
Melatonin: Regulation of Prion Protein Phase Separation in Cancer Multidrug Resistance



Abstract
:1. Introduction
2. Liquid–Liquid Phase Separation May Regulate Prion Conversion and Propagation
2.1. Melatonin May Modulate Stress-Induced Prion Conversion
2.2. The Intrinsically Disordered Region in Prions Is Requisite for Liquid–Liquid Phase Separation, Cytoplasmic Inheritance, and Modulation of Pathological Conversion
2.2.1. The Role of ATP and RNA in Prion Phase Separation
2.2.2. RNA- and Copper-Binding Modulate the Conversion of PrPC to PrPSc
2.3. The Role of Melatonin in the Regulation of Liquid–Liquid Phase Separation and ROS-Induced Cleavage in Prions
2.3.1. The Role of Melatonin in PrPC LLPS and Amyloid Beta Binding
2.3.2. Is the N-1 Fragment from the Intrinsically Disordered N-1 Domain Necessary and Sufficient for LLPS?
2.3.3. Changing pH and/or Crossing Isoelectric Points Can Drive Phase Separation of Prion N2 Fragments
2.3.4. Copper Chelation by Melatonin in Prion Phase Separation May Ameliorate Prion-Induced Multidrug Resistance
3. Melatonin May Promote PrP Physiological Functions and Inhibit Pathological Effects via Global Modulation of the Tumor Microenvironment to Enhance Cancer Drug Efficacy
3.1. Melatonin May Attenuate Prion Propagation and Cancer Multidrug Resistance by Increasing Extracellular pH
3.2. PrPC Protective Physiological Responses and Ligand-Binding May Become Pathological Liabilities in the Tumor Microenvironment
3.3. Interactions between PrPC, Iron, and Heme May Enhance Aggressive Drug Resistance in Tumors
3.3.1. Iron and Heme Facilitate Increased Energy Production in Cancer Cells
3.3.2. PrPC Regulates Heme Synthesis and Export to Modulate Glucose and Antioxidant Homeostasis in Cancer
3.3.3. Upregulation of Hemoglobin Synthesis by Hemin-Bound PrPC May Increase Cancer Multidrug Resistance
3.4. Melatonin Maintains Hemoglobin Redox Balance by Protecting CYB5R3 and Band 3 Protein in an Antioxidant-Independent Manner
3.5. Melatonin Increases O2 Saturation to Reduce TME Hypoxic Stress by Protecting Band 3 Protein
3.5.1. Hypoxia in TME Is Modulated by Fluctuations in Red Blood Cell Flux
3.5.2. Hypoxia Prolongs Deoxygenation and Elevates Hemin Release to Damage RBC Membrane Integrity and Band 3 Proteins
3.5.3. Oxygen Saturation and Transport Are Directly Modulated by Heme Redox Balance
3.5.4. The Role of Membrane Lipids and Lipid Rafts in Prion Physiological Function and Pathological Propagation
3.6. Melatonin May Prevent PrPC Pathological Conversion from Phase Separation Caused by Mutations
4. The Effects of Melatonin on Lipid Phase Transition, Lipid Composition, and Prion Propagation in Cancer Multidrug Resistance
4.1. Melatonin Maintains Lipid Raft Integrity and Prion Physiological Functions by Modulating Cholesterol and Lipid Phase Transitions
4.2. Melatonin May Preserve Band 3 Interactions with Membrane Lipids in Antioxidant-Dependent and -Independent Manners
4.3. The Pleiotropic Effects of Melatonin in the Regulation of Prions in Cancer Multidrug Resistance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3OHM | 3-hydroxymelatonin |
| Aβ | β-amyloid peptide |
| Aβo | amyloid-β oligomers |
| Akt | protein kinase B |
| ATP | adenosine triphosphate |
| COX | cytochrome c oxidase |
| CYB5R3 | NADH-cytochrome b5 reductase 3 |
| DNA | deoxyribonucleic acid |
| ER | endoplasmic reticulum |
| ES | embryonic stem |
| G6P | glucose 6-phosphate |
| G6PD | glucose-6-phosphate-dehydrogenase |
| Ga | giga annum (billion years) |
| GLUT1 | glucose transporter 1 |
| GOE | great oxidation event |
| H+ | hydrogen proton |
| H2O2 | hydrogen peroxide |
| IDR | intrinsically disordered region |
| Ld | liquid disordered |
| Lo | liquid ordered |
| LLPS | liquid–liquid phase separation |
| mM | millimolar |
| μM | micromolar |
| MD | molecular dynamics |
| MetHb | methemoglobin |
| MLO | membraneless organelle |
| MSC | mesenchymal stem cell |
| NAD+ | nicotinamide adenine dinucleotide |
| NADH | nicotinamide adenine dinucleotide hydrogen |
| NLRP3 | NLR pyrin domain containing 3 (inflammasome) |
| nM | nanomolar |
| •OH | hydroxyl radical |
| •OOH | hydroperoxyl radical |
| OXPHOS | oxidative phosphorylation |
| pHe | extracellular pH |
| pHi | intracellular pH |
| PI3K | phosphoinositide 3-kinase |
| POPE | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine |
| PTM | post-translational modification |
| RBC | red blood cell |
| RCF | red cell flux |
| Redox | oxidation-reduction |
| RNA | ribonucleic acid |
| RNP | ribonucleoprotein |
| ROS | reactive oxygen species |
| UPS | ubiquitin-protease system |
| UVR | ultraviolet radiation |
| VDA | vascular disrupting agent |
| WT | wild-type |
References
- Zabel, M.D.; Reid, C. A Brief History of Prions. Pathog. Dis. 2015, 73, ftv087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Novel Proteinaceous Infectious Particles Cause Scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Riek, R.; Hornemann, S.; Wider, G.; Glockshuber, R.; Wüthrich, K. NMR Characterization of the Full-Length Recombinant Murine Prion Protein, mPrP(23-231). FEBS Lett. 1997, 413, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Castle, A.R.; Gill, A.C. Physiological Functions of the Cellular Prion Protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Peralta, O.A.; Huckle, W.R.; Eyestone, W.H. Expression and Knockdown of Cellular Prion Protein (PrPC) in Differentiating Mouse Embryonic Stem Cells. Differentiation 2011, 81, 68–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, A.; Ramos-Ibeas, P.; Pericuesta, E.; Ramirez, M.A.; Gutierrez-Adan, A. The Role of Prion Protein in Stem Cell Regulation. Reproduction 2013, 146, R91–R99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Baskakov, I.V. The Cellular Form of the Prion Protein Guides the Differentiation of Human Embryonic Stem Cells into Neuron-, Oligodendrocyte-, and Astrocyte-Committed Lineages. Prion 2014, 8, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, D.M.; Campbell, E.A.; Jakobson, C.M.; Tsuchiya, M.; Shaw, E.A.; DiNardo, A.L.; Kaeberlein, M.; Jarosz, D.F. A Prion Accelerates Proliferation at the Expense of Lifespan. Elife 2021, 10, e60917. [Google Scholar] [CrossRef] [PubMed]
- Málaga-Trillo, E.; Solis, G.P.; Schrock, Y.; Geiss, C.; Luncz, L.; Thomanetz, V.; Stuermer, C.A.O. Regulation of Embryonic Cell Adhesion by the Prion Protein. PLoS Biol. 2009, 7, e55. [Google Scholar] [CrossRef]
- Bremer, J.; Baumann, F.; Tiberi, C.; Wessig, C.; Fischer, H.; Schwarz, P.; Steele, A.D.; Toyka, K.V.; Nave, K.-A.; Weis, J.; et al. Axonal Prion Protein Is Required for Peripheral Myelin Maintenance. Nat. Neurosci. 2010, 13, 310–318. [Google Scholar] [CrossRef]
- Tobler, I.; Gaus, S.E.; Deboer, T.; Achermann, P.; Fischer, M.; Rülicke, T.; Moser, M.; Oesch, B.; McBride, P.A.; Manson, J.C. Altered Circadian Activity Rhythms and Sleep in Mice Devoid of Prion Protein. Nature 1996, 380, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Cagampang, F.R.; Whatley, S.A.; Mitchell, A.L.; Powell, J.F.; Campbell, I.C.; Coen, C.W. Circadian Regulation of Prion Protein Messenger RNA in the Rat Forebrain: A Widespread and Synchronous Rhythm. Neuroscience 1999, 91, 1201–1204. [Google Scholar] [CrossRef]
- Liebert, A.; Bicknell, B.; Adams, R. Prion Protein Signaling in the Nervous System—A Review and Perspective. Sign. Transduct. Insights 2014, 3, STI.S12319. [Google Scholar] [CrossRef] [Green Version]
- Ashok, A.; Singh, N. Prion Protein Modulates Glucose Homeostasis by Altering Intracellular Iron. Sci. Rep. 2018, 8, 6556. [Google Scholar] [CrossRef] [PubMed]
- Strom, A.; Wang, G.-S.; Scott, F.W. Impaired Glucose Tolerance in Mice Lacking Cellular Prion Protein. Pancreas 2011, 40, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.D.; Jackson, G.S.; Altmann, D.M. The Role of the Cellular Prion Protein in the Immune System. Clin. Exp. Immunol. 2006, 146, 1–8. [Google Scholar] [CrossRef]
- Aucouturier, P.; Carp, R.I.; Carnaud, C.; Wisniewski, T. Prion Diseases and the Immune System. Clin. Immunol. 2000, 96, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Rachidi, W.; Vilette, D.; Guiraud, P.; Arlotto, M.; Riondel, J.; Laude, H.; Lehmann, S.; Favier, A. Expression of Prion Protein Increases Cellular Copper Binding and Antioxidant Enzyme Activities but Not Copper Delivery. J. Biol. Chem. 2003, 278, 9064–9072. [Google Scholar] [CrossRef] [Green Version]
- Alfaidy, N.; Chauvet, S.; Donadio-Andrei, S.; Salomon, A.; Saoudi, Y.; Richaud, P.; Aude-Garcia, C.; Hoffmann, P.; Andrieux, A.; Moulis, J.-M.; et al. Prion Protein Expression and Functional Importance in Developmental Angiogenesis: Role in Oxidative Stress and Copper Homeostasis. Antioxid. Redox Signal. 2013, 18, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Haldar, S.; Tripathi, A.; Qian, J.; Beserra, A.; Suda, S.; McElwee, M.; Turner, J.; Hopfer, U.; Singh, N. Prion Protein Promotes Kidney Iron Uptake via Its Ferrireductase Activity. J. Biol. Chem. 2015, 290, 5512–5522. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion Protein Regulates Iron Transport by Functioning as a Ferrireductase. J. Alzheimers Dis. 2013, 35, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kong, Q.; Luo, X.; Petersen, R.B.; Meyerson, H.; Singh, N. Prion Protein (PrP) Knock-out Mice Show Altered Iron Metabolism: A Functional Role for PrP in Iron Uptake and Transport. PLoS ONE 2009, 4, e6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, K.; Kandel, E.R. The Role of Functional Prion-Like Proteins in the Persistence of Memory. Cold Spring Harb. Perspect. Biol. 2016, 8, a021774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, H. Prion Proteins May Store Memories. Nature 2003. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PRNP Prion Protein [Homo Sapiens (Human)]-Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/5621 (accessed on 12 December 2021).
