1. Introduction
Dengue fever is an acute infectious disease caused by the dengue virus and is transmitted mainly through mosquito vectors. After infection with the dengue virus, fever, muscle and joint pain, gastrointestinal bleeding, hemorrhagic shock, and even death can occur [
1]. Today, the spread of dengue fever seriously threatens public health security, with around tens of millions of infections worldwide [
2]. The dengue virus is mainly divided into four serotypes, and there are still no effective treatment drugs and vaccines [
3]. Therefore, it is urgent to discover and develop new treatments or drugs against DNEV.
The dengue virus is a single-stranded positive-strand RNA (+ssRNA) virus, which initiates infection of a permissive cell via clathrin-mediated endocytosis and then releases its genomic RNA into the cytosol after fusing with the late endosome [
4]. Various cells could be infected, such as the skin’s resident dendritic cells, monocytes and macrophages, megakaryocytes, erythroid precursor cells, liver cells, and endothelial cells. Several studies have revealed the relationship between lipid metabolism and viral replication during viral replication in cells [
5,
6,
7]. We indicated the role of the VEGFR2/AMPK pathway in regulating cellular lipids [
8], indicating the role of lipids in dengue viral infection. First, the enveloped virions need lipids as structural membrane components, with nearly 20% of the weight of the dengue virion being lipids [
9]. Second, viral replication expression, replication, and even assembly always occur in cellular lipid bilayers to escape the host immune surveillance [
10]. Third, β-oxidation is a normal way to provide energy for DENV replication in two ways, increasing autophagy and de novo synthesis [
11]. Despite the numerous roles of autophagy in regulating cellular homeostasis, its regulation of lipid metabolism is a significant contributor to robust DENV replication [
11,
12]. DENV-induced autophagy stimulates the delivery of lipids to lysosomal compartments, releasing free fatty acids, which undergo β-oxidation in the mitochondria to generate ATP, producing a metabolically favorable environment for viral replication [
11]. Lipid de novo synthesis is another way to provide energy, and it is reported that fatty acid synthase (FAS) re-localized to the replication complex by interaction with DENV NS3 protein, which increases the de novo synthesis of fatty acids during DENV infection [
13]. Considering the above reasons, drugs targeting lipid metabolism are a promising way to inhibit DENV proliferation. Indeed, several small molecules regulating autophagy and lipid metabolism have been reported. Autophagy inhibitor 3-methyladenine (3MA) or siRNAs targeting autophagy gene expression compromised viral infection [
11]. In addition, a small molecule inhibitor of FAS (C75) was reported to inhibit DENV replication in cell culture [
14]. However, another FAS inhibitor (Orlistat) has weak antiviral activity and weak interaction between FAS and NS3 [
15]. In addition, the small molecular compound PF-429242, known as an S1P inhibitor, is a promising candidate against DENV [
5] and anti-Zika virus. Considering these reasons, it is reasonable to consider whether modulation of the upstream FAS pathway could inhibit dengue virus infection.
Fatty acid synthase action is started from palmitic acid, a 16-carbon fatty acid, and longer saturated fatty acids are synthesized by an elongation enzyme system based on palmitic acid [
15,
16]. Additional carbons are added in 2-carbon units (CO
2 released) using malonyl-coenzyme A (malonyl CoA) as the carbon donor and different elongation enzymes. Malonyl CoA is generated from acetyl-CoA carboxylase (ACC), not only a substrate for de novo lipogenesis but also an inhibitor of mitochondrial fatty acid β-oxidation through inhibition of carnitine-palmitoyl transferase I (CPT-1), responsible for the transport of long-chain fatty acyl-CoAs across the mitochondrial membrane. ACC has two isoforms, ACC1 located primarily in the liver and adipose tissue and ACC2 dominant in skeletal and heart muscle. ACC inhibitors are hoped to inhibit de novo lipogenesis and increase the β-oxidation of long-chain fatty acids with the potential to treat of type 2 diabetes [
16] and cancers [
17]. The key regulatory role of ACC in fatty acid synthesis and oxidation pathways makes it an attractive target for various metabolic diseases [
18]. In particular, the combination of ACC inhibitors with other drugs is a new strategy for the treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis [
19]. Expanding the clinical indications for ACC inhibitors will be one of the main research directions in the future.
