
Cytotoxic and Luminescent Properties of Novel Organotin Complexes with Chelating Antioxidant Ligand

 ,
,  , , ,
, , ,  and
and 
Abstract
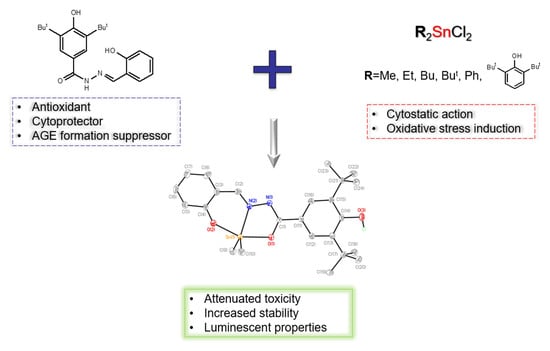
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
Molecular Structure
2.2. Studies of Antioxidant Activity
2.2.1. DPPH-Test
2.2.2. NBT-Test
2.2.3. Lipoxygenase Inhibitory Activity
2.2.4. Study of Antiglycation Activity
2.3. Studies of Biological Activity
2.3.1. Stability Investigation
2.3.2. MTT-Assay
2.4. Luminescence Studies
3. Materials and Methods
3.1. General Synthesis of Ligand (L)
Di-tert-butyl-4-hydroxy-N′-(2-hydroxybenzylidene)-benzohydrazine (1)
3.2. General Synthesis of Complexes (2–7)
3.2.1. Me2SnL (2)
3.2.2. Et2SnL (3)
3.2.3. Bu2SnL (4)
3.2.4. But2SnL (5)
3.2.5. Ph2SnL (6)
3.2.6. (2,6-Di-tert-butylphenol)2SnL (7)
3.3. Crystallographic Data Collection and Structure Determination
3.4. Antioxidant Assay
3.4.1. DPPH Assay
3.4.2. Enzymatic Generation of the Superoxide Radical Anion О2−• in the Xanthine—Xanthine Oxidase System (NBT Assay)
3.4.3. Inhibition of Lipoxygenase (LOX 1-B)
3.4.4. Study of Antiglycation Activity
3.5. Biological Studies
3.5.1. Stability Studies
3.5.2. MTT Assay
3.6. Luminescence Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Nath, M.; Saini, P.K. Chemistry and applications of organotin(IV) complexes of Schiff bases. Dalton Trans. 2011, 40, 7077–7121. [Google Scholar] [CrossRef]
- Ahmad Shah, S.S.; Ashfaq, M.; Waseem, A.; Ahmed, M.M.; Najam, T.; Shaheen, S.; Rivera, G. Synthesis and Biological Activities of Organotin(IV) Complexes as Antitumoral and Antimicrobial Agents. A Review. Mini. Rev. Med. Chem. 2015, 15, 406–426. [Google Scholar] [CrossRef]
- Hunt, D.W. Rostaporfin (Miravant Medical Technologies). IDrugs Investig. Drugs J. 2002, 5, 180–186. [Google Scholar]
- Sen Sarma, M. Cytotoxic activity of organotin(IV) complexes—A short review. Prajnan-O-Sadhona-A Sci. Annu. 2015, 2, 99–115. [Google Scholar]
- Milaeva, E.R.; Shpakovsky, D.B.; Gracheva, Y.A.; Antonenko, T.A.; Ksenofontova, T.D.; Nikitin, E.A.; Berseneva, D.A. Novel selective anticancer agents based on Sn and Au complexes. Mini-review. Pure Appl. Chem. 2020, 92, 1201. [Google Scholar] [CrossRef]
- Gennari, A.; Viviani, B.; Galli, C.L.; Marinovich, M.; Pieters, R.; Corsini, E. Organotins induce apoptosis by disturbance of [Ca2+] and mitochondrial activity, causing oxidative stress and activation of caspases in rat thymocytes. Toxicol. Appl. Pharmacol. 2000, 169, 185–190. [Google Scholar] [CrossRef]
- Jensen, K.G.; Onfelt, A.; Wallin, M.; Lidums, V.; Andersen, O. Effects of organotin compounds on mitosis, spindle structure, toxicity and in vitro microtubule assembly. Mutagenesis 1991, 6, 409–416. [Google Scholar] [CrossRef]