- Faris, R.; Moore, R.A.; Ward, A.; Race, B.; Dorward, D.W.; Hollister, J.R.; Fischer, E.R.; Priola, S.A. Cellular Prion Protein Is Present in Mitochondria of Healthy Mice. Sci. Rep. 2017, 7, 41556. [Google Scholar] [CrossRef] [PubMed]
- Masison, D.C.; Wickner, R.B. Prion-Inducing Domain of Yeast Ure2p and Protease Resistance of Ure2p in Prion-Containing Cells. Science 1995, 270, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickner, R.B. [URE3] as an Altered URE2 Protein: Evidence for a Prion Analog in Saccharomyces Cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Patino, M.M.; Liu, J.J.; Glover, J.R.; Lindquist, S. Support for the Prion Hypothesis for Inheritance of a Phenotypic Trait in Yeast. Science 1996, 273, 622–626. [Google Scholar] [CrossRef] [Green Version]
- True, H.L.; Lindquist, S.L. A Yeast Prion Provides a Mechanism for Genetic Variation and Phenotypic Diversity. Nature 2000, 407, 477–483. [Google Scholar] [CrossRef] [PubMed]
- True, H.L.; Berlin, I.; Lindquist, S.L. Epigenetic Regulation of Translation Reveals Hidden Genetic Variation to Produce Complex Traits. Nature 2004, 431, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J.; Lindquist, S. Prions as Adaptive Conduits of Memory and Inheritance. Nat. Rev. Genet. 2005, 6, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Wiltzius, J.J.W.; Landau, M.; Nelson, R.; Sawaya, M.R.; Apostol, M.I.; Goldschmidt, L.; Soriaga, A.B.; Cascio, D.; Rajashankar, K.; Eisenberg, D. Molecular Mechanisms for Protein-Encoded Inheritance. Nat. Struct. Mol. Biol. 2009, 16, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Tuite, M.F.; Serio, T.R. The Prion Hypothesis: From Biological Anomaly to Basic Regulatory Mechanism. Nat. Rev. Mol. Cell Biol. 2010, 11, 823–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halfmann, R.; Jarosz, D.F.; Jones, S.K.; Chang, A.; Lancaster, A.K.; Lindquist, S. Prions Are a Common Mechanism for Phenotypic Inheritance in Wild Yeasts. Nature 2012, 482, 363–368. [Google Scholar] [CrossRef] [Green Version]
- Wickner, R.B.; Edskes, H.K.; Bateman, D.A.; Kelly, A.C.; Gorkovskiy, A.; Dayani, Y.; Zhou, A. Amyloids and Yeast Prion Biology. Biochemistry 2013, 52, 1514–1527. [Google Scholar] [CrossRef]
- Harvey, Z.H.; Chen, Y.; Jarosz, D.F. Protein-Based Inheritance: Epigenetics beyond the Chromosome. Mol. Cell 2018, 69, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Harvey, Z.H.; Chakravarty, A.K.; Futia, R.A.; Jarosz, D.F. A Prion Epigenetic Switch Establishes an Active Chromatin State. Cell 2020, 180, 928–940.e14. [Google Scholar] [CrossRef]
- Manjrekar, J. Epigenetic Inheritance, Prions and Evolution. J. Genet. 2017, 96, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kaufman, S.K.; Holmes, B.B.; Diamond, M.I. Prions and Protein Assemblies That Convey Biological Information in Health and Disease. Neuron 2016, 89, 433–448. [Google Scholar] [CrossRef] [Green Version]
- Newby, G.A.; Lindquist, S. Blessings in Disguise: Biological Benefits of Prion-like Mechanisms. Trends Cell Biol. 2013, 23, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halfmann, R.; Alberti, S.; Lindquist, S. Prions, Protein Homeostasis, and Phenotypic Diversity. Trends Cell Biol. 2010, 20, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.M.; Jarosz, D.F. Rebels with a Cause: Molecular Features and Physiological Consequences of Yeast Prions. FEMS Yeast Res. 2014, 14, 136–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, H.; Wiegmans, A.P.; Wei, M.Q.; Chernoff, Y.O.; Khanna, K.K.; Munn, A.L. Potential Roles for Prions and Protein-Only Inheritance in Cancer. Cancer Metastasis Rev. 2012, 31, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senft, D.; Ronai, Z.E.A. Adaptive Stress Responses during Tumor Metastasis and Dormancy. Trends Cancer Res. 2016, 2, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Easwaran, H.; Tsai, H.-C.; Baylin, S.B. Cancer Epigenetics: Tumor Heterogeneity, Plasticity of Stem-like States, and Drug Resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oamen, H.P.; Lau, Y.; Caudron, F. Prion-like Proteins as Epigenetic Devices of Stress Adaptation. Exp. Cell Res. 2020, 396, 112262. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.F.; Chambers, A.F.; Hill, R.P.; Ling, V. Metastatic Variants Are Generated Spontaneously at a High Rate in Mouse KHT Tumor. Proc. Natl. Acad. Sci. USA 1982, 79, 5547–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Bellot, E.; Guillemet, E.; Cullin, C. The Yeast Prion [URE3] Can Be Greatly Induced by a Functional Mutated URE2 Allele. EMBO J. 2000, 19, 3215–3222. [Google Scholar] [CrossRef] [Green Version]
- Quintana, E.; Shackleton, M.; Foster, H.R.; Fullen, D.R.; Sabel, M.S.; Johnson, T.M.; Morrison, S.J. Phenotypic Heterogeneity among Tumorigenic Melanoma Cells from Patients That Is Reversible and Not Hierarchically Organized. Cancer Cell 2010, 18, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Derkatch, I.L.; Bradley, M.E.; Zhou, P.; Chernoff, Y.O.; Liebman, S.W. Genetic and Environmental Factors Affecting the de Novo Appearance of the [PSI+] Prion in Saccharomyces Cerevisiae. Genetics 1997, 147, 507–519. [Google Scholar] [CrossRef]
- Liang, J.; Pan, Y.L.; Ning, X.X.; Sun, L.J.; Lan, M.; Hong, L.; Du, J.P.; Liu, N.; Liu, C.J.; Qiao, T.D.; et al. Overexpression of PrPC and Its Antiapoptosis Function in Gastric Cancer. Tumour Biol. 2006, 27, 84–91. [Google Scholar] [CrossRef]
- Mehrpour, M.; Codogno, P. Prion Protein: From Physiology to Cancer Biology. Cancer Lett. 2010, 290, 1–23. [Google Scholar] [CrossRef]
- Dalai, W.; Matsuo, E.; Takeyama, N.; Kawano, J.; Saeki, K. Increased Expression of Prion Protein Gene Is Accompanied by Demethylation of CpG Sites in a Mouse Embryonal Carcinoma Cell Line, P19C6. J. Vet. Med. Sci. 2017, 79, 644–648. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-C.; Won, S.-Y.; Jeong, B.-H. Identification of Prion Disease-Related Somatic Mutations in the Prion Protein Gene (PRNP) in Cancer Patients. Cells 2020, 9, 1480. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Pan, Y.; Zhang, D.; Guo, C.; Shi, Y.; Wang, J.; Chen, Y.; Wang, X.; Liu, J.; Guo, X.; et al. Cellular Prion Protein Promotes Proliferation and G1/S Transition of Human Gastric Cancer Cells SGC7901 and AGS. FASEB J. 2007, 21, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Bai, F.; Luo, G.; Wang, J.; Liu, J.; Ge, F.; Pan, Y.; Yao, L.; Du, R.; Li, X.; et al. Hypoxia Induced Overexpression of PrP(C) in Gastric Cancer Cell Lines. Cancer Biol. Ther. 2007, 6, 769–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, Y.H.-Y.; Say, Y.-H. Resistance against Apoptosis by the Cellular Prion Protein Is Dependent on Its Glycosylation Status in Oral HSC-2 and Colon LS 174T Cancer Cells. Cancer Lett. 2011, 306, 111–119. [Google Scholar] [CrossRef]
- Wang, Y.-M.; Jin, B.-Z.; Ai, F.; Duan, C.-H.; Lu, Y.-Z.; Dong, T.-F.; Fu, Q.-L. The Efficacy and Safety of Melatonin in Concurrent Chemotherapy or Radiotherapy for Solid Tumors: A Meta-Analysis of Randomized Controlled Trials. Cancer Chemother. Pharmacol. 2012, 69, 1213–1220. [Google Scholar] [CrossRef]
- Wang, Q.; Qian, J.; Wang, F.; Ma, Z. Cellular Prion Protein Accelerates Colorectal Cancer Metastasis via the Fyn-SP1-SATB1 Axis. Oncol. Rep. 2012, 28, 2029–2034. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Rao, G.; Wang, H.; Li, B.; Tian, W.; Cui, J.; He, L.; Laffin, B.; Tian, X.; Hao, C.; et al. CD44-Positive Cancer Stem Cells Expressing Cellular Prion Protein Contribute to Metastatic Capacity in Colorectal Cancer. Cancer Res. 2013, 73, 2682–2694. [Google Scholar] [CrossRef] [Green Version]
- Martin-Lannerée, S.; Hirsch, T.Z.; Hernandez-Rapp, J.; Halliez, S.; Vilotte, J.-L.; Launay, J.-M.; Mouillet-Richard, S. PrP(C) from Stem Cells to Cancer. Front. Cell Dev. Biol 2014, 2, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsaro, A.; Bajetto, A.; Thellung, S.; Begani, G.; Villa, V.; Nizzari, M.; Pattarozzi, A.; Solari, A.; Gatti, M.; Pagano, A.; et al. Cellular Prion Protein Controls Stem Cell-like Properties of Human Glioblastoma Tumor-Initiating Cells. Oncotarget 2016, 7, 38638–38657. [Google Scholar] [CrossRef] [PubMed]
- Go, G.; Lee, S.H. The Cellular Prion Protein: A Promising Therapeutic Target for Cancer. Int. J. Mol. Sci. 2020, 21, 9208. [Google Scholar] [CrossRef]
- Ke, J.; Wu, G.; Zhang, J.; Li, H.; Gao, S.; Shao, M.; Gao, Z.; Sy, M.-S.; Cao, Y.; Yang, X.; et al. Melanoma Migration Is Promoted by Prion Protein via Akt-hsp27 Signaling Axis. Biochem. Biophys. Res. Commun. 2020, 523, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Bing, T.; Wang, J.; Shen, L.; Liu, X.; Shangguan, D. Prion Protein Targeted by a Prostate Cancer Cell Binding Aptamer, a Potential Tumor Marker? ACS Appl. Bio Mater. 2020, 3, 2658–2665. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Sin, M.J.; Kim, M.J.; Kim, H.J.; Kim, Y.S.; Choi, E.K.; Kim, M.Y. Involvement of Cellular Prion Protein in Invasion and Metastasis of Lung Cancer by Inducing Treg Cell Development. Biomolecules 2021, 11, 285. [Google Scholar] [CrossRef]
- Mouillet-Richard, S.; Ghazi, A.; Laurent-Puig, P. The Cellular Prion Protein and the Hallmarks of Cancer. Cancers 2021, 13, 5032. [Google Scholar] [CrossRef]
- Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Giambelluca, M.A.; Morelli, L.; Frati, A.; Fornai, F. The Role of Cellular Prion Protein in Promoting Stemness and Differentiation in Cancer. Cancers 2021, 13, 170. [Google Scholar] [CrossRef]
- Chernoff, Y.O. Stress and Prions: Lessons from the Yeast Model. FEBS Lett. 2007, 581, 3695–3701. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, G.; Shimazu, N.; Tanaka, M. A Yeast Prion, Mod5, Promotes Acquired Drug Resistance and Cell Survival under Environmental Stress. Science 2012, 336, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Déry, M.-A.; Jodoin, J.; Ursini-Siegel, J.; Aleynikova, O.; Ferrario, C.; Hassan, S.; Basik, M.; LeBlanc, A.C. Endoplasmic Reticulum Stress Induces PRNP Prion Protein Gene Expression in Breast Cancer. Breast Cancer Res. 2013, 15, R22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinton, C.; Antony, H.; Hashimi, S.M.; Munn, A.; Wei, M.Q. Significance of Prion and Prion-like Proteins in Cancer Development, Progression and Multi-Drug Resistance. Curr. Cancer Drug Targets 2013, 13, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Pan, Y.; Shi, Y.; Guo, C.; Jin, X.; Sun, L.; Liu, N.; Qiao, T.; Fan, D. Overexpression and Significance of Prion Protein in Gastric Cancer and Multidrug-Resistant Gastric Carcinoma Cell Line SGC7901/ADR. Int. J. Cancer 2005, 113, 213–220. [Google Scholar] [CrossRef]
- Li, Q.-Q.; Cao, X.-X.; Xu, J.-D.; Chen, Q.; Wang, W.-J.; Tang, F.; Chen, Z.-Q.; Liu, X.-P.; Xu, Z.-D. The Role of P-Glycoprotein/cellular Prion Protein Interaction in Multidrug-Resistant Breast Cancer Cells Treated with Paclitaxel. Cell. Mol. Life Sci. 2009, 66, 504–515. [Google Scholar] [CrossRef]
- Ryskalin, L.; Busceti, C.L.; Biagioni, F.; Limanaqi, F.; Familiari, P.; Frati, A.; Fornai, F. Prion Protein in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5107. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Yun, C.W.; Lee, S.H. Cellular Prion Protein Enhances Drug Resistance of Colorectal Cancer Cells via Regulation of a Survival Signal Pathway. Biomol. Ther. 2018, 26, 313–321. [Google Scholar] [CrossRef]
- Meslin, F.; Hamaï, A.; Gao, P.; Jalil, A.; Cahuzac, N.; Chouaib, S.; Mehrpour, M. Silencing of Prion Protein Sensitizes Breast Adriamycin-Resistant Carcinoma Cells to TRAIL-Mediated Cell Death. Cancer Res. 2007, 67, 10910–10919. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Yun, S.; Lee, J.H.; Han, Y.-S.; Yoon, Y.M.; An, D.; Lee, S.H. Silencing Prion Protein in HT29 Human Colorectal Cancer Cells Enhances Anticancer Response to Fucoidan. Anticancer Res. 2016, 36, 4449–4458. [Google Scholar] [CrossRef] [Green Version]
- Meslin, F.; Conforti, R.; Mazouni, C.; Morel, N.; Tomasic, G.; Drusch, F.; Yacoub, M.; Sabourin, J.C.; Grassi, J.; Delaloge, S.; et al. Efficacy of Adjuvant Chemotherapy according to Prion Protein Expression in Patients with Estrogen Receptor-Negative Breast Cancer. Ann. Oncol. 2007, 18, 1793–1798. [Google Scholar] [CrossRef]
- Wickner, R.B.; Kelly, A.C. Prions Are Affected by Evolution at Two Levels. Cell. Mol. Life Sci. 2016, 73, 1131–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Chen, Y.; Dubrulle, J.; Stossi, F.; Putluri, V.; Sreekumar, A.; Putluri, N.; Baluya, D.; Lai, S.Y.; Sandulache, V.C. Cisplatin Generates Oxidative Stress Which Is Accompanied by Rapid Shifts in Central Carbon Metabolism. Sci. Rep. 2018, 8, 4306. [Google Scholar] [CrossRef] [Green Version]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Pérez, J.M. Biochemical Mechanisms of Cisplatin Cytotoxicity. Anticancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of Doxorubicin on Apoptosis and Oxidative Stress in Breast Cancer Cell Lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Barciszewska, A.-M.; Gurda, D.; Głodowicz, P.; Nowak, S.; Naskręt-Barciszewska, M.Z. A New Epigenetic Mechanism of Temozolomide Action in Glioma Cells. PLoS ONE 2015, 10, e0136669. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, C.J.; Kawamata, F.; Liu, C.; Ham, S.; Győrffy, B.; Munn, A.L.; Wei, M.Q.; Möller, A.; Whitehall, V.; Wiegmans, A.P. EGFR and Prion Protein Promote Signaling via FOXO3a-KLF5 Resulting in Clinical Resistance to Platinum Agents in Colorectal Cancer. Mol. Oncol. 2019, 13, 725–737. [Google Scholar] [CrossRef] [Green Version]