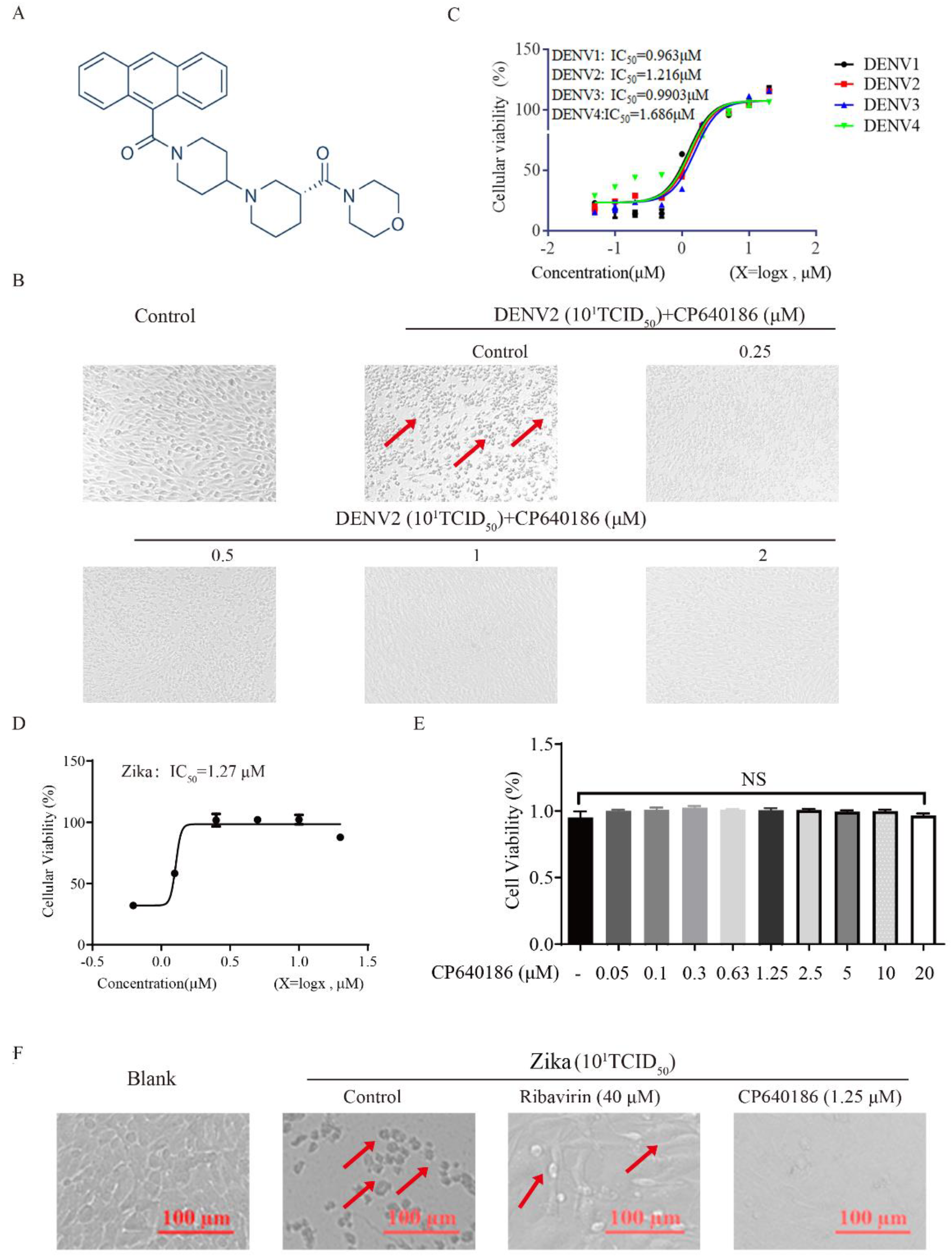
CP640186 is a reported potent inhibitor of mammalian ACCs (
Figure 1A), which was discovered by researchers at Pfizer Inc. with IC
50 values of about 55 nM [
20]. CP640186 could reduce tissue malonyl-CoA levels, inhibit fatty acid biosynthesis, and stimulate fatty acid oxidation. In addition, CP640186 could reduce body fat mass and body weight and improve insulin sensitivity. It inhibited both isozymes with an IC
50 of 55 nM in inhibiting HepG2 cell fatty acid and TG synthesis. CP640186 also stimulated fatty acid oxidation in C2C12 cells (ACC2) and in rat epitrochlearis muscle strips with an EC
50 of 57 nM and 1.3 μM. Kinetic studies showed that CP640186 is noncompetitive versus the acetyl-CoA substrate but may function at the active site of the CT domain [
20]. At present, a variety of ACC inhibitors based on the structure of CP640186 have been developed for the treatment of metabolic diseases, so we mainly use CP640186 in our research. Recently, two groups have reported the role of ACC in West Nile virus (WMV) replication, and ACC inhibitors (PF-05175157, PF-05206574, PF-06256254, and 5-(tetradecyloxy)-2-furoic acid (TOFA)) were shown to have an impressive effect against WMV in vitro and in vivo [
21,
22]. Teresa et al. reported the effect of the ACC inhibitor 5-(tetradecyloxy)-2-furoic acid (TOFA) on infection by WNV. Treatment with TOFA significantly reduced the cellular content of multiple lipids and inhibited the multiplication of WNV in a dose-dependent manner. They also found that another ACC inhibitor, 3,3,14,14-tetramethylhexadecanedioic acid (MEDICA 16), also inhibited WNV infection, pointing to the ACC as a druggable cellular target suitable for antiviral development against West Nile virus (WNV) [
22]. Nereida et al. reported the effect of three small-molecule ACC inhibitors (PF-05175157, PF-05206574, and PF-06256254) on the infection of WNV, dengue virus, and Zika virus. They found that PF-05175157 induced a reduction in the viral load in serum and kidney in WNV-infected mice, unveiling its therapeutic potential for treating chronic kidney disease associated with persistent WNV infection [
21]. Another group reported that the cholesterol-enriched cellular environment is crucial for viral replication, and they found that metformin and lovastatin (HMGCR inhibitor) inhibited the dengue virus proliferation [
23]. These results support the repositioning of metabolic inhibitors as broad-spectrum antivirals [
21,
23,
24]. However, the detailed mechanism of ACC against DENV need further study.
The above study indicated that ACC might participate in the DENV proliferation process. However, whether CP640186 could suppress DENV proliferation in vivo still needs to be studied. In this study, we applied CP640186 and DENV New Guinea C strain (NGC) as tools to disclose the antiviral ability between ACC and dengue viral proliferation, and also to evaluate the antiviral ability of CP640186 against DENV in vivo.
