
Tandem Pd-Catalyzed Cyclization/Coupling of Non-Terminal Acetylenic Activated Methylenes with (Hetero)Aryl Bromides †


Abstract
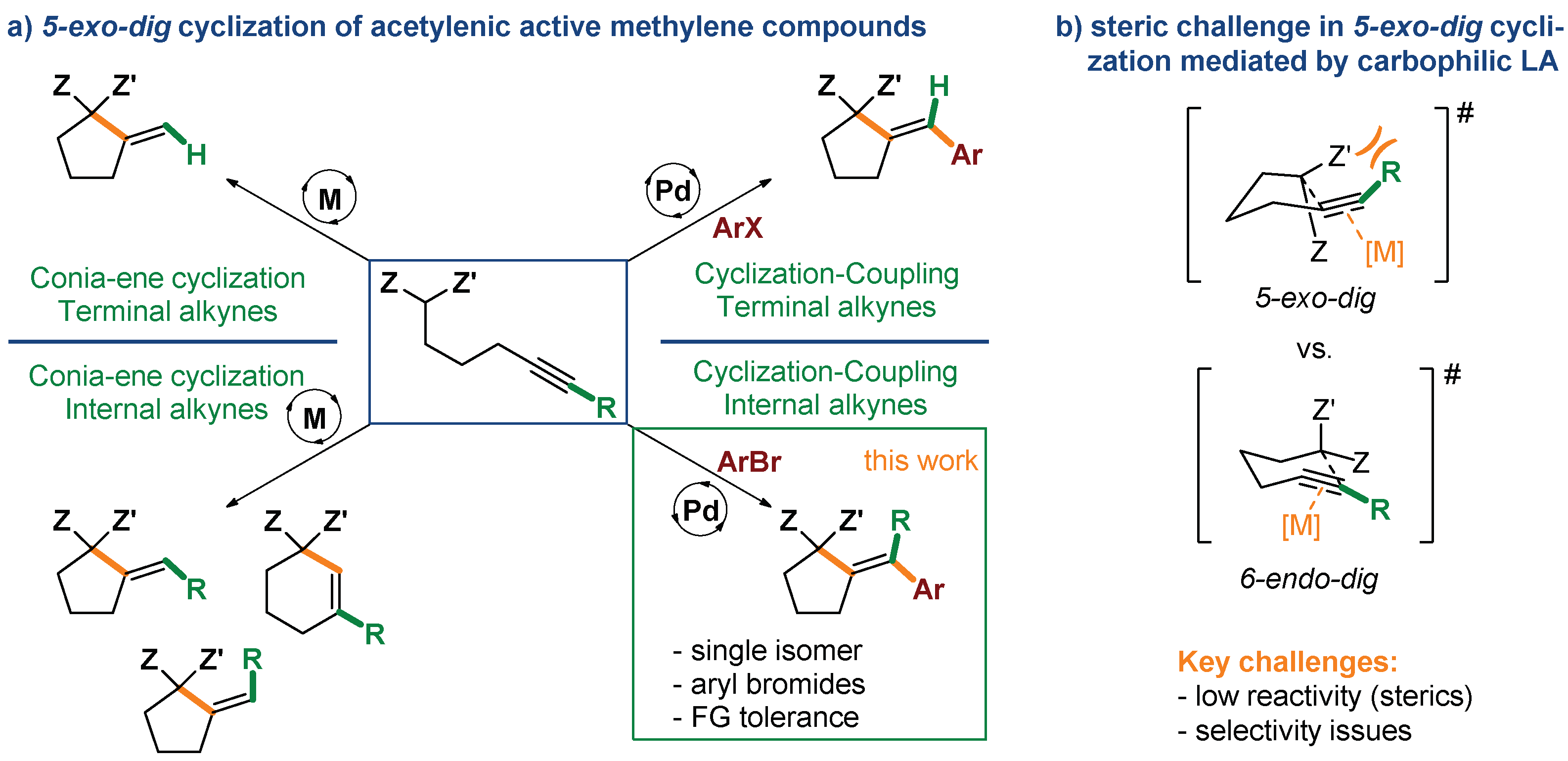
:1. Introduction
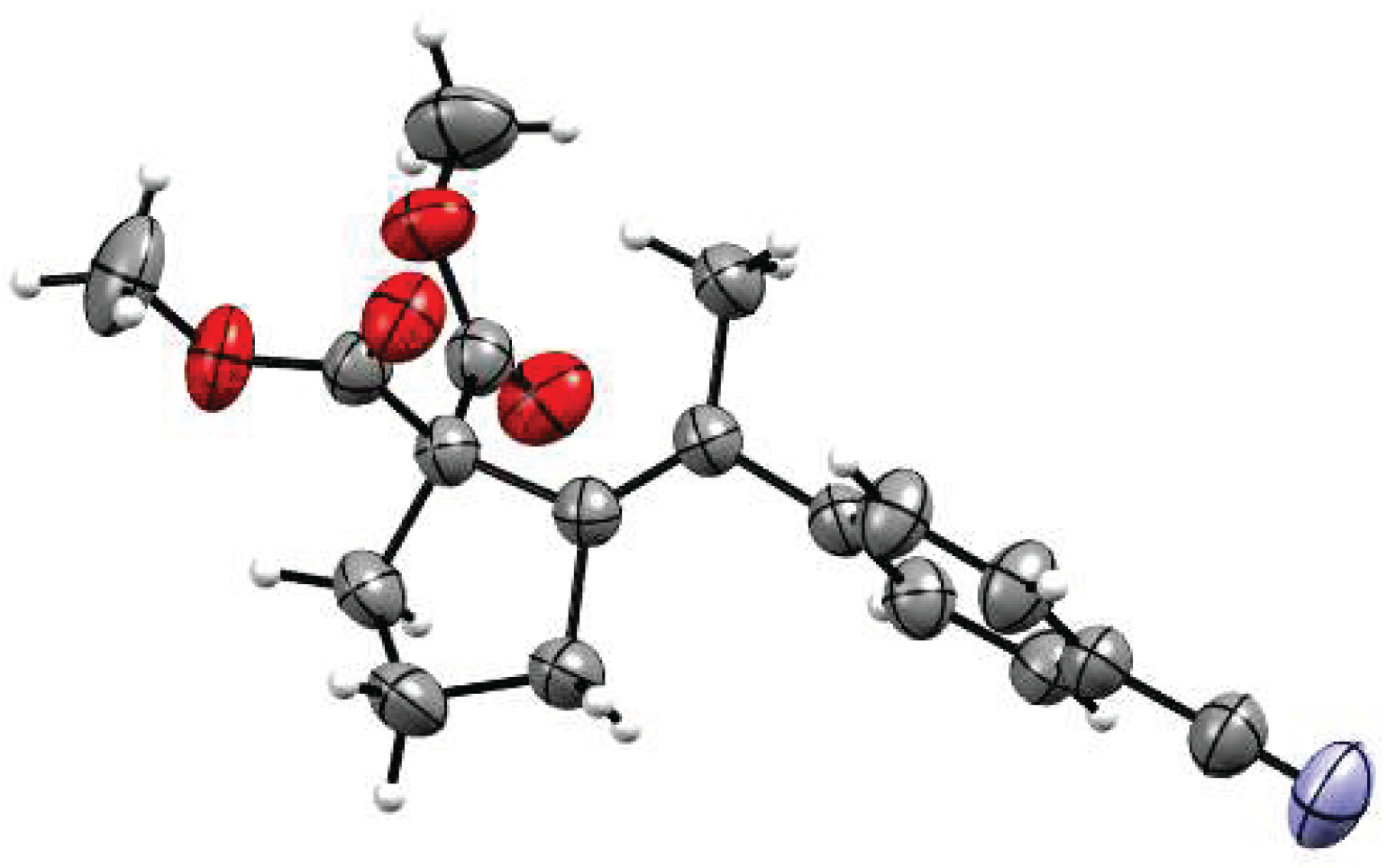
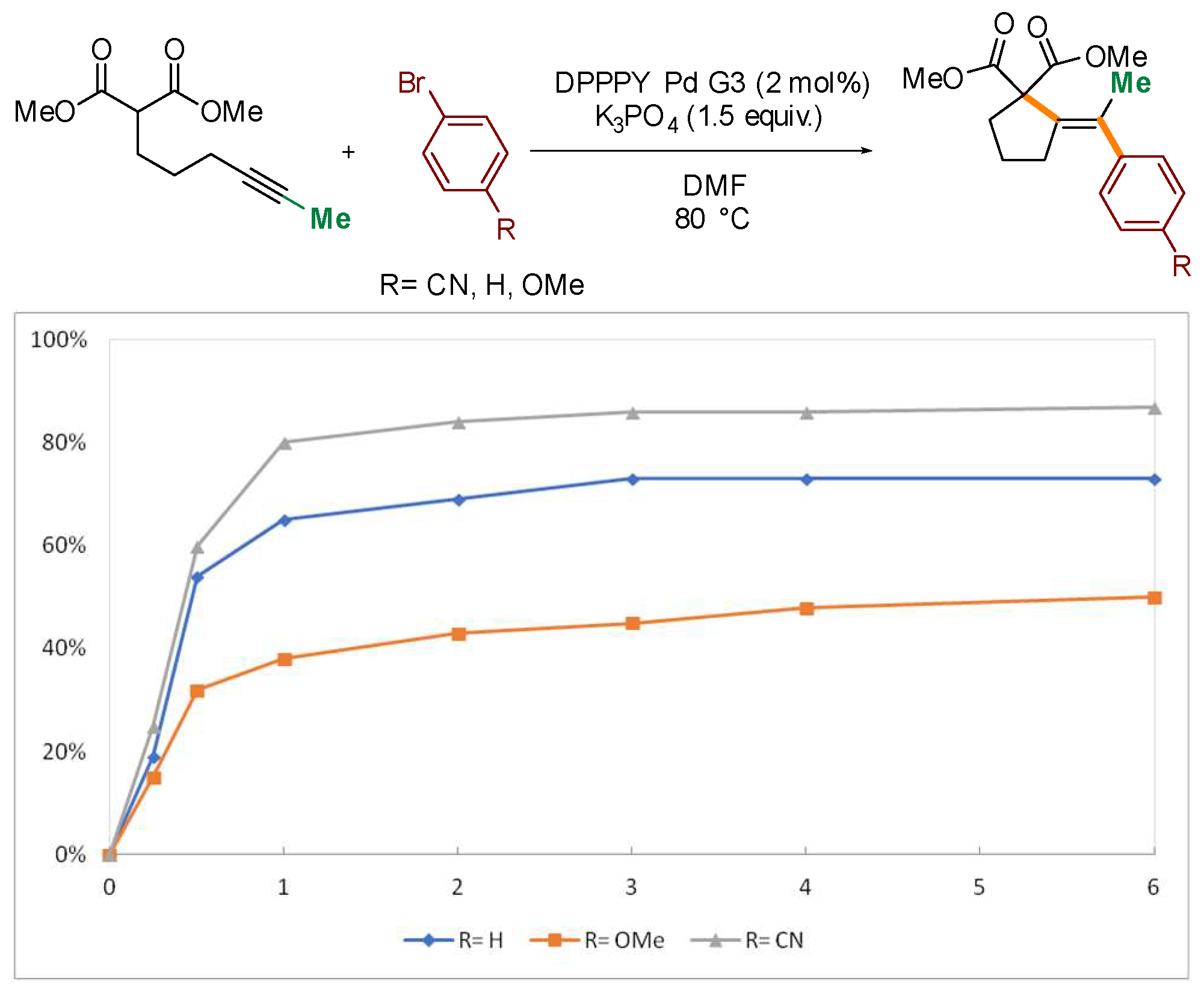
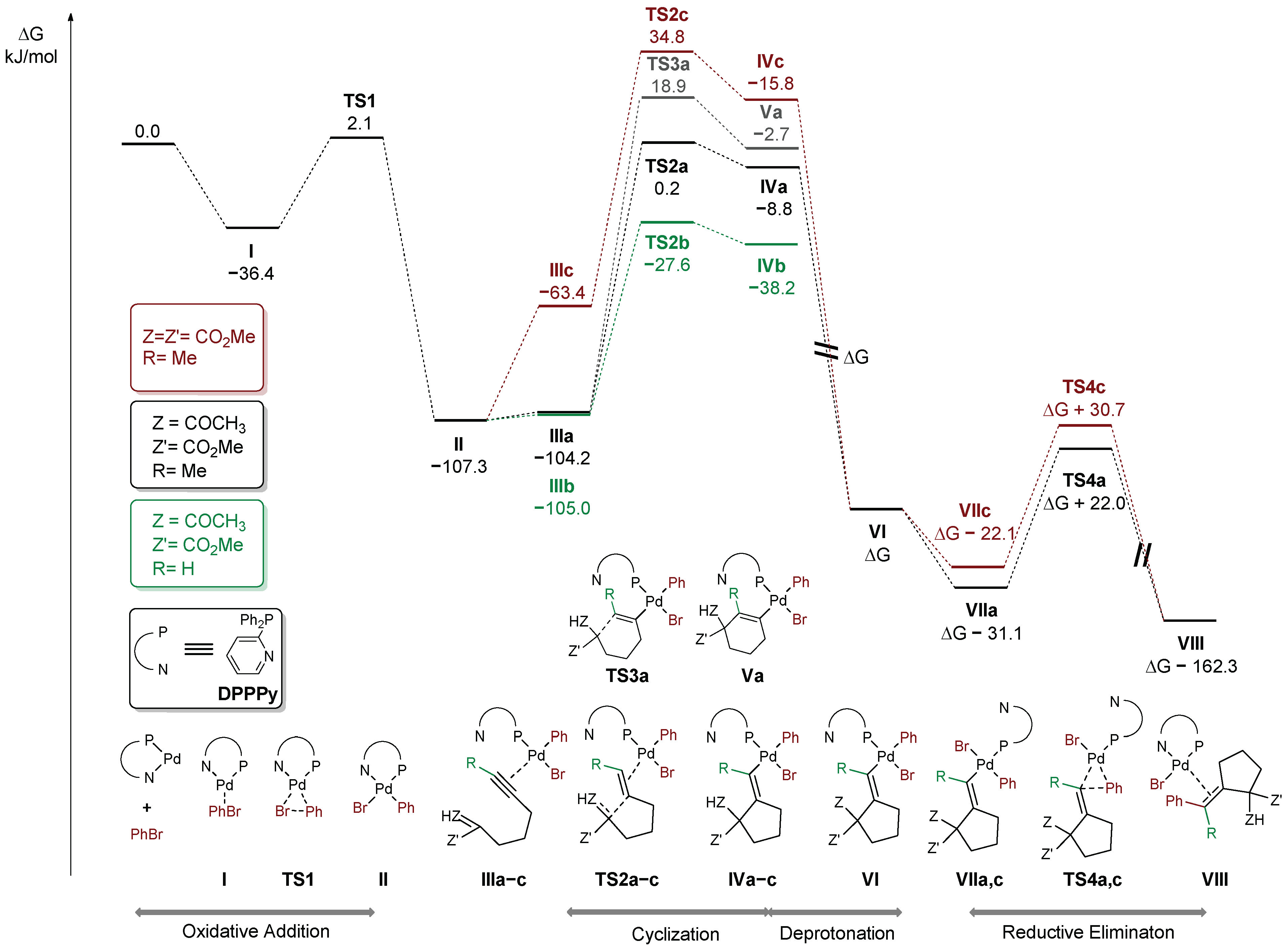
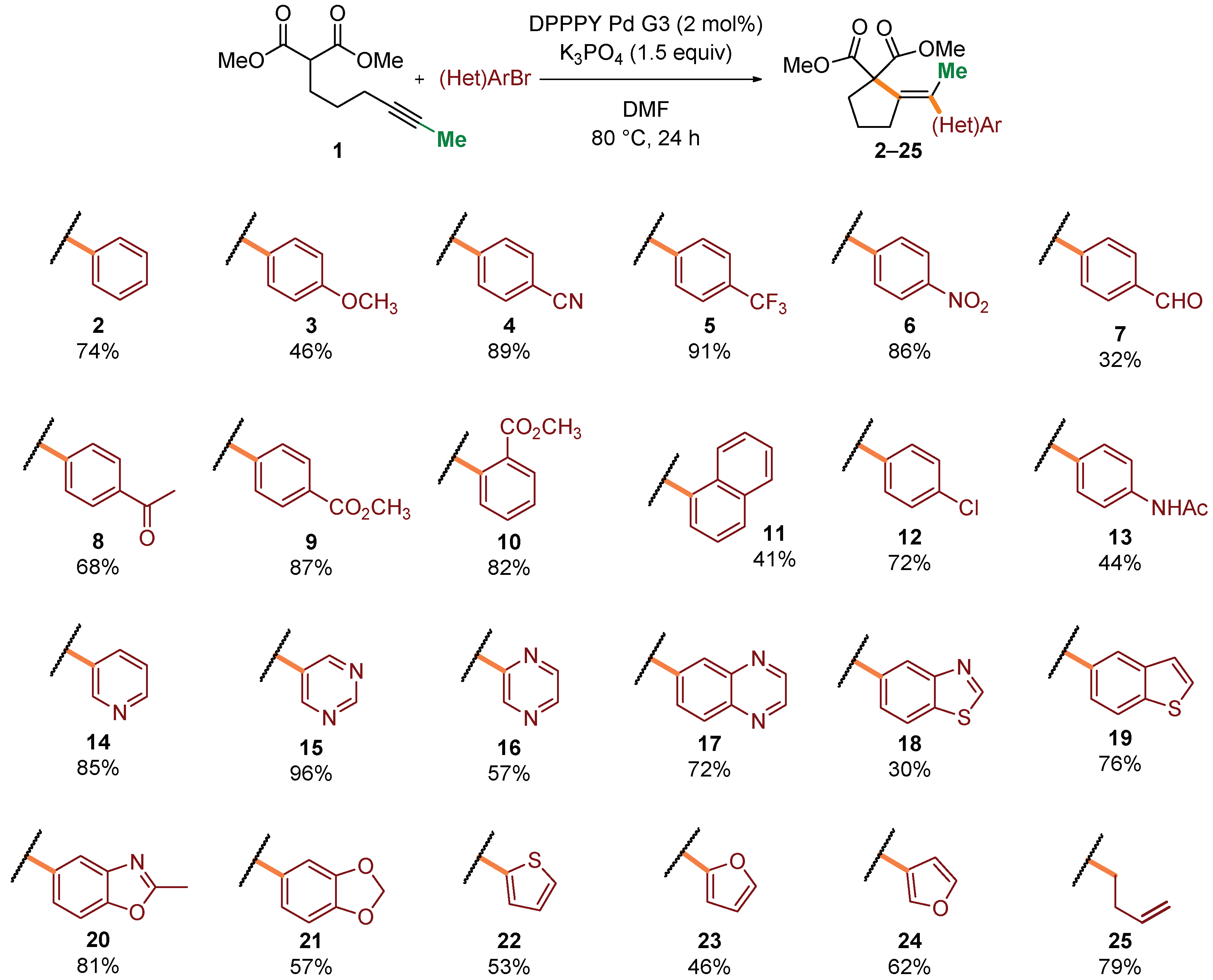
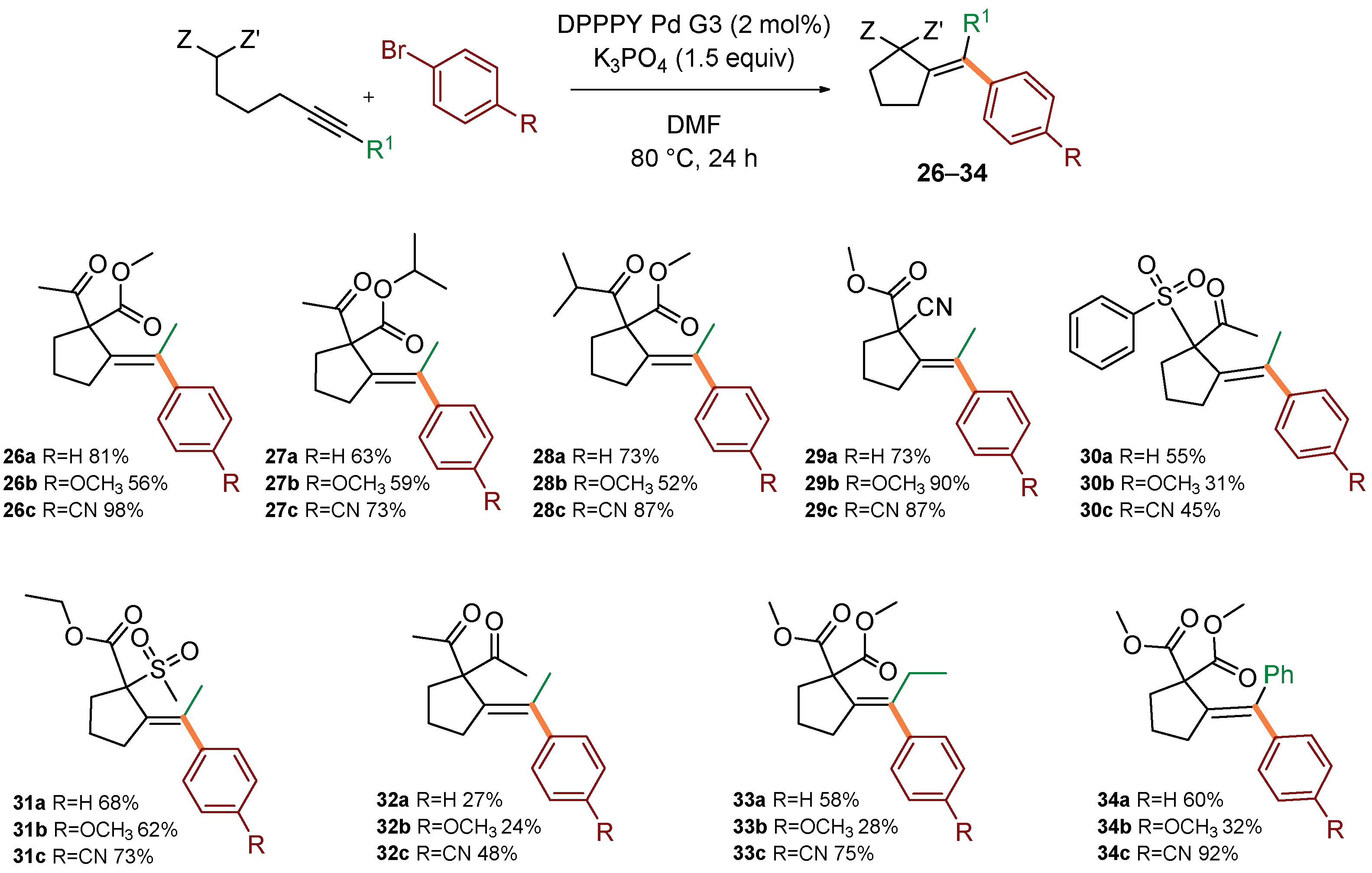
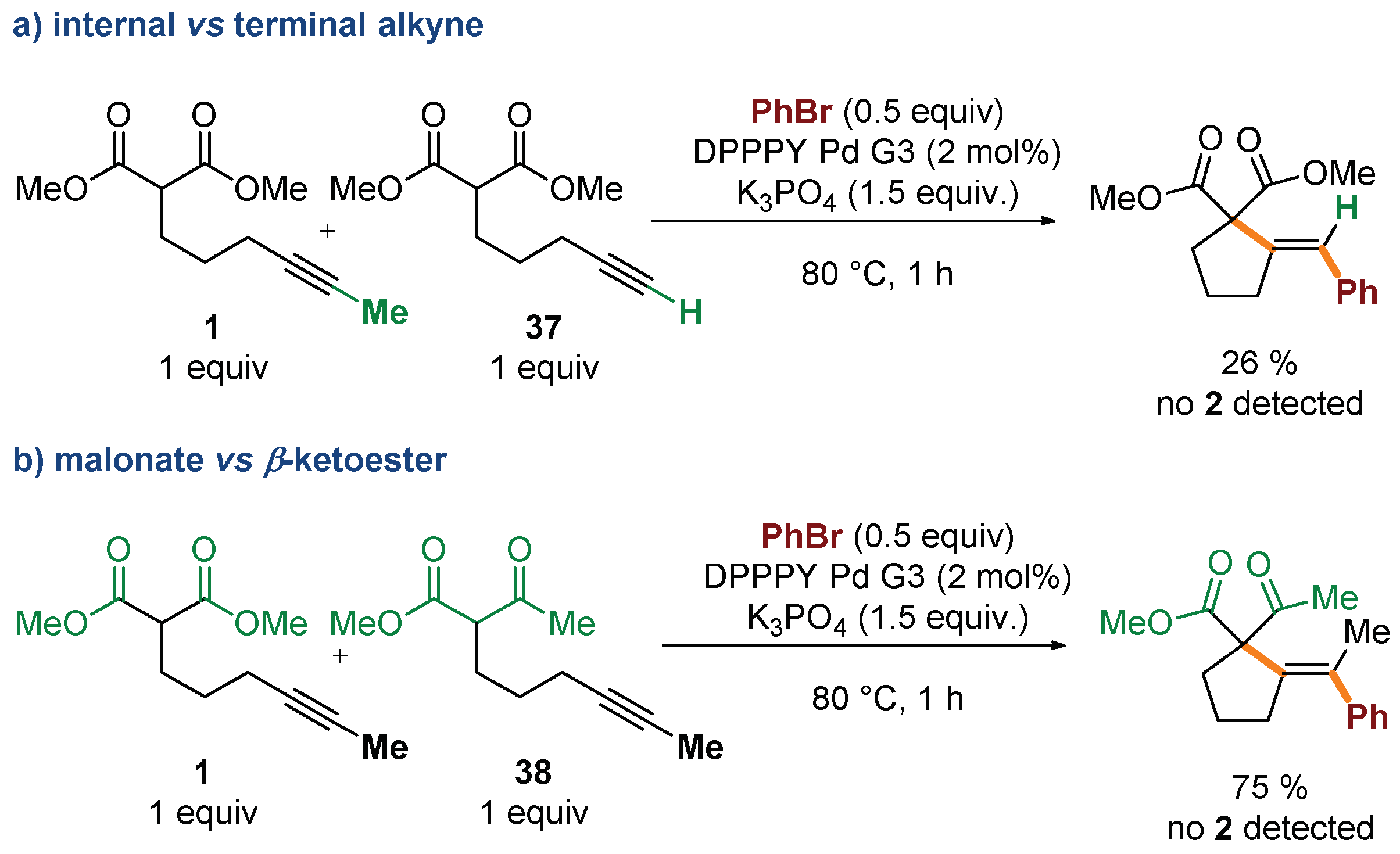
2. Results and Discussion
3. Conclusions
4. Experimental Section
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balme, G.; Bossharth, E.; Monteiro, N. Pd-Assisted Multicomponent Synthesis of Heterocycles. Eur. J. Org. Chem. 2003, 2003, 4101–4111. [Google Scholar] [CrossRef]
- Balme, G.; Bouyssi, D.; Lomberget, T.; Monteiro, N. Cyclisations Involving Attack of Carbo- and Heteronucleophiles on Carbon-Carbon π-Bonds Activated by Organopalladium Complexes. Synthesis 2003, 2003, 2115–2134. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-Metal-Catalyzed Addition of Heteroatom−Hydrogen Bonds to Alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef]
- Balme, G.; Bouyssi, D.; Monteiro, N. Palladium-Mediated Cascade or Multicomponent Reactions: A New Route to Carbo- and Heterocyclic Compounds. Pure Appl. Chem. 2006, 78, 231–239. [Google Scholar] [CrossRef]
