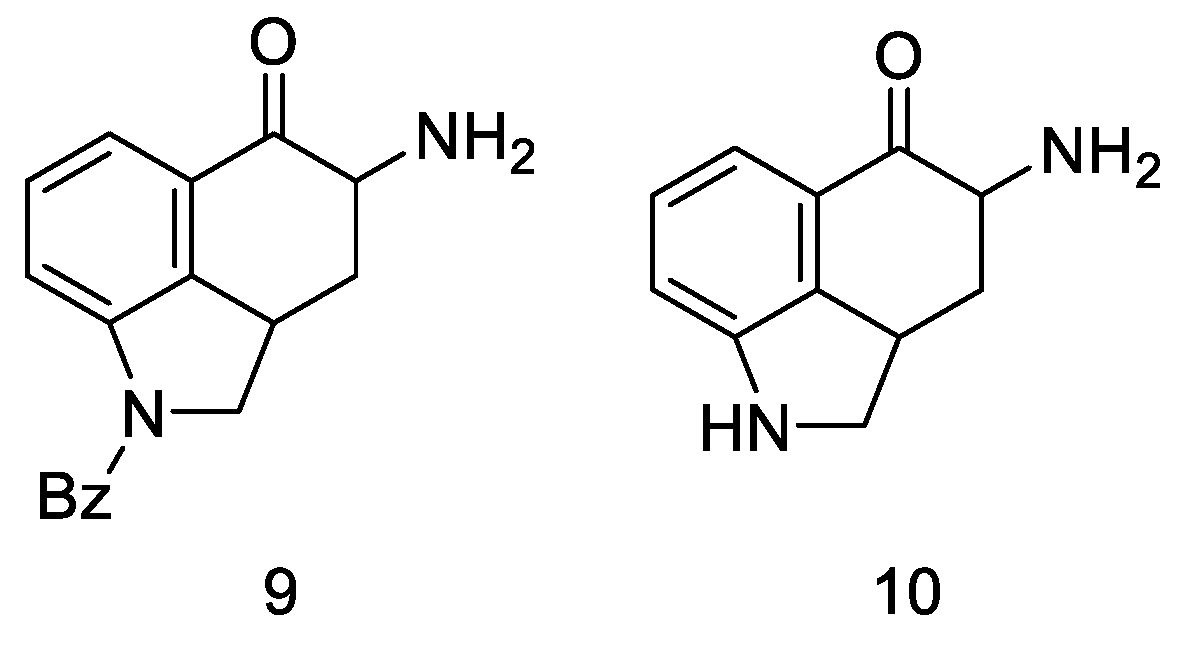{kind=link}
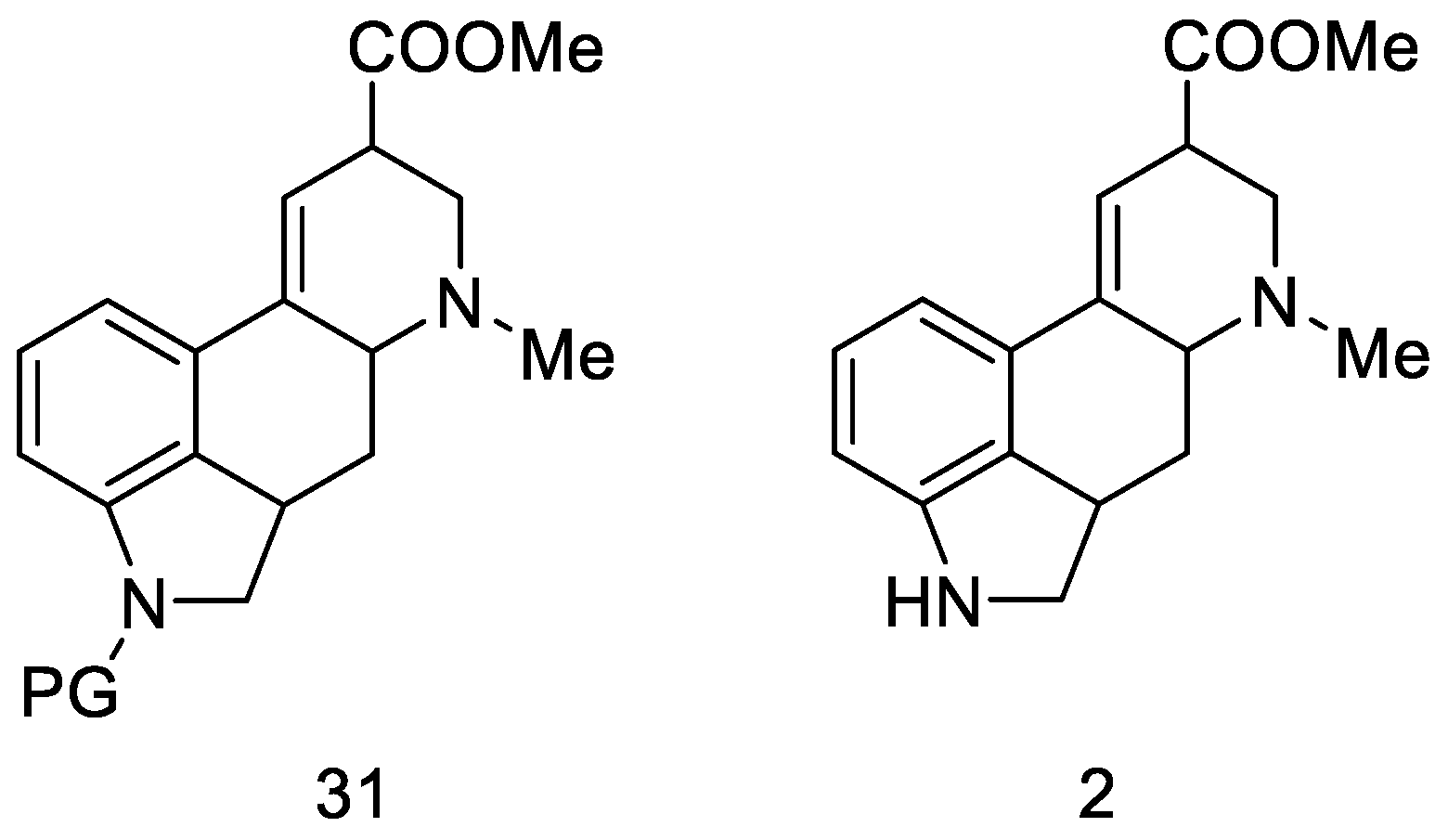{kind=link}
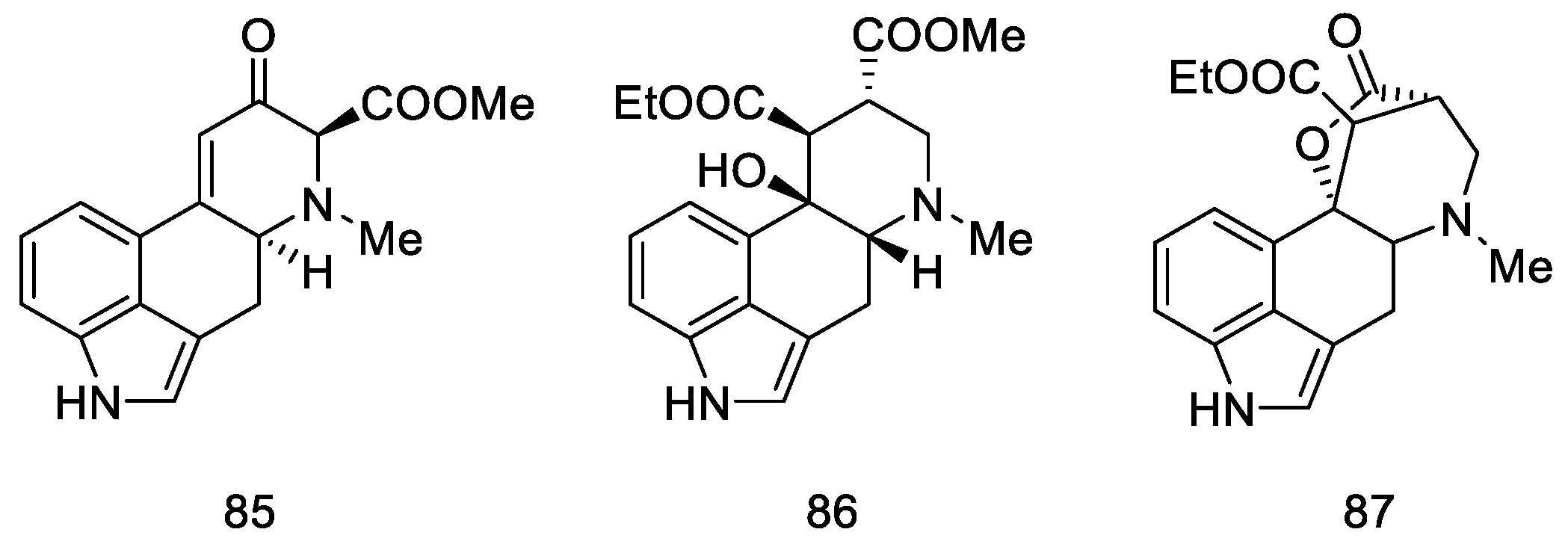{kind=link}
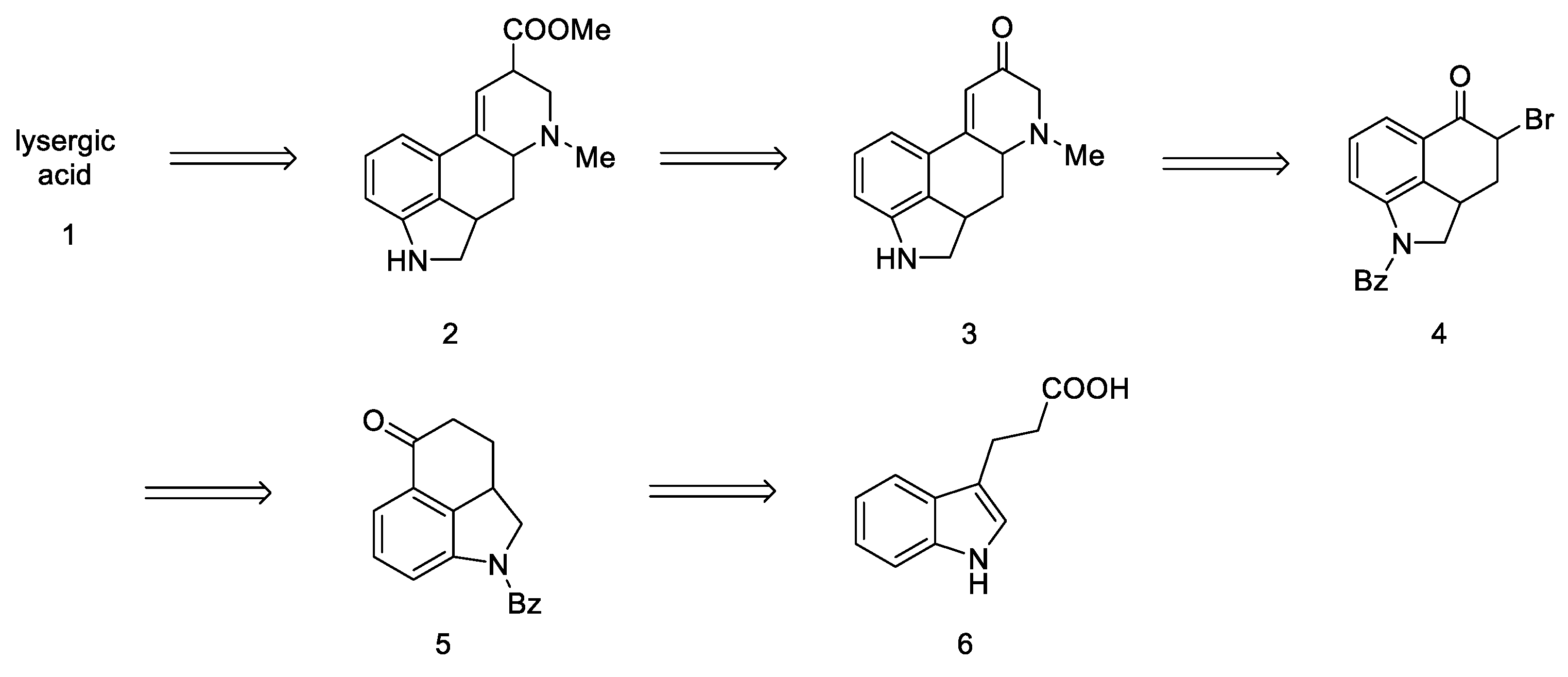{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
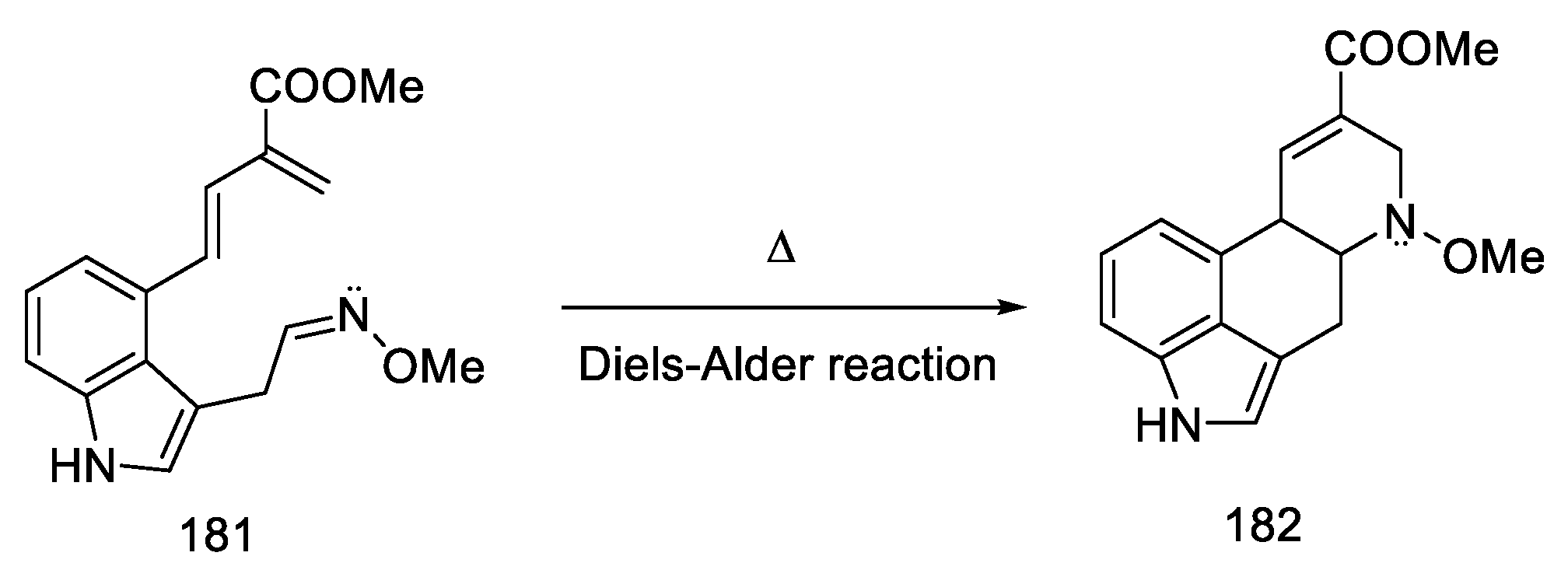{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lysergic Acid Derivatives | Ergotism | Uterotonic Contraction | Vasoconstriction |
|---|---|---|---|
| Ergotamine | Very strong effect | Strong effect | Very strong effect |
| Dihydroergotamine | Moderate effect | Moderate effect | Strong effect |