- Tang, L.; Li, Y.-l.; Ge, R.; Li, Q.-S. Oxidative stress in di-n-butyl-di-(4-chlorobenzohydroxamato)tin (IV)-induced hepatotoxicity determined by proteomic profiles. Toxicol. Lett. 2012, 213, 167–173. [Google Scholar] [CrossRef]
- Preedy, V.R. Aging: Oxidative Stress and Dietary Antioxidants, 1st ed.; Academic Press: Cambridge, MA, USA, 2014; 301p. [Google Scholar]
- Shpakovsky, D.B.; Banti, C.N.; Beaulieu-Houle, G.; Kourkoumelis, N.; Manoli, M.; Manos, M.J.; Tasiopoulos, A.J.; Hadjikakou, S.K.; Milaeva, E.R.; Charalabopoulos, K.; et al. Synthesis, structural characterization and in vitro inhibitory studies against human breast cancer of the bis-(2,6-di-tert-butylphenol)tin(IV) dichloride and its complexes. Dalton Trans. 2012, 41, 14568–14582. [Google Scholar] [CrossRef]
- Milaeva, E.R.; Shpakovsky, D.B.; Gracheva, Y.A.; Orlova, S.I.; Maduar, V.V.; Tarasevich, B.N.; Meleshonkova, N.N.; Dubova, L.G.; Shevtsova, E.F. Metal complexes with functionalised 2,2′-dipicolylamine ligand containing an antioxidant 2,6-di-tert-butylphenol moiety: Synthesis and biological studies. Dalton Trans. 2013, 42, 6817–6828. [Google Scholar] [CrossRef]
- Rychlý, J.; Mosnáčková, K.; Rychlá, L.; Fiedlerová, A.; Kasza, G.; Nádor, A.; Osváth, Z.; Stumphauser, T.; Szarka, G.; Czaníková, K.; et al. Comparison of the UV stabilisation effect of commercially available processing stabilizers Irganox HP 136 and Irganox 1010. Polym. Degrad. Stab. 2015, 118, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Carocho, M.; Morales, P.; Ferreira, I.C.F.R. Antioxidants: Reviewing the chemistry, food applications, legislation and role as preservatives. Trends Food Sci. Technol. 2018, 71, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Burd, S.G.; Lebedeva, A.V.; Pantina, N.V.; Rubleva, Y.V.; Pizova, N.V.; Vasil’ev, S.V.; Belova, A.N.; Vorob’eva, O.V.; Emel’yanova, V.V.; Zhadnov, V.A.; et al. Clinical results and prospects for the use of phenosanic acid in patients with focal epilepsy. Zh. Nevrol. Psikhiatr. Im S. S. Korsakova 2021, 121, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Shpakovsky, D.B.; Banti, C.N.; Mukhatova, E.M.; Gracheva, Y.A.; Osipova, V.P.; Berberova, N.T.; Albov, D.V.; Antonenko, T.A.; Aslanov, L.A.; Milaeva, E.R.; et al. Synthesis, antiradical activity and in vitro cytotoxicity of novel organotin complexes based on 2,6-di-tert-butyl-4-mercaptophenol. Dalton Trans. 2014, 43, 6880–6890. [Google Scholar] [CrossRef] [PubMed]
- Antonenko, T.A.; Shpakovsky, D.B.; Vorobyov, M.A.; Gracheva, Y.A.; Kharitonashvili, E.V.; Dubova, L.G.; Shevtsova, E.F.; Tafeenko, V.A.; Aslanov, L.A.; Iksanova, A.G.; et al. Antioxidative vs cytotoxic activities of organotin complexes bearing 2,6-di-tert-butylphenol moieties. Appl. Organomet. Chem. 2018, 32, e4381. [Google Scholar] [CrossRef]