3. Discussion
Cellular lipid metabolism is closely related to dengue virus infection, as shown our and other groups’ reports [
5,
6,
8,
22,
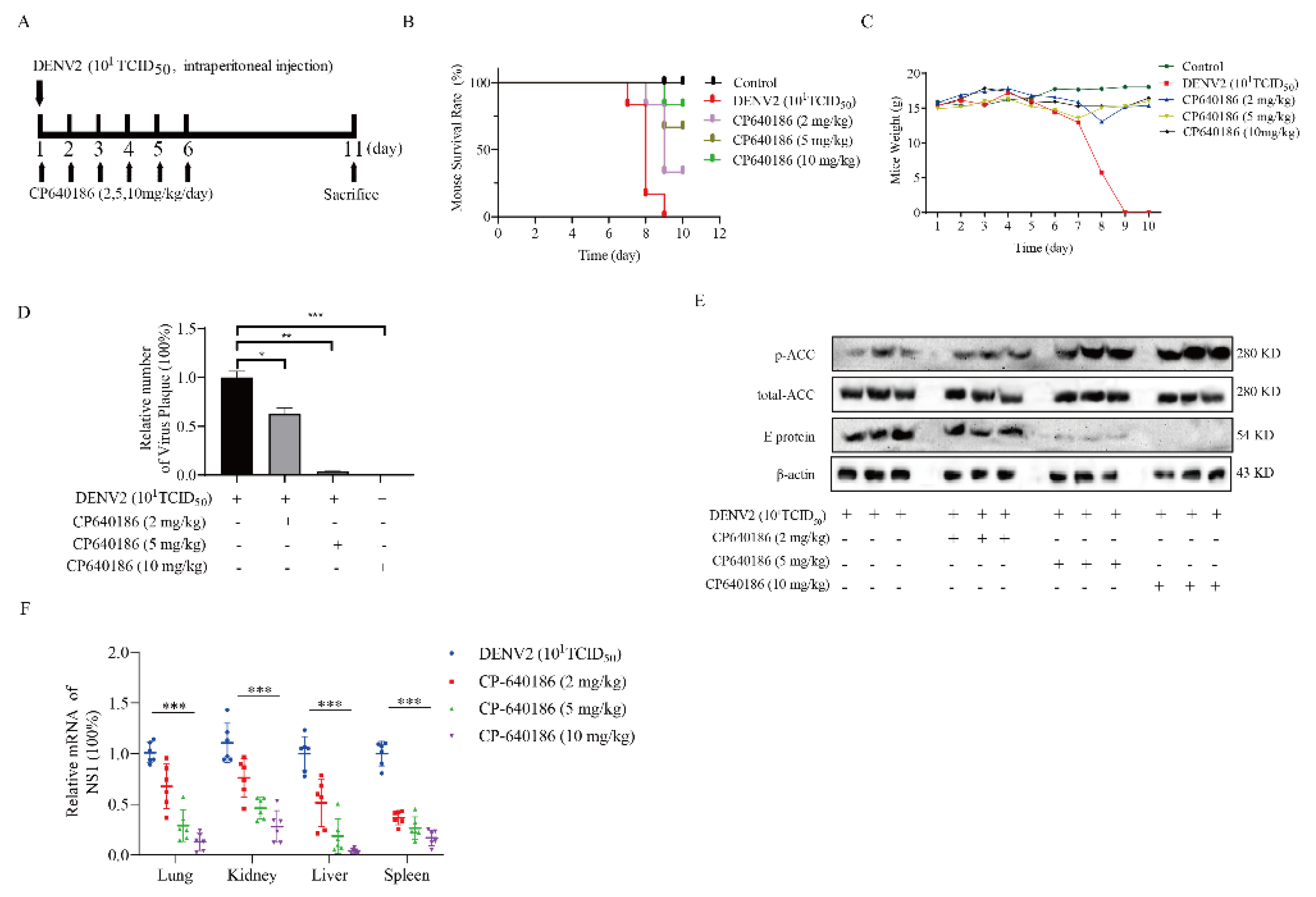
24,
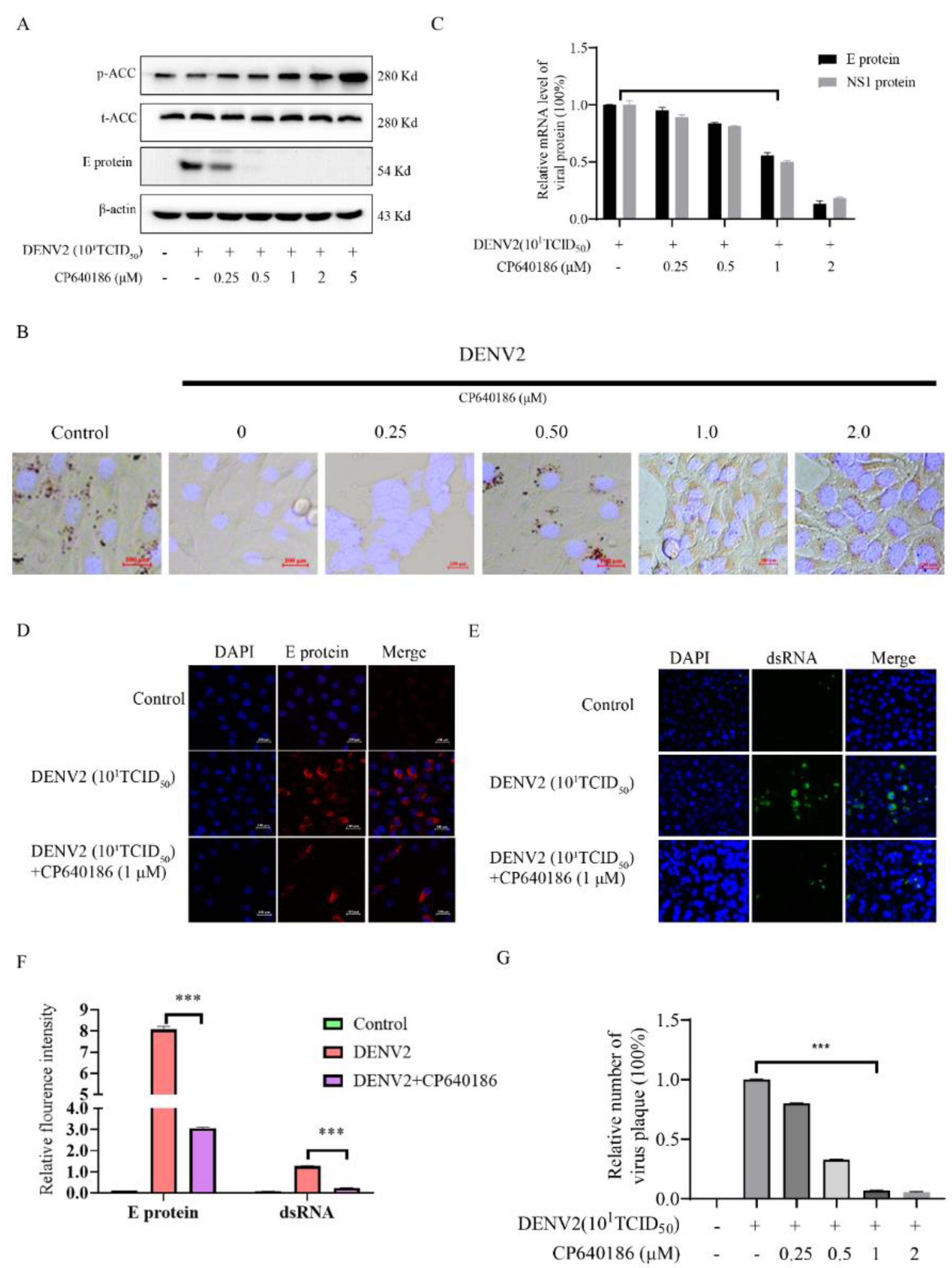
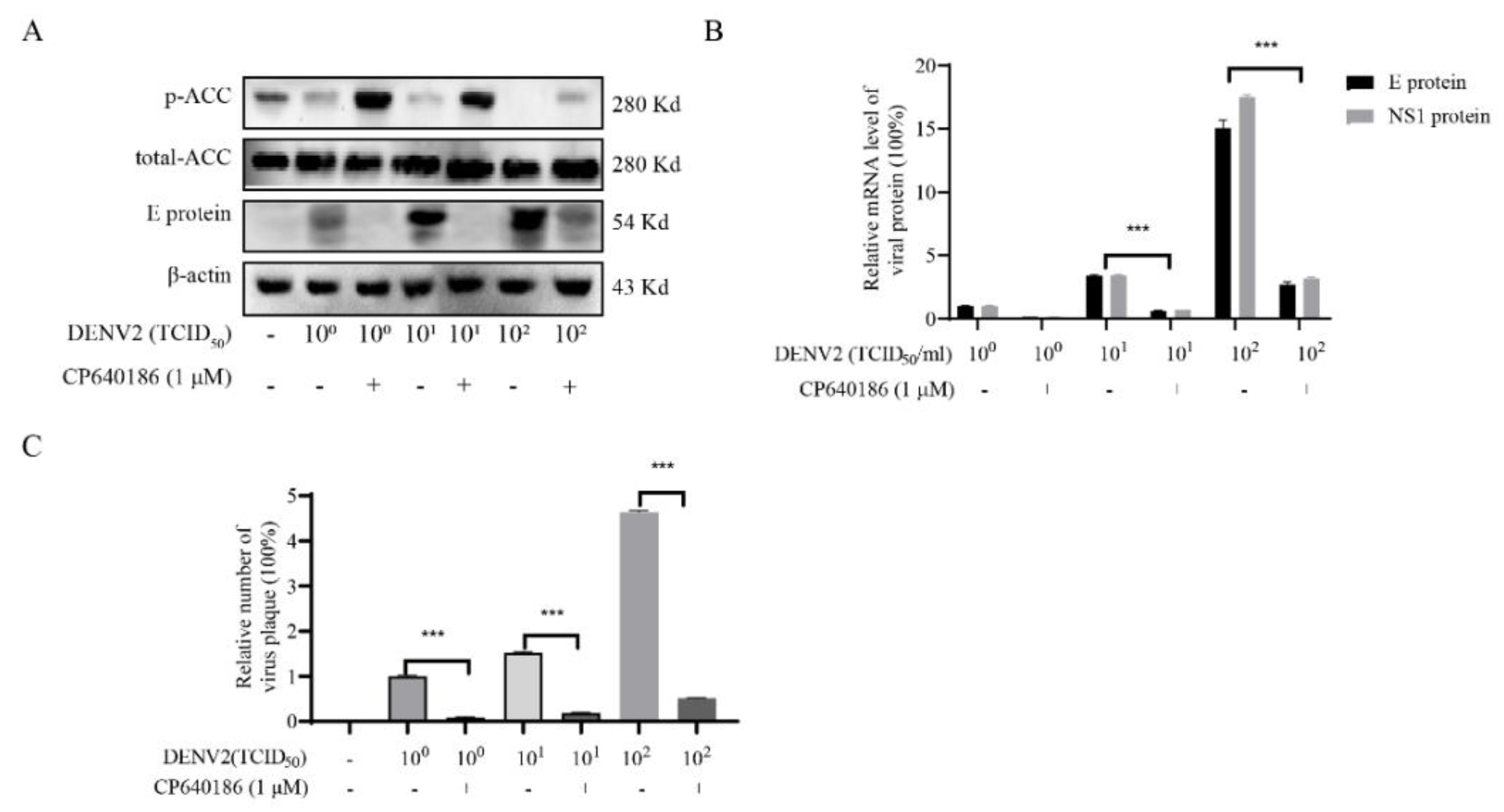
26]. Since ACC plays a rate-limiting role in lipid synthesis, we think ACC inhibitors could be promising antivirals against DENV. However, there are currently no in vivo data reported about the ACC inhibitors against dengue virus. The main reason is that the antiviral effect of the reported ACC inhibitor is not good enough, and there is also a lack of in vivo models. To address this issue, we found CP640186, with good antiviral activity and low toxicity. DENV2 proliferation could reduce by about 84.42% with CP640186 treatment at 1 μM with an EC
50 of 0.5 µM, which is basically the lowest effective concentration in cell experiments. At the same time, we validated the antiviral effect of CP640186 in knock-out mice (B6 background mice). It is worth noting that CP640186 showed good antiviral activity in vivo, which could resist death with viral infection at a dosage of 10 mg/kg/day orally administrated. Our study comprehensively demonstrated the antiviral effect of CP640186 against the dengue virus in vivo and in vitro, showing that CP640186 is a promising compound and could be used in clinical applications. In a word, this is the first report of an ACC inhibitor with in vivo anti-DENV activity.