- Chieng, C.K.-L.; Say, Y.-H. Cellular Prion Protein Contributes to LS 174T Colon Cancer Cell Carcinogenesis by Increasing Invasiveness and Resistance against Doxorubicin-Induced Apoptosis. Tumour Biol. 2015, 36, 8107–8120. [Google Scholar] [CrossRef] [PubMed]
- Klamt, F.; Dal-Pizzol, F.; Conte da Frota, M.L., Jr.; Walz, R.; Andrades, M.E.; da Silva, E.G.; Brentani, R.R.; Izquierdo, I.; Fonseca Moreira, J.C. Imbalance of Antioxidant Defense in Mice Lacking Cellular Prion Protein. Free Radic. Biol. Med. 2001, 30, 1137–1144. [Google Scholar] [CrossRef]
- Watt, N.T.; Routledge, M.N.; Wild, C.P.; Hooper, N.M. Cellular Prion Protein Protects against Reactive-Oxygen-Species-Induced DNA Damage. Free Radic. Biol. Med. 2007, 43, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Arnould, H.; Baudouin, V.; Baudry, A.; Ribeiro, L.W.; Ardila-Osorio, H.; Pietri, M.; Caradeuc, C.; Soultawi, C.; Williams, D.; Alvarez, M.; et al. Loss of Prion Protein Control of Glucose Metabolism Promotes Neurodegeneration in Model of Prion Diseases. PLoS Pathog. 2021, 17, e1009991. [Google Scholar] [CrossRef] [PubMed]
- Viles, J.H.; Cohen, F.E.; Prusiner, S.B.; Goodin, D.B.; Wright, P.E.; Dyson, H.J. Copper Binding to the Prion Protein: Structural Implications of Four Identical Cooperative Binding Sites. Proc. Natl. Acad. Sci. USA 1999, 96, 2042–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegmans, A.P.; Saunus, J.M.; Ham, S.; Lobb, R.; Kutasovic, J.R.; Dalley, A.J.; Miranda, M.; Atkinson, C.; Foliaki, S.T.; Ferguson, K.; et al. Secreted Cellular Prion Protein Binds Doxorubicin and Correlates with Anthracycline Resistance in Breast Cancer. JCI Insight 2019, 5, e124092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, D.; Liu, Y.; Mao, Y.; Gao, L.; Zhang, H.; Luan, S.; Huang, F.; Li, Q. TMZ-Induced PrPc/par-4 Interaction Promotes the Survival of Human Glioma Cells. Int. J. Cancer 2012, 130, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shao, F.; Sun, J.; Du, K.; Sun, Y.; Feng, F. Enhanced Copper-Temozolomide Interactions by Protein for Chemotherapy against Glioblastoma Multiforme. ACS Appl. Mater. Interfaces 2019, 11, 41935–41945. [Google Scholar] [CrossRef]
- Odeh, L.H.; Talib, W.H.; Basheti, I.A. Synergistic Effect of Thymoquinone and Melatonin against Breast Cancer Implanted in Mice. J. Cancer Res. Ther. 2018, 14, 324–330. [Google Scholar] [CrossRef]
- Zhelev, Z.; Ivanova, D.; Bakalova, R.; Aoki, I.; Higashi, T. Synergistic Cytotoxicity of Melatonin and New-Generation Anticancer Drugs Against Leukemia Lymphocytes But Not Normal Lymphocytes. Anticancer Res. 2017, 37, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Hoffmann, K.; Gao, C.; Petrulionis, M.; Herr, I.; Schemmer, P. Melatonin Promotes Sorafenib-Induced Apoptosis through Synergistic Activation of JNK/c-Jun Pathway in Human Hepatocellular Carcinoma. J. Pineal Res. 2017, 62, e12398. [Google Scholar] [CrossRef]
- Gurunathan, S.; Qasim, M.; Kang, M.-H.; Kim, J.-H. Role and Therapeutic Potential of Melatonin in Various Type of Cancers. Onco. Targets. Ther. 2021, 14, 2019–2052. [Google Scholar] [CrossRef]
- Talib, W.H.; Alsayed, A.R.; Abuawad, A.; Daoud, S.; Mahmod, A.I. Melatonin in Cancer Treatment: Current Knowledge and Future Opportunities. Molecules 2021, 26, 2506. [Google Scholar] [CrossRef]
- Schettig, R.; Sears, T.; Klein, M.; Tan-Lim, R.; Matthias, R.; Aussems, C.; Hummel, M.; Sears, R.; Poteet, Z.; Warren, D.; et al. Melatonin: A Powerful Integrative Adjunctive Agent for Oncology. J. Cancer Ther. 2020, 11, 571–596. [Google Scholar] [CrossRef]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.-X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.-F.; Xu, K. Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef] [Green Version]
- Su, S.-C.; Hsieh, M.-J.; Yang, W.-E.; Chung, W.-H.; Reiter, R.J.; Yang, S.-F. Cancer Metastasis: Mechanisms of Inhibition by Melatonin. J. Pineal Res. 2017, 62, e12370. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, G.; Mascia, F.; Gualano, L.; Di Bella, L. Melatonin Anticancer Effects: Review. Int. J. Mol. Sci. 2013, 14, 2410–2430. [Google Scholar] [CrossRef] [Green Version]
- Mediavilla, M.D.; Sanchez-Barcelo, E.J.; Tan, D.X.; Manchester, L.; Reiter, R.J. Basic Mechanisms Involved in the Anti-Cancer Effects of Melatonin. Curr. Med. Chem. 2010, 17, 4462–4481. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Barceló, E.J.; Mediavilla, M.D.; Tan, D.X.; Reiter, R.J. Clinical Uses of Melatonin: Evaluation of Human Trials. Curr. Med. Chem. 2010, 17, 2070–2095. [Google Scholar] [CrossRef] [PubMed]
- Sainz, R.M.; Mayo, J.C.; Rodriguez, C.; Tan, D.X.; Lopez-Burillo, S.; Reiter, R.J. Melatonin and Cell Death: Differential Actions on Apoptosis in Normal and Cancer Cells. Cell. Mol. Life Sci. 2003, 60, 1407–1426. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.-X.; Sainz, R.M.; Mayo, J.C.; Lopez-Burillo, S. Melatonin: Reducing the Toxicity and Increasing the Efficacy of Drugs. J. Pharm. Pharmacol. 2002, 54, 1299–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayalaxmi; Thomas, C.R., Jr.; Reiter, R.J.; Herman, T.S. Melatonin: From Basic Research to Cancer Treatment Clinics. J. Clin. Oncol. 2002, 20, 2575–2601. [Google Scholar] [CrossRef]
- Panzer, A.; Viljoen, M. The Validity of Melatonin as an Oncostatic Agent. J. Pineal Res. 1997, 22, 184–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, R.J. Melatonin Suppression by Static and Extremely Low Frequency Electromagnetic Fields: Relationship to the Reported Increased Incidence of Cancer. Rev. Environ. Health 1994, 10, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yoon, Y.M.; Han, Y.-S.; Jung, S.K.; Lee, S.H. Melatonin Protects Mesenchymal Stem Cells from Autophagy-Mediated Death under Ischaemic ER-Stress Conditions by Increasing Prion Protein Expression. Cell Prolif. 2019, 52, e12545. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Kim, S.; Lee, J.H.; Lee, S.H. Melatonin Promotes Apoptosis of Colorectal Cancer Cells via Superoxide-Mediated ER Stress by Inhibiting Cellular Prion Protein Expression. Anticancer Res. 2018, 38, 3951–3960. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yun, C.W.; Han, Y.-S.; Kim, S.; Jeong, D.; Kwon, H.Y.; Kim, H.; Baek, M.-J.; Lee, S.H. Melatonin and 5-Fluorouracil Co-Suppress Colon Cancer Stem Cells by Regulating Cellular Prion Protein-Oct4 Axis. J. Pineal Res. 2018, 65, e12519. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoon, Y.M.; Han, Y.-S.; Yun, C.W.; Lee, S.H. Melatonin Promotes Apoptosis of Oxaliplatin-Resistant Colorectal Cancer Cells Through Inhibition of Cellular Prion Protein. Anticancer Res. 2018, 38, 1993–2000. [Google Scholar] [CrossRef]
- Salvesen, Ø.; Reiten, M.R.; Espenes, A.; Bakkebø, M.K.; Tranulis, M.A.; Ersdal, C. LPS-Induced Systemic Inflammation Reveals an Immunomodulatory Role for the Prion Protein at the Blood-Brain Interface. J. Neuroinflamm. 2017, 14, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Han, Y.-S.; Lee, S.H. Potentiation of Biological Effects of Mesenchymal Stem Cells in Ischemic Conditions by Melatonin via Upregulation of Cellular Prion Protein Expression. J. Pineal Res. 2017, 62, e12385. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.-X.; Zhang, Z.-H.; Ye, Q.-L.; Xu, S.; Hong, Q.; Xing, W.-Y.; Chen, L.; Yu, D.-X.; Xu, D.-X.; Xie, D.-D. Melatonin Inhibits Migration and Invasion in LPS-Stimulated and -Unstimulated Prostate Cancer Cells Through Blocking Multiple EMT-Relative Pathways. J. Inflamm. Res. 2021, 14, 2253–2265. [Google Scholar] [CrossRef]
- Agarwal, A.; Rai, S.K.; Avni, A.; Mukhopadhyay, S. An Intrinsically Disordered Pathological Prion Variant Y145Stop Converts into Self-Seeding Amyloids via Liquid-Liquid Phase Separation. Proc. Natl. Acad. Sci. USA 2021, 118, e2100968118. [Google Scholar] [CrossRef] [PubMed]
- Tange, H.; Ishibashi, D.; Nakagaki, T.; Taguchi, Y.; Kamatari, Y.O.; Ozawa, H.; Nishida, N. Liquid-Liquid Phase Separation of Full-Length Prion Protein Initiates Conformational Conversion in Vitro. J. Biol. Chem. 2021, 296, 100367. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.; Oamen, H.P.; Caudron, F. Protein Phase Separation during Stress Adaptation and Cellular Memory. Cells 2020, 9, 1302. [Google Scholar] [CrossRef]
- Matos, C.O.; Passos, Y.M.; do Amaral, M.J.; Macedo, B.; Tempone, M.H.; Bezerra, O.C.L.; Moraes, M.O.; Almeida, M.S.; Weber, G.; Missailidis, S.; et al. Liquid-Liquid Phase Separation and Fibrillation of the Prion Protein Modulated by a High-Affinity DNA Aptamer. FASEB J. 2020, 34, 365–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nüske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase Separation of a Yeast Prion Protein Promotes Cellular Fitness. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, D.; Reiter, R.J. Melatonin: Regulation of Biomolecular Condensates in Neurodegenerative Disorders. Antioxidants 2021, 10, 1483. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F. Cyclic 3-Hydroxymelatonin: A Melatonin Metabolite Generated as a Result of Hydroxyl Radical Scavenging. Biol. Signals Recept. 1999, 8, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. Melatonin: Lowering the High Price of Free Radicals. News Physiol. Sci. 2000, 15, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F.; Limson, J.; Weintraub, S.T.; Qi, W. Melatonin Directly Scavenges Hydrogen Peroxide: A Potentially New Metabolic Pathway of Melatonin Biotransformation. Free Radic. Biol. Med. 2000, 29, 1177–1185. [Google Scholar] [CrossRef]
- de Almeida, E.A.; Martinez, G.R.; Klitzke, C.F.; de Medeiros, M.H.G.; Di Mascio, P. Oxidation of Melatonin by Singlet Molecular Oxygen (O2(1deltag)) Produces N1-Acetyl-N2-Formyl-5-Methoxykynurenine. J. Pineal Res. 2003, 35, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Matuszak, Z.; Bilska, M.A.; Reszka, K.J.; Chignell, C.F.; Bilski, P. Interaction of Singlet Molecular Oxygen with Melatonin and Related Indoles. Photochem. Photobiol. 2003, 78, 449–455. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.-X.; Terron, M.P.; Flores, L.J.; Czarnocki, Z. Melatonin and Its Metabolites: New Findings Regarding Their Production and Their Radical Scavenging Actions. Acta Biochim. Pol. 2007, 54, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Noda, Y.; Mori, A.; Liburdy, R.; Packer, L. Melatonin and Its Precursors Scavenge Nitric Oxide. J. Pineal Res. 1999, 27, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Reiter, R.J. Melatonin and Its Metabolites vs Oxidative Stress: From Individual Actions to Collective Protection. J. Pineal Res. 2018, 65, e12514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purushothaman, A.; Sheeja, A.A.; Janardanan, D. Hydroxyl Radical Scavenging Activity of Melatonin and Its Related Indolamines. Free Radic. Res. 2020, 54, 373–383. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin, Its Metabolites and Their Interference with Reactive Nitrogen Compounds. Molecules 2021, 26, 4105. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.B.; Case, J.D.; Takahashi, Y.; Lee, T.H.; Mori, W. Isolation of melatonin, the pineal gland factor that lightens melanocytes. J. Am. Chem. Soc. 1958, 80, 2587. [Google Scholar] [CrossRef]
- He, C.; Wang, J.; Zhang, Z.; Yang, M.; Li, Y.; Tian, X.; Ma, T.; Tao, J.; Zhu, K.; Song, Y.; et al. Mitochondria Synthesize Melatonin to Ameliorate Its Function and Improve Mice Oocyte’s Quality under in Vitro Conditions. Int. J. Mol. Sci. 2016, 17, 939. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.-X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef] [PubMed]
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual Role of Mitochondria in Producing Melatonin and Driving GPCR Signaling to Block Cytochrome c Release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a Mitochondria-Targeted Antioxidant: One of Evolution’s Best Ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881. [Google Scholar] [CrossRef]
- Zhao, D.; Yu, Y.; Shen, Y.; Liu, Q.; Zhao, Z.; Sharma, R.; Reiter, R.J. Melatonin Synthesis and Function: Evolutionary History in Animals and Plants. Front. Endocrinol. 2019, 10, 249. [Google Scholar] [CrossRef]
- Tan, D.-X.; Hardeland, R.; Back, K.; Manchester, L.C.; Alatorre-Jimenez, M.A.; Reiter, R.J. On the Significance of an Alternate Pathway of Melatonin Synthesis via 5-Methoxytryptamine: Comparisons across Species. J. Pineal Res. 2016, 61, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.-X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and Chloroplasts as the Original Sites of Melatonin Synthesis: A Hypothesis Related to Melatonin’s Primary Function and Evolution in Eukaryotes. J. Pineal Res. 2013, 54, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Coon, S.L.; Klein, D.C. Evolution of Arylalkylamine N-Acetyltransferase: Emergence and Divergence. Mol. Cell. Endocrinol. 2006, 252, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Arnao, M.B.; Hernández-Ruiz, J. Growth Conditions Determine Different Melatonin Levels in Lupinus albus, L. J. Pineal Res. 2013, 55, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.; Nieves-Cordones, M.; Lopez-Delacalle, M.; Rodenas, R.; Mestre, T.C.; Garcia-Sanchez, F.; Rubio, F.; Nortes, P.A.; Mittler, R.; Rivero, R.M. Tolerance to Stress Combination in Tomato Plants: New Insights in the Protective Role of Melatonin. Molecules 2018, 23, 535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byeon, Y.; Back, K. Melatonin Synthesis in Rice Seedlings in Vivo Is Enhanced at High Temperatures and under Dark Conditions due to Increased Serotonin N-Acetyltransferase and N-Acetylserotonin Methyltransferase Activities. J. Pineal Res. 2014, 56, 189–195. [Google Scholar] [CrossRef]
