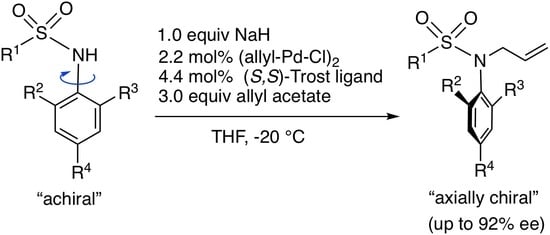
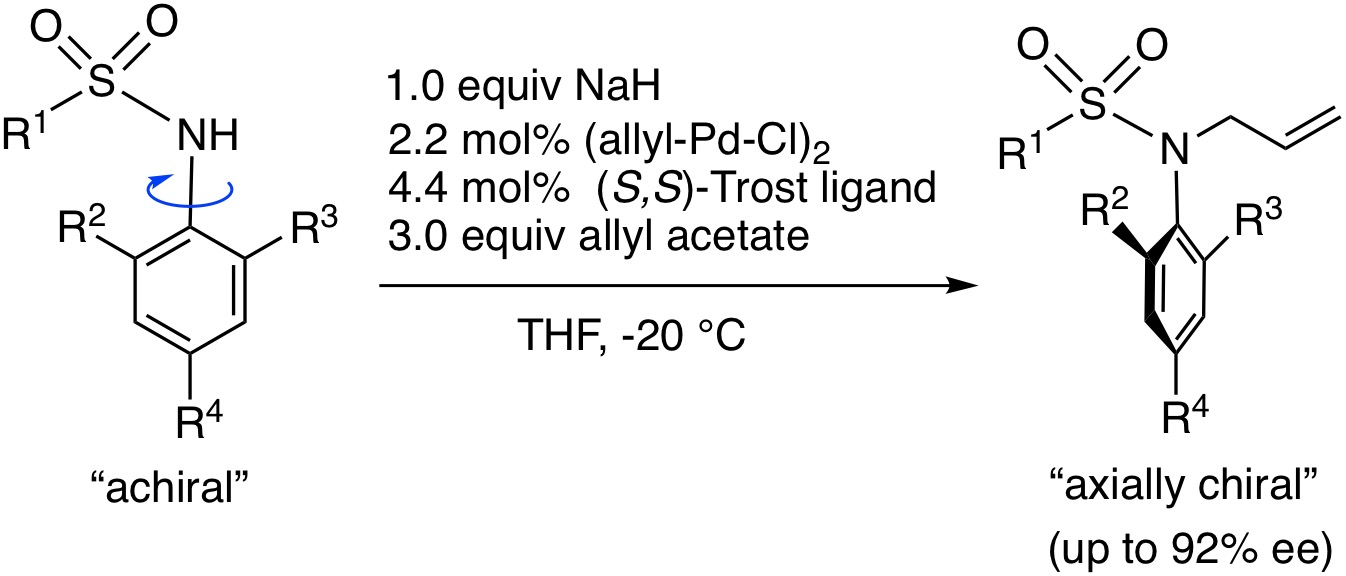
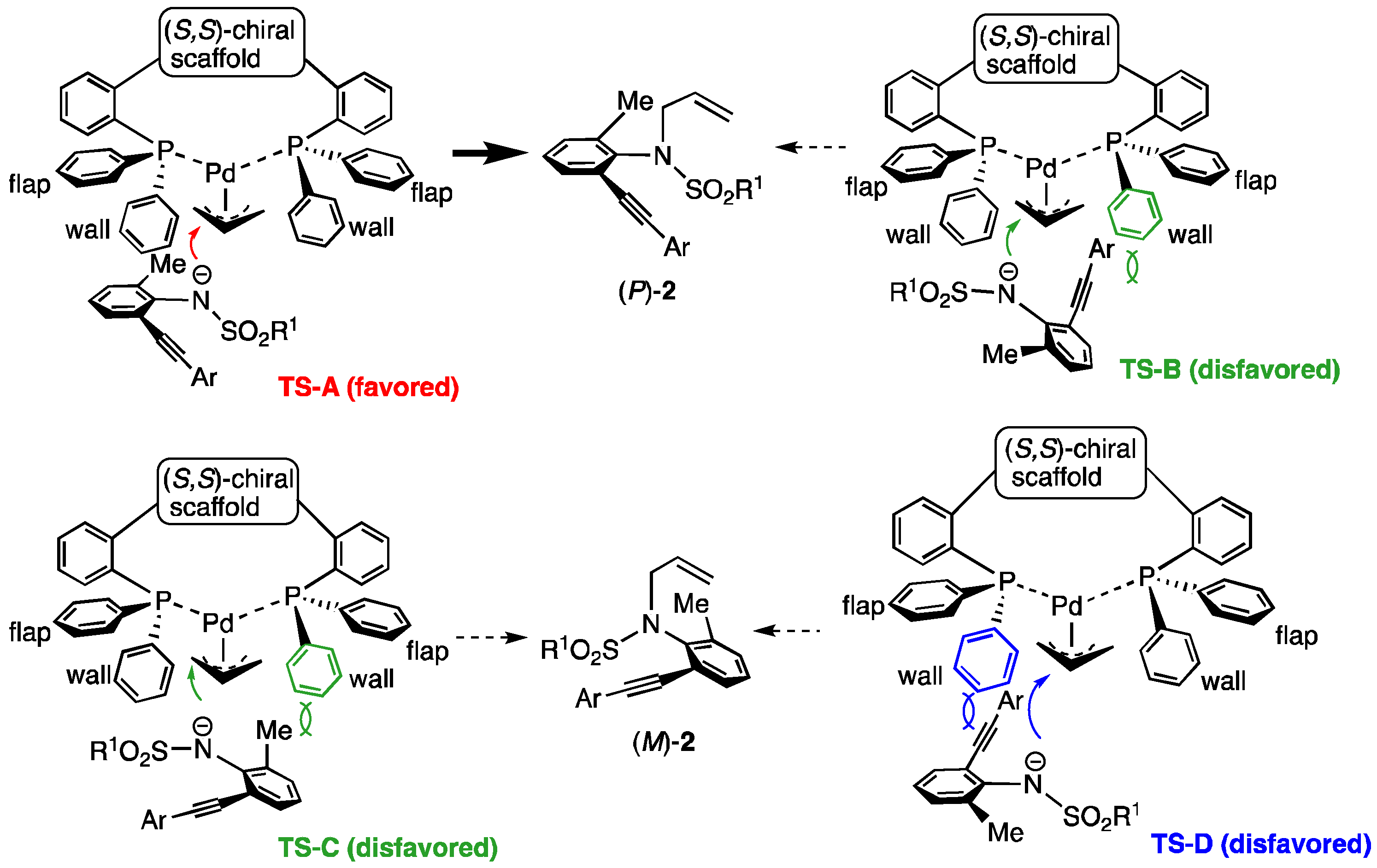
3.2. Synthesis of Substrates 1 and Asymmetric N-Allylation with 1
N-(2-tert-Butyl-6-methylphenyl)-4-methylbenzenesulfonamide (1a). Under N2 atmosphere, to 2-tert-butyl-6-methylaniline (488 mg, 3.0 mmol, commercially available) and pyridine (0.36 mL, 4.5 mmol) in CH2Cl2 (4.0 mL) was added 4-tosyl chloride (631 mg, 3.3 mmol), and then the mixture was stirred for 22 h at 0 °C–rt. The mixture was poured into water and extracted with AcOEt. The AcOEt extracts were washed with brine, dried over MgSO4, and evaporated to dryness. Hexane was added to the residue and the mixture was filtered in vacuo. After washing the residue with hexane, 1a was obtained (417 mg, 44%). 1a: white solid; mp 157–160 °C; IR (neat) 3268, 1323, 1155 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.68 (2H, d, J = 8.1 Hz), 7.34 (1H, d, J = 8.1 Hz), 7.27 (2H, d, J = 8.1 Hz), 7.14 (1H, t, J = 7.6 Hz), 7.00 (1H, d, J = 7.1 Hz), 6.30 (1H, s), 2.43 (3H, s), 1.91 (3H, s), 1.43 (9H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 148.6, 143.2, 138.9, 138.6, 132.3, 129.4, 129.0, 127.4, 127.0, 126.4, 36.1, 32.4, 21.5, 20.1; MS (ESI-TOF) m/z: [M + Na]+ 340; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H23NNaO2S 340.1347; Found 340.1347.
N-((2-Iodo-4-methyl-6-phenyl)phenyl)-4-methylbenzenesulfonamide (1b). Under N2 atmosphere, to phenylboronic acid (438 mg, 3.6 mmol) and potassium carbonate (1.66 g, 12.0 mmol) in H2O (10 mL) were added bis(triphenylphosphine)palladium(II) chloride (107 mg, 0.15 mmol) and 2,6-diiodo-4-methylaniline (1.04 g, 2.9 mmol). The mixture was stirred for 3 h at rt. The mixture was poured into water and extracted with AcOEt. The AcOEt extracts were washed with brine, dried over MgSO4, and evaporated to dryness. Purification of the residue by column chromatography (hexane/AcOEt = 150) gave 2-iodo-4-methyl-6-phenyl aniline (358 mg, 40%). Under N2 atmosphere, to 2-iodo-4-methyl-6-phenylaniline (610 mg, 2.0 mmol) and pyridine (0.24 mL, 3.0 mmol) in CH2Cl2 (6.0 mL) was added 4-tosyl chloride (414 mg, 2.2 mmol), and then the mixture was stirred for 22 h at 0 °C–rt. The mixture was poured into water and extracted with AcOEt. The AcOEt extracts were washed with brine, dried over MgSO4, and evaporated to dryness. Purification of the residue by column chromatography (hexane/AcOEt = 120 and then 5) gave 1b (428 mg, 47%). 1b: white solid; mp 135–136 °C; IR (neat) 3258, 1331, 1155 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.67 (1H, m), 7.26 (2H, dt, J = 8.5, 1.9 Hz), 7.19–7.22 (3H, m), 7.14–7.17 (2H, m), 7.06 (1H, d, J = 1.4 Hz), 7.01 (2H, d, J = 7.6 Hz), 6.42 (1H, s), 2.37 (3H, s), 2.31 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.0, 141.8, 139.6, 139.5, 137.2, 132.6, 132.0, 129.2, 128.8, 128.0, 127.2, 127.0, 102.1, 21.5, 20.4; MS (ESI-TOF) m/z: [M + Na]+ 486; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C20H18INNaO2S 486.0001; Found 485.9972.
N-((2-Bromo-4-methyl-6-phenyl)phenyl)-4-methylbenzenesulfonamide (1c). In accordance with the experimental procedure for the synthesis of 1b, 1c was prepared from 2-bromo-4-methyl-6-phenylaniline (318 mg, 1.2 mmol, commercially available) and 4-tosyl chloride (280 mg, 1.4 mmol). The reaction was conducted for 18 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 50 and then 3) gave 1c (213 mg, 42%). 1c: white solid; mp 149–152 °C; IR (neat) 3250, 1337, 1163 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.38 (1H, dd, J = 1.9, 1.0 Hz), 7.29 (2H, dt, J = 8.4, 1.9 Hz), 7.21–7.24 (5H, m), 7.06 (1H, d, J = 1.4 Hz), 7.03 (2H, d, J = 8.1 Hz), 6.42 (1H, s), 2.37 (3H, s), 2.33 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.1, 142.5, 139.3, 139.2, 137.0, 132.7, 131.7, 129.2, 129.04, 128.97, 128.0, 127.1, 124.6, 21.5, 20.7; MS (ESI-TOF) m/z: [M + Na]+ 440; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C20H1881BrNNaO2S 440.0119; Found 440.0102.