- Zeni, G.; Larock, R.C. Synthesis of Heterocycles via Palladium-Catalyzed Oxidative Addition. Chem. Rev. 2006, 106, 4644–4680. [Google Scholar] [CrossRef] [PubMed]
- Dénès, F.; Pérez-Luna, A.; Chemla, F. Addition of Metal Enolate Derivatives to Unactivated Carbon−Carbon Multiple Bonds. Chem. Rev. 2010, 110, 2366–2447. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. Chemicals from Alkynes with Palladium Catalysts. Chem. Rev. 2014, 114, 1783–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, H. Recent Advances in the Construction of Polycyclic Compounds by Palladium-Catalyzed Atom-Economical Cascade Reactions. Asian J. Org. Chem. 2013, 2, 18–28. [Google Scholar] [CrossRef]
- Chen, L.; Chen, K.; Zhu, S. Transition-Metal-Catalyzed Intramolecular Nucleophilic Addition of Carbonyl Groups to Alkynes. Chem 2018, 4, 1208–1262. [Google Scholar] [CrossRef] [Green Version]
- Albano, G.; Aronica, L.A. From Alkynes to Heterocycles through Metal-Promoted Silylformylation and Silylcarbocyclization Reactions. Catalysts 2020, 10, 1012. [Google Scholar] [CrossRef]
- Rouessac, F.; Conia, J.-M. La cyclisation thermique des cetones εζ-ethyleniques. Tetrahedron Lett. 1965, 6, 3313–3318. [Google Scholar] [CrossRef]
- Conia, J.M.; Perchec, P.L. The Thermal Cyclisation of Unsaturated Carbonyl Compounds. Synthesis 1975, 1975, 1–19. [Google Scholar] [CrossRef]