- Sun, C.; Liu, L.; Wang, L.; Li, B.; Jin, C.; Lin, X. Melatonin: A Master Regulator of Plant Development and Stress Responses. J. Integr. Plant Biol. 2021, 63, 126–145. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.F.; McIntyre, I.M. Stress-Induced Synthesis of Melatonin: Possible Involvement of the Endogenous Monoamine Oxidase Inhibitor (tribulin). Life Sci. 1985, 37, 1743–1746. [Google Scholar] [CrossRef]
- Stokkan, K.A.; Nonaka, K.O.; Lerchl, A.; Vaughan, M.K.; Reiter, R.J. Low Temperature Stimulates Pineal Activity in Syrian Hamsters. J. Pineal Res. 1991, 10, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Stokkan, K.A.; Reiter, R.J.; Nonaka, K.O.; Lerchl, A.; Yu, B.P.; Vaughan, M.K. Food Restriction Retards Aging of the Pineal Gland. Brain Res. 1991, 545, 66–72. [Google Scholar] [CrossRef]
- Uzun, A.; Baltaci, A.K.; Kilic, M.; Mogulkoc, R. The Effect of Acute Swimming Exercise on Plasma Melatonin Levels in Rats. Bratisl. Lek. Listy 2012, 113, 64–66. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.-X.; Manchester, L.C.; Esteban-Zubero, E.; Zhou, Z.; Reiter, R.J. Melatonin as a Potent and Inducible Endogenous Antioxidant: Synthesis and Metabolism. Molecules 2015, 20, 18886–18906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.-X.; Hardeland, R. The Reserve/Maximum Capacity of Melatonin’s Synthetic Function for the Potential Dimorphism of Melatonin Production and Its Biological Significance in Mammals. Molecules 2021, 26, 7302. [Google Scholar] [CrossRef]
- Carrillo-Vico, A.; Calvo, J.R.; Abreu, P.; Lardone, P.J.; García-Mauriño, S.; Reiter, R.J.; Guerrero, J.M. Evidence of Melatonin Synthesis by Human Lymphocytes and Its Physiological Significance: Possible Role as Intracrine, Autocrine, And/or Paracrine Substance. FASEB J. 2004, 18, 537–539. [Google Scholar] [CrossRef]
- Morcillo-Parra, M.Á.; Beltran, G.; Mas, A.; Torija, M.-J. Effect of Several Nutrients and Environmental Conditions on Intracellular Melatonin Synthesis in Saccharomyces Cerevisiae. Microorganisms 2020, 8, 853. [Google Scholar] [CrossRef]
- Morcillo-Parra, M.Á.; Valera, M.J.; Beltran, G.; Mas, A.; Torija, M.-J. Glycolytic Proteins Interact With Intracellular Melatonin in Saccharomyces Cerevisiae. Front. Microbiol. 2019, 10, 2424. [Google Scholar] [CrossRef] [PubMed]
- Germann, S.M.; Baallal Jacobsen, S.A.; Schneider, K.; Harrison, S.J.; Jensen, N.B.; Chen, X.; Stahlhut, S.G.; Borodina, I.; Luo, H.; Zhu, J.; et al. Glucose-Based Microbial Production of the Hormone Melatonin in Yeast Saccharomyces Cerevisiae. Biotechnol. J. 2016, 11, 717–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunyer-Figueres, M.; Vázquez, J.; Mas, A.; Torija, M.-J.; Beltran, G. Transcriptomic Insights into the Effect of Melatonin in Saccharomyces Cerevisiae in the Presence and Absence of Oxidative Stress. Antioxidants 2020, 9, 947. [Google Scholar] [CrossRef] [PubMed]
- Bisquert, R.; Muñiz-Calvo, S.; Guillamón, J.M. Protective Role of Intracellular Melatonin Against Oxidative Stress and UV Radiation in Saccharomyces Cerevisiae. Front. Microbiol. 2018, 9, 318. [Google Scholar] [CrossRef] [Green Version]
- Bellinger-Kawahara, C.; Cleaver, J.E.; Diener, T.O.; Prusiner, S.B. Purified Scrapie Prions Resist Inactivation by UV Irradiation. J. Virol. 1987, 61, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Alper, T.; Cramp, W.A.; Haig, D.A.; Clarke, M.C. Does the Agent of Scrapie Replicate without Nucleic Acid? Nature 1967, 214, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Bernardino-Sgherri, J.; Siberchicot, C.; Auvré, F.; Busso, D.; Brocas, C.; El Masri, G.; Lioutsko, A.; Ferri, F.; Radicella, J.P.; Romeo, P.-H.; et al. Tumor Resistance to Radiotherapy Is Triggered by an ATM/TAK1-Dependent-Increased Expression of the Cellular Prion Protein. Oncogene 2021, 40, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Vijayalaxmi; Reiter, R.J.; Tan, D.-X.; Herman, R.S.; Thomas, C.R., Jr. Melatonin as a Radioprotective Agent: A Review. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 639–653. [Google Scholar] [CrossRef]
- Farhood, B.; Goradel, N.H.; Mortezaee, K.; Khanlarkhani, N.; Salehi, E.; Nashtaei, M.S.; Mirtavoos-Mahyari, H.; Motevaseli, E.; Shabeeb, D.; Musa, A.E.; et al. Melatonin as an Adjuvant in Radiotherapy for Radioprotection and Radiosensitization. Clin. Transl. Oncol. 2019, 21, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Alonso-González, C.; González, A.; Menéndez-Menéndez, J.; Martínez-Campa, C.; Cos, S. Melatonin as a Radio-Sensitizer in Cancer. Biomedicines 2020, 8, 247. [Google Scholar] [CrossRef]
- Gehrmann, M.; Specht, H.M.; Bayer, C.; Brandstetter, M.; Chizzali, B.; Duma, M.; Breuninger, S.; Hube, K.; Lehnerer, S.; van Phi, V.; et al. Hsp70—A Biomarker for Tumor Detection and Monitoring of Outcome of Radiation Therapy in Patients with Squamous Cell Carcinoma of the Head and Neck. Radiat. Oncol. 2014, 9, 131. [Google Scholar] [CrossRef] [Green Version]
- Rothammer, A.; Sage, E.K.; Werner, C.; Combs, S.E.; Multhoff, G. Increased Heat Shock Protein 70 (Hsp70) Serum Levels and Low NK Cell Counts after Radiotherapy—Potential Markers for Predicting Breast Cancer Recurrence? Radiat. Oncol. 2019, 14, 78. [Google Scholar] [CrossRef]
- Allen, K.D.; Wegrzyn, R.D.; Chernova, T.A.; Müller, S.; Newnam, G.P.; Winslett, P.A.; Wittich, K.B.; Wilkinson, K.D.; Chernoff, Y.O. Hsp70 Chaperones as Modulators of Prion Life Cycle: Novel Effects of Ssa and Ssb on the Saccharomyces Cerevisiae Prion [PSI+]. Genetics 2005, 169, 1227–1242. [Google Scholar] [CrossRef] [Green Version]
- Roh, B.H.; Kim, D.H.; Cho, M.K.; Park, Y.L.; Whang, K.U. Expression of Heat Shock Protein 70 in Human Skin Cells as a Photoprotective Function after UV Exposure. Ann. Dermatol. 2008, 20, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.M.; Reikerstorfer, A.; Schwarz, A.; Krone, C.; Luger, T.A.; Jäättelä, M.; Schwarz, T. Heat Shock Protein 70 Overexpression Affects the Response to Ultraviolet Light in Murine Fibroblasts. Evidence for Increased Cell Viability and Suppression of Cytokine Release. J. Clin. Investig. 1995, 95, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, M.; Hoshino, T.; Yamashita, Y.; Tanaka, K.-I.; Maji, D.; Sato, K.; Adachi, H.; Sobue, G.; Ihn, H.; Funasaka, Y.; et al. Prevention of UVB Radiation-Induced Epidermal Damage by Expression of Heat Shock Protein 70. J. Biol. Chem. 2010, 285, 5848–5858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, T.W.; Sweatman, T.W.; Semak, I.; Sayre, R.M.; Wortsman, J.; Slominski, A. Constitutive and UV-Induced Metabolism of Melatonin in Keratinocytes and Cell-Free Systems. FASEB J. 2006, 20, 1564–1566. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.W.; Kleszczyński, K.; Hardkop, L.H.; Kruse, N.; Zillikens, D. Melatonin Enhances Antioxidative Enzyme Gene Expression (CAT, GPx, SOD), Prevents Their UVR-Induced Depletion, and Protects against the Formation of DNA Damage (8-Hydroxy-2′-Deoxyguanosine) in Ex Vivo Human Skin. J. Pineal Res. 2013, 54, 303–312. [Google Scholar] [CrossRef]
- Kleszczyński, K.; Zwicker, S.; Tukaj, S.; Kasperkiewicz, M.; Zillikens, D.; Wolf, R.; Fischer, T.W. Melatonin Compensates Silencing of Heat Shock Protein 70 and Suppresses Ultraviolet Radiation-Induced Inflammation in Human Skin Ex Vivo and Cultured Keratinocytes. J. Pineal Res. 2015, 58, 117–126. [Google Scholar] [CrossRef]
- Mellatyar, H.; Talaei, S.; Pilehvar-Soltanahmadi, Y.; Barzegar, A.; Akbarzadeh, A.; Shahabi, A.; Barekati-Mowahed, M.; Zarghami, N. Targeted Cancer Therapy through 17-DMAG as an Hsp90 Inhibitor: Overview and Current State of the Art. Biomed. Pharmacother. 2018, 102, 608–617. [Google Scholar] [CrossRef]
- Mays, C.E.; Armijo, E.; Morales, R.; Kramm, C.; Flores, A.; Tiwari, A.; Bian, J.; Telling, G.C.; Pandita, T.K.; Hunt, C.R.; et al. Prion Disease Is Accelerated in Mice Lacking Stress-Induced Heat Shock Protein 70 (HSP70). J. Biol. Chem. 2019, 294, 13619–13628. [Google Scholar] [CrossRef]
- Herbst, M.; Wanker, E.E. Small Molecule Inducers of Heat-Shock Response Reduce polyQ-Mediated Huntingtin Aggregation. A Possible Therapeutic Strategy. Neurodegener. Dis. 2007, 4, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-L.; Shen, H.-H.; Cheng, P.-Y.; Chu, Y.-J.; Hwang, H.-R.; Lam, K.-K.; Lee, Y.-M. 17-DMAG, an HSP90 Inhibitor, Ameliorates Multiple Organ Dysfunction Syndrome via Induction of HSP70 in Endotoxemic Rats. PLoS ONE 2016, 11, e0155583. [Google Scholar] [CrossRef] [Green Version]
- Shyu, W.-C.; Harn, H.-J.; Saeki, K.; Kubosaki, A.; Matsumoto, Y.; Onodera, T.; Chen, C.-J.; Hsu, Y.-D.; Chiang, Y.-H. Molecular Modulation of Expression of Prion Protein by Heat Shock. Mol. Neurobiol. 2002, 26, 1–12. [Google Scholar] [CrossRef]
- Redecke, L.; Binder, S.; Elmallah, M.I.Y.; Broadbent, R.; Tilkorn, C.; Schulz, B.; May, P.; Goos, A.; Eich, A.; Rübhausen, M.; et al. UV-Light-Induced Conversion and Aggregation of Prion Proteins. Free Radic. Biol. Med. 2009, 46, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Webber, C.J.; Lei, S.; Wolozin, B. Chapter Five-The Pathophysiology of Neurodegenerative Disease: Disturbing the Balance between Phase Separation and Irreversible Aggregation. In Progress in Molecular Biology and Translational Science; Uversky, V.N., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 174, pp. 187–223. [Google Scholar] [CrossRef]
- Feng, Z.; Chen, X.; Wu, X.; Zhang, M. Formation of Biological Condensates via Phase Separation: Characteristics, Analytical Methods, and Physiological Implications. J. Biol. Chem. 2019, 294, 14823–14835. [Google Scholar] [CrossRef] [Green Version]
- Ning, W.; Guo, Y.; Lin, S.; Mei, B.; Wu, Y.; Jiang, P.; Tan, X.; Zhang, W.; Chen, G.; Peng, D.; et al. DrLLPS: A Data Resource of Liquid–liquid Phase Separation in Eukaryotes. Nucleic Acids Res. 2019, 48, D288–D295. [Google Scholar] [CrossRef]
- Gomes, E.; Shorter, J. The Molecular Language of Membraneless Organelles. J. Biol. Chem. 2019, 294, 7115–7127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azaldegui, C.A.; Vecchiarelli, A.G.; Biteen, J.S. The Emergence of Phase Separation as an Organizing Principle in Bacteria. Biophys. J. 2021, 120, 1123–1138. [Google Scholar] [CrossRef]
- Ladouceur, A.-M.; Parmar, B.S.; Biedzinski, S.; Wall, J.; Tope, S.G.; Cohn, D.; Kim, A.; Soubry, N.; Reyes-Lamothe, R.; Weber, S.C. Clusters of Bacterial RNA Polymerase Are Biomolecular Condensates That Assemble through Liquid-Liquid Phase Separation. Proc. Natl. Acad. Sci. USA 2020, 117, 18540–18549. [Google Scholar] [CrossRef] [PubMed]
- Salvador-Castell, M.; Demé, B.; Oger, P.; Peters, J. Lipid Phase Separation Induced by the Apolar Polyisoprenoid Squalane Demonstrates Its Role in Membrane Domain Formation in Archaeal Membranes. Langmuir 2020, 36, 7375–7382. [Google Scholar] [CrossRef]
- Price, R.E.; Lesniewski, R.; Nitzsche, K.S.; Meyerdierks, A.; Saltikov, C.; Pichler, T.; Amend, J.P. Archaeal and Bacterial Diversity in an Arsenic-Rich Shallow-Sea Hydrothermal System Undergoing Phase Separation. Front. Microbiol. 2013, 4, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riback, J.A.; Katanski, C.D.; Kear-Scott, J.L.; Pilipenko, E.V.; Rojek, A.E.; Sosnick, T.R.; Drummond, D.A. Stress-Triggered Phase Separation Is an Adaptive, Evolutionarily Tuned Response. Cell 2017, 168, 1028–1040.e19. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase Transitions in the Assembly of Multivalent Signalling Proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef]
- Su, X.; Ditlev, J.A.; Hui, E.; Xing, W.; Banjade, S.; Okrut, J.; King, D.S.; Taunton, J.; Rosen, M.K.; Vale, R.D. Phase Separation of Signaling Molecules Promotes T Cell Receptor Signal Transduction. Science 2016, 352, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Kimura, W. Roles of Phase Separation for Cellular Redox Maintenance. Front. Genet. 2021, 12, 691946. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, G.; Mekhail, K. Biomolecular Condensates as Arbiters of Biochemical Reactions inside the Nucleus. Commun. Biol. 2020, 3, 773. [Google Scholar] [CrossRef]
- Franzmann, T.M.; Alberti, S. Prion-like Low-Complexity Sequences: Key Regulators of Protein Solubility and Phase Behavior. J. Biol. Chem. 2019, 294, 7128–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzmann, T.M.; Alberti, S. Protein Phase Separation as a Stress Survival Strategy. Cold Spring Harb. Perspect. Biol. 2019, 11, a034058. [Google Scholar] [CrossRef] [Green Version]
- Lawson, V.A.; Priola, S.A.; Meade-White, K.; Lawson, M.; Chesebro, B. Flexible N-Terminal Region of Prion Protein Influences Conformation of Protease-Resistant Prion Protein Isoforms Associated with Cross-Species Scrapie Infection in Vivo and in Vitro. J. Biol. Chem. 2004, 279, 13689–13695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankenfield, K.N.; Powers, E.T.; Kelly, J.W. Influence of the N-Terminal Domain on the Aggregation Properties of the Prion Protein. Protein Sci. 2005, 14, 2154–2166. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, A.K.; Jarosz, D.F. More than Just a Phase: Prions at the Crossroads of Epigenetic Inheritance and Evolutionary Change. J. Mol. Biol. 2018, 430, 4607–4618. [Google Scholar] [CrossRef]