N-(2-Iodo-4,6-dimethylphenyl)-4-methylbenzenesulfonamide (1d). In accordance with the experimental procedure for the synthesis of 1b, 1d was prepared from 2-iodo-4,6-dimethylaniline (494 mg, 2.0 mmol, commercially available) and 4-tosyl chloride (419 mg, 2.2 mmol). The reaction was conducted for 17 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 10 and then 3) gave 1d (804 mg, quant). 1d: White solid; mp 161–163 °C; IR (neat) 3275, 1331, 1157 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.56 (2H, dt, J = 8.1, 1.9 Hz), 7.37 (1H, d, J = 1.4 Hz), 7.23 (2H, d, J = 8.1 Hz), 7.04 (1H, d, J = 1.4 Hz), 6.14 (1H, s), 2.45 (3H, s), 2.42 (3H, s), 2.24 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.9, 139.4, 139.3, 137.5, 137.2, 133.2, 132.7, 129.5, 127.9, 100.2, 21.6, 20.7, 20.3; MS (ESI-TOF) m/z: [M + Na]+ 424; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C15H16127INNaO2S 423.9844; Found 423.9816.
(
E)-
N-(2,4-Dimethyl-6-styrylphenyl)-4-methylbenzenesulfonamide (
1e). In accordance with the experimental procedure for the synthesis of
1b,
1e was prepared from 2,4-dimethyl-6-styrylaniline (218 mg, 1.0 mmol) [
40] and 4-tosyl chloride (279 mg, 1.5 mmol). The reaction was conducted for 22 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 5) gave
1e (323 mg, 88%).
1e: white solid; mp 150–152 °C; IR (neat) 3242, 1325, 1157 cm
−1;
1H NMR (400 MHz, CDCl
3)
δ: 7.61 (2H, dt,
J = 8.1, 1.9 Hz), 7.21–7.30 (4H, m), 7.16 (2H, dd,
J = 8.1, 1.9 Hz), 7.06 (2H, d,
J = 7.6 Hz), 6.99 (1H, d,
J = 1.9 Hz), 6.83 (1H, d,
J = 16.6 Hz), 6.74 (1H, d,
J = 16.6 Hz), 6.30 (1H, s), 2.33 (3H, s), 2.30 (3H, s), 2.18 (3H, s);
13C{
1H} NMR (100 MHz, CDCl
3)
δ: 143.6, 138.8, 137.9, 137.4, 137.0, 136.3, 131.3, 129.9, 129.6, 129.2, 128.3, 127.6, 127.1, 126.6, 124.3, 124.2, 21.3, 21.1, 19.0; MS (ESI-TOF)
m/
z: [M + Na]
+ 400; HRMS (ESI-TOF)
m/
z: [M + Na]
+ Calcd for C
23H
23NNaO
2S 400.1347; Found 400.1330.
N-(2-Iodo-4-methyl-6-(p-tolylethynyl)phenyl])-4-methylbenzenesulfonamide (1f). Under N2 atmosphere, to 2,6-diiodo-4-methylaniline (1.08 g, 3.0 mmol, commercially available), copper iodide(I) (11.4 mg, 0.060 mol) and bis(triphenylphosphine)palladium(II)dichloride (42 mg, 0.060 mmol) in triethylamine (15 mL) was added 4-ethynyltoluene (384 mg, 3.3 mmol), and then the mixture was stirred for 19 h at rt. The mixture was poured into 2N HCl aqueous solution and extracted with AcOEt. The AcOEt extracts were washed with brine, dried over MgSO4, and evaporated to dryness. Purification of residue by column chromatography (hexane/AcOEt = 200) gave 2-iodo-4-methyl-6-(4-tolylethynyl)aniline (454 mg, 44%). In accordance with the experimental procedure for the synthesis of 1b, 1f was prepared from 2-iodo-4-methyl-6-(4-tolylethynyl)aniline (355 mg, 1.0 mmol) and 4-tosyl chloride (211 mg, 1.1 mmol). The reaction was conducted for 24 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 20 and then 5) gave 1f (181 mg, 35%). 1f: White solid; mp 208–211 °C; IR (neat) 3231, 2216, 1337, 1165 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.66 (1H, d, J = 1.4 Hz), 7.62 (2H, dt, J = 8.1, 1.9 Hz), 7.22–7.28 (3H, m), 7.12 (2H, d, J = 8.1 Hz), 7.08 (2H, d, J = 8.1 Hz), 6.49 (1H, s), 2.37 (3H, s), 2.28 (3H, s), 2.25 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.6, 140.7, 139.1, 138.9, 137.6, 135.9, 133.6, 131.6, 129.5, 128.9, 127.6, 123.5, 119.3, 99.8, 94.8, 85.0, 21.6, 21.5, 20.3; MS (ESI-TOF) m/z: [M + Na]+ 524; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H20127INNaO2S 524.0157; Found 524.0130.
N-(2-Bromo-4-methyl-6-(p-tolylethynyl)phenyl)-4-methylbenzenesulfonamide (1g). Under N2 atmosphere, to 2-bromo-4-methyl-6-iodoaniline (469 mg, 1.5 mmol, commercially available), copper iodide(I) (5.7 mg, 0.030 mmol) and bis(triphenylphosphine)palladium(II) dichloride (21 mg 0.030 mmol) in triethylamine (7.5 mL) was added 4-ethynyltoluene (192 mg, 1.7 mmol), and then the mixture was stirred for 22 h at rt. The mixture was poured into 2N HCl aqueous solution and extracted with AcOEt. The AcOEt extracts were washed with brine, dried over MgSO4, and evaporated to dryness. Purification of residue by column chromatography (hexane/AcOEt = 30) gave 2-bromo-4-methyl-6-(4-tolylethynyl)aniline (353 mg, 78%). In accordance with the experimental procedure for the synthesis of 1b, 1g was prepared from 2-bromo-4-methyl-6-(4-tolylethynyl)aniline (903 mg, 3.0 mmol) and 4-tosyl chloride (636 mg, 3.3 mmol). The reaction was conducted for 24 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 20 and then 3) gave 1g (618 mg, 45%). 1g: white solid; mp 191–197 °C; IR (neat) 3229, 2218, 1339, 1165 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.65 (2H, dt, J = 8.5, 1.9 Hz), 7.37 (1H, d, J = 1.4 Hz), 7.26–7.28 (3H, m), 7.09–7.13 (4H, m), 6.43 (1H, s), 2.37 (3H, s), 2.30 (3H, s), 2.27 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.6, 138.92, 138.85, 137.6, 134.0, 132.8, 132.7, 131.7, 129.5, 128.9, 127.5, 124.5, 123.5, 119.4, 95.0, 85.0, 21.6, 21.5, 20.6; MS (ESI-TOF) m/z: [M + Na]+ 478; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H2081BrNNaO2S 478.0275; Found 478.0249.
N-(2-Chloro-6-(p-tolylethynyl)phenyl)-4-methylbenzenesulfonamide (1h). Under N2 atmosphere, to 2-chloro-6-iodoaniline (507 mg, 2.0 mmol, commercially available), copper iodide(I) (7.5 mg, 0.040 mmol) and bis(triphenylphosphine)palladium(II) dichloride (29 mg, 0.041 mol) in triethylamine (10 mL) was added 4-ethynyltoluene (255 mg, 2.2 mmol), and then the mixture was stirred for 19 h at rt. The mixture was poured into 2N HCl aqueous solution and extracted with AcOEt. The AcOEt extracts were washed with brine, dried over MgSO4, and evaporated to dryness. Purification of residue by column chromatography (hexane/AcOEt = 30) gave 2-chloro-6-(4-tolylethynyl)aniline (398 mg, 82%). In accordance with the experimental procedure for the synthesis of 1b, 1h was prepared from 2-chloro-6-(4-tolylethynyl)aniline (214 mg, 0.9 mmol) and 4-tosyl chloride (189 mg, 1.0 mmol). The reaction was conducted for 24 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 30 and then 5) gave 1h (121 mg, 34%). 1h: white solid; mp 192–193 °C; IR (neat) 3229, 2209, 1337, 1165 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.67 (2H, d, J = 8.1 Hz), 7.40 (1H, dd, J = 7.6, 1.4 Hz), 7.36 (1H, dd, J = 8.1, 1.4 Hz), 7.30 (2H, d, J = 8.1 Hz), 7.10–7.19 (5H, m), 6.52 (1H, s), 2.38 (3H, s), 2.29 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.7, 139.1, 137.4, 134.2, 133.3, 131.7, 131.3, 130.2, 129.5, 129.0, 128.0, 127.4, 124.7, 119.2, 95.7, 84.5, 21.6, 21.5; MS (ESI-TOF) m/z: [M + Na]+ 418; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H1835ClNNaO2S 418.0645; Found 418.0615.
N-(2,4-Dimethyl-6-(p-tolylethynyl)phenyl)-4-methylbenzenesulfonamide (1i). Under N2 atmosphere, to 2,4-dimethyl-6-iodoaniline (494 mg, 2.0 mmol, commercially available), copper iodide(I) (7.6 mg, 0.040 mmol) and bis(triphenylphosphine)palladium(II) dichloride (28 mg, 0.040 mmol) in triethylamine (10 mL) was added 4-ethynyltoluene (254 mg, 2.2 mmol), and then the mixture was stirred for 19 h at rt. The mixture was poured into 2N HCl aqueous solution and extracted with AcOEt. The AcOEt extracts were washed with brine, dried over MgSO4, and evaporated to dryness. Purification of residue by column chromatography (hexane/AcOEt = 15) gave 2-bromo-4-methyl-6-(4-tolylethynyl)aniline (468 mg, 99%). In accordance with the experimental procedure for the synthesis of 1b, 1i was prepared from 2,4-dimethyl-6-(4-tolylethynyl)aniline (708 mg, 3.0 mmol) and 4-tosyl chloride (629 mg, 3.3 mmol). The reaction was conducted for 27 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 15 and then 5) gave 1i (918 mg, 78%). 1i: white solid; mp 149–152 °C; IR (neat) 3248, 2205, 1331, 1163 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.50 (2H, dt, J = 8.5, 1.9 Hz), 7.11–7.16 (4H, m), 7.06 (1H, s), 7.03 (1H, s), 6.99 (2H, d, J = 8.1), 6.44 (1H, s), 2.51 (3H, s), 2.38 (3H, s), 2.27 (3H, s), 2.23 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.3, 138.6, 137.9, 136.9, 136.5, 132.7, 132.3, 131.3, 130.4, 129.3, 128.9, 127.4, 121.5, 119.4, 94.0, 84.8, 21.5, 21.4, 20.7, 19.4; MS (ESI-TOF) m/z: [M + Na]+ 412; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C24H23NNaO2S 412.1347; Found 412.1332.