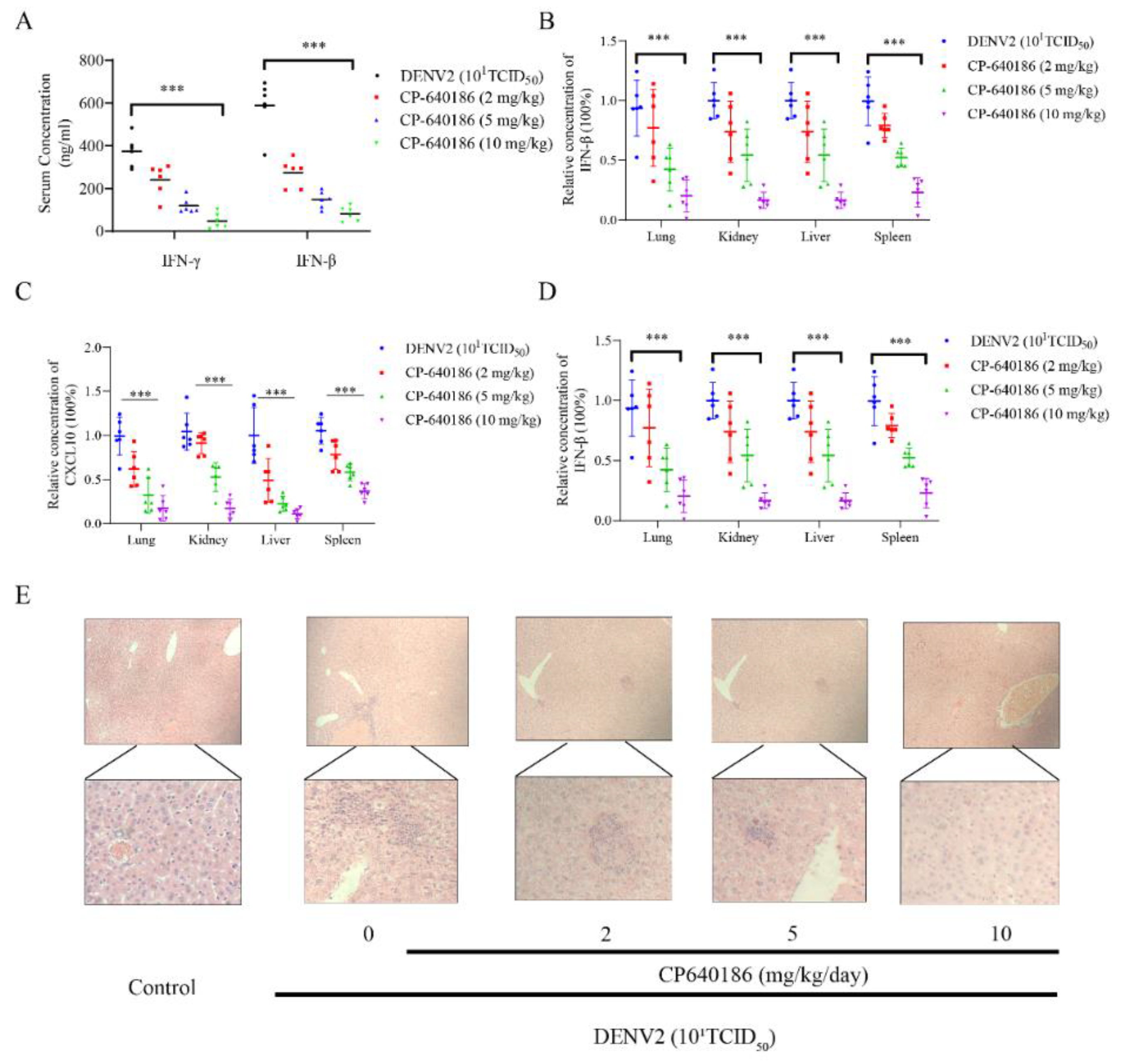
Although several studies disclosed the antiviral effect of an ACC inhibitor against flaviviruses, particularly West Nile virus, its underlying mechanism is still unclear. However, our results could explain the above phenomenon to a certain extent. Under normal circumstances, ACC inhibition will result in a decrease in intracellular lipids. However, we found that CP640186 retained cellular lipids during dengue virus infection. In other words, dengue virus infection consumed cell lipid droplets, and the CP640186 inhibited this process. This phenomenon raised two possibilities: firstly, that CP640186 inhibited dengue virus replication, which in turn affected dengue virus consumption of lipids; and secondly, that ACC inhibition leads to the loss of a key lipid in the process of dengue replication, which in turn inhibits the consumption of lipids by dengue. Based on our results, a large amount of cellular lipids was consumed, indicated by the oil red stain, especially when the inoculum of the virus (number) was increased. These results support the latter hypothesis, that CP640186 treatment leads to the loss of a key lipid in the process of dengue replication. As for the detailed mechanism of CP640186, we currently believe that it is mainly due to the inactivation of AMPK during dengue virus infection [
8], which thus decreased the phosphorylation of ACC and increased the activity of ACC. Meanwhile, inhibition of ACC could increase the phosphorylation of ACC and reduce the activity of ACC. We added these descriptions to the discussion section.