- Chakrabortee, S.; Byers, J.S.; Jones, S.; Garcia, D.M.; Bhullar, B.; Chang, A.; She, R.; Lee, L.; Fremin, B.; Lindquist, S.; et al. Intrinsically Disordered Proteins Drive Emergence and Inheritance of Biological Traits. Cell 2016, 167, 369–381.e12. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Edskes, H.K.; Khamar, H.J.; Winchester, C.-L.; Greenler, A.J.; Zhou, A.; McGlinchey, R.P.; Gorkovskiy, A.; Wickner, R.B. Sporadic Distribution of Prion-Forming Ability of Sup35p from Yeasts and Fungi. Genetics 2014, 198, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Edskes, H.K.; Engel, A.; McCann, L.M.; Brachmann, A.; Tsai, H.-F.; Wickner, R.B. Prion-Forming Ability of Ure2 of Yeasts Is Not Evolutionarily Conserved. Genetics 2011, 188, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Basile, W.; Salvatore, M.; Bassot, C.; Elofsson, A. Why Do Eukaryotic Proteins Contain More Intrinsically Disordered Regions? PLoS Comput. Biol. 2019, 15, e1007186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Uversky, V.N.; Kurgan, L. Disordered Nucleiome: Abundance of Intrinsic Disorder in the DNA- and RNA-Binding Proteins in 1121 Species from Eukaryota, Bacteria and Archaea. Proteomics 2016, 16, 1486–1498. [Google Scholar] [CrossRef]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally Abundant Exceptions: Comprehensive Characterization of Intrinsic Disorder in All Domains of Life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly Order in Protein Intrinsic Disorder Distribution: Disorder in 3500 Proteomes from Viruses and the Three Domains of Life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef]
- Ito, Y.; Mikawa, T.; Smith, B.O. In-Cell NMR of Intrinsically Disordered Proteins in Prokaryotic Cells. In Intrinsically Disordered Protein Analysis: Volume 1, Methods and Experimental Tools; Uversky, V.N., Dunker, A.K., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 19–31. [Google Scholar] [CrossRef]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Archaic Chaos: Intrinsically Disordered Proteins in Archaea. BMC Syst. Biol. 2010, 4 (Suppl S1), S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, B.S.; Dignon, G.L.; Tang, W.S.; Kelley, F.M.; Ranganath, A.K.; Jahnke, C.N.; Simpkins, A.G.; Regy, R.M.; Hammer, D.A.; Good, M.C.; et al. Identifying Sequence Perturbations to an Intrinsically Disordered Protein That Determine Its Phase-Separation Behavior. Proc. Natl. Acad. Sci. USA 2020, 117, 11421–11431. [Google Scholar] [CrossRef]
- Kamimura, Y.R.; Kanai, M. Chemical Insights into Liquid-Liquid Phase Separation in Molecular Biology. BCSJ 2021, 94, 1045–1058. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlers, J.; Adams, E.M.; Bader, V.; Pezzotti, S.; Winklhofer, K.F.; Tatzelt, J.; Havenith, M. The Key Role of Solvent in Condensation: Mapping Water in Liquid-Liquid Phase-Separated FUS. Biophys. J. 2021, 120, 1266–1275. [Google Scholar] [CrossRef]
- Hondele, M.; Heinrich, S.; De Los Rios, P.; Weis, K. Membraneless Organelles: Phasing out of Equilibrium. Emerg. Top. Life Sci. 2020, 4, 331–342. [Google Scholar] [CrossRef]
- Riback, J.A.; Zhu, L.; Ferrolino, M.C.; Tolbert, M.; Mitrea, D.M.; Sanders, D.W.; Wei, M.-T.; Kriwacki, R.W.; Brangwynne, C.P. Composition-Dependent Thermodynamics of Intracellular Phase Separation. Nature 2020, 581, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-Y.; Wu, J.-J.; Li, Y.-M. Regulation of Liquid-Liquid Phase Separation with Focus on Post-Translational Modifications. Chem. Commun. 2021, 57, 13275–13287. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Lawrence Zipursky, S.; Matsudaira, P.; Baltimore, D.; Darnell, J. Biochemical Energetics; W. H. Freeman and Company: New York, NY, USA, 2000. [Google Scholar]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active Liquid-like Behavior of Nucleoli Determines Their Size and Shape in Xenopus Laevis Oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, R.H.G.; Le Dily, F.; Beato, M. ATP, Mg2+, Nuclear Phase Separation, and Genome Accessibility. Trends Biochem. Sci. 2019, 44, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a Biological Hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef]
- Hayes, M.H.; Peuchen, E.H.; Dovichi, N.J.; Weeks, D.L. Dual Roles for ATP in the Regulation of Phase Separated Protein Aggregates in Xenopus Oocyte Nucleoli. Elife 2018, 7, e35224. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lim, L.; Song, J. ATP Enhances at Low Concentrations but Dissolves at High Concentrations Liquid-Liquid Phase Separation (LLPS) of ALS/FTD-Causing FUS. Biochem. Biophys. Res. Commun. 2018, 504, 545–551. [Google Scholar] [CrossRef]
- Sridharan, S.; Kurzawa, N.; Werner, T.; Günthner, I.; Helm, D.; Huber, W.; Bantscheff, M.; Savitski, M.M. Proteome-Wide Solubility and Thermal Stability Profiling Reveals Distinct Regulatory Roles for ATP. Nat. Commun. 2019, 10, 1155. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Paul, S. ATP Controls the Aggregation of Aβ16-22 Peptides. J. Phys. Chem. B 2020, 124, 210–223. [Google Scholar] [CrossRef]
- Song, J. Adenosine Triphosphate Energy-Independently Controls Protein Homeostasis with Unique Structure and Diverse Mechanisms. Protein Sci. 2021, 30, 1277–1293. [Google Scholar] [CrossRef]
- Mehringer, J.; Do, T.-M.; Touraud, D.; Hohenschutz, M.; Khoshsima, A.; Horinek, D.; Kunz, W. Hofmeister versus Neuberg: Is ATP Really a Biological Hydrotrope? Cell Rep. Phys. Sci. 2021, 2, 100343. [Google Scholar] [CrossRef]
- Mandl, I.; Grauer, A.; Neuberg, C. Solubilization of Insoluble Matter in Nature; I. The Part Played by Salts of Adenosinetriphosphate. Biochim. Biophys. Acta 1952, 8, 654–663. [Google Scholar] [CrossRef]
- Diaz-Espinoza, R.; Mukherjee, A.; Soto, C. Kosmotropic Anions Promote Conversion of Recombinant Prion Protein into a PrPSc-like Misfolded Form. PLoS ONE 2012, 7, e31678. [Google Scholar] [CrossRef] [PubMed]
- Yeh, V.; Broering, J.M.; Romanyuk, A.; Chen, B.; Chernoff, Y.O.; Bommarius, A.S. The Hofmeister Effect on Amyloid Formation Using Yeast Prion Protein. Protein Sci. 2010, 19, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Usluer, S.; Zhang, F.; Lenard, A.J.; Bourgeois, B.M.R.; Madl, T. ATP Regulates RNA-Driven Cold Inducible RNA Binding Protein Phase Separation. Protein Sci. 2021, 30, 1438–1453. [Google Scholar] [CrossRef]
- Garcia-Jove Navarro, M.; Kashida, S.; Chouaib, R.; Souquere, S.; Pierron, G.; Weil, D.; Gueroui, Z. RNA Is a Critical Element for the Sizing and the Composition of Phase-Separated RNA-Protein Condensates. Nat. Commun. 2019, 10, 3230. [Google Scholar] [CrossRef] [Green Version]
- Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; Chen, C.C.-H.; Eckmann, C.R.; Myong, S.; Brangwynne, C.P. The Disordered P Granule Protein LAF-1 Drives Phase Separation into Droplets with Tunable Viscosity and Dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 7189–7194. [Google Scholar] [CrossRef] [Green Version]
- Conn, G.L.; Gittis, A.G.; Lattman, E.E.; Misra, V.K.; Draper, D.E. A Compact RNA Tertiary Structure Contains a Buried Backbone-K+ Complex. J. Mol. Biol. 2002, 318, 963–973. [Google Scholar] [CrossRef]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225.e24. [Google Scholar] [CrossRef]
- Gotor, N.L.; Armaos, A.; Calloni, G.; Torrent Burgas, M.; Vabulas, R.M.; De Groot, N.S.; Tartaglia, G.G. RNA-Binding and Prion Domains: The Yin and Yang of Phase Separation. Nucleic Acids Res. 2020, 48, 9491–9504. [Google Scholar] [CrossRef]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. RNA Molecules Stimulate Prion Protein Conversion. Nature 2003, 425, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Saá, P.; Sferrazza, G.F.; Ottenberg, G.; Oelschlegel, A.M.; Dorsey, K.; Lasmézas, C.I. Strain-Specific Role of RNAs in Prion Replication. J. Virol. 2012, 86, 10494–10504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, M.P.B.; Millen, T.A.; Ferreira, P.S.; e Silva, N.L.C.; Vieira, T.C.R.G.; Almeida, M.S.; Silva, J.L.; Cordeiro, Y. Prion Protein Complexed to N2a Cellular RNAs through Its N-Terminal Domain Forms Aggregates and Is Toxic to Murine Neuroblastoma Cells. J. Biol. Chem. 2008, 283, 19616–19625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conaway, R.C.; Conaway, J.W. ATP Activates Transcription Initiation from Promoters by RNA Polymerase II in a Reversible Step prior to RNA Synthesis. J. Biol. Chem. 1988, 263, 2962–2968. [Google Scholar] [CrossRef]
- Grummt, I.; Grummt, F. Control of Nucleolar RNA Synthesis by the Intracellular Pool Sizes of ATP and GTP. Cell 1976, 7, 447–453. [Google Scholar] [CrossRef]
- Joyce, G.F. RNA Evolution and the Origins of Life. Nature 1989, 338, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Joyce, G.F. The Antiquity of RNA-Based Evolution. Nature 2002, 418, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.P.; Joyce, G.F. The Origins of the RNA World. Cold Spring Harb. Perspect. Biol. 2012, 4, a003608. [Google Scholar] [CrossRef]
- White, H.B., 3rd. Coenzymes as Fossils of an Earlier Metabolic State. J. Mol. Evol. 1976, 7, 101–104. [Google Scholar] [CrossRef]
- Goldman, A.D.; Kacar, B. Cofactors Are Remnants of Life’s Origin and Early Evolution. J. Mol. Evol. 2021, 89, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Jheeta, S.; Chatzitheodoridis, E.; Devine, K.; Block, J. The Way Forward for the Origin of Life: Prions and Prion-Like Molecules First Hypothesis. Life 2021, 11, 872. [Google Scholar] [CrossRef] [PubMed]
- Zajkowski, T.; Lee, M.D.; Mondal, S.S.; Carbajal, A.; Dec, R.; Brennock, P.D.; Piast, R.W.; Snyder, J.E.; Bense, N.B.; Dzwolak, W.; et al. The Hunt for Ancient Prions: Archaeal Prion-Like Domains Form Amyloid-Based Epigenetic Elements. Mol. Biol. Evol. 2021, 38, 2088–2103. [Google Scholar] [CrossRef]
- Maury, C.P.J. Amyloid and the Origin of Life: Self-Replicating Catalytic Amyloids as Prebiotic Informational and Protometabolic Entities. Cell. Mol. Life Sci. 2018, 75, 1499–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maury, C.P.J. Self-Propagating Beta-Sheet Polypeptide Structures as Prebiotic Informational Molecular Entities: The Amyloid World. Orig. Life Evol. Biosph. 2009, 39, 141–150. [Google Scholar] [CrossRef]
- Lupi, O.; Dadalti, P.; Cruz, E.; Sanberg, P.R.; Cryopraxis’ Task Force for Prion Research. Are Prions Related to the Emergence of Early Life? Med. Hypotheses 2006, 67, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Kandola, T.S.; Wu, J.; Venkatesan, S.; Ketter, E.; Lange, J.J.; Rodríguez Gama, A.; Box, A.; Unruh, J.R.; Cook, M.; et al. Quantifying Nucleation In Vivo Reveals the Physical Basis of Prion-like Phase Behavior. Mol. Cell 2018, 71, 155–168.e7. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Qian, J.; Xu, Z.; Yin, S.; Zhou, L.; Zheng, S.; Zhang, W. Emerging Roles of Liquid–Liquid Phase Separation in Cancer: From Protein Aggregation to Immune-Associated Signaling. Front. Cell Dev. Biol. 2021, 9, 1465. [Google Scholar] [CrossRef]
- Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Passos, Y.M.; Mota, M.F.; Jakobus, B.; de Sousa, G.D.S.; Pereira da Costa, F.; Felix, A.L.; Ferretti, G.D.S.; et al. Phase Separation of p53 Precedes Aggregation and Is Affected by Oncogenic Mutations and Ligands. Chem. Sci. 2021, 12, 7334–7349. [Google Scholar] [CrossRef]
- de Oliveira, G.A.P.; Cordeiro, Y.; Silva, J.L.; Vieira, T.C.R.G. Chapter Nine-Liquid-Liquid Phase Transitions and Amyloid Aggregation in Proteins Related to Cancer and Neurodegenerative Diseases. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 118, pp. 289–331. [Google Scholar] [CrossRef]
- Ahn, J.H.; Davis, E.S.; Daugird, T.A.; Zhao, S.; Quiroga, I.Y.; Uryu, H.; Li, J.; Storey, A.J.; Tsai, Y.-H.; Keeley, D.P.; et al. Phase Separation Drives Aberrant Chromatin Looping and Cancer Development. Nature 2021, 595, 591–595. [Google Scholar] [CrossRef]
- Zeigler, T.M.; Chung, M.C.; Narayan, O.P.; Guan, J. Protein Phase Separation: Physical Models and Phase-Separation- Mediated Cancer Signaling. Adv. Phys. X 2021, 6, 1936638. [Google Scholar] [CrossRef]
- Taniue, K.; Akimitsu, N. Aberrant Phase Separation and Cancer. FEBS J. 2021, 289, 17–39. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Young, R.A. Biomolecular Condensates and Cancer. Cancer Cell 2021, 39, 174–192. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, R.-S.; Yamamoto, T.; Takahashi, M.; Tachiwana, H.; Maruyama, R.; Hirota, T.; Saitoh, N. Nuclear Microenvironment in Cancer: Control through Liquid-Liquid Phase Separation. Cancer Sci. 2020, 111, 3155–3163. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, R.; Tones, J.; Liu, M.; Dilley, R.L.; Chenoweth, D.M.; Greenberg, R.A.; Lampson, M.A. Nuclear Body Phase Separation Drives Telomere Clustering in ALT Cancer Cells. Mol. Biol. Cell 2020, 31, 2048–2056. [Google Scholar] [CrossRef]
- Jiang, S.; Fagman, J.B.; Chen, C.; Alberti, S.; Liu, B. Protein Phase Separation and Its Role in Tumorigenesis. Elife 2020, 9, e60264. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, Y.; Xu, A.; Cai, M.; Cao, J.; Zhu, H.; Yang, B.; Shao, X.; Ying, M.; He, Q. Protein Phase Separation: A Novel Therapy for Cancer? Br. J. Pharmacol. 2020, 177, 5008–5030. [Google Scholar] [CrossRef]