N-(2-((4-Methoxyphenyl)ethynyl)-4,6-dimethylphenyl)-4-methylbenzenesulfonamide (1j). Under N2 atmosphere, to 2,4-dimethyl-6-iodoaniline (619 mg, 2.5 mmol, commercially available), copper iodide(I) (10 mg, 0.053 mmol) and bis(triphenylphosphine)palladium(II) dichloride (35 mg, 0.050 mmol) in triethylamine (10 mL) was added 4-ethynylanisole (363 mg, 2.7 mmol), and then the mixture was stirred for 17 h at rt. The mixture was poured into 2N HCl aqueous solution and extracted with AcOEt. The AcOEt extracts were washed with brine, dried over MgSO4, and evaporated to dryness. Purification of residue by column chromatography (hexane/AcOEt = 30) gave 2,4-dimerthyl-6-((4-tmethoxyphenyl)ethynyl)aniline (402 mg, 64%). In accordance with the experimental procedure for the synthesis of 1b, 1j was prepared from 2,4-dimethyl-6-((4-methoxyphenyl)ethynyl)aniline (402 mg, 1.6 mmol) and 4-tosyl chloride (336 mg, 1.8 mmol). The reaction was conducted for 19 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 10 and then 5) gave 1j (384 mg, 59%). 1j: white solid; mp 149–150 °C; IR (neat) 3264, 2203, 1329, 1157 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.50 (2H, dt, J = 8.5, 1.9 Hz), 7.19 (2H, dt, J = 9.0, 2.4 Hz), 7.05 (1H, s), 7.01 (1H, s), 7.00 (2H, d, J = 7.6 Hz), 6.84 (2H, dt, J = 9.0, 2.4 Hz), 6.41 (1H, s), 3.84 (3H, s), 2.49 (3H, s), 2.27 (3H, s), 2.25 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 159.7, 143.4, 137.9, 136.9, 136.6, 132.9, 132.6, 132.3, 130.3, 129.3, 127.5, 121.6, 114.7, 113.8, 93.9, 84.2, 55.3, 21.5, 20.7, 19.5; MS (ESI-TOF) m/z: [M + Na]+ 428; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C24H23NNaO3S 428.1296; Found 428.1282.
N-(2,4-Dimethyl-6-(phenylethynyl)phenyl)-4-methylbenzenesulfonamide (1k). In accordance with the experimental procedure for the synthesis of 1b, 1k was prepared from 2,4-dimethyl-6-(4-phenyl)ethynyl)aniline (379 mg, 1.7 mmol, commercially available) and 4-tosyl chloride (357 mg, 1.9 mmol). The reaction was conducted for 22 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 20 and then 10) gave racemic 1k (582 mg, 90%). white solid; mp 186–188 °C; IR (neat) 3241, 2212, 1331, 1165 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.51 (2H, dt, J = 8.1, 2.4 Hz), 7.24–7.34 (5H, m), 7.08 (1H, s), 7.05 (1H, s), 6.98 (2H, d, J = 7.6 Hz), 6.42 (1H, s), 2.51 (3H, s), 2.28 (3H, s), 2.22 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.4, 138.0, 137.0, 136.6, 132.9, 132.4, 131.4, 130.5, 129.3, 128.5, 128.1, 127.4, 122.6, 121.3, 93.7, 85.5, 21.4, 20.7, 19.4; MS (ESI-TOF) m/z: [M + Na]+ 398; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H21NNaO2S 398.1191; Found 398.1179.
N-(2,4-Dimethyl-6-((trimethylsilyl)ethynyl)phenyl)-4-methylbenzenesulfonamide (
1l). In accordance with the experimental procedure for the synthesis of
1b,
1l was prepared from 2,4-dimethyl-6-(4-trimethylsilyl)ethynyl)aniline (434 mg, 2.0 mmol) [
41] and 4-tosyl chloride (419 mg, 2.2 mmol). The reaction was conducted for 22 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 20 and then 5) gave
1l (587 mg, 79%).
1l: white solid; mp 112–113 °C; IR (neat) 3225, 2158, 1335, 1165 cm
−1;
1H NMR (400 MHz, CDCl
3)
δ: 7.47 (2H, dt,
J = 8.5, 1.9 Hz), 7.17 (2H, d,
J = 8.5 Hz), 7.05 (1H, s), 6.98 (1H, s), 6.37 (1H, s), 2.47 (3H, s), 2.40 (3H, s), 2.24 (3H, s), 0.13 (9H, m);
13C{
1H} NMR (100 MHz, CDCl
3)
δ: 143.4, 137.3, 136.6, 136.4, 133.2, 132.8, 130.7, 129.3, 127.8, 120.9, 100.6, 99.6, 21.6, 20.7, 19.5, −0.17; MS (ESI-TOF)
m/
z: [M + Na]
+ 394; HRMS (ESI-TOF)
m/
z: [M + Na]
+ Calcd for C
20H
25NNaO
2S
28Si 394.1273; Found 394.1257.
N-(2-(Hex-1-yn-1-yl)-4,6-dimethylphenyl)-4-methylbenzenesulfonamide (1m). In accordance with the experimental procedure for the synthesis of 1b, 1m was prepared from 2,4-dimethyl-6-(2-hex-1-yn-1-yl)aniline (363 mg, 1.8 mmol, commercially available) and 4-tosyl chloride (378 mg, 2.0 mmol). The reaction was conducted for 26 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 50 and then 30) gave 1m (498 mg, 78%). 1m: orange solid; mp 71–73 °C; IR (neat) 3233, 2228, 1333, 1165 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.50 (2H, d, J = 8.1 Hz), 7.18 (2H, d, J = 8.1 Hz), 7.00 (1H, s), 6.89 (1H, s), 6.32 (1H, s), 2.47 (3H, s), 2.39 (3H, s), 2.23 (3H, s), 2.06 (2H, t, J = 6.6 Hz), 1.28–1.41 (4H, m), 0.91 (3H, t, J = 7.1 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.3, 137.5, 136.8, 136.7, 132.4, 132.1, 130.3, 129.1, 127.6, 121.8, 95.2, 76.4, 30.5, 22.0, 21.5, 20.7, 19.5, 19.1, 13.6; MS (ESI-TOF) m/z: [M + Na]+ 378; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C21H25NNaO2S 378.1504; Found 378.1475.
N-(2,4-Dimethyl-6-(p-tolylethynyl)phenyl)-4-methoxybenzenesulfonamide (1n). In accordance with the experimental procedure for the synthesis of 1b, 1n was prepared from 2,4-dimethyl-6-(4-tolylethynyl)aniline (401 mg, 1.7 mmol) and 4-methoxybenzenesulfonyl chloride (386 mg, 1.9 mmol). The reaction was conducted for 20 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 20 and then 5) gave 1n (536 mg, 78%). 1n: white solid; mp 162–163 °C; IR (neat) 3239, 2207, 1335, 1155 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.53 (2H, dt, J = 9.0, 2.0 Hz), 7.18 (2H, d, J = 8.1 Hz), 7.12 (2H, d, J = 8.1 Hz), 7.06 (1H, s), 7.02 (1H, s), 6.66 (2H, dt, J = 9.0, 2.0 Hz), 6.41 (1H, s), 3.66 (3H, s), 2.50 (3H, s), 2.37 (3H, s), 2.27 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 162.8, 138.7, 137.9, 136.9, 132.7, 132.4, 131.4, 131.1, 130.4, 129.6, 129.0, 121.5, 119.5, 113.8, 93.9, 84.9, 55.3, 21.5, 20.7, 19.4; MS (ESI-TOF) m/z: [M + Na]+ 428; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C24H23NNaO3S 428.1296; Found 428.1270.
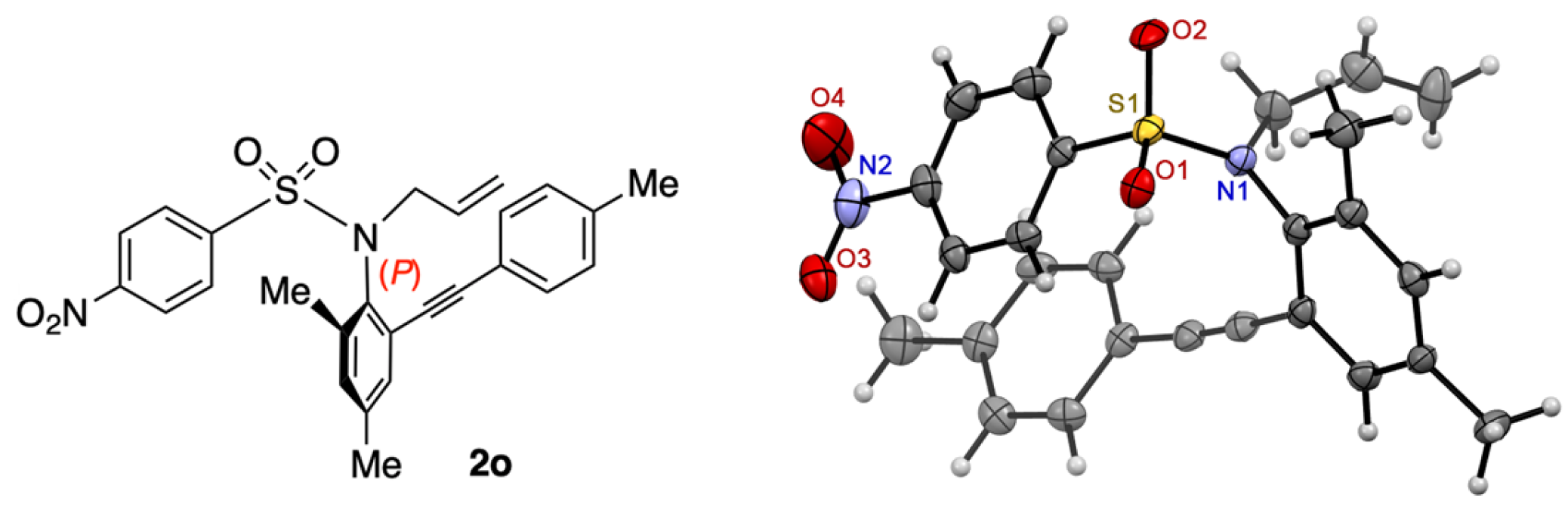
N-(2,4-Dimethyl-6-(p-tolylethynyl)phenyl)-4-nitrobenzenesulfonamide (1o). In accordance with the experimental procedure for the synthesis of 1a, 1o was prepared from 2,4-dimethyl-6-(4-tolylethynyl)aniline (489 mg, 2.1 mmol) and 4-nitrobenzenesulfonyl chloride (507 mg, 2.3 mmol). The reaction was conducted for 22 h at 0 °C–rt. Hexane was added to the residue and the mixture was filtered in vacuo. After washing the residue by hexane, 1o was obtained (400 mg, 46%). 1o: yellow solid; mp 203–204 °C; IR (neat) 3242, 2209, 1522, 1341, 1167 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.95 (2H, dt, J = 9.0, 2.0 Hz), 7.76 (2H, dt, J = 9.0, 2.0 Hz), 7.05–7.11 (3H, m), 7.03–7.05 (3H, m), 6.53 (1H, s), 2.54 (3H, s), 2.38 (3H, s), 2.30 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 149.8, 145.2, 139.4, 138.5, 138.0, 133.0, 131.2, 131.1, 130.7, 129.2, 128.7, 123.8, 121.6, 118.8, 94.3, 84.7, 21.5, 20.8, 19.4; MS (ESI-TOF) m/z: [M + Na]+ 443; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H20N2NaO4S 443.1042; Found 443.1015.