Lipids are widely used during dengue virus proliferation and viral assembly [
14]. Therefore, we attempted to clarify which process was affected by CP640186. Previous results indicated that lipid inhibition impaired membrane rearrangements associated with viral replication. In addition, lipid rafts combined with a cellular receptor are related to the entry of the dengue virus into cells [
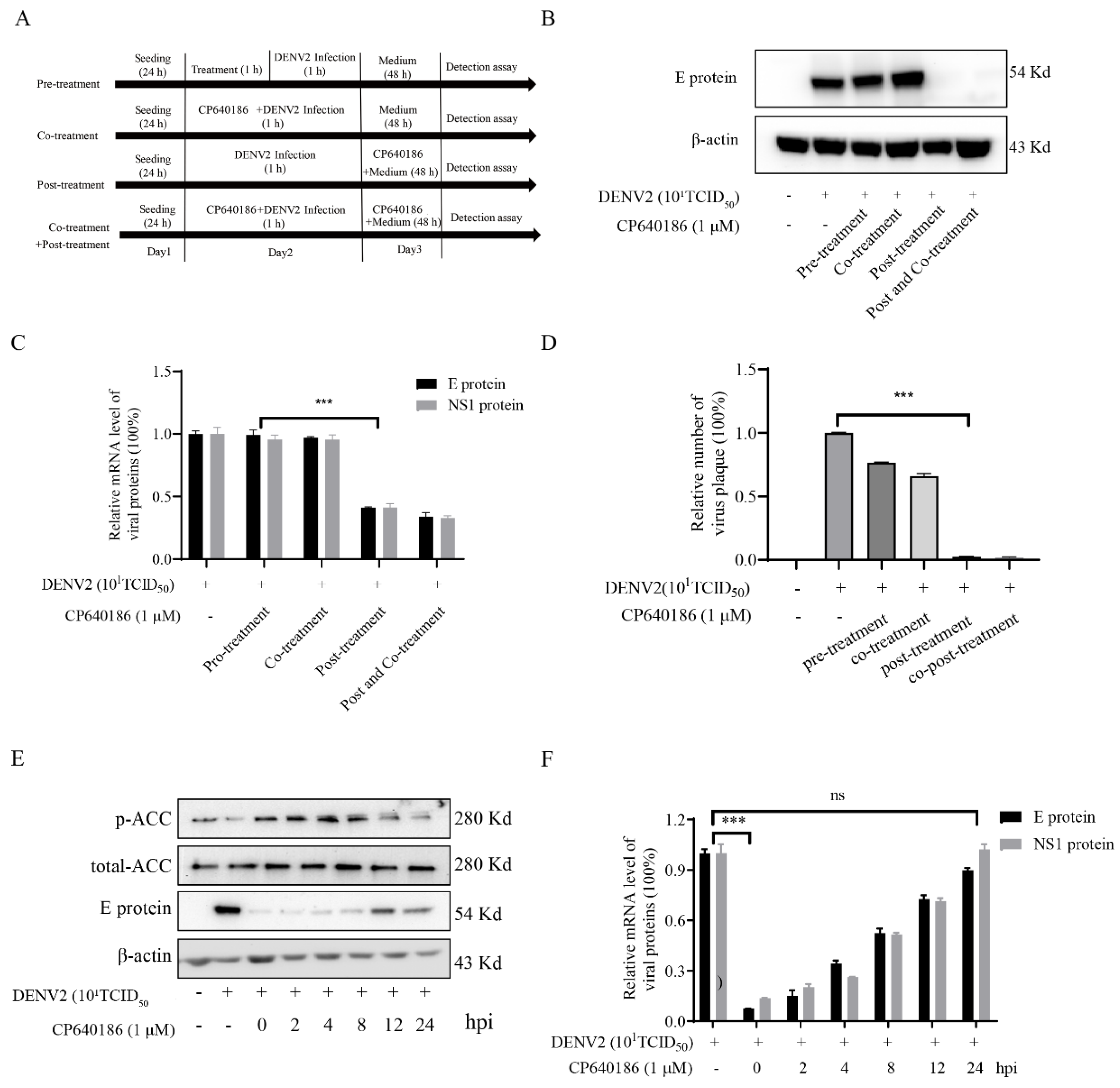
10]. However, we found that giving CP640186 treatment before virus infection did not inhibit viral replication, so this hypothesis was ruled out. Next, there are reports indicating that cellular lipids are related to viral immune escape. In this study, we found that CP640186 cannot change the innate immune pathway no matter when the virus infected or with CP640186 acting alone. These results indicated that ACC did not affect the natural immune response pathway, and it could not enhance the innate immune response, nor immune escape. In addition, lipid inhibition could alter the viral envelope composition and impair viral particle assembly. In fact, we found that CP640186 played a role in the early stages of viral replication. In addition, dysfunction of lipid synthesis could cause the generation of an unfavorable metabolic environment for virus replication. We did observe that CP640186 affected virus replication in the early stages of viral infection (1–4 hpi), suggesting that CP640186 might create an unfavorable environment for virus replication, which thus affected the viral assembly.