- Daniel, W.A. Mechanisms of cellular distribution of psychotropic drugs. Significance for drug action and interactions. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Liu, C.; Li, X.; Zhu, H.; Wang, K.; Zhu, B. Recent advances in chemotherapy-based organic small molecule theranostic reagents. Coord. Chem. Rev. 2022, 473, 214808. [Google Scholar] [CrossRef]
- Shende, P.; Gandhi, S. Current strategies of radiopharmaceuticals in theranostic applications. J. Drug Deliv. Sci. Technol. 2021, 64, 102594. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, C.; Su, M.; Rong, X.; Zhang, Y.; Wang, X.; Wang, K.; Li, X.; Yu, Y.; Zhang, X.; et al. Recent advances in 4-hydroxy-1,8-naphthalimide-based small-molecule fluorescent probes. Coord. Chem. Rev. 2021, 448, 214153. [Google Scholar] [CrossRef]
- Yang, K.; Qi, S.; Yu, X.; Bai, B.; Zhang, X.; Mao, Z.; Huang, F.; Yu, G. A Hybrid Supramolecular Polymeric Nanomedicine for Cascade-Amplified Synergetic Cancer Therapy. Angew. Chem. Int. Ed. 2022, 61, e202203786. [Google Scholar] [CrossRef]
- Chen, X.; Lee, K.-A.; Ren, X.; Ryu, J.-C.; Kim, G.; Ryu, J.-H.; Lee, W.-J.; Yoon, J. Synthesis of a highly HOCl-selective fluorescent probe and its use for imaging HOCl in cells and organisms. Nat. Protoc. 2016, 11, 1219–1228. [Google Scholar] [CrossRef]
- Zav’yalov, I.A.; Polyakova, O.V.; Milaeva, E.R.; Prokof’ev, A.I. Synthesis and ESR study of Co and Ni hydrazides and hydroxamates containing 2,6-di-tert-butylphenol moiety in the ligands. Russ. Chem. Bull. 1995, 44, 1725–1728. [Google Scholar] [CrossRef]
- Yang, Y.; Hong, M.; Xu, L.; Cui, J.; Chang, G.; Li, D.; Li, C.-Z. Organotin(IV) complexes derived from Schiff base N’-[(1E)-(2-hydroxy-3-methoxyphenyl)methylidene]pyridine-3-carbohydrazone: Synthesis, in vitro cytotoxicities and DNA/BSA interaction. J. Organomet. Chem. 2016, 804, 48–58. [Google Scholar] [CrossRef]
- Holmes, R.R. Five-Coordinated Structures. In Progress in Inorganic Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1984; pp. 119–235. [Google Scholar]
- Lee, S.M.; Tan, Y.S.; Tiekink, E.R.T. Crystal structure of {N-(3-ethoxy-2-oxidobenzylidene)-4-fluorobenzohydrazonato-κ3O,N,O′}dimethyltin(IV), C18H19FN2O3Sn. Z. Krist.-New Cryst. Struct. 2018, 233, 335–337. [Google Scholar] [CrossRef] [Green Version]
- Bondet, V.; Brand-Williams, W.; Berset, C. Kinetics and Mechanisms of Antioxidant Activity using the DPPH. Free Radical Method. Food Sci. Technol. 1997, 30, 609–615. [Google Scholar] [CrossRef]