- Hack, D.; Blümel, M.; Chauhan, P.; Philipps, A.R.; Enders, D. Catalytic Conia-Ene and Related Reactions. Chem. Soc. Rev. 2015, 44, 6059–6093. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A.; Davies, P.W. Catalytic Carbophilic Activation: Catalysis by Platinum and Gold π Acids. Angew. Chem. Int. Ed. 2007, 46, 3410–3449. [Google Scholar] [CrossRef] [PubMed]
- Kennedy-Smith, J.J.; Staben, S.T.; Toste, F.D. Gold(I)-Catalyzed Conia-Ene Reaction of β-Ketoesters with Alkynes. J. Am. Chem. Soc. 2004, 126, 4526–4527. [Google Scholar] [CrossRef]
- Staben, S.T.; Kennedy-Smith, J.J.; Toste, F.D. Gold(I)-Catalyzed 5-Endo-Dig Carbocyclization of Acetylenic Dicarbonyl Compounds. Angew. Chem. Int. Ed. 2004, 43, 5350–5352. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ma, X.; Zheng, Z.; Ma, S. Controllable Cyclization Reactions of 2-(2′,3′-Allenyl)Acetylacetates Catalyzed by Gold and Palladium Affording Substituted Cyclopentene and 4,5-Dihydrofuran Derivatives with Distinct Selectivity. Chem. Eur. J. 2008, 14, 8572–8578. [Google Scholar] [CrossRef] [PubMed]
- Gou, F.-R.; Bi, H.-P.; Guo, L.-N.; Guan, Z.-H.; Liu, X.-Y.; Liang, Y.-M. New Insight into Ni(II)-Catalyzed Cyclization Reactions of Propargylic Compounds with Soft Nucleophiles: Novel Indenes Formation. J. Org. Chem. 2008, 73, 3837–3841. [Google Scholar] [CrossRef] [PubMed]