Given that inhibition of ACC activity could alter lipid metabolism, CP640186 was commonly used in studies of metabolic diseases such as hypertension, diabetes, visceral obesity, hyperlipidemia, cancer, fungal and bacterial infections, and herbicide exposure [
20]. ACC is an enzyme that controls the rate-limiting reaction of long-chain fatty acid synthesis and mitochondrial fatty acid oxidation [
26,
27]. CP640186 inhibits the synthesis of fatty acids and the oxidation of fatty acids, so that various lipids are lacking during the replication of DENV, which in turn affects the generation of progeny viruses. Due to the rapid onset of dengue fever and the short duration of medication, the side effects of the drug can be ignored, because these side effects are often caused by long-term medication.
The doses of CP640186 used in vivo were different, which inhibited fatty acid synthesis in rats, CD1 mice, and ob/ob mice with ED
50 of 13, 11, and 4 mg/kg, and stimulated rat whole-body fatty acid oxidation with an ED
50 of 30 mg/kg. Taken together, the concentration used in this study was consistent with the previous report. Recently, several groups have disclosed the relationship between lipid metabolism and flavivirus infection [
21,
23,
24]. Our research provided a new ACC inhibitor, CP640186, and studied the details of the antiviral effect in vitro and in vivo.
4. Materials and Methods
4.1. Materials
Baby Syrian hamster kidney cell line (BHK-21) was cultured in RPMI-1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal calf serum (Thermo Fisher Scientific, Waltham, MA, USA) in a 37 °C incubator with 5% CO2. C6/36 mosquito larva cells and DENV2 (New Guinea C derivative strain) were provided by Professor Xiaoguang Chen. DENV2 was amplified in C6/36 cells and stored at −80 °C until use. Professor Wei Zhao from Southern Medical University provided DENV1, 3, and 4. CP640186 were purchased from SuperLan chemical (Shanghai, China). MTT was purchased from Beyotime Biotechnology (Shanghai, China) (CAS: 97062-376, purity 98%).
4.2. The Cytopathic Effect (CPE) Assay
BHK-21 cells were incubated in a 96-well plate (5 × 10
4 cells/well) overnight, washed twice with PBS, and DENV (10
1TCID
50) was seeded with 100 μL/well and incubated at 37 °C for 1 h. Then, the virus was removed, and the cells were washed twice with PBS. RPMI1640 containing CP640186 hydrochloride (0–2 µM) was added and the culture was continued for 4 days. The CPE on the cells was observed under a microscope (Nikon, Tokyo, Japan) [
23]. The cellular viability was analyzed by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl) assay.
4.3. Quantitative Real-Time PCR (RT-qPCR)
BHK-21 cells were first cultured in a 12-well plate (105 cells/well). The cells were then infected with DENV2 (101TCID50) for 1 h. Cellular RNA was extracted and purified using a viral RNA extraction kit (Qiagen, Dusseldorf, Germany). Then, cDNA was synthesized and amplified with a SuperScript-III kit (Takara, DaLian, China). The primer sequences are as follows. GAPDH primers were used as a reference.
DENV2 E protein Forward: 5′-GAGGGGAGCGAAGAGAATGG-3′
DENV2 E protein Reverse: 5′-GCCCCATAGATTGCTCCGAA-3′
GAPDH Forward: 5′-TGACCTCAACTACATGGTCTACA-3′
GAPDH Reverse: 5′-CTTCCCATTCTCGGCCTTG-3′
ISG56 Forward: 5′-ACACCTGAAAGGCCAGAATGAGGA-3′
ISG56 Reverse: 5′-TGCCAGTCTGCCCATGTGGTAATA-3′
DENV2 NS1 Forward: 5′-GGCATTTGTGGAATCCGCTC-3′
DENV2 NS1 Reverse: 5′-AGAGCATTTTCGCTTTGCCC-3′
IFN-β Forward: 5′-TTGTTGAGAACCTCCTGGCT-3′
IFN-β Reverse: 5′-CAGGTAATGCAGAATCCTCCCA-3′
IL-1β Forward: 5′-GTGTGGATCCCAAACAATACCC-3′
IL-1β Reverse: 5′-AAGACAGGTCTGTGCTCTGC-3′
IFITM1 Forward: 5′-GTTACTGGTATTCGGCTCTG-3′
IFITM1 Reverse: 5′-GGTGTGTGGGTATAAACTGC-3′
ISG15 Forward: 5′-GGTGTCCGTGACTAACTCCAT-3′
ISG15 Reverse: 5′-TGGAAAGGGTAAGACCGTCCT-3′
ISG54 Forward: 5′-GACACGGTTAAAGTGTGGAG-3′
ISG54 Reverse: 5′-GGTACTGGTTGTCAGGATTC-3′
CXCL10 Forward: 5′-CCAAGTGCTCCGTTTTTC-3′
CXCL10 Reverse: 5′-GGCTCGCAGGGATTTCAA-3′
IFITM3 Forward: 5′-ATGTCTCTGCCGTC-3′
IFITM3 Reverse: 5′-GTCATGAGGATGCCCAGAAT-3′
4.4. Western Blotting
E antibody was purchased from Neo bioscience Company (GTX127277, Beijing, China), and J2 (English & Scientific Consulting Kft, Budapest, Hungary), IFR3(11904S), p-IRF3 (29047S), β-actin (4970S), p-STAT1 (9167S), STAT1 (14994S), p-STAT2 (88410S), and STAT2 (72604S) were from Cell Signaling Technology (Boston, MA, USA). Briefly, cell lysate was separated by SDS-PAGE and transferred into the PVDF membrane (Amersham Biosciences, Buckinghamshire, UK). After incubation with corresponding primary antibodies overnight at 4 °C, the membrane was incubated with PBST with 3% BSA diluted secondary antibody (goat anti-mouse or goat-anti-rabbit IgG-HRP (1:5000)) for 0.5 h at room temperature. Finally, the membranes were visualized using the Dura detection system (Thermo Fisher Scientific, Waltham, MA, USA).