N-(2,4-Dimethyl-6-(phenylethynyl)phenyl)-4-nitrobenzenesulfonamide (1p). In accordance with the experimental procedure for the synthesis of 1b, 1p was prepared from 2,4-dimethyl-6-(phenylethynyl)aniline (426 mg, 1.9 mmol) and 4-nitrobenzenesulfonyl chloride (488 mg, 2.2 mmol). The reaction was conducted for 5 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 5) gave 1p (575 mg, 73%). 1p: white solid; mp 201–203 °C; IR (neat) 3231, 1522, 1344, 1167 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.95 (2H, d, J = 8.5 Hz), 7.77 (2H, d, J = 8.5 Hz), 7.29–7.38 (3H, m), 7.13–7.16 (3H, m), 7.06 (1H, s), 6.53 (1H, s), 2.54 (3H, s), 2.31 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 149.8, 145.2, 138.6, 138.1, 133.2, 131.23, 131.18, 130.8, 129.1, 128.7, 128.5, 123.8, 121.9, 121.5, 94.0, 85.3, 20.8, 19.4; MS (ESI-TOF) m/z: [M + Na]+ 429; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H18N2NaO4S 429.0885; Found 429.0869.
N-(2,4-dimethyl-6-(phenylethynyl)phenyl)benzenesulfonamide (1q). In accordance with the experimental procedure for the synthesis of 1b, 1q was prepared from 2,4-dimethyl-6-(phenylethynyl)aniline (289 mg, 1.3 mmol) and benzenesulfonyl chloride (255 mg, 1.4 mmol). The reaction was conducted for 18 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 15 and then 10) gave 1q (417 mg, 88%). 1q: white solid; mp 154–155 °C; IR (neat) 3248, 2211, 1327, 1159 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.62–7.64 (2H, m), 7.20–7.41 (8H, m), 7.08 (1H, s), 7.04 (1H, s), 6.48 (1H, s), 2.50 (3H, s), 2.28 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 139.3, 137.9, 137.1, 132.9, 132.7, 132.2, 131.5, 130.4, 128.6, 128.5, 128.2, 127.5, 122.4, 121.4, 93.9, 85.3, 20.7, 19.4; MS (ESI-TOF) m/z: [M + Na]+ 384; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H19NNaO2S 384.1034; Found 384.1011.
N-(2,4-Dimethyl-6-(p-tolylethynyl)phenyl)methanesulfonamide (1r). In accordance with the experimental procedure for the synthesis of 1b, 1r was prepared from 2,4-dimethyl-6-(4-tolylethynyl)aniline (457 mg, 1.9 mmol) and methanesulfonyl chloride (253 mg, 2.2 mmol). The reaction was conducted for 23 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 20 and then 5) gave 1r (430 mg, 71%). 1r: white solid; mp 171–177 °C; IR (neat) 3246, 2199, 1316, 1157 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.42 (2H, d, J = 8.1 Hz), 7.24 (1H, s), 7.19 (2H, d, J = 8.1 Hz), 7.09 (1H, s), 6.35 (1H, s), 3.08 (3H, s), 2.47 (3H, s), 2.39 (3H, s), 2.32 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 139.3, 138.5, 137.6, 133.0, 132.2, 131.3, 130.7, 129.4, 121.4, 119.0, 95.0, 85.7, 40.5, 21.5, 20.7, 19.3; MS (ESI-TOF) m/z: [M + Na]+ 336; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H19NNaO2S 336.1034; Found 336.1017.
N-(2,4-Dimethyl-6-(p-tolylethynyl)phenyl)-2,4,6-trimethylbenzenesulfonamide (1s). In accordance with the experimental procedure for the synthesis of 1b, 1r was prepared from 2,4-dimethyl-6-(4-tolylethynyl)aniline (339 mg, 1.4 mmol) and 2,4,6-trimethylbenzenesulfonyl chloride (313 mg, 1.4 mmol). The reaction was conducted for 16 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 30 and then 20) gave 1s (443 mg, 74%). 1s: white solid; mp 161–162 °C; IR (neat) 3277, 2207, 1323, 1161 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.18 (2H, d, J = 8.1 Hz), 7.12 (2H, d, J = 8.1 Hz), 7.09 (1H, s), 7.03 (1H, s), 6.70 (2H, s), 6.43 (1H, s), 2.38 (3H, s), 2.36 (6H, s), 2.34 (3H, s), 2.27 (3H, s), 2.17 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 141.9, 139.2, 138.6, 137.9, 137.0, 135.2, 132.6, 132.4, 131.9, 131.4, 130.7, 128.8, 122.1, 119.6, 93.6, 84.8, 23.5, 21.5, 20.8, 20.7, 19.2; MS (ESI-TOF) m/z: [M + Na]+ 440; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C26H27NNaO2S 440.1660; Found 440.1674.
N-Allyl-N-[(2-bromo-4-methyl-6-phenyl)phenyl]-4-methylbenzenesulfonamide (2c). Under N2 atmosphere, to 1c (125 mg, 0.3 mmol) in THF (2.5 mL) was added NaH (60% assay, 12 mg, 0.3 mmol) at 0 °C, and the mixture was stirred for 20 min at −20 °C. (Allyl-Pd-Cl)2 (2.5 mg, 0.0068 mmol), (S,S)-Trost ligand (10.5 mg, 0.0133 mmol) and allyl acetate (98 μL, 0.9 mmol) in THF (2.0 mL) were added to the reaction mixture, and then the mixture was stirred for 10 h at −20 °C. The mixture was poured into 1N HCl solution and extracted with AcOEt. The AcOEt extracts were washed with brine, dried over MgSO4, and evaporated to dryness. Purification of the residue by column chromatography (hexane/AcOEt = 10) gave 2c (133 mg, 97%). The ee (38% ee) of 2c was determined by HPLC analysis using a chiral column (CHIRALCEL OD-3) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2c (major); tR = 8.0 min, (-)-2c (minor); tR = 9.5 min). 2c: white solid; mp 113–115 °C (38% ee); IR (neat) 1333, 1150 cm−1; [α]D = +18.6° (38% ee, CHCl3, c = 1.00); 1H NMR (400 MHz, CDCl3) δ: 7.52 (2H, dt, J = 8.6, 1.9 Hz), 7.32–7.45 (6H, m), 7.17 (2H, d, J = 8.1 Hz), 7.07 (1H, d, J = 2.9 Hz), 5.68 (1H, ddt, J = 17.1, 10.0, 7.1 Hz), 5.08 (1H, dd, J = 17.1, 1.4 Hz), 5.01 (1H, dd, J = 10.2, 1.4 Hz), 4.19 (1H, dd, J = 14.2, 7.1 Hz), 3.96 (1H, dd, J = 14.2, 7.1 Hz), 2.40 (3H, s), 2.35 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 146.2, 142.9, 139.6, 139.4, 137.6, 133.7, 133.6, 132.5, 131.7, 129.5, 129.0, 128.0, 127.6, 127.4, 125.9, 119.1, 53.6, 21.4, 20.6; MS (ESI-TOF) m/z: [M + Na]+ 480; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H2281BrNNaO2S 480.0432; Found 480.0417.
N-Allyl-N-(2-(tert-butyl)-6-methylphenyl)-4-methylbenzenesulfonamide (2a). In accordance with the experimental procedure for the synthesis of 2c, 2a was prepared from 1a (96 mg, 0.3 mmol). The reaction was conducted for 21 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 30) gave 2a (62 mg, 58%). The ee (10% ee) of 2a was determined by HPLC analysis using a chiral column (CHIRALPAK AS-H) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (-)-2a (major); tR = 12.8 min, (+)-2a (minor); tR = 8.7 min). 2a: white solid; mp 105–107 °C (10% ee); IR (neat) 1339, 1159 cm−1; [α]D = −5.2° (10% ee, CHCl3, c = 0.83); 1H NMR (400 MHz, CDCl3) δ: 7.63 (2H, d, J = 7.6 Hz), 7.44–7.46 (1H, m), 7.29 (2H, d, J = 7.6 Hz), 7.17 (1H, t, J = 7.6 Hz), 6.87 (1H, dd, J = 7.6, 0.9 Hz), 5.66 (1H, dddd, J = 16.6, 10.4, 7.6, 6.2 Hz), 5.21 (1H, dd, J = 16.6, 1.4 Hz), 5.04 (1H, dd, J = 10.4, 1.4 Hz), 4.50 (1H, ddt, J = 13.2, 6.2, 1.4 Hz), 4.14 (1H, dd, J = 13.2, 7.6 Hz), 2.43 (3H, s), 1.55 (9H, s), 1.47 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 151.7, 143.2, 138.9, 137.3, 133.7, 131.5, 129.5, 128.7, 128.6, 127.9, 127.7, 119.3, 53.8, 37.2, 33.3, 21.5, 19.4; MS (ESI-TOF) m/z: [M + Na]+ 380; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C21H27NNaO2S 380.1660; Found 380.1641.
N-Allyl-N-((2-iodo-4-methyl-6-phenyl)phenyl)-4-methylbenzenesulfonamide (2b). In accordance with the experimental procedure for the synthesis of 2c, 2b was prepared from 1b (93 mg, 0.2 mmol). The reaction was conducted for 27 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 10) gave 2b (10 mg, 10%). The ee (43% ee) of 2b was determined by HPLC analysis using a chiral column (CHIRALCEL OD-3) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2b (major); tR = 9.0 min, (-)-2b (minor); tR = 10.0 min). 2b: colorless oil; IR (neat) 1344, 1157 cm−1; [α]D = +32.8° (48% ee, CHCl3, c = 1.01); 1H NMR (400 MHz, CDCl3) δ: 7.76 (1H, d, J = 1.4 Hz), 7.53 (2H, d, 8.1 Hz), 7.28–7.36 (5H, m), 7.16 (2H, d, J = 8.1 Hz), 7.07 (1H, d, J = 1.4 Hz), 5.77 (1H, ddt, J = 17.1, 10.0, 7.1 Hz), 5.14 (1H, dd, J = 17.1, 1.4 Hz), 5.04 (1H, dd, J = 10.0, 1.4 Hz), 4.26 (1H, dd, J = 14.5, 6.9 Hz), 3.99 (1H, dd, J = 14.5, 7.1 Hz), 2.40 (3H, s), 2.32 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 145.7, 142.9, 140.5, 139.7, 139.6, 137.9, 137.3, 132.7, 129.4, 129.0, 128.1, 127.43, 127.38, 119.1, 102.5, 54.0, 21.4, 20.3; MS (ESI-TOF) m/z: [M +Na]+ 526; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H22127INNaO2S 526.0314; Found 526.0294.
N-Allyl-N-(2-iodo-4,6-dimethylphenyl)-4-methylbenzenesulfonamide (2d). In accordance with the experimental procedure for the synthesis of 2c, 2d was prepared from 1d (120 mg, 0.3 mmol). The reaction was conducted for 6 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 5) gave 2d (124 mg, 94%). The ee (65% ee) of 2d was determined by HPLC analysis using a chiral column (CHIRALCEL OD-3) (25 cm × 0.46 cm i.d.; 3% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2d (major); tR = 11.1min, (-)-2d (minor); tR = 10.5 min). 2d: white solid; mp 67–69 °C (65% ee); IR (neat) 1343, 1159 cm−1; [α]D = +40.9° (65% ee, CHCl3, c = 1.00); 1H NMR (400 MHz, CDCl3) δ: 7.74 (2H, d, J = 8.1 Hz), 7.50 (1H, d, J = 1.4 Hz), 7.29 (2H, d, J = 8.1 Hz), 7.01 (1H, d, J = 1.4 Hz), 5.97 (1H, m), 5.02–5.08 (2H, m), 4.32 (1H, dd, J = 14.5, 6.6 Hz), 4.12 (1H, dd, J = 14.5, 7.8 Hz), 2.42 (3H, s), 2.28 (3H, s), 2.23 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.3, 141.8, 139.8, 138.7, 138.3, 137.5, 132.8, 132.1, 129.4, 128.0, 119.3, 101.1, 53.6, 21.5, 20.5, 20.3; MS (ESI-TOF) m/z: [M + Na]+ 464; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H20127INNaO2S 464.0157; Found 464.0128.