- Nikitin, E.A.; Shpakovsky, D.B.; Tyurin, V.Y.; Kazak, A.A.; Gracheva, Y.A.; Vasilichin, V.A.; Pavlyukov, M.S.; Mironova, E.M.; Gontcharenko, V.E.; Lyssenko, K.A.; et al. Novel organotin complexes with phenol and imidazole moieties for optimized antitumor properties. J. Organomet. Chem. 2022, 959, 122212. [Google Scholar] [CrossRef]
- Kubo, I.; Masuoka, N.; Ha, T.J.; Tsujimoto, K. Antioxidant activity of anacardic acids. Food Chem. 2006, 99, 555–562. [Google Scholar] [CrossRef]
- Pidgeon, G.P.; Lysaght, J.; Krishnamoorthy, S.; Reynolds, J.V.; O’Byrne, K.; Nie, D.; Honn, K.V. Lipoxygenase metabolism: Roles in tumor progression and survival. Cancer Metastasis Rev. 2007, 26, 503–524. [Google Scholar] [CrossRef] [PubMed]
- Andreou, A.; Feussner, I. Lipoxygenases—Structure and reaction mechanism. Phytochemistry 2009, 70, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Inoue, T.; Kawasaki, N.; Miyataka, H.; Matsumoto, H.; Taguchi, T.; Inagaki, N.; Nagai, H.; Satoh, T. Synthesis and biological activities of novel antiallergic agents with 5-lipoxygenase inhibiting action. Bioorg. Med. Chem. 2000, 8, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Werz, O.; Steinhilber, D. Therapeutic options for 5-lipoxygenase inhibitors. Pharmacol. Therapeut. 2006, 112, 701–718. [Google Scholar] [CrossRef]
- Pontiki, E.; Hadjipavlou-Litina, D.; Litinas, K.; Nicolotti, O.; Carotti, A. Design, synthesis and pharmacobiological evaluation of novel acrylic acid derivatives acting as lipoxygenase and cyclooxygenase-1 inhibitors with antioxidant and anti-inflammatory activities. Eur. J. Med. Chem. 2011, 46, 191–200. [Google Scholar] [CrossRef]
- Legaard, G.E.; Feineis, C.S.; Johansen, M.Y.; Hansen, K.B.; Vaag, A.A.; Larsen, E.L.; Poulsen, H.E.; Almdal, T.P.; Karstoft, K.; Pedersen, B.K.; et al. Effects of an exercise-based lifestyle intervention on systemic markers of oxidative stress and advanced glycation endproducts in persons with type 2 diabetes: Secondary analysis of a randomised clinical trial. Free Radic. Biol. Med. 2022, 188, 328–336. [Google Scholar] [CrossRef]
- Reddy Addi, U.; Jakhotia, S.; Reddy, S.S.; Reddy, G.B. Age-related neuronal damage by advanced glycation end products through altered proteostasis. Chem. Biol. Interact. 2022, 355, 109840. [Google Scholar] [CrossRef] [PubMed]
- Milaeva, E.R.; Shpakovsky, D.B.; Gracheva, Y.A.; Antonenko, T.A.; Osolodkin, D.I.; Palyulin, V.A.; Shevtsov, P.N.; Neganova, M.E.; Vinogradova, D.V.; Shevtsova, E.F. Some insight into the mode of cytotoxic action of organotin compounds with protective 2,6-di-tert-butylphenol fragments. J. Organomet. Chem. 2015, 782, 96–102. [Google Scholar] [CrossRef]
- Niks, M.; Otto, M. Towards an optimized MTT assay. J. Immunol. Methods 1990, 130, 149–151. [Google Scholar] [CrossRef]