4.5. Determination of Viral Dosage (Plaque Focus Forming Assay, FFA)
BHK-21 (1 × 10
5 cells/well) was incubated in a 96-well plate overnight. The virus was diluted, and the cells were infected for 1 h. The cells were fixed with 1.2% CMC and incubated for 3–4 days. After observing the lesion under a microscope, cells were fixed by adding 1% polyoxymethylene. The cells were perforated with PBS with 0.1% Triton X–100 (PBST). It was then blocked with PBST with 3% BSA for 1 h. The primary antibody (Millipore, MAB10216 (1:5000)) was incubated for 1 h at 37 °C, followed by incubation with PBST 3% BSA diluted secondary antibody (goat anti-mouse IgG-HRP (1:5000)) for 1 h at 37 °C. After washing with PBST 3 times, the cells were incubated with TMB on a shaker for 20 min, and the plate was washed and observed under a microscope to calculate the number of spots [
8].
4.6. Confocal Microscopy
BHK-21 cells were plated at 5 × 104 cell/mL on confocal dishes and were treated with different concentrations of CP640186 for 48 h. Then, the cells were fixed with 4% (w/v paraformaldehyde and were treated with 0.1% PBS (v/v) of Triton X-100 and were flow stained with primary antibody (2 μg/mL) at 4 °C overnight. Then, these cells were washed 3 times with PBS, fluorescein isothiocyanate or tetramethylrhodamine isothiocyanate-conjugated secondary antibody (Sigma, St. Louis, MO, USA) was added and incubated at 25 °C for 2 h. Then the cell nucleus were stained with HOCHEST33285 for 20 min. After washing 3 times with PBS, the image was obtained by a confocal microscope (Zeiss, Jena, Germany).
4.7. Oil Red Stain
An amount of 0.5 g of ORO (s-O0625, purchased from Sigma–Aldrich Company, Darmstadt, Germany) was added to 120 mL of 99% (
v/
v) isopropanol, and then 80 mL of H
2O was added. The solution was then filtered through a 0.45 μM filter to remove the precipitates after stirring. The cells were washed twice with PBS and fixed with 4% paraformaldehyde (DF0135, purchased from LEAGEN, Shanghai, China) for 20 min. Then, the paraformaldehyde was washed away with PBS. The ethanol was then sucked out, and oil red dye solution was directly added, enough to cover the orifice, at room temperature for 30 min. The cells were washed with PBS 3 times after the red dye solution evaporated. Then, the cells were incubated with Hoechst (40729ES10) from Yeasen Biotechnology Co., Ltd., (Shanghai, China) for 20 min, and the nuclei were stained (PBS containing 3% BSA). The staining was observed under the confocal microscope as described in the previous report [
8] (Zeiss, Jena, Germany).
4.8. Drug- and Time-Addition Assay
BHK-21 cells were incubated in 12-well plates (10
5 cells/mL) overnight. As a drug-addition assay, the cells were infected with DENV2 (10
1TCID
50) for 1 h. As in our previous report [
27,
28,
29,
30], dengue virus was incubated with CP640186 in different models, and the viral protein was analyzed. As a time-addition assay, 10 µM of CP640186 was added to the infected cells at 0, 4, 8, and 12 h post infection (hpi). After 24 hpi, cellular RNA or protein was analyzed.
4.9. ELISA
The levels of IFN-β and IFN-γ in mouse serum were evaluated by IFN-β (EMC016, NEOBIOSCIENCE, Beijing, China) and IFN-γ (EK0375, BOSTER, Pleasanton, CA, USA) ELISA kit. Standards and samples were measured in triplicate. The optical density of the final color-developed sample was measured at 450 nm using the Infinite M1000 Pro (Tecan, Männedorf, Switzerland). The parameters of the “one site–total binding” equation were fitted to the 8-point standard curve to obtain the cytokine concentration using GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA).
4.10. Animal Experiments
This study was approved by the Animal Care and Use Committee of Southern Medical University (2019054) and conducted according to the care, use, and protocol of experimental animals. CP640186 was diluted in PBS at the appropriate concentration. To investigate the effects of CP640186 on the DENV2 infection, IFNAR−/−C57Bl/6 (B6) mice, gifts from Prof Jincun Zhao, were infected with DENV2 (106 plaque-forming units) by intraperitoneal injection. The mice were divided into four groups of six. Mice in each group were orally administered 2, 5, or 10 mg/kg CP640186 or PBS once daily for six days. The mice were sacrificed at the end of the experiment and tissues were obtained for further analysis.
4.11. Statistical Analysis
Differences between treatments and control groups were evaluated using the Prism 5 software (GraphPad Software, San Diego, CA, USA) In all cases, parametric or nonparametric tests and the appropriate post hoc test were applied. If data conformed to the normality and equivariance (parametric) of the analysis of variance (ANOVA) hypothesis, a one-way ANOVA was performed followed by a Holm–Sidak multiple comparison post hoc test. Instead, multiple comparisons were performed by a Kruskal–Wallis one-way ANOVA on ranks followed by Dunnett multiple comparisons post hoc test for the data that did not meet ANOVA assumptions (nonparametric). In addition, the Student’s t-test was conducted for some cases. All the data are expressed as the mean ± standard deviation (SD), and p < 0.05 was regarded statistically significant. Values are presented as follows: ns p > 0.05, * p < 0.05, ** p < 0.01, and *** p < 0.001.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