(E)-N-Allyl-N-(2,4-dimethyl-6-styrylphenyl)-4-methylbenzenesulfonamide (2e). In accordance with the experimental procedure for the synthesis of 2c, 2e was prepared from 1e (113 mg, 0.3 mmol). The reaction was conducted for 23 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 5) gave 2e (114 mg, 91%). The ee (65% ee) of 2e was determined by HPLC analysis using a chiral column (CHIRALCEL OD-3) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2e (major); tR = 7.6 min, (-)-2e (minor); tR = 6.7 min). 2e: white oil; IR (neat) 1343, 1157 cm−1; [α]D = +132.8° (65% ee, CHCl3, c = 1.01); 1H NMR (400 MHz, CDCl3) δ: 7.74 (2H, dd, J = 8.5, 1.9 Hz), 7.32 (1H, s), 7.20–7.27 (5H, m), 6.99–7.04 (3H, m), 6.86 (1H, d, J = 16.1 Hz), 6.50 (1H, d, J = 16.1 Hz), 5.90 (1H, dddd, J = 16.6, 10.0, 7.6, 6.2 Hz), 4.99–5.04 (2H, m), 4.32 (1H, ddt, J = 14.8, 6.2, 1.0 Hz), 3.97 (1H, dd, J = 14.8, 7.6 Hz), 2.39 (3H, s), 2.35 (3H, s), 2.30 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.4, 140.5, 138.2, 138.1, 137.0, 136.5, 133.8, 132.8, 131.6, 129.9, 129.7, 128.4, 127.6, 127.5, 126.5, 125.2, 124.2, 119.2, 54.4, 21.4, 21.2, 19.6; MS (ESI-TOF) m/z: [M + Na]+ 440; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C26H27NNaO2S 440.1660; Found 440.1648.
N-Allyl-N-(2-iodo-4-methyl-6-(p-tolylethynyl)phenyl)-4-methylbenzenesulfonamide (2f). In accordance with the experimental procedure for the synthesis of 2c, 2f was prepared from 1f (94 mg, 0.19 mmol). The reaction was conducted for 5 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 10) gave 2f (84 mg, 82%). The ee (73% ee) of 2f was determined by HPLC analysis using a chiral column (CHIRALCEL OD-3) (25 cm × 0.46 cm i.d.; 3% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2f (major); tR = 24.1 min, (-)-2f (minor); tR = 27.7 min). 2f: colorless oil; IR (neat) 2205, 1344, 1157 cm−1; [α]D = +71.1° (74% ee, CHCl3, c = 0.83); 1H NMR (400 MHz, CDCl3) δ: 7.74 (2H, d, J = 8.1 Hz), 7.72 (1H, m), 7.28 (1H, d, J = 1.4 Hz),7.04–7.08 (6H, m), 6.12 (1H, dddd, J = 16.1, 10.0, 8.1, 6.2 Hz), 5.10 (1H, d, J = 17.1 Hz), 5.03 (1H, d, J = 10.0 Hz), 4.48 (1H, dd, J = 14.2, 6.2 Hz), 4.38 (1H, dd, J = 14.2, 8.1 Hz), 2.35 (3H, s), 2.28 (3H, s), 2.10 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.2, 140.8, 139.63, 139.58, 138.7, 138.0, 134.2, 132.8, 131.3, 129.3, 128.7, 128.0, 125.2, 119.3, 119.2, 105.2, 94.5, 85.9, 53.1, 21.5, 21.2, 20.2; MS (ESI-TOF) m/z: [M + Na]+ 564; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C26H24INNaO2S 564.0470; Found 564.0442.
N-Allyl-N-(2-bromo-4-methyl-6-(p-tolylethynyl)phenyl)-4-methylbenzenesulfonamide (2g). In accordance with the experimental procedure for the synthesis of 2c, 2g was prepared from 1g (91 mg, 0.2 mmol). The reaction was conducted for 7 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 5) gave 2g (101 mg, quant). The ee (72% ee) of 2g was determined by HPLC analysis using a chiral column (CHIRALCEL OD-3) (25 cm × 0.46 cm i.d.; 5% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2g (major); tR = 16.0 min, (-)-2g (minor); tR = 13.5 min). 2g: white oil; IR (neat) 2212, 1343, 1155 cm−1; [α]D = +61.5° (69% ee, CHCl3, c = 0.68); 1H NMR (400 MHz, CDCl3) δ: 7.77 (2H, d, J = 8.1 Hz), 7.42 (1H, d, J = 1.9 Hz), 7.27 (1H, d, J = 1.9 Hz), 7.07–7.12 (6H, m), 6.06 (1H, dddd, J = 17.1, 10.0, 7.6, 6.6 Hz), 5.08 (1H, dd, J = 17.1, 1.0 Hz), 5.02 (1H, dd, J = 10.0, 1.0 Hz), 4.40 (1H, dd, J = 14.2, 6.6 Hz), 4.36 (1H, dd, J = 14.2, 7.6 Hz), 2.36 (3H, s), 2.31 (3H, s), 2.15 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.1, 139.5, 138.7, 138.0, 136.4, 134.1, 133.0, 132.8, 131.3, 129.2, 128.7, 127.8, 127.4, 126.5, 119.3, 119.0, 94.6, 85.9, 52.9, 21.4, 21.2, 20.5; MS (ESI-TOF) m/z: [M + Na]+ 518; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C26H2481BrNNaO2S 518.0588; Found 518.0592.
N-Allyl-N-(2-chloro-6-(p-tolylethynyl)phenyl)-4-methylbenzenesulfonamide (2h). In accordance with the experimental procedure for the synthesis of 2c, 2h was prepared from 1h (119 mg, 0.3 mmol). The reaction was conducted for 5 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 10) gave 2h (124 mg, 95%). The ee (79% ee) of 2h was determined by HPLC analysis using a chiral column (CHIRALPAK AS-H) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2h (major); tR = 24.7 min, (-)-2h (minor); tR = 18.6 min). 2h: yellow solid; mp 101–104 °C (76% ee); IR (neat) 2224, 1346, 1155 cm−1; [α]D = +103.8° (76% ee, CHCl3, c = 1.00); 1H NMR (400 MHz, CDCl3) δ: 7.78 (2H, d, J = 8.1 Hz), 7.40–7.44 (2H, m), 7.23 (1H, t, J = 7.6 Hz), 7.09–7.16 (6H, m), 6.01 (1H, ddt, J = 17.1, 10.0, 7.1 Hz), 5.05 (1H, d, J = 17.1 Hz), 5.01 (2H, d, J = 6.6 Hz), 4.01 (1H, d, J = 10.0 Hz), 2.36 (3H, s), 2.19 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.2, 138.9, 138.0, 137.6, 137.2, 132.8, 131.7, 131.5, 130.2, 129.3, 129.0, 128.8, 127.9, 127.4, 119.3, 119.1, 95.1, 85.8, 52.8, 21.5, 21.3; MS (ESI-TOF) m/z: [M + Na]+ 458; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H2235ClNNaO2S 458.0958; Found 458.0951.
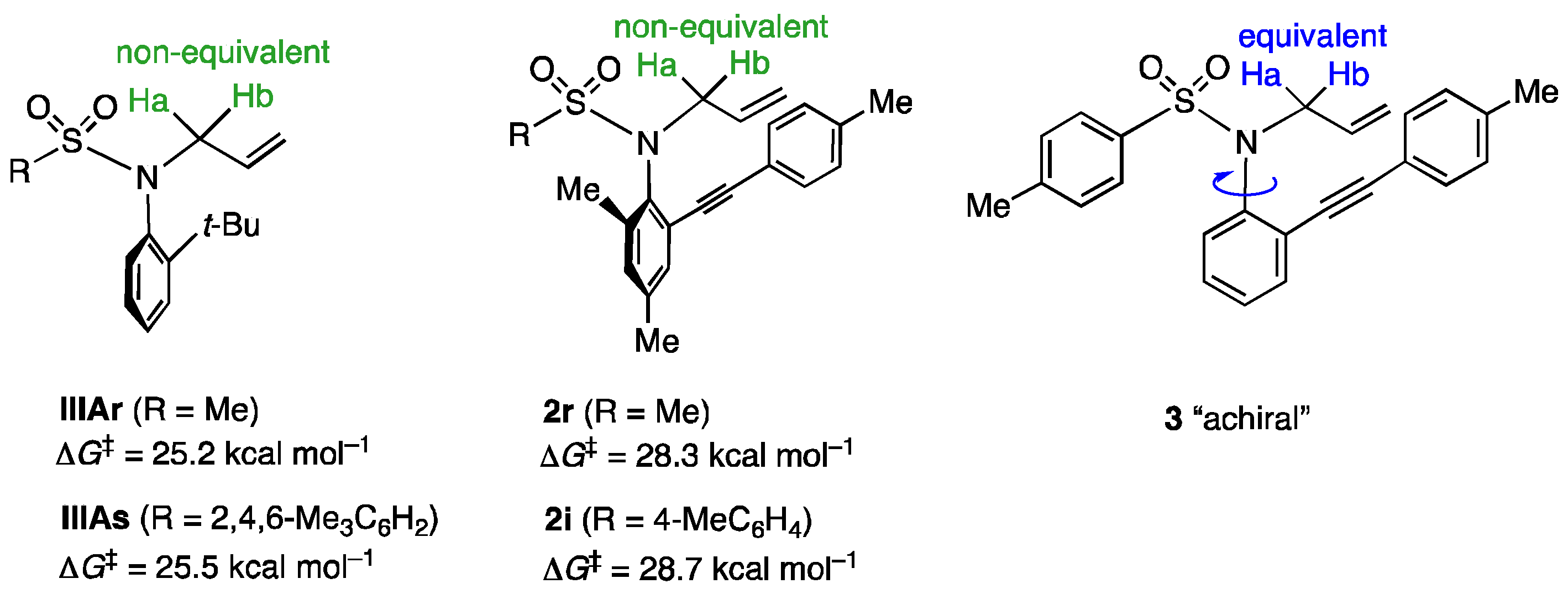
N-Allyl-N-(2,4-dimethyl-6-(p-tolylethynyl)phenyl)-4-methylbenzenesulfonamide (2i). In accordance with the experimental procedure for the synthesis of 2c, 2i was prepared from 1i (117 mg, 0.3 mmol). The reaction was conducted for 6 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 20) gave 2i (109 mg, 88%). The ee (86% ee) of 2i was determined by HPLC analysis using a chiral column (CHIRALPAK AD-H) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2i (major); tR = 9.9 min, (-)-2i (minor); tR = 12.1 min). 2i: yellow oil; IR (neat) 2205, 1341, 1155 cm−1; [α]D = +180.7° (80% ee, CHCl3, c = 1.01); 1H NMR (400 MHz, CDCl3) δ: 7.71 (2H, dt, J = 8.5, 1.9 Hz), 7.14 (1H, d, J = 1.9 Hz), 7.02–7.07 (5H, m), 6.95 (2H, d, J = 8.1 Hz), 5.98 (1H, dddd, J = 17.2, 10.4, 8.5, 5.7 Hz), 5.07 (1H, dd, J = 17.2, 1.4 Hz), 5.04 (1H, d, J = 10.4 Hz), 4.48 (1H, ddt, J = 14.2, 5.7, 1.4 Hz), 4.25 (1H, dd, J = 14.2, 8.5 Hz), 2.45 (3H, s), 2.35 (3H, s), 2.30 (3H, s), 2.08 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.0, 141.2, 138.3, 137.83, 137.78, 136.2, 133.2, 132.2, 131.8, 131.1, 129.3, 128.6, 127.8, 123.3, 119.7, 119.0, 93.3, 86.8, 53.0, 21.4, 21.2, 20.8, 19.6; MS (ESI-TOF) m/z: [M + Na]+ 452; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C27H27NNaO2S 452.1660; Found 452.1631.