- Montaignac, B.; Vitale, M.R.; Ratovelomanana-Vidal, V.; Michelet, V. Cooperative Copper(I) and Primary Amine Catalyzed Room-Temperature Carbocyclization of Formyl Alkynes. Eur. J. Org. Chem. 2011, 2011, 3723–3727. [Google Scholar] [CrossRef]
- Pei, T.; Wang, X.; Widenhoefer, R.A. Palladium-Catalyzed Intramolecular Oxidative Alkylation of Unactivated Olefins. J. Am. Chem. Soc. 2003, 125, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Widenhoefer, R.A. Mechanism of the Palladium-Catalyzed Intramolecular Hydroalkylation of 7-Octene-2,4-Dione. J. Am. Chem. Soc. 2003, 125, 2056–2057. [Google Scholar] [CrossRef]
- Reeves, R.D.; Phelps, A.M.; Raimbach, W.A.T.; Schomaker, J.M. Diastereoselective Au-Catalyzed Allene Cycloisomerizations to Highly Substituted Cyclopentenes. Org. Lett. 2017, 19, 3394–3397. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Lu, X. Palladium(II)-Catalyzed Coupling Reactions of Alkynes and Allylic Compounds Initiated by Intramolecular Carbopalladation of Alkynes. Tetrahedron Lett. 2002, 43, 6791–6794. [Google Scholar] [CrossRef]
- Fujino, D.; Yorimitsu, H.; Oshima, K. Palladium-Catalyzed Intramolecular Carboacetoxylation of 4-Pentenyl-Substituted Malonate Esters with Iodobenzene Diacetate. Chem. Asian J. 2010, 5, 1758–1760. [Google Scholar] [CrossRef] [PubMed]