- Hadjikakou, S.K.; Hadjiliadis, N. Antiproliferative and anti-tumor activity of organotin compounds. Coord. Chem. Rev. 2009, 253, 235–249. [Google Scholar] [CrossRef]
- Hennighausen, G.; Lange, P.; Merkord, J. The Relationship Between the Length of the Alkyl Chain of Dialkyltin Compounds and their Effects on Thymus and Bile Ducts in Mice. In Further Studies in the Assessment of Toxic Actions; Springer: Berlin/Heidelberg, Germany, 1980; pp. 175–178. [Google Scholar]
- Banti, C.N.; Hadjikakou, S.K.; Sismanoglu, T.; Hadjiliadis, N. Anti-proliferative and antitumor activity of organotin(IV) compounds. An overview of the last decade and future perspectives. J. Inorg. Biochem. 2019, 194, 114–152. [Google Scholar] [CrossRef]
- Antonenko, T.A.; Shpakovsky, D.B.; Berseneva, D.А.; Gracheva, Y.A.; Dubova, L.G.; Shevtsov, P.N.; Redkozubova, O.M.; Shevtsova, E.F.; Tafeenko, V.A.; Aslanov, L.A.; et al. Cytotoxic activity of organotin carboxylates based on synthetic phenolic antioxidants and polycyclic bile acids. J. Organomet. Chem. 2020, 909, 121089. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Nikitin, E.A.; Shpakovsky, D.B.; Pryakhin, A.D.; Antonenko, T.A.; Tyurin, V.Y.; Kazak, A.A.; Ulyanov, A.N.; Tafeenko, V.A.; Aslanov, L.A.; Dubova, L.G.; et al. Antioxidant activity of modified 2,6-Di-tert-butylphenols with pyridine moiety. Pharm. Pharmacol. Int. J. 2020, 8, 122–134. [Google Scholar] [CrossRef]
- Milaeva, E.R.; Tyurin, V.Y.; Shpakovsky, D.B.; Moiseeva, A.A.; Gracheva, Y.A.; Antonenko, T.A.; Maduar, V.V.; Osolodkin, D.I.; Palyulin, V.A.; Shevtsova, E.F. Redox-active metal complexes with 2,2′-dipicolylamine containing ferrocenyl moiety: Synthesis, electrochemical behavior and biological activity. J. Organomet. Chem. 2017, 839, 60–70. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Quantity of Reduced DPPH after 20 h, % | I (NBT-Test), % | I (LOX Inhibition Assay), % |
|---|---|---|---|
| 1 | - * | 0.5 ± 0.1 | 51.7 ± 4.2 |
| 2 | 35.2 ± 1.9 | 6.3 ± 1.8 | 57.1 ± 4.7 |
| 3 | 41.3 ± 2.1 | 7.3 ± 2.0 | 61.1 ± 5.1 |
| 4 | 50.5 ± 2.8 | −4.6 ± 1.2 | 47.5 ± 3.8 |
| 5 | 26.5 ± 1.7 | 5.9 ± 1.5 | 61.4 ± 5.3 |
| 6 | 47.4 ± 2.7 | −28.0 ± 3.6 | 44.9 ± 3.7 |
| 7 | - * | −40.5 ± 4.2 | 66.0 ± 5.9 |
| Compound | Concentration, µM | Activity, %, M ± SEM | IC50, µM |
|---|---|---|---|
| 1 | 1000 | 70.2 ± 8.0 | 2.8 |
| 300 | 73.2 ± 1.9 | ||
| 100 | 61.6 ± 0.5 | ||
| 30 | 62.0 ± 0.7 | ||
| 10 | 60.1 ± 1.1 | ||
| 3 | 53.9 ± 2.5 | ||
| 1 | 0.6 ± 2.3 | ||
| 0.3 | −1.9 ± 1.4 | ||
| Aminoguanidine | 10,000 | 62.0 ± 2.0 | 5166.5 |
| 3000 | 39.5 ± 2.2 | ||
| 1000 | 21.6 ± 1.8 |
| Compound | IC50, µM | |||
|---|---|---|---|---|
| HCT116 | MCF-7 | A549 | WI-38 | |