- Park, S.-K.; Park, S.; Pentek, C.; Liebman, S.W. Tumor Suppressor Protein p53 Expressed in Yeast Can Remain Diffuse, Form a Prion, or Form Unstable Liquid-like Droplets. iScience 2021, 24, 102000. [Google Scholar] [CrossRef] [PubMed]
- Kamagata, K.; Kanbayashi, S.; Honda, M.; Itoh, Y.; Takahashi, H.; Kameda, T.; Nagatsugi, F.; Takahashi, S. Liquid-like Droplet Formation by Tumor Suppressor p53 Induced by Multivalent Electrostatic Interactions between Two Disordered Domains. Sci. Rep. 2020, 10, 580. [Google Scholar] [CrossRef]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of Tumor Suppressor p53 and Its Intrinsically Disordered N-Terminal Transactivation Domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamps, J.; Lin, Y.-H.; Oliva, R.; Bader, V.; Winter, R.; Winklhofer, K.F.; Tatzelt, J. The N-Terminal Domain of the Prion Protein Is Required and Sufficient for Liquid-Liquid Phase Separation: A Crucial Role of the Aβ-Binding Domain. J. Biol. Chem. 2021, 297, 100860. [Google Scholar] [CrossRef]
- Polido, S.A.; Kamps, J.; Tatzelt, J. Biological Functions of the Intrinsically Disordered N-Terminal Domain of the Prion Protein: A Possible Role of Liquid–Liquid Phase Separation. Biomolecules 2021, 11, 1201. [Google Scholar] [CrossRef]
- Posey, A.E.; Holehouse, A.S.; Pappu, R.V. Phase Separation of Intrinsically Disordered Proteins. Methods Enzymol. 2018, 611, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.W.; Harmon, T.S.; Hopkins, J.B.; Chakravarthy, S.; Incicco, J.J.; Schuck, P.; Soranno, A.; Mittag, T. A Multi-Step Nucleation Process Determines the Kinetics of Prion-like Domain Phase Separation. Nat. Commun. 2021, 12, 4513. [Google Scholar] [CrossRef]
- Kovachev, P.S.; Gomes, M.P.B.; Cordeiro, Y.; Ferreira, N.C.; Valadão, L.P.F.; Ascari, L.M.; Rangel, L.P.; Silva, J.L.; Sanyal, S. RNA Modulates Aggregation of the Recombinant Mammalian Prion Protein by Direct Interaction. Sci. Rep. 2019, 9, 12406. [Google Scholar] [CrossRef]
- Naslavsky, N.; Stein, R.; Yanai, A.; Friedlander, G.; Taraboulos, A. Characterization of Detergent-Insoluble Complexes Containing the Cellular Prion Protein and Its Scrapie Isoform. J. Biol. Chem. 1997, 272, 6324–6331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, V.; Hooper, N.M. The Role of Lipid Rafts in Prion Protein Biology. Front. Biosci. 2011, 16, 151–168. [Google Scholar] [CrossRef] [Green Version]
- Beaudoin, S.; Vanderperre, B.; Grenier, C.; Tremblay, I.; Leduc, F.; Roucou, X. A Large Ribonucleoprotein Particle Induced by Cytoplasmic PrP Shares Striking Similarities with the Chromatoid Body, an RNA Granule Predicted to Function in Posttranscriptional Gene Regulation. Biochim. Biophys. Acta 2009, 1793, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, S. Historical Survey on Chromatoid Body Research. Acta Histochem. Cytochem. 2008, 41, 65–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.; Taraboulos, A. Scrapie-like Prion Protein Accumulates in Aggresomes of Cyclosporin A-Treated Cells. EMBO J. 2003, 22, 404–417. [Google Scholar] [CrossRef]
- Goggin, K.; Beaudoin, S.; Grenier, C.; Brown, A.-A.; Roucou, X. Prion Protein Aggresomes Are poly(A)+ Ribonucleoprotein Complexes That Induce a PKR-Mediated Deficient Cell Stress Response. Biochim. Biophys. Acta 2008, 1783, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Mironov, A., Jr.; Latawiec, D.; Wille, H.; Bouzamondo-Bernstein, E.; Legname, G.; Williamson, R.A.; Burton, D.; DeArmond, S.J.; Prusiner, S.B.; Peters, P.J. Cytosolic Prion Protein in Neurons. J. Neurosci. 2003, 23, 7183–7193. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Montalban, N.; Lee, Y.J.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Changes in Prion Replication Environment Cause Prion Strain Mutation. FASEB J. 2013, 27, 3702–3710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alred, E.J.; Lodangco, I.; Gallaher, J.; Hansmann, U.H.E. Mutations Alter RNA-Mediated Conversion of Human Prions. ACS Omega 2018, 3, 3936–3944. [Google Scholar] [CrossRef] [PubMed]
- Morillas, M.; Vanik, D.L.; Surewicz, W.K. On the Mechanism of Alpha-Helix to Beta-Sheet Transition in the Recombinant Prion Protein. Biochemistry 2001, 40, 6982–6987. [Google Scholar] [CrossRef]
- Wang, X.; Cui, M.; Zhao, C.; He, L.; Zhu, D.; Wang, B.; Du, W. Regulation of Aggregation Behavior and Neurotoxicity of Prion Neuropeptides by Platinum Complexes. Inorg. Chem. 2014, 53, 5044–5054. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-J.; Li, X.-N.; Liu, W.-L.; Yuan, H.-Y.; Gao, Y.; Wang, K.; Tang, B.; Pang, D.-W.; Chen, J.; Liang, Y. Neutralizing Mutations Significantly Inhibit Amyloid Formation by Human Prion Protein and Decrease Its Cytotoxicity. J. Mol. Biol. 2020, 432, 828–844. [Google Scholar] [CrossRef]
- Wang, J.H.; Du, J.P.; Li, S.J.; Zhai, L.P.; Yang, X.Y.; Wang, Z.H.; Wu, Z.T.; Han, Y. Octarepeat Peptides of Prion Are Essential for Multidrug Resistance in Gastric Cancer Cells. J. Dig. Dis. 2012, 13, 143–152. [Google Scholar] [CrossRef]
- Zahn, R. The Octapeptide Repeats in Mammalian Prion Protein Constitute a pH-Dependent Folding and Aggregation Site. J. Mol. Biol. 2003, 334, 477–488. [Google Scholar] [CrossRef]
- Haigh, C.L.; Drew, S.C.; Boland, M.P.; Masters, C.L.; Barnham, K.J.; Lawson, V.A.; Collins, S.J. Dominant Roles of the Polybasic Proline Motif and Copper in the PrP23-89-Mediated Stress Protection Response. J. Cell Sci. 2009, 122 Pt 10, 1518–1528. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, M.; Walter, E.D.; Newell, D.J.; Jackson, P.J.; Aronoff-Spencer, E.; Peisach, J.; Gerfen, G.J.; Bennett, B.; Antholine, W.E.; Millhauser, G.L. The Octarepeat Domain of the Prion Protein Binds Cu(II) with Three Distinct Coordination Modes at pH 7.4. J. Am. Chem. Soc. 2005, 127, 12647–12656. [Google Scholar] [CrossRef] [Green Version]
- Stöckel, J.; Safar, J.; Wallace, A.C.; Cohen, F.E.; Prusiner, S.B. Prion Protein Selectively Binds copper(II) Ions. Biochemistry 1998, 37, 7185–7193. [Google Scholar] [CrossRef] [PubMed]
- Ridge, P.G.; Zhang, Y.; Gladyshev, V.N. Comparative Genomic Analyses of Copper Transporters and Cuproproteomes Reveal Evolutionary Dynamics of Copper Utilization and Its Link to Oxygen. PLoS ONE 2008, 3, e1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassallo, N.; Herms, J. Cellular Prion Protein Function in Copper Homeostasis and Redox Signalling at the Synapse. J. Neurochem. 2003, 86, 538–544. [Google Scholar] [CrossRef]
- Majumder, S.; Chatterjee, S.; Pal, S.; Biswas, J.; Efferth, T.; Choudhuri, S.K. The Role of Copper in Drug-Resistant Murine and Human Tumors. Biometals 2009, 22, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting Copper in Cancer Therapy: “Copper That Cancer”. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef]
- Dong, S.-L.; Cadamuro, S.A.; Fiorino, F.; Bertsch, U.; Moroder, L.; Renner, C. Copper Binding and Conformation of the N-Terminal Octarepeats of the Prion Protein in the Presence of DPC Micelles as Membrane Mimetic. Biopolymers 2007, 88, 840–847. [Google Scholar] [CrossRef]
- Wong, B.S.; Vénien-Bryan, C.; Williamson, R.A.; Burton, D.R.; Gambetti, P.; Sy, M.S.; Brown, D.R.; Jones, I.M. Copper Refolding of Prion Protein. Biochem. Biophys. Res. Commun. 2000, 276, 1217–1224. [Google Scholar] [CrossRef]
- Evans, E.G.B.; Pushie, M.J.; Markham, K.A.; Lee, H.-W.; Millhauser, G.L. Interaction between Prion Protein’s Copper-Bound Octarepeat Domain and a Charged C-Terminal Pocket Suggests a Mechanism for N-Terminal Regulation. Structure 2016, 24, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Alsiary, R.A.; Alghrably, M.; Saoudi, A.; Al-Ghamdi, S.; Jaremko, L.; Jaremko, M.; Emwas, A.-H. Using NMR Spectroscopy to Investigate the Role Played by Copper in Prion Diseases. Neurol. Sci. 2020, 41, 2389–2406. [Google Scholar] [CrossRef] [PubMed]
- M Passos, Y.; J do Amaral, M.; C Ferreira, N.; Macedo, B.; Chaves, J.A.P.; E de Oliveira, V.; P B Gomes, M.; L Silva, J.; Cordeiro, Y. The Interplay between a GC-Rich Oligonucleotide and Copper Ions on Prion Protein Conformational and Phase Transitions. Int. J. Biol. Macromol. 2021, 173, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.L.; Pan, J.; Singh, R.R.P. A Mechanism for Copper Inhibition of Infectious Prion Conversion. Biophys. J. 2006, 91, L11–L13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giese, A.; Levin, J.; Bertsch, U.; Kretzschmar, H. Effect of Metal Ions on de Novo Aggregation of Full-Length Prion Protein. Biochem. Biophys. Res. Commun. 2004, 320, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Bocharova, O.V.; Breydo, L.; Salnikov, V.V.; Baskakov, I.V. Copper(II) Inhibits in Vitro Conversion of Prion Protein into Amyloid Fibrils. Biochemistry 2005, 44, 6776–6787. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.-F.; Harischandra, D.S.; Kanthasamy, A.; Sivasankar, S. Copper-Induced Structural Conversion Templates Prion Protein Oligomerization and Neurotoxicity. Sci. Adv. 2016, 2, e1600014. [Google Scholar] [CrossRef] [Green Version]
- Miura, T.; Hori-i, A.; Takeuchi, H. Metal-Dependent Alpha-Helix Formation Promoted by the Glycine-Rich Octapeptide Region of Prion Protein. FEBS Lett. 1996, 396, 248–252. [Google Scholar] [CrossRef] [Green Version]
- Cinar, H.; Winter, R. The Effects of Cosolutes and Crowding on the Kinetics of Protein Condensate Formation Based on Liquid-Liquid Phase Separation: A Pressure-Jump Relaxation Study. Sci. Rep. 2020, 10, 17245. [Google Scholar] [CrossRef]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef]
- Cao, Z.; Fang, Y.; Lu, Y.; Tan, D.; Du, C.; Li, Y.; Ma, Q.; Yu, J.; Chen, M.; Zhou, C.; et al. Melatonin Alleviates Cadmium-Induced Liver Injury by Inhibiting the TXNIP-NLRP3 Inflammasome. J. Pineal Res. 2017, 62, e12389. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Grailer, J.J.; Wang, N.; Wang, M.; Yao, J.; Zhong, R.; Gao, G.F.; Ward, P.A.; Tan, D.-X.; et al. Melatonin Alleviates Acute Lung Injury through Inhibiting the NLRP3 Inflammasome. J. Pineal Res. 2016, 60, 405–414. [Google Scholar] [CrossRef]
- Mediavilla, M.D.; Cos, S.; Sánchez-Barceló, E.J. Melatonin Increases p53 and p21WAF1 Expression in MCF-7 Human Breast Cancer Cells in Vitro. Life Sci. 1999, 65, 415–420. [Google Scholar] [CrossRef]
- Santoro, R.; Marani, M.; Blandino, G.; Muti, P.; Strano, S. Melatonin Triggers p53Ser Phosphorylation and Prevents DNA Damage Accumulation. Oncogene 2012, 31, 2931–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proietti, S.; Cucina, A.; Dobrowolny, G.; D’Anselmi, F.; Dinicola, S.; Masiello, M.G.; Pasqualato, A.; Palombo, A.; Morini, V.; Reiter, R.J.; et al. Melatonin down-Regulates MDM2 Gene Expression and Enhances p53 Acetylation in MCF-7 Cells. J. Pineal Res. 2014, 57, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.-X.; Halfmann, R.; Chen, Z.J. Prion-like Polymerization Underlies Signal Transduction in Antiviral Immune Defense and Inflammasome Activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef] [Green Version]
- Samir, P.; Kanneganti, T.-D. DDX3X Sits at the Crossroads of Liquid-Liquid and Prionoid Phase Transitions Arbitrating Life and Death Cell Fate Decisions in Stressed Cells. DNA Cell Biol. 2020, 39, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; De Moura Gallo, C.V.; Costa, D.C.F.; Rangel, L.P. Prion-like Aggregation of Mutant p53 in Cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef]
- Shim, D.-W.; Lee, K.-H. Posttranslational Regulation of the NLR Family Pyrin Domain-Containing 3 Inflammasome. Front. Immunol. 2018, 9, 1054. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Li, T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front. Immunol. 2018, 9, 2305. [Google Scholar] [CrossRef] [Green Version]
- Akther, M.; Haque, M.E.; Park, J.; Kang, T.-B.; Lee, K.-H. NLRP3 Ubiquitination-A New Approach to Target NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2021, 22, 8780. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Post-Translational Modification of p53 in Tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Rodriguez, M.S.; Desterro, J.M.; Lain, S.; Lane, D.P.; Hay, R.T. Multiple C-Terminal Lysine Residues Target p53 for Ubiquitin-Proteasome-Mediated Degradation. Mol. Cell. Biol. 2000, 20, 8458–8467. [Google Scholar] [CrossRef] [Green Version]
- Kahyo, T.; Nishida, T.; Yasuda, H. Involvement of PIAS1 in the Sumoylation of Tumor Suppressor p53. Mol. Cell 2001, 8, 713–718. [Google Scholar] [CrossRef]
- Kim, Y.; Myong, S. RNA Remodeling Activity of DEAD Box Proteins Tuned by Protein Concentration, RNA Length, and ATP. Mol. Cell 2016, 63, 865–876. [Google Scholar] [CrossRef] [Green Version]
- Otvos, L., Jr.; Cudic, M. Post-Translational Modifications in Prion Proteins. Curr. Protein Pept. Sci. 2002, 3, 643–652. [Google Scholar] [CrossRef]
- Dear, D.V.; Young, D.S.; Kazlauskaite, J.; Meersman, F.; Oxley, D.; Webster, J.; Pinheiro, T.J.T.; Gill, A.C.; Bronstein, I.; Lowe, C.R. Effects of Post-Translational Modifications on Prion Protein Aggregation and the Propagation of Scrapie-like Characteristics in Vitro. Biochim. Biophys. Acta 2007, 1774, 792–802. [Google Scholar] [CrossRef] [Green Version]