- Fujino, D.; Yorimitsu, H.; Oshima, K. Synthesis of Alkylidenecyclopropanes by Palladium-Catalyzed Reaction of Propargyl-Substituted Malonate Esters with Aryl Halides by Anti-Carbopalladation Pathway. J. Am. Chem. Soc. 2011, 133, 9682–9685. [Google Scholar] [CrossRef]
- Fujino, D.; Yorimitsu, H.; Osuka, A. Synthesis of 1,2-Disubstituted Cyclopentenes by Palladium-Catalyzed Reaction of Homopropargyl-Substituted Dicarbonyl Compounds with Organic Halides via 5-Endo-Dig Cyclization. Org. Lett. 2012, 14, 2914–2917. [Google Scholar] [CrossRef]
- Hess, W.; Burton, J.W. Palladium-Catalysed Cyclisation of N-Alkynyl Aminomalonates. Chem. Eur. J. 2010, 16, 12303–12306. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.-N.; Duan, X.-H.; Bi, H.-P.; Liu, X.-Y.; Liang, Y.-M. Synthesis of Indenes via Palladium-Catalyzed Carboannulation of Diethyl 2-(2-(1-Alkynyl)Phenyl)Malonate and Organic Halides. J. Org. Chem. 2006, 71, 3325–3327. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Ballav, T.; Biswas, K.; Ghosh, S.; Ganesh, V. Exploiting the Versatility of Palladium Catalysis: A Modern Toolbox for Cascade Reactions. Eur. J. Org. Chem. 2021, 2021, 4566–4602. [Google Scholar] [CrossRef]
- Fournet, G.; Balme, G.; Van Hemelryck, B.; Gore, J. Stereospecific Synthesis of Arylidene and Allylidene Cyclopentanes by a Palladium-Catalyzed Cylisation. Tetrahedron Lett. 1990, 31, 5147–5150. [Google Scholar] [CrossRef]