| Lysergic acid amides 1 | Moderate effect | Very strong effect | Moderate effect |
| LSD | Strong effect | Strong effect | Moderate effect |
| Name of Strategy | Year | Key Intermediate | Optical Rotation | Steps * | Yield [%] ** | Ref |
|---|---|---|---|---|---|---|
| Woodward strategy |  | (±) | ||||
| Woodward | 1956 | 15 | 0.8 | [31] | ||
| Julia (Baillarge group) | 1969 | 11 | - | [47] | ||
| Ramage | 1976 | 19 | 1.5 | [32] | ||
| Kiguchi and Ninomiya | 1982 | 20 | 0.03 | [59] | ||
| Rebek | 1983 | 14 | 4.4 | [55,56] | ||
| Hendrickson strategy |  | (±) | ||||
| Hendrickson | 2004 | 8 | 10.6 | [64] | ||
| Yigang (Snieckus group) | 2010 | 9 | - | [67,69] | ||
| Beaundry | 2020 | 10 | - | [68] | ||
| Szantay strategy | - |  | (+) | |||
| Szantay | 2004 | 15 | 0.7 | [70] | ||
| Garner | 2021 | 20 | 0.21 | [74] | ||
| Closing the C/D-ring using the Heck method | ||||||
| Ortar | 1986 | (±) | 14 | 2.0 | [79] | |
| Kurihara | 1988 | (±) | 12 | 1.3 | [80,81] | |
| Fukuyama | 2009 | (+) | 34 | 0.9 | [80] | |
| Fukuyama | 2009 | (+) | 24 | 0.08 | [86] | |
| Fukuyama | 2013 | (+) | 19 | 12 | [88] | |