- Callender, J.A.; Sevillano, A.M.; Soldau, K.; Kurt, T.D.; Schumann, T.; Pizzo, D.P.; Altmeppen, H.; Glatzel, M.; Esko, J.D.; Sigurdson, C.J. Prion Protein Post-Translational Modifications Modulate Heparan Sulfate Binding and Limit Aggregate Size in Prion Disease. Neurobiol. Dis. 2020, 142, 104955. [Google Scholar] [CrossRef]
- Aguilar-Calvo, P.; Xiao, X.; Bett, C.; Eraña, H.; Soldau, K.; Castilla, J.; Nilsson, K.P.R.; Surewicz, W.K.; Sigurdson, C.J. Post-Translational Modifications in PrP Expand the Conformational Diversity of Prions in vivo. Sci. Rep. 2017, 7, 43295. [Google Scholar] [CrossRef]
- Valette, N.M.; Radford, S.E.; Harris, S.A.; Warriner, S.L. Phosphorylation as a Tool to Modulate Aggregation Propensity and to Predict Fibril Architecture. ChemBioChem 2012, 13, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portz, B.; Shorter, J. Biochemical Timekeeping Via Reentrant Phase Transitions. J. Mol. Biol. 2021, 433, 166794. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, A.R.; Zeng, F.; Hooper, N.M. The N-Terminal Region of the Prion Protein Ectodomain Contains a Lipid Raft Targeting Determinant. J. Biol. Chem. 2003, 278, 37241–37248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.R.; Hooper, N.M. The Prion Protein and Lipid Rafts. Mol. Membr. Biol. 2006, 23, 89–99. [Google Scholar] [CrossRef]
- Linden, R.; Martins, V.R.; Prado, M.A.M.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the Prion Protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Lipid Rafts as Signaling Hubs in Cancer Cell Survival/death and Invasion: Implications in Tumor Progression and Therapy: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Yang, C.; Zheng, W.; Huang, L.; Hong, Y.; Wang, L.; Sha, Y. Effects of Lipid Composition and Phase on the Membrane Interaction of the Prion Peptide 106-126 Amide. Biophys. J. 2009, 96, 4610–4621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, N.; Robinson, M.; Davis, J.H.; Leonenko, Z. Melatonin Alters Fluid Phase Co-Existence in POPC/DPPC/cholesterol Membranes. Biophys. J. 2020, 119, 2391–2402. [Google Scholar] [CrossRef]
- Severcan, F.; Sahin, I.; Kazanci, N. Melatonin Strongly Interacts with Zwitterionic Model Membranes—Evidence from Fourier Transform Infrared Spectroscopy and Differential Scanning Calorimetry. Biochim. Biophys. Acta 2005, 1668, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolmatov, D.; McClintic, W.T.; Taylor, G.; Stanley, C.B.; Do, C.; Collier, C.P.; Leonenko, Z.; Lavrentovich, M.O.; Katsaras, J. Deciphering Melatonin-Stabilized Phase Separation in Phospholipid Bilayers. Langmuir 2019, 35, 12236–12245. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Morantes, C.Y.; Wille, H. The Structure of Human Prions: From Biology to Structural Models-Considerations and Pitfalls. Viruses 2014, 6, 3875–3892. [Google Scholar] [CrossRef]
- Zahn, R.; Liu, A.; Lührs, T.; Riek, R.; von Schroetter, C.; López García, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wüthrich, K. NMR Solution Structure of the Human Prion Protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Riesner, D. Biochemistry and Structure of PrP(C) and PrP(Sc). Br. Med. Bull. 2003, 66, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Hill, A.F.; Antoniou, M.; Collinge, J. Protease-Resistant Prion Protein Produced in Vitro Lacks Detectable Infectivity. J. Gen. Virol. 1999, 80 Pt 1, 11–14. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E. Conversion of Alpha-Helices into Beta-Sheets Features in the Formation of the Scrapie Prion Proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoneau, S.; Rezaei, H.; Salès, N.; Kaiser-Schulz, G.; Lefebvre-Roque, M.; Vidal, C.; Fournier, J.-G.; Comte, J.; Wopfner, F.; Grosclaude, J.; et al. In Vitro and in vivo Neurotoxicity of Prion Protein Oligomers. PLoS Pathog. 2007, 3, e125. [Google Scholar] [CrossRef]
- Lau, A.L.; Yam, A.Y.; Michelitsch, M.M.D.; Wang, X.; Gao, C.; Goodson, R.J.; Shimizu, R.; Timoteo, G.; Hall, J.; Medina-Selby, A.; et al. Characterization of Prion Protein (PrP)-Derived Peptides That Discriminate Full-Length PrPSc from PrPC. Proc. Natl. Acad. Sci. USA 2007, 104, 11551–11556. [Google Scholar] [CrossRef] [Green Version]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the Conformation of the Pathologic Isoform of the Prion Protein Enciphering and Propagating Prion Diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.Q.; Sweeting, B.; Mulligan, V.K.; Arslan, P.E.; Cashman, N.R.; Pai, E.F.; Chakrabartty, A. Prion Disease Susceptibility Is Affected by Beta-Structure Folding Propensity and Local Side-Chain Interactions in PrP. Proc. Natl. Acad. Sci. USA 2010, 107, 19808–19813. [Google Scholar] [CrossRef] [Green Version]
- Kuwata, K.; Nishida, N.; Matsumoto, T.; Kamatari, Y.O.; Hosokawa-Muto, J.; Kodama, K.; Nakamura, H.K.; Kimura, K.; Kawasaki, M.; Takakura, Y.; et al. Hot Spots in Prion Protein for Pathogenic Conversion. Proc. Natl. Acad. Sci. USA 2007, 104, 11921–11926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguzzi, A.; Falsig, J. Prion Propagation, Toxicity and Degradation. Nat. Neurosci. 2012, 15, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Abskharon, R.N.N.; Giachin, G.; Wohlkonig, A.; Soror, S.H.; Pardon, E.; Legname, G.; Steyaert, J. Probing the N-Terminal β-Sheet Conversion in the Crystal Structure of the Human Prion Protein Bound to a Nanobody. J. Am. Chem. Soc. 2014, 136, 937–944. [Google Scholar] [CrossRef]
- Kostylev, M.A.; Tuttle, M.D.; Lee, S.; Klein, L.E.; Takahashi, H.; Cox, T.O.; Gunther, E.C.; Zilm, K.W.; Strittmatter, S.M. Liquid and Hydrogel Phases of PrPC Linked to Conformation Shifts and Triggered by Alzheimer’s Amyloid-β Oligomers. Mol. Cell 2018, 72, 426–443.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rösener, N.S.; Gremer, L.; Reinartz, E.; König, A.; Brener, O.; Heise, H.; Hoyer, W.; Neudecker, P.; Willbold, D. A D-Enantiomeric Peptide Interferes with Heteroassociation of Amyloid-β Oligomers and Prion Protein. J. Biol. Chem. 2018, 293, 15748–15764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieznanski, K.; Choi, J.-K.; Chen, S.; Surewicz, K.; Surewicz, W.K. Soluble Prion Protein Inhibits Amyloid-β (Aβ) Fibrillization and Toxicity. J. Biol. Chem. 2012, 287, 33104–33108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viola, K.L.; Klein, W.L. Amyloid β Oligomers in Alzheimer’s Disease Pathogenesis, Treatment, and Diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious Effects of Amyloid Beta Oligomers Acting as an Extracellular Scaffold for mGluR5. Neuron 2010, 66, 739–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, L.T.; Strittmatter, S.M. Oligomers of Amyloid β Prevent Physiological Activation of the Cellular Prion Protein-Metabotropic Glutamate Receptor 5 Complex by Glutamate in Alzheimer Disease. J. Biol. Chem. 2016, 291, 17112–17121. [Google Scholar] [CrossRef] [Green Version]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic Targeting by Alzheimer’s-Related Amyloid Beta Oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic Glutamate Receptor 5 Is a Coreceptor for Alzheimer Aβ Oligomer Bound to Cellular Prion Protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef] [Green Version]
- De Mario, A.; Castellani, A.; Peggion, C.; Massimino, M.L.; Lim, D.; Hill, A.F.; Sorgato, M.C.; Bertoli, A. The Prion Protein Constitutively Controls Neuronal Store-Operated Ca(2+) Entry through Fyn Kinase. Front. Cell. Neurosci. 2015, 9, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J. Calcium Regulation of Neural Rhythms, Memory and Alzheimer’s Disease. J. Physiol. 2014, 592, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Petrillo, F.; Nasso, R.; Ferretti, G. Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions. Int. J. Mol. Sci. 2020, 21, 4444. [Google Scholar] [CrossRef]
- Saito, Y.D.; Jensen, A.R.; Salgia, R.; Posadas, E.M. Fyn: A Novel Molecular Target in Cancer. Cancer 2010, 116, 1629–1637. [Google Scholar] [CrossRef]
- Elias, D.; Ditzel, H.J. Fyn Is an Important Molecule in Cancer Pathogenesis and Drug Resistance. Pharmacol. Res. 2015, 100, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, Z.; Wei, Z.; Wu, J.; OuYang, J.; Huang, W.; He, Y.; Zhang, C. FYN Promotes Gastric Cancer Metastasis by Activating STAT3-Mediated Epithelial-Mesenchymal Transition. Transl. Oncol. 2020, 13, 100841. [Google Scholar] [CrossRef]
- Hsieh, M.-C.; Ho, Y.-C.; Lai, C.-Y.; Chou, D.; Wang, H.-H.; Chen, G.-D.; Lin, T.-B.; Peng, H.-Y. Melatonin Impedes Tet1-Dependent mGluR5 Promoter Demethylation to Relieve Pain. J. Pineal Res. 2017, 63, e12436. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin Regulates Aβ Production/clearance Balance and Aβ Neurotoxicity: A Potential Therapeutic Molecule for Alzheimer’s Disease. Biomed. Pharmacother. 2020, 132, 110887. [Google Scholar] [CrossRef]
- König, A.S.; Rösener, N.S.; Gremer, L.; Tusche, M.; Flender, D.; Reinartz, E.; Hoyer, W.; Neudecker, P.; Willbold, D.; Heise, H. Structural Details of Amyloid β Oligomers in Complex with Human Prion Protein as Revealed by Solid-State MAS NMR Spectroscopy. J. Biol. Chem. 2021, 296, 100499. [Google Scholar] [CrossRef]
- Nochebuena, J.; Quintanar, L.; Vela, A.; Cisneros, G.A. Structural and Electronic Analysis of the Octarepeat Region of Prion Protein with Four Cu2+ by Polarizable MD and QM/MM Simulations. Phys. Chem. Chem. Phys. 2021, 23, 21568–21578. [Google Scholar] [CrossRef]
- Surewicz, W.K.; Apostol, M.I. Prion Protein and Its Conformational Conversion: A Structural Perspective. In Prion Proteins; Tatzelt, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 135–167. [Google Scholar] [CrossRef]
- Perera, W.S.; Hooper, N.M. Ablation of the Metal Ion-Induced Endocytosis of the Prion Protein by Disease-Associated Mutation of the Octarepeat Region. Curr. Biol. 2001, 11, 519–523. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.E.; Klewpatinond, M.; Abdelraheim, S.R.; Brown, D.R.; Viles, J.H. Probing copper2+ Binding to the Prion Protein Using Diamagnetic nickel2+ and 1H NMR: The Unstructured N Terminus Facilitates the Coordination of Six copper2+ Ions at Physiological Concentrations. J. Mol. Biol. 2005, 346, 1393–1407. [Google Scholar] [CrossRef] [PubMed]
- Salzano, G.; Brennich, M.; Mancini, G.; Tran, T.H.; Legname, G.; D’Angelo, P.; Giachin, G. Deciphering Copper Coordination in the Mammalian Prion Protein Amyloidogenic Domain. Biophys. J. 2020, 118, 676–687. [Google Scholar] [CrossRef]
- Jones, C.E.; Abdelraheim, S.R.; Brown, D.R.; Viles, J.H. Preferential Cu2+ Coordination by His96 and His111 Induces Beta-Sheet Formation in the Unstructured Amyloidogenic Region of the Prion Protein. J. Biol. Chem. 2004, 279, 32018–32027. [Google Scholar] [CrossRef] [Green Version]
- Jackson, G.S.; Murray, I.; Hosszu, L.L.; Gibbs, N.; Waltho, J.P.; Clarke, A.R.; Collinge, J. Location and Properties of Metal-Binding Sites on the Human Prion Protein. Proc. Natl. Acad. Sci. USA 2001, 98, 8531–8535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Acqua, S.; Massardi, E.; Monzani, E.; Di Natale, G.; Rizzarelli, E.; Casella, L. Interaction between Hemin and Prion Peptides: Binding, Oxidative Reactivity and Aggregation. Int. J. Mol. Sci. 2020, 21, 7553. [Google Scholar] [CrossRef]
- Hooper, N.M.; Taylor, D.R.; Watt, N.T. Mechanism of the Metal-Mediated Endocytosis of the Prion Protein. Biochem. Soc. Trans. 2008, 36 Pt 6, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Watt, N.T.; Perera, W.S.S.; Hooper, N.M. Assigning Functions to Distinct Regions of the N-Terminus of the Prion Protein That Are Involved in Its Copper-Stimulated, Clathrin-Dependent Endocytosis. J. Cell Sci. 2005, 118 Pt 21, 5141–5153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunyach, C.; Jen, A.; Deng, J.; Fitzgerald, K.T.; Frobert, Y.; Grassi, J.; McCaffrey, M.W.; Morris, R. The Mechanism of Internalization of Glycosylphosphatidylinositol-Anchored Prion Protein. EMBO J. 2003, 22, 3591–3601. [Google Scholar] [CrossRef]
- Shyng, S.L.; Moulder, K.L.; Lesko, A.; Harris, D.A. The N-Terminal Domain of a Glycolipid-Anchored Prion Protein Is Essential for Its Endocytosis via Clathrin-Coated Pits. J. Biol. Chem. 1995, 270, 14793–14800. [Google Scholar] [CrossRef] [Green Version]
- Harmey, J.H.; Doyle, D.; Brown, V.; Rogers, M.S. The Cellular Isoform of the Prion Protein, PrPc, Is Associated with Caveolae in Mouse Neuroblastoma (N2a) Cells. Biochem. Biophys. Res. Commun. 1995, 210, 753–759. [Google Scholar] [CrossRef]
- Altmeppen, H.C.; Puig, B.; Dohler, F.; Thurm, D.K.; Falker, C.; Krasemann, S.; Glatzel, M. Proteolytic Processing of the Prion Protein in Health and Disease. Am. J. Neurodegener. Dis. 2012, 1, 15–31. [Google Scholar]
- McMahon, H.E.; Mangé, A.; Nishida, N.; Créminon, C.; Casanova, D.; Lehmann, S. Cleavage of the Amino Terminus of the Prion Protein by Reactive Oxygen Species. J. Biol. Chem. 2001, 276, 2286–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posadas, Y.; López-Guerrero, V.E.; Segovia, J.; Perez-Cruz, C.; Quintanar, L. Dissecting the Copper Bioinorganic Chemistry of the Functional and Pathological Roles of the Prion Protein: Relevance in Alzheimer’s Disease and Cancer. Curr. Opin. Chem. Biol. 2021, 66, 102098. [Google Scholar] [CrossRef]