- Fournet, G.; Balme, G.; Gore, J. Synthese de (e)-Arylidene et Allylidene Cyclopentanes Par Une Annelation Catalysee Par Un Complexe de Palladium(0). Tetrahedron 1991, 47, 6293–6304. [Google Scholar] [CrossRef]
- Fournet, G.; Balme, G.; Gore, J. New Palladium Mediated Cyclopentanation of Alkenes Bearing a δ Nucleophilic Substituent. Tetrahedron Lett. 1989, 30, 69–70. [Google Scholar] [CrossRef]
- Chaładaj, W.; Domański, S. Mild and Functional Group Tolerant Method for Tandem Palladium-Catalyzed Carbocyclization–Coupling of ε-Acetylenic β-Ketoesters with Aryl Bromides and Chlorides. Adv. Synth. Catal. 2016, 358, 1820–1825. [Google Scholar] [CrossRef]
- Błocka, A.; Woźnicki, P.; Stankevič, M.; Chaładaj, W. Pd-Catalyzed Intramolecular Addition of Active Methylene Compounds to Alkynes with Subsequent Cross-Coupling with (Hetero)Aryl Halides. RSC Adv. 2019, 9, 40152–40167. [Google Scholar] [CrossRef] [Green Version]
- Kołodziejczyk, A.; Domański, S.; Chaładaj, W. Tandem Palladium-Catalyzed 6-Exo-Dig Oxocyclization Coupling of δ-Acetylenic β-Ketoesters with Aryl Bromides and Chlorides: Route to Substituted Dihydropyrans. J. Org. Chem. 2018, 83, 12887–12896. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Makida, Y.; Ochida, A.; Ohmiya, H.; Sawamura, M. Cyclization of Nonterminal Alkynic β-Keto Esters Catalyzed by Gold(I) Complex with a Semihollow, End-Capped Triethynylphosphine Ligand. Org. Lett. 2008, 10, 5051–5054. [Google Scholar] [CrossRef]