N-Allyl-N-(2-((4-methoxyphenyl)ethynyl)-4,6-dimethylphenyl)-4-methylbenzenesulfonamide (2j). In accordance with the experimental procedure for the synthesis of 2c, 2j was prepared from 1j (122 mg, 0.3 mmol). The reaction was conducted for 23 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 20) gave 2j (131 mg, 98%). The ee (88% ee) of 2j was determined by HPLC analysis using a chiral column (CHIRALPAK AD-H) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2j (major); tR = 17.7 min, (-)-2j (minor); tR = 23.6 min). 2j: white solid; mp 78–80 °C (87% ee); IR (neat) 2211, 1341, 1159 cm−1; [α]D = +193.3° (87% ee, CHCl3, c = 1.00); 1H NMR (400 MHz, CDCl3) δ: 7.72 (2H, d, J = 8.1 Hz), 7.13 (1H, s), 7.06 (1H, s), 7.05 (2H, d, J = 8.1 Hz), 7.00 (2H, dt, J = 8.5, 1.9 Hz), 6.77 (2H, dt, J = 8.5, 2.8 Hz), 5.98 (1H, dddd, J = 18.0, 10.4, 8.5, 5.7 Hz), 5.07 (1H, d, J = 18.0 Hz), 5.04 (1H, d, J = 10.4 Hz), 4.47 (1H, dd, J = 14.2, 5.7 Hz), 4.25 (1H, dd, J = 14.2, 8.5 Hz), 3.81 (3H, s), 2.44 (3H, s), 2.29 (3H, s), 2.11 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 159.4, 142.9, 141.1, 137.9, 137.7, 136.0, 133.1, 132.6, 131.9, 131.6, 129.2, 127.7, 123.4, 118.9, 114.9, 113.5, 93.2, 86.1, 55.2, 52.9, 21.2, 20.7, 19.5; MS (ESI-TOF) m/z: [M + Na]+ 468; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C27H27NNaO3S 468.1609; Found 468.1615.
N-Allyl-N-(2,4-dimethyl-6-(phenylethynyl)phenyl)-4-methylbenzenesulfonamide (2k). In accordance with the experimental procedure for the synthesis of 2c, 2j was prepared from 1j (113 mg, 0.3 mmol). The reaction was conducted for 21 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 20) gave 2j (114 mg, 92%). The ee (89% ee) of 2j was determined by HPLC analysis using a chiral column (CHIRALPAK AD-H) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2k (major); tR = 11.8 min, (-)-2k (minor); tR = 16.4 min). 2k: white solid; mp 87–89 °C (90% ee); IR (neat) 1339, 1155 cm−1; [α]D = +196.7° (90% ee, CHCl3, c = 0.79); 1H NMR (400 MHz, CDCl3) δ: 7.73 (2H, dt, J = 8.5, 1.9 Hz), 7.23–7.29 (3H, m), 7.16 (1H, d, J = 2.4 Hz), 7.06–7.09 (3H, m), 7.03 (2H, d, J = 8.1 Hz), 6.00 (1H, dddd, J = 17.1, 10.0, 8.5, 5.7 Hz), 5.09 (1H, d, J = 17.1 Hz), 5.07 (1H, d, J = 10.0 Hz), 4.51 (1H, ddt, J = 14.2, 5.7, 1.4 Hz), 4.28 (1H, dd, J = 14.2, 8.5 Hz), 2.46 (3H, s), 2.31 (3H, s), 2.07 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.0, 141.2, 137.8, 136.2, 133.1, 132.3, 131.9, 131.1, 129.3, 128.1, 127.8, 127.7, 123.1, 122.7, 119.0, 93.0, 87.3, 53.0, 21.1, 20.7, 19.5; MS (ESI-TOF) m/z: [M + Na]+ 438; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C26H25NNaO2S 438.1504; Found 438.1484.
N-Allyl-N-(2,4-dimethyl-6-((trimethylsilyl)ethynyl)phenyl)-4-methylbenzenesulfonamide (2l). In accordance with the experimental procedure for the synthesis of 2c, 2l was prepared from 1l (112 mg, 0.3 mmol). The reaction was conducted for 9 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 5) gave 2l (86 mg, 70%). The ee (75% ee) of 2l was determined by HPLC analysis using a chiral column (CHIRALPAK AD-H) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2l (major); tR = 5.3 min, (-)-2l (minor); tR = 6.1 min). 2l: yellow oil; IR (neat) 2153, 1344, 1159 cm−1; [α]D = +129.7° (75% ee, CHCl3, c = 0.83); 1H NMR (400 MHz, CDCl3) δ: 7.68 (2H, dt, J = 8.5, 1.9 Hz), 7.25 (2H, d, J = 8.5 Hz), 7.10 (1H, d, J = 1.4 Hz), 7.05 (1H, m), 5.93 (1H, dddd, J = 17.1, 10.0, 8.1, 5.7 Hz), 5.04 (1H, dt, J = 17.1, 1.4 Hz), 5.01 (1H, d, J = 10.0 Hz), 4.34 (1H, ddt, J = 14.7, 5.7, 1.4 Hz), 4.18 (1H, dd, J = 14.7, 8.1 Hz), 2.41 (3H, s), 2.36 (3H, s), 2.26 (3H, s), 0.02–0.03 (9H, m); 13C{1H} NMR (100 MHz, CDCl3) δ: 142.8, 141.0, 138.1, 137.7, 136.3, 133.3, 132.6, 129.4, 128.1, 123.3, 118.8, 102.7, 98.4, 52.8, 21.6, 20.7, 19.6, −0.38; MS (ESI-TOF) m/z: [M + Na]+ 434; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H29NNaO2S28Si 434.1586; Found 434.1560.
N-Allyl-N-(2-(hex-1-yn-1-yl)-4,6-dimethylphenyl)-4-methylbenzenesulfonamide (2m). In accordance with the experimental procedure for the synthesis of 2c, 2m was prepared from 1m (107 mg, 0.3 mmol). The reaction was conducted for 25 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 20) gave 2m (114 mg, 96%). The ee (77% ee) of 2m was determined by HPLC analysis using a chiral column (CHIRALPAK AS-H) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2m (major); tR = 8.5 min, (-)-2m (minor); tR = 6.6 min). 2m: yellow oil; IR (neat) 2230, 1344, 1159 cm−1; [α]D = +110.6° (77% ee, CHCl3, c = 0.87); 1H NMR (400 MHz, CDCl3) δ: 7.71 (2H, dt, J = 8.5, 1.9 Hz), 7.26 (2H, d, J = 8.5 Hz), 7.00 (1H, s), 6.99 (1H, s), 5.91 (1H, dddd, J = 17.1, 10.0, 8.5, 5.7 Hz), 4.99–5.05 (2H, m), 4.40 (1H, ddt, J =14.2, 5.7, 1.0 Hz), 4.12 (1H, dd, J = 14.2, 8.5 Hz), 2.41 (3H, s), 2.40 (3H, s), 2.25 (3H, s), 1.80–1.92 (2H, m), 1.19–1.34 (4H, m), 0.87 (3H, t, J = 7.1 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ: 142.6, 141.0, 138.3, 137.6, 136.1, 133.3, 131.9, 131.5, 129.1, 128.0, 123.7, 118.7, 94.4, 78.2, 52.9, 30.4, 22.0, 21.4, 20.7, 19.6, 18.9, 13.5; MS (ESI-TOF) m/z: [M + Na]+ 418; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C24H29NNaO2S 418.1817; Found 418.1843.
N-Allyl-N-(2,4-dimethyl-6-(p-tolylethynyl)phenyl)-4-methoxybenzenesulfonamide (2n). In accordance with the experimental procedure for the synthesis of 2c, 2n was prepared from 1n (122 mg, 0.3 mmol). The reaction was conducted for 22 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 20 and then 10) gave 2n (117 mg, 88%). The ee (85% ee) of 2n was determined by HPLC analysis using a chiral column (CHIRALPAK AS-H) (25 cm × 0.46 cm i.d.; 5% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2n (major); tR = 29.5 min, (-)-2j (minor); tR = 24.9 min). 2n: white solid; mp 124–132 °C (84% ee); IR (neat) 2207, 1344, 1150 cm−1; [α]D = +194.8° (84% ee, CHCl3, c = 0.99); 1H NMR (400 MHz, CDCl3) δ: 7.75 (2H, d, J = 8.5 Hz), 7.13 (1H, s), 7.07 (1H, s), 7.05 (2H, d, J = 8.1 Hz), 6.97 (2H, d, J = 8.1 Hz), 6.68 (2H, d, J = 8.5 Hz), 5.97 (1H, dddd, J = 17.1, 10.0, 8.5, 5.7 Hz), 5.07 (1H, d, J = 18.5 Hz), 5.04 (1H, d, J = 10.4 Hz), 4.46 (1H, dd, J = 14.5, 5.7 Hz), 4.24 (1H, dd, J = 14.5, 8.1 Hz), 3.52 (3H, s), 2.44 (3H, s), 2.35 (3H, s), 2.30 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 162.4, 141.3, 138.3, 137.8, 136.3, 133.3, 132.5, 132.2, 131.8, 131.1, 129.9, 128.7, 123.3, 119.8, 119.0, 113.8, 93.2, 86.9, 55.0, 53.0, 21.5, 20.8, 19.6; MS (ESI-TOF) m/z: [M + Na]+ 468; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C27H27NNaO3S 468.1609; Found 468.1581.