| Fukuyama | 2018 | (+) | 30 | 0.07 | [91] | |
| Jia and Liu | 2011 | (+) | 20 | 5.1 | [93] | |
| Jia and Liu | 2013 | (+) | 12 | 1.0 | [94] | |
| Other | ||||||
| Oppolzer | 1981 | (±) | 17 | 0.9 | [85] | |
| Fuji and Ohno | 2008 | (±) | 21 | 3.1 | [98] | |
| Fuji and Ohno | 2011 | (+) | 16 | 5.9 | [99] | |
| Fuji and Ohno | 2011 | (+) | 17 | 1.8 | [100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jastrzębski, M.K.; Kaczor, A.A.; Wróbel, T.M. Methods of Lysergic Acid Synthesis—The Key Ergot Alkaloid. Molecules 2022, 27, 7322. https://doi.org/10.3390/molecules27217322
Jastrzębski MK, Kaczor AA, Wróbel TM. Methods of Lysergic Acid Synthesis—The Key Ergot Alkaloid. Molecules. 2022; 27(21):7322. https://doi.org/10.3390/molecules27217322
Chicago/Turabian StyleJastrzębski, Michał K., Agnieszka A. Kaczor, and Tomasz M. Wróbel. 2022. "Methods of Lysergic Acid Synthesis—The Key Ergot Alkaloid" Molecules 27, no. 21: 7322. https://doi.org/10.3390/molecules27217322
APA StyleJastrzębski, M. K., Kaczor, A. A., & Wróbel, T. M. (2022). Methods of Lysergic Acid Synthesis—The Key Ergot Alkaloid. Molecules, 27(21), 7322. https://doi.org/10.3390/molecules27217322