- Chaładaj, W.; Kołodziejczyk, A.; Domański, S. Gold(I)-Catalyzed Conia-Ene Cyclization of Internal ϵ-Acetylenic β-Ketoesters under High Pressure. ChemCatChem 2017, 9, 4334–4339. [Google Scholar] [CrossRef]
- Montel, S.; Bouyssi, D.; Balme, G. An Efficient and General Microwave-Assisted Copper-Catalyzed Conia-Ene Reaction of Terminal and Internal Alkynes Tethered to a Wide Variety of Carbonucleophiles. Adv. Synth. Catal. 2010, 352, 2315–2320. [Google Scholar] [CrossRef]
- See Supporting Information for Details.
- Bruno, N.C.; Tudge, M.T.; Buchwald, S.L. Design and Preparation of New Palladium Precatalysts for C–C and C–N Cross-Coupling Reactions. Chem. Sci. 2013, 4, 916–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordwell PKa Table. Available online: https://organicchemistrydata.org/hansreich/resources/pka/ (accessed on 9 November 2021).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Solvent, Base | Temp | Yield 2 |
|---|---|---|---|---|
| 1 | XPhos Pd G3 | DMF, K3PO4 | 60 °C | 31% |
| 2 | XPhos Pd G3 | DMF, K3PO4 | 80 °C | 41% |
| 3 | RuPhos Pd G3 | DMF, K3PO4 | 60 °C | 0 % |
| 4 | BINAP Pd G3 | DMF, K3PO4 | 60 °C | 38% |
| 5 | DPPE Pd G3 | DMF, K3PO4 | 60 °C | 16% |
| 6 | DPPPY Pd G3 | DMF, K3PO4 | 60 °C | 77% |
| 7 | DPPPY Pd G3 | DMF, K3PO4 | 80 °C | 89% |