N-Allyl-N-(2,4-dimethyl-6-(p-tolylethynyl)phenyl)-4-nitrobenzenesulfonamide (2o). In accordance with the experimental procedure for the synthesis of 2c, 2o was prepared from 1o (126 mg, 0.3 mmol). The reaction was conducted for 6 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 10) gave 2o (137 mg, quant). The ee (89% ee) of 2o was determined by HPLC analysis using a chiral column (CHIRALCEL OD-3) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2o (major); tR = 9.9 min, (-)-2o (minor); tR = 8.0 min). 2o: yellow solid; mp 102–104 °C (97% ee); IR (neat) 2209, 1524, 1344, 1159 cm−1; [α]D = +201.0° (97% ee, CHCl3, c = 0.59); 1H NMR (400 MHz, CDCl3) δ: 7.96 (2H, dd, J = 6.9, 2.4 Hz), 7.91 (2H, dd, J = 6.9, 2.4 Hz), 7.11 (1H, s), 7.10 (1H, s), 6.99 (2H, d, J = 8.1 Hz), 6.86 (2H, d, J = 8.1 Hz), 5.98 (1H, dddd, J = 17.1, 10.0, 8.5, 5.7 Hz), 5.13 (1H, d, J = 17.1 Hz), 5.11 (1H, d, J = 10.0 Hz), 4.54 (1H, dd, J = 14.2, 5.7 Hz), 4.27 (1H, dd, J = 14.2, 8.5 Hz), 2.46 (3H, s), 2.33 (3H, s), 2.31 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 149.3, 146.3, 141.2, 139.4, 138.5, 135.5, 132.5, 132.4, 131.8, 130.7, 128.9, 128.8, 123.8, 122.8, 120.0, 118.9, 93.5, 86.6, 53.5, 21.4, 20.8, 19.6; MS (ESI-TOF) m/z: [M + Na]+ 483; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C26H24N2NaO4S 483.1355; Found 483.1346.
N-Allyl-N-(2,4-dimethyl-6-(phenylethynyl)phenyl)-4-nitrobenzenesulfonamide (2p). In accordance with the experimental procedure for the synthesis of 2c, 2p was prepared from 1p (122 mg, 0.3 mmol). The reaction was conducted for 7 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 20) gave 2p (132 mg, 98%). The ee (92% ee) of 2p was determined by HPLC analysis using a chiral column (CHIRALCEL OD-3) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2p (major); tR = 13.1 min, (-)-2p (minor); tR = 9.9 min). 2p: yellow oil; IR (neat) 1520, 1344, 1165 cm−1; [α]D = +193.1° (90% ee, CHCl3, c = 1.07); 1H NMR (400 MHz, CDCl3) δ: 7.98 (2H, d, J = 8.5 Hz), 7.93 (2H, d, J = 8.5 Hz), 7.27–7.29 (1H, m), 7.21 (2H, t, J = 7.6 Hz), 7.13 (1H, s), 7.12 (1H, s), 6.98 (2H, d, J = 8.5 Hz), 5.98 (1H, dddd, J = 16.6, 9.5, 7.1, 5.2 Hz), 5.14 (1H, d, J = 17.1 Hz), 5.11 (1H, d, J = 10.0 Hz), 4.56 (1H, dd, J = 14.2, 5.7 Hz), 4.28 (1H, dd, J = 14.2, 8.5 Hz), 2.46 (3H, s), 2.32 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 149.2, 146.3, 141.2, 138.5, 135.4, 132.7, 132.3, 131.9, 130.7, 128.9, 128.8, 128.2, 123.8, 122.6, 121.9, 119.9, 93.2, 87.1, 53.5, 20.7, 19.5; MS (ESI-TOF) m/z: [M + Na]+ 469; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H22N2NaO4S 469.1198; Found 469.1170.
N-Allyl-N-(2,4-dimethyl-6-(phenylethynyl)phenyl)benzenesulfonamide (2q). In accordance with the experimental procedure for the synthesis of 2c, 2q was prepared from 1q (108 mg, 0.3 mmol). The reaction was conducted for 7 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 10) gave 2q (127 mg, quant). The ee (86% ee) of 2q was determined by HPLC analysis using a chiral column (CHIRALPAK AD-H) (25 cm × 0.46 cm i.d.; 15% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2q (major); tR = 9.1 min, (-)-2q (minor); tR = 10.3 min). 2q: yellow oil; IR (neat) 1343, 1157 cm−1; [α]D = +172.1° (85% ee, CHCl3, c = 0.97); 1H NMR (400 MHz, CDCl3) δ: 7.84–7.86 (2H, m), 7.22–7.31 (6H, m), 7.17 (1H, d, J = 1.9 Hz), 7.07–7.10 (3H, m), 6.00 (1H, dddd, J = 17.1, 10.4, 8.5, 5.7 Hz), 5.09 (1H, dd, J = 17.1, 1.4 Hz), 5.05 (1H, d, J = 10.4 Hz), 4.51 (1H, ddt, J = 14.2, 5.7, 1.4 Hz), 4.29 (1H, dd, J = 14.2, 8.5 Hz), 2.43 (3H, s), 2.31 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 141.0, 140.7, 137.9, 136.0, 133.0, 132.4, 132.2, 131.8, 131.3, 128.6, 128.2, 127.9, 127.7, 123.3, 122.6, 119.1, 93.1, 87.3, 53.2, 20.7, 19.5; MS (ESI-TOF) m/z: [M + Na]+ 424; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H23NNaO2S 424.1347; Found 424.1347.
N-Allyl-N-(2,4-dimethyl-6-(p-tolylethynyl)phenyl)methanesulfonamide (2r). In accordance with the experimental procedure for the synthesis of 2c, 2r was prepared from 1r (94 mg, 0.3 mmol). The reaction was conducted for 6 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 20) gave 2r (83 mg, 78%). The ee (87% ee) of 2r was determined by HPLC analysis using a chiral column (CHIRALPACK AS-H) (25 cm × 0.46 cm i.d.; 5% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2r (major); tR = 32.9 min, (-)-2r (minor); tR = 27.9 min). 2r: yellow oil; IR (neat) 2207, 1335, 1150 cm−1; [α]D = +7.7° (84% ee, CHCl3, c = 0.57); 1H NMR (400 MHz, CDCl3) δ: 7.39 (2H, d, J = 8.1 Hz), 7.25 (1H, d, J = 1.4 Hz), 7.19 (2H, d, J = 8.1 Hz), 7.08 (1H, d, J = 1.4 Hz), 6.00 (1H, dddd, J = 17.1, 10.0, 8.1, 5.7 Hz), 5.14 (1H, dd, J = 17.1, 1.0 Hz), 5.09 (1H, d, J = 10.0 Hz), 4.40 (1H, dd, J = 14.2, 5.7 Hz), 4.34 (1H, dd, J = 14.2, 8.1 Hz), 3.13 (3H, s), 2.39 (6H, s), 2.31 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 140.9, 139.1, 138.2, 136.2, 133.1, 132.5, 131.8, 131.1, 129.4, 123.1, 119.5, 119.3, 93.8, 87.3, 53.7, 41.0, 21.5, 20.8, 19.4; MS (ESI-TOF) m/z: [M + Na]+ 376; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C21H23NNaO2S 376.1347; Found 376.1342.
N-Allyl-N-(2,4-dimethyl-6-(p-tolylethynyl)phenyl)-2,4,6-trimethylbenzenesulfonamide (2s). In accordance with the experimental procedure for the synthesis of 2c, 2s was prepared from 1s (125 mg, 0.3 mmol). The reaction was conducted for 23 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 30) gave 2s (117 mg, 85%). The ee (63% ee) of 2s was determined by HPLC analysis using a chiral column (CHIRALCEL OD-3) (25 cm × 0.46 cm i.d.; 1% i-PrOH in hexane; flow rate, 0.8 mL/min; (+)-2s (major); tR = 13.2 min, (-)-2s (minor); tR = 10.1 min). 2s: white solid; mp 171–172 °C (62% ee); IR (neat) 2212, 1337, 1155 cm−1; [α]D = +155.4° (62% ee, CHCl3, c = 1.00); 1H NMR (400 MHz, CDCl3) δ: 7.14 (1H, d, J = 2.4 Hz), 7.05–7.08 (3H, m), 7.02 (2H, d, J = 8.5 Hz), 6.73 (2H, s), 6.00 (1H, dddd, J = 17.1, 10.0, 8.5, 5.7 Hz), 5.07 (1H, dd, J = 17.1, 1.4 Hz), 5.04 (1H, d, J = 10.0 Hz), 4.63 (1H, ddt, J = 14.2, 5.7, 1.4 Hz), 4.42 (1H, dd, J = 14.2, 8.5 Hz), 2.44 (6H, s), 2.40 (3H, s), 2.36 (3H, s), 2.29 (3H, s), 2.09 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 141.5, 141.1, 139.5, 138.3, 137.7, 136.0, 135.8, 133.3, 132.2, 132.1, 131.6, 131.1, 128.7, 124.4, 119.9, 119.0, 93.2, 86.6, 53.1, 24.2, 21.5, 20.8, 20.7, 19.6; MS (ESI-TOF) m/z: [M + Na]+ 480; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C29H31NNaO2S 480.1973; Found 480.1960.
N-(2-(p-Tolylethynyl)phenyl)-4-methylbenzenesulfonamide (6). In accordance with the experimental procedure for the synthesis of 1b, 6 was prepared from 2-(4-tolylethynyl)-4methylaniline (558 mg, 2.7 mmol, commercially available) and 4-tosyl chloride (567 mg, 3.0 mmol). The reaction was conducted for 1 h at 0 °C–rt. Purification of the residue by column chromatography (hexane/AcOEt = 20) gave 6 (835 mg, 86%). 6: white solid; mp 126–128 °C; IR (neat) 3239, 2212, 1335, 1159 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.68 (2H, dd, J = 6.6, 1.9 Hz), 7.63 (1H, dd, J = 8.5, 0.9 Hz), 7.35–7.38 (3H, m), 7.24–7.30 (2H, m), 7.20 (2H, d, J = 7.6 Hz), 7.17 (2H, d, J = 8.1 Hz), 7.06 (1H, td, J = 7.6, 0.9 Hz), 2.40 (3H, s), 2.33 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 144.0, 139.3, 137.3, 135.9, 131.8, 131.4, 129.5, 129.4, 129.3, 127.2, 124.5, 120.2, 118.8, 114.8, 96.3, 83.0, 21.6, 21.5; MS (ESI-TOF) m/z: [M + Na]+ 384; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H19NNaO2S 384.1034; Found 384.1063.
N-Allyl-N-(2-(p-tolylethynyl)phenyl)-4-methylbenzenesulfonamide (3). In accordance with the experimental procedure for the synthesis of 2c, 3 was prepared from 6 (109 mg, 0.3 mmol). The reaction was conducted for 20 h at −20 °C. Purification of the residue by column chromatography (hexane/AcOEt = 20) gave 3 (150 mg, quant). 3: white solid; mp 66–69 °C; IR (neat) 2214, 1344, 1159 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.64 (2H, d, J = 8.5 Hz), 7.48 (1H, m), 7.27–7.34 (3H, m), 7.09–7.17 (6H, m), 5.87 (1H, ddt, J = 17.1, 10.0, 6.6 Hz), 5.10 (1H, dd, J = 17.1, 0.9 Hz), 5.05 (1H, dd, J = 10.0, 0.9 Hz), 4.38 (2H, d, J = 6.6 Hz), 2.38 (3H, s), 2.23 (3H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 143.1, 139.6, 138.7, 137.1, 133.28, 133.26, 132.4, 131.3, 129.4, 128.9, 128.6, 128.0, 127.6, 123.8, 119.7, 118.6, 94.4, 85.7, 53.1, 21.5, 21.3; MS (ESI-TOF) m/z: [M + Na]+ 424; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C25H23NNaO2S 424.1347; Found 424.1374.