| 1 | 1.7 ± 0.4 | 4.7 ± 0.8 | 2.2 ± 0.4 | 6.6 ± 1.2 |
| 2 | 2.4 ± 0.8 | 5.2 ± 1.0 | 2.2 ± 0.7 | 9.5 ± 3.1 |
| 3 | 1.4 ± 0.5 | 3.7 ± 0.5 | 1.5 ± 0.5 | 5.8 ± 1.5 |
| 4 | 10.0 ± 2.3 | 1.01 ± 0.4 | 14.5 ± 4.5 | 10.8 ± 3.5 |
| 5 | 34.3 ± 14.0 | 50.4 ± 12.6 | 43 ± 10.0 | 26.3 ± 11.3 |
| 6 | 20.0 ± 11.0 | 26.3 ± 3.5 | 20.2 ± 4.0 | 52.6 ± 14.6 |
| 7 | 20.5 ± 6.4 | 32.7 ± 9.5 | 31.9 ± 5.7 | 10.6 ± 3.5 |
| Cisplatin | 8.3 ± 3.5 | 15.5 ± 3.0 | 10.4 ± 1.5 | 16.3 ± 1.9 |
| Compound | Quantum Yield, % | ε, (λmax, nm), M−1·cm−1 | ε, (λ = 365 nm), M−1·cm−1 | Luminosity (λmax), M−1·cm−1 | Luminosity (λ = 365 nm), M−1·cm−1 | |||
|---|---|---|---|---|---|---|---|---|
| Powders * | DMSO Solution | |||||||
| 1000 µM | 100 µM | 10 µM | ||||||
| 2 | 14 ± 2 | 39 ± 3 | 56 ± 1 | 17 ± 3 | 13,070, (403) | 8617 | 771,130, (403) | 508,403 |
| 3 | 14 ± 2 | 30 ± 2 | 45 ± 1 | 17 ± 2 | 14,020, (410) | 4489 | 658,940, (410) | 210,983 |
| 4 * | 4 ± 3 | 36 ± 3 | 66 ± 2 | 41 ± 3 | 5950, (409) | 2181 | 398,650, (409) | 146,127 |
| 5 | 8 ± 1 | 29 ± 2 | 37 ± 1 | 20 ± 2 | 9920, (412) | 3080 | 664,640, (412) | 206,360 |
| 6 | 6 ± 2 | 26 ± 2 | 67 ± 2 | 25 ± 2 | 13,450, (393) | 9010 | 497,650, (393) | 333,370 |
| 7 | 20 ± 2 | 32 ± 2 | 42 ± 1 | 21 ± 2 | 13,270, (402) | 7002 | 557,340, (402) | 294,084 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikitin, E.; Mironova, E.; Shpakovsky, D.; Gracheva, Y.; Koshelev, D.; Utochnikova, V.; Lyssenko, K.; Oprunenko, Y.; Yakovlev, D.; Litvinov, R.; et al. Cytotoxic and Luminescent Properties of Novel Organotin Complexes with Chelating Antioxidant Ligand. Molecules 2022, 27, 8359. https://doi.org/10.3390/molecules27238359
Nikitin E, Mironova E, Shpakovsky D, Gracheva Y, Koshelev D, Utochnikova V, Lyssenko K, Oprunenko Y, Yakovlev D, Litvinov R, et al. Cytotoxic and Luminescent Properties of Novel Organotin Complexes with Chelating Antioxidant Ligand. Molecules. 2022; 27(23):8359. https://doi.org/10.3390/molecules27238359
Chicago/Turabian StyleNikitin, Evgeny, Ekaterina Mironova, Dmitry Shpakovsky, Yulia Gracheva, Daniil Koshelev, Valentina Utochnikova, Konstantin Lyssenko, Yury Oprunenko, Dmitry Yakovlev, Roman Litvinov, and et al. 2022. "Cytotoxic and Luminescent Properties of Novel Organotin Complexes with Chelating Antioxidant Ligand" Molecules 27, no. 23: 8359. https://doi.org/10.3390/molecules27238359
APA StyleNikitin, E., Mironova, E., Shpakovsky, D., Gracheva, Y., Koshelev, D., Utochnikova, V., Lyssenko, K., Oprunenko, Y., Yakovlev, D., Litvinov, R., Seryogina, M., Spasov, A., & Milaeva, E. (2022). Cytotoxic and Luminescent Properties of Novel Organotin Complexes with Chelating Antioxidant Ligand. Molecules, 27(23), 8359. https://doi.org/10.3390/molecules27238359