| 8 | DPPPY Pd G3 | DMF, MeOK | 80 °C | 0% |
| 9 | DPPPY Pd G3 | DMF, KHMDS | 80 °C | 28% |
| 10 | DPPPY Pd G3 | DMF, NaOH | 80 °C | 40% |
| 11 | DPPPY Pd G3 | THF, K3PO4 | 80 °C | 20% |
| 11 | DPPPY Pd G3 | Toluene, K3PO4 | 80 °C | 0% |

| Entry | R | ΔG‡ (cycl.) | ΔG‡ (RE) |
|---|---|---|---|
| 1 | H | 107.5 kJ/mol | 53.1 kJ/mol |
| 2 | CN | 101.3 kJ/mol | 38.8 kJ/mol |
| 3 | OMe | 107.9 kJ/mol | 50.6 kJ/mol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Błocka, A.; Chaładaj, W. Tandem Pd-Catalyzed Cyclization/Coupling of Non-Terminal Acetylenic Activated Methylenes with (Hetero)Aryl Bromides. Molecules 2022, 27, 630. https://doi.org/10.3390/molecules27030630
Błocka A, Chaładaj W. Tandem Pd-Catalyzed Cyclization/Coupling of Non-Terminal Acetylenic Activated Methylenes with (Hetero)Aryl Bromides. Molecules. 2022; 27(3):630. https://doi.org/10.3390/molecules27030630
Chicago/Turabian StyleBłocka, Aleksandra, and Wojciech Chaładaj. 2022. "Tandem Pd-Catalyzed Cyclization/Coupling of Non-Terminal Acetylenic Activated Methylenes with (Hetero)Aryl Bromides" Molecules 27, no. 3: 630. https://doi.org/10.3390/molecules27030630
APA StyleBłocka, A., & Chaładaj, W. (2022). Tandem Pd-Catalyzed Cyclization/Coupling of Non-Terminal Acetylenic Activated Methylenes with (Hetero)Aryl Bromides. Molecules, 27(3), 630. https://doi.org/10.3390/molecules27030630