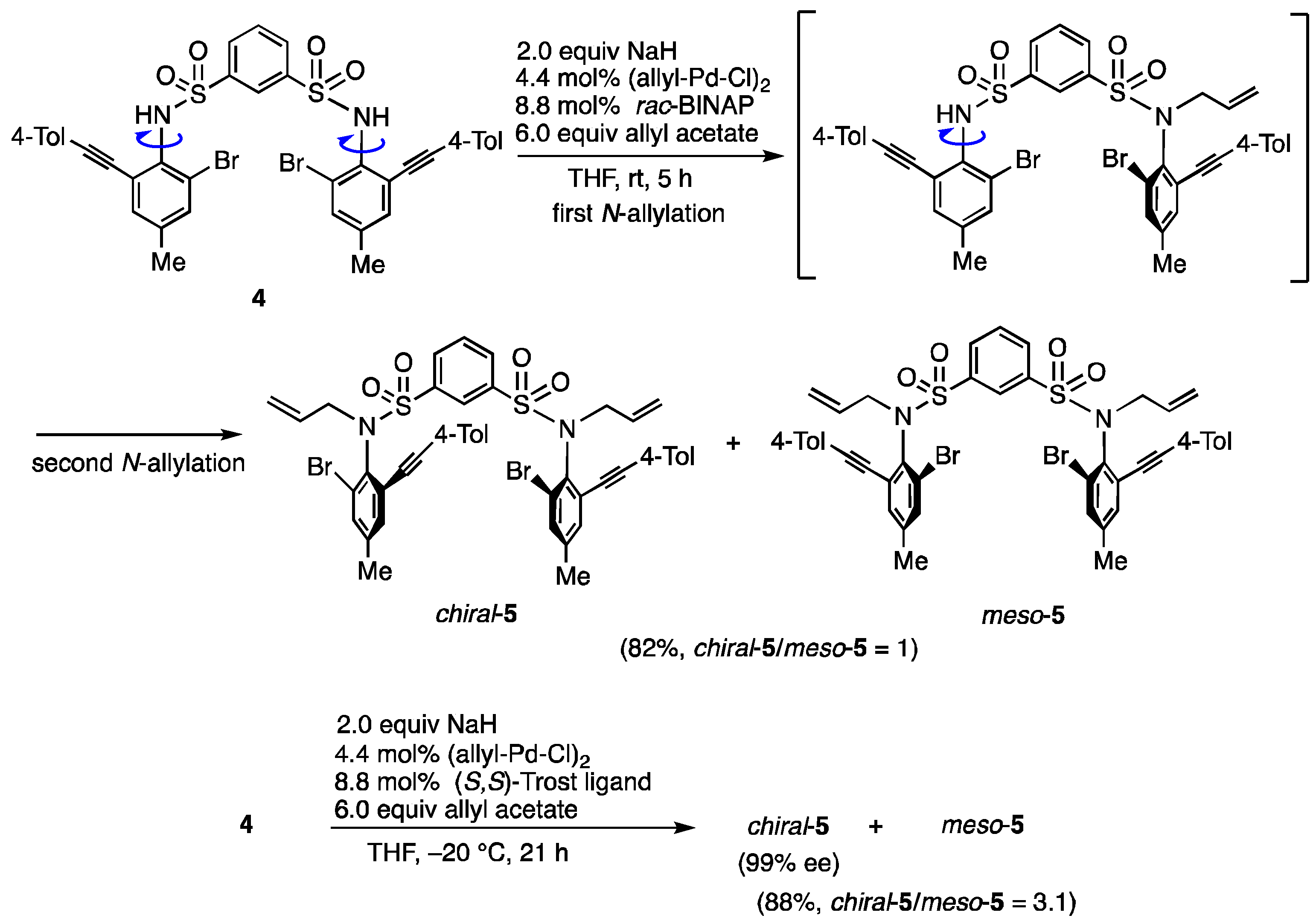
N,N-Bis(2-bromo-4-methyl-6-(4-tolylethynyl)phenyl)benzene-1,3-disulfonamide (4). In accordance with the experimental procedure for the synthesis of 1b, 4 was prepared from 2-bromo-4methyl-6-(4-tolylethynyl)aniline (1.694 g, 5.6 mmol) and benzene-1,3-disulfonyl chloride (704 mg, 2.6 mmol). Purification of the residue by column chromatography (hexane/AcOEt = 15 and then 5) gave 4 (266 mg, 13%). 4: yellow solid; mp 268−270 °C; IR (neat) 3246, 2211, 1346, 1161 cm−1; 1H NMR (400 MHz, (CD3)2SO) δ: 10.23 (2H, s), 8.07 (1H, s), 7.84 (2H, d, J = 7.3 Hz), 7.44–7.48 (3H, m), 7.38 (2H, s), 7.30 (4H, d, J = 7.9 Hz), 7.20 (4H, d, J = 7.9 Hz), 2.32 (6H, s), 2.27 (6H, s); 13C{1H} NMR (100 MHz, (CD3)2SO) δ: 143.2, 139.6, 138.9, 133.8, 132.9, 132.5, 131.5, 130.3, 130.1, 129.2, 126.0, 124.7, 124.6, 119.1, 94.0, 86.2, 21.2, 19.9; MS (ESI-TOF) m/z: [M + Na]+ 827; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C38H30Br2N2NaO4S2 826.98705; Found: 826.98759.
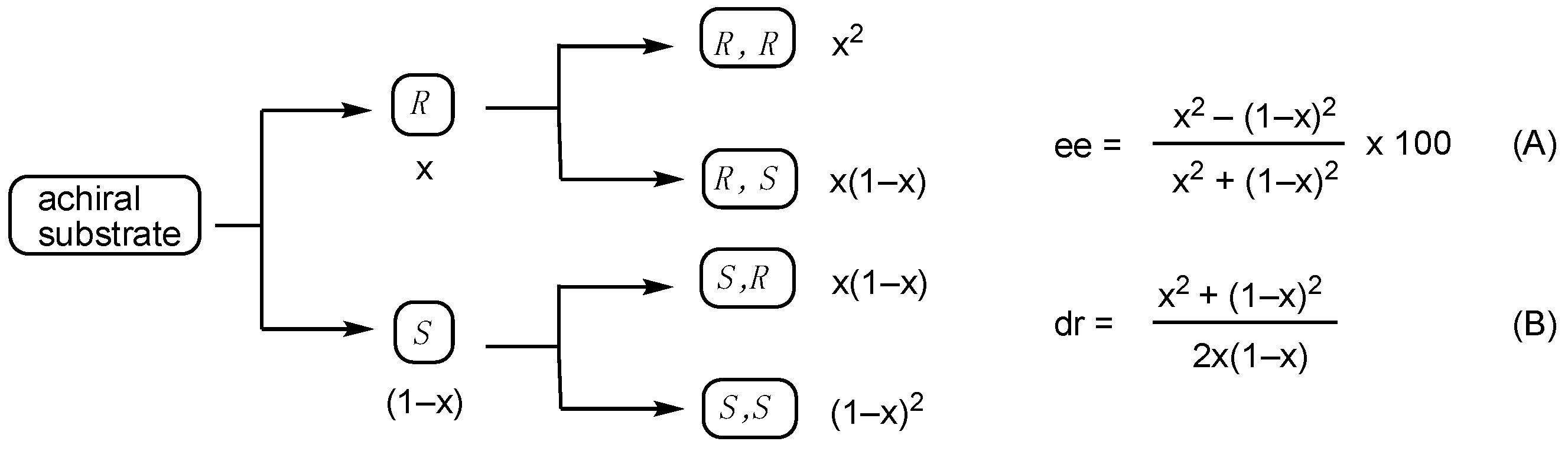
N,N-Diallyl-N,N-bis(2-bromo-4-methyl-6-(4-tolylethynyl)phenyl)benzene-1,3-disulfonamide (chiral-5 and meso-5). Under N2 atmosphere, to 4 (241 mg, 0.3 mmol) in THF (2.5 mL) was added NaH (60% assay, 24 mg, 0.6 mmol) at 0 °C, and the mixture was stirred for 20 min at −20 °C. (Allyl-Pd-Cl)2 (4.8 mg, 0.044 mmol), (S,S)-Trost ligand (21 mg, 0.088 mmol) and allyl acetate (194 μL, 1.8 mmol) in THF (1.5 mL) were added to the reaction mixture, and then the mixture was stirred for 21 h at −20 °C. The mixture was poured into 1N HCl solution and extracted with AcOEt. The AcOEt extracts were washed with brine, dried over MgSO4, and evaporated to dryness. Purification of the residue by column chromatography (hexane/AcOEt = 10) gave the mixture of chiral-5 and meso-5 (232 mg, 88%). The ratio (3.1:1) of chiral-5 and meso-5 was determined by 1H NMR. MPLC of the mixture gave chiral-5 (147 mg, less polar) and meso-5 (45 mg, more polar). The ee (99% ee) of chiral-5 was determined by HPLC analysis using a chiral column (CHIRALPAK AD-H) (25 cm × 0.46 cm i.d.; 10% i-PrOH in hexane; flow rate, 0.8 mL/min; (–)-chiral-5 (major); tR = 14.1 min, (+)-chiral-5 (minor); tR = 17.0 min). chiral-5: white solid; mp 93–95 °C (99% ee), 89−94 °C (racemate); [α]D25 = −18.8 (99% ee, CHCl3, c 1.00); IR (neat) 2212, 1354, 1088 cm−1; 1H NMR (400 MHz, CDCl3) δ: 8.44 (1H, t, J = 1.8 Hz), 7.78 (2H, dd, J = 7.3, 1.2 Hz), 7.40 (2H, d, J = 1.2 Hz), 7.28 (2H, d, J = 1.8 Hz), 7.06–7.20 (9H, m), 6.00 (2H, ddt, J = 17.1, 9.8, 7.0 Hz), 5.08 (2H, dd, J = 17.1, 1.2 Hz), 5.03 (2H, dd, J = 9.8, 1.2 Hz), 4.36 (2H, dd, J = 14.0, 7.3 Hz), 4.30 (2H, dd, J = 15.9, 6.7 Hz), 2.34 (6H, s), 2.31 (6H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 141.9, 140.0, 139.1, 136.0, 134.2, 133.0, 132.3, 131.5, 131.4, 129.3, 129.0, 127.4, 127.0, 126.7, 119.7, 118.9, 95.0, 85.8, 53.3, 21.6, 20,6; MS (ESI-TOF) m/z: [M + Na]+ 907; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C44H38Br2N2NaO4S2 907.04965; Found: 907.04749. meso-5: white solid; mp 89−94 °C; IR (neat) 2211, 1348, 1159 cm−1; 1H NMR (400 MHz, CDCl3) δ: 8.38 (1H, s), 7.76 (2H, dd, J = 7.9, 1.8 Hz), 7.41 (2H, d, J = 1.2 Hz), 7.28 (2H, d, J = 1.8 Hz), 7.07–7.17 (9H, m), 6.01 (2H, ddt, J = 17.1, 9.8, 6.7 Hz), 5.08 (2H, d, J = 17.1 Hz), 5.03 (2H, d, J = 9.8 Hz), 4.34 (4H, d, J = 6.7 Hz), 2.34 (6H, s), 2.31 (6H, s); 13C{1H} NMR (100 MHz, CDCl3) δ: 141.8, 140.0, 139.1, 135.9, 134.2, 133.0, 132.3, 131.4, 129.3, 129.0, 127.5, 126.9, 126.8, 119.7, 118.9, 95.0, 85.7, 53.3, 21.6, 20,6; MS (ESI-TOF) m/z: [M + Na]+ 907; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C44H38Br2N2NaO4S2 907.04965; Found: 907.04712.





 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