- Boland, M.P.; Hatty, C.R.; Separovic, F.; Hill, A.F.; Tew, D.J.; Barnham, K.J.; Haigh, C.L.; James, M.; Masters, C.L.; Collins, S.J. Anionic Phospholipid Interactions of the Prion Protein N Terminus Are Minimally Perturbing and Not Driven Solely by the Octapeptide Repeat Domain. J. Biol. Chem. 2010, 285, 32282–32292. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Kong, Q. α-Cleavage of Cellular Prion Protein. Prion 2012, 6, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Mangé, A.; Béranger, F.; Peoc’h, K.; Onodera, T.; Frobert, Y.; Lehmann, S. Alpha- and Beta- Cleavages of the Amino-Terminus of the Cellular Prion Protein. Biol. Cell 2004, 96, 125–132. [Google Scholar] [CrossRef]
- Harris, D.A.; Huber, M.T.; van Dijken, P.; Shyng, S.L.; Chait, B.T.; Wang, R. Processing of a Cellular Prion Protein: Identification of N- and C-Terminal Cleavage Sites. Biochemistry 1993, 32, 1009–1016. [Google Scholar] [CrossRef]
- Jarosz-Griffiths, H.H.; Corbett, N.J.; Rowland, H.A.; Fisher, K.; Jones, A.C.; Baron, J.; Howell, G.J.; Cowley, S.A.; Chintawar, S.; Cader, M.Z.; et al. Proteolytic Shedding of the Prion Protein via Activation of Metallopeptidase ADAM10 Reduces Cellular Binding and Toxicity of Amyloid-β Oligomers. J. Biol. Chem. 2019, 294, 7085–7097. [Google Scholar] [CrossRef] [Green Version]
- Hooper, N.M. Roles of Proteolysis and Lipid Rafts in the Processing of the Amyloid Precursor Protein and Prion Protein. Biochem. Soc. Trans. 2005, 33 Pt 2, 335–338. [Google Scholar] [CrossRef] [Green Version]
- Yadavalli, R.; Guttmann, R.P.; Seward, T.; Centers, A.P.; Williamson, R.A.; Telling, G.C. Calpain-Dependent Endoproteolytic Cleavage of PrPSc Modulates Scrapie Prion Propagation. J. Biol. Chem. 2004, 279, 21948–21956. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.K.; Yokoyama, T.; Takata, M.; Iwamaru, Y.; Imamura, M.; Ushiki, Y.K.; Shinagawa, M. The N-Terminal Cleavage Site of PrPSc from BSE Differs from that of PrPSc from Scrapie. Biochem. Biophys. Res. Commun. 2005, 328, 1024–1027. [Google Scholar] [CrossRef]
- Linsenmeier, L.; Altmeppen, H.C.; Wetzel, S.; Mohammadi, B.; Saftig, P.; Glatzel, M. Diverse Functions of the Prion Protein-Does Proteolytic Processing Hold the Key? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2128–2137. [Google Scholar] [CrossRef]
- McDonald, A.J.; Dibble, J.P.; Evans, E.G.B.; Millhauser, G.L. A New Paradigm for Enzymatic Control of α-Cleavage and β-Cleavage of the Prion Protein. J. Biol. Chem. 2014, 289, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, V.; Hill, A.F.; Haigh, C.L.; Klug, G.M.; Masters, C.L.; Lawson, V.A.; Collins, S.J. Increased Proportions of C1 Truncated Prion Protein Protect against Cellular M1000 Prion Infection. J. Neuropathol. Exp. Neurol. 2009, 68, 1125–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, A.J.; Millhauser, G.L. PrP Overdrive: Does Inhibition of α-Cleavage Contribute to PrP(C) Toxicity and Prion Disease? Prion 2014, 8, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Watt, N.T.; Taylor, D.R.; Gillott, A.; Thomas, D.A.; Perera, W.S.S.; Hooper, N.M. Reactive Oxygen Species-Mediated Beta-Cleavage of the Prion Protein in the Cellular Response to Oxidative Stress. J. Biol. Chem. 2005, 280, 35914–35921. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.J.; Tumpach, C.; Groveman, B.R.; Drew, S.C.; Haigh, C.L. Prion Protein Cleavage Fragments Regulate Adult Neural Stem Cell Quiescence through Redox Modulation of Mitochondrial Fission and SOD2 Expression. Cell. Mol. Life Sci. 2018, 75, 3231–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.-C.; Zeniou, M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef]
- Bruschini, S.; Ciliberto, G.; Mancini, R. The Emerging Role of Cancer Cell Plasticity and Cell-Cycle Quiescence in Immune Escape. Cell Death Dis. 2020, 11, 471. [Google Scholar] [CrossRef]
- Schilling, K.M.; Tao, L.; Wu, B.; Kiblen, J.T.M.; Ubilla-Rodriguez, N.C.; Pushie, M.J.; Britt, R.D.; Roseman, G.P.; Harris, D.A.; Millhauser, G.L. Both N-Terminal and C-Terminal Histidine Residues of the Prion Protein Are Essential for Copper Coordination and Neuroprotective Self-Regulation. J. Mol. Biol. 2020, 432, 4408–4425. [Google Scholar] [CrossRef]
- Hopkins, E.; Sanvictores, T.; Sharma, S. Physiology, Acid Base Balance. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nüske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous Interactions and Protein Disaggregases Determine the Material State of Stress-Inducible RNP Granules. Elife 2015, 4, e06807. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Triandafillou, C.; Drummond, D.A. Cellular Sensing by Phase Separation: Using the Process, Not Just the Products. J. Biol. Chem. 2019, 294, 7151–7159. [Google Scholar] [CrossRef] [Green Version]
- Munder, M.C.; Midtvedt, D.; Franzmann, T.; Nüske, E.; Otto, O.; Herbig, M.; Ulbricht, E.; Müller, P.; Taubenberger, A.; Maharana, S.; et al. A pH-Driven Transition of the Cytoplasm from a Fluid- to a Solid-like State Promotes Entry into Dormancy. Elife 2016, 5, e09347. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Biring, S. A Quantitative Characterization of Interaction between Prion Protein with Nucleic Acids. Biochem. Biophys. Rep. 2018, 14, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Nandi, P.K. Nucleic Acid Induced Unfolding of Recombinant Prion Protein Globular Fragment Is pH Dependent. Protein Sci. 2014, 23, 1780–1788. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, W.H.; Puckridge, J. The Dissociation of Proteins by Chaotropic Salts. J. Biol. Chem. 1973, 248, 8429–8433. [Google Scholar] [CrossRef]
- Apetri, A.C.; Surewicz, W.K. Atypical Effect of Salts on the Thermodynamic Stability of Human Prion Protein. J. Biol. Chem. 2003, 278, 22187–22192. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, K.M.; Baldwin, R.L. Charged Histidine Affects Alpha-Helix Stability at All Positions in the Helix by Interacting with the Backbone Charges. Proc. Natl. Acad. Sci. USA 1993, 90, 11337–11340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabryelczyk, B.; Cai, H.; Shi, X.; Sun, Y.; Swinkels, P.J.M.; Salentinig, S.; Pervushin, K.; Miserez, A. Hydrogen Bond Guidance and Aromatic Stacking Drive Liquid-Liquid Phase Separation of Intrinsically Disordered Histidine-Rich Peptides. Nat. Commun. 2019, 10, 5465. [Google Scholar] [CrossRef] [Green Version]
- Van Lindt, J.; Bratek-Skicki, A.; Nguyen, P.N.; Pakravan, D.; Durán-Armenta, L.F.; Tantos, A.; Pancsa, R.; Van Den Bosch, L.; Maes, D.; Tompa, P. A Generic Approach to Study the Kinetics of Liquid-Liquid Phase Separation under near-Native Conditions. Commun. Biol. 2021, 4, 77. [Google Scholar] [CrossRef]
- Cleaves, H.J.J. Isoelectric Point. In Encyclopedia of Astrobiology; Gargaud, M., Amils, R., Quintanilla, J.C., Cleaves, H.J.J., Irvine, W.M., Pinti, D.L., Viso, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; p. 858. [Google Scholar] [CrossRef]
- Adame-Arana, O.; Weber, C.A.; Zaburdaev, V.; Prost, J.; Jülicher, F. Liquid Phase Separation Controlled by pH. Biophys. J. 2020, 119, 1590–1605. [Google Scholar] [CrossRef]
- Shaw, K.L.; Grimsley, G.R.; Yakovlev, G.I.; Makarov, A.A.; Pace, C.N. The Effect of Net Charge on the Solubility, Activity, and Stability of Ribonuclease Sa. Protein Sci. 2001, 10, 1206–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorini, M.; Bongianni, M.; Zanusso, G. Molecular Signature in Human and Animal Prion Disorders. In Prion; Tutar, Y., Ed.; IntechOpen: Rijeka, Yugoslavia, 2017. [Google Scholar] [CrossRef] [Green Version]
- Miura, T.; Hori-i, A.; Mototani, H.; Takeuchi, H. Raman Spectroscopic Study on the copper(II) Binding Mode of Prion Octapeptide and Its pH Dependence. Biochemistry 1999, 38, 11560–11569. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The Cellular Prion Protein Binds Copper in Vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Gaetke, L.M.; Chow, C.K. Copper Toxicity, Oxidative Stress, and Antioxidant Nutrients. Toxicology 2003, 189, 147–163. [Google Scholar] [CrossRef]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological Relevance and Mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.C. Ceruloplasmin and Other Copper Binding Components of Blood Plasma and Their Functions: An Update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef]
- Yim, J.; Kwon, S.B.; Han, J.S.; Kim, J.-H.; Lee, E.H.; Lee, S.-G. Total and Exchangeable Copper Assay Using Inductively Coupled Plasma Mass Spectrometry and Establishment of a Pediatric Reference Interval. Arch. Pathol. Lab. Med. 2021, 145, 877–882. [Google Scholar] [CrossRef]
- El Balkhi, S.; Poupon, J.; Trocello, J.-M.; Leyendecker, A.; Massicot, F.; Galliot-Guilley, M.; Woimant, F. Determination of Ultrafiltrable and Exchangeable Copper in Plasma: Stability and Reference Values in Healthy Subjects. Anal. Bioanal. Chem. 2009, 394, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Kirsipuu, T.; Zadorožnaja, A.; Smirnova, J.; Friedemann, M.; Plitz, T.; Tõugu, V.; Palumaa, P. Copper(II)-Binding Equilibria in Human Blood. Sci. Rep. 2020, 10, 5686. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.L.; Kratzin, H.D.; Schmidt, B.; Römer, A.; Windl, O.; Liemann, S.; Hornemann, S.; Kretzschmar, H. Prion Protein Binds Copper within the Physiological Concentration Range. J. Biol. Chem. 2001, 276, 16711–16719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper Metabolism as a Unique Vulnerability in Cancer. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef] [PubMed]
- Blockhuys, S.; Zhang, X.; Wittung-Stafshede, P. Single-Cell Tracking Demonstrates Copper Chaperone Atox1 to Be Required for Breast Cancer Cell Migration. Proc. Natl. Acad. Sci. USA 2020, 117, 2014–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.R.; Clive, C.; Haswell, S.J. Antioxidant Activity Related to Copper Binding of Native Prion Protein. J. Neurochem. 2001, 76, 69–76. [Google Scholar] [CrossRef]
- Varela-Nallar, L.; Toledo, E.M.; Larrondo, L.F.; Cabral, A.L.B.; Martins, V.R.; Inestrosa, N.C. Induction of Cellular Prion Protein Gene Expression by Copper in Neurons. Am. J. Physiol. Cell Physiol. 2006, 290, C271–C281. [Google Scholar] [CrossRef] [Green Version]
- Qin, K.; Zhao, L.; Ash, R.D.; McDonough, W.F.; Zhao, R.Y. ATM-Mediated Transcriptional Elevation of Prion in Response to Copper-Induced Oxidative Stress. J. Biol. Chem. 2009, 284, 4582–4593. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.A.; Groveman, B.R.; Williams, K.; Moore, R.; Race, B.; Haigh, C.L. Prion Protein N1 Cleavage Peptides Stimulate Microglial Interaction with Surrounding Cells. Sci. Rep. 2020, 10, 6654. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.-S.; Lalucque, H.; Malagnac, F.; Silar, P. Chapter 5-Prions and Prion-Like Phenomena in Epigenetic Inheritance. In Handbook of Epigenetics, 2nd ed.; Tollefsbol, T.O., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 61–72. [Google Scholar] [CrossRef]
- Limson, J.; Nyokong, T.; Daya, S. The Interaction of Melatonin and Its Precursors with Aluminium, Cadmium, Copper, Iron, Lead, and Zinc: An Adsorptive Voltammetric Study. J. Pineal Res. 1998, 24, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Ramos, E.; de Los Ríos, C.; Egea, J.; Del Pino, J.; Reiter, R.J. A Review of Metal-Catalyzed Molecular Damage: Protection by Melatonin. J. Pineal Res. 2014, 56, 343–370. [Google Scholar] [CrossRef]
- Cao, Y.-Y.; Qi, C.-D.; Li, S.; Wang, Z.; Wang, X.; Wang, J.; Ren, S.; Li, X.; Zhang, N.; Guo, Y.-D. Melatonin Alleviates Copper Toxicity via Improving Copper Sequestration and ROS Scavenging in Cucumber. Plant. Cell Physiol. 2019, 60, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, X.; He, Y.; Jia, L.; Yang, C.S.; Reiter, R.J.; Zhang, J. Antioxidant and Pro-Oxidant Activities of Melatonin in the Presence of Copper and Polyphenols In Vitro and In Vivo. Cells 2019, 8, 903. [Google Scholar] [CrossRef] [Green Version]
- Galano, A.; Medina, M.E.; Tan, D.X.; Reiter, R.J. Melatonin and Its Metabolites as Copper Chelating Agents and Their Role in Inhibiting Oxidative Stress: A Physicochemical Analysis. J. Pineal Res. 2015, 58, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, S.; Percival, S.L. EDTA: An Antimicrobial and Antibiofilm Agent for Use in Wound Care. Adv. Wound Care 2015, 4, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Cervantes, E.; García-Revilla, M.A.; Soto-Arredondo, K.; Villaseñor-Granados, T.; Martínez-Alfaro, M.; Robles, J. Computational Study of Metal Complexes Formed with EDTA, Melatonin, and Its Main Metabolites: Implications in Lead Intoxication and Clues to a Plausible Alternative Treatment. J. Mol. Model. 2019, 25, 18. [Google Scholar] [CrossRef]
- Reina, M.; Martínez, A. A New Free Radical Scavenging Cascade Involving Melatonin and Three of Its Metabolites (3OHM, AFMK and AMK). Comput. Theor. Chem. 2018, 1123, 111–118. [Google Scholar] [CrossRef]
- Abadi, S.H.M.H.; Shirazi, A.; Alizadeh, A.M.; Changizi, V.; Najafi, M.; Khalighfard, S.; Nosrati, H. The Effect of Melatonin on Superoxide Dismutase and Glutathione Peroxidase Activity, and Malondialdehyde Levels in the Targeted and the Non-Targeted Lung and Heart Tissues after Irradiation in Xenograft Mice Colon Cancer. Curr. Mol. Pharmacol. 2018, 11, 326–335. [Google Scholar] [CrossRef] [PubMed]
- de Fortunato Miranda, N. The Dual Role of Cellular Prion Protein in Colorectal Cancer. Ph.D. Thesis, The University of Melbourne, Melbourne, Australia, 2021. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perona, R.; Serrano, R. Increased pH and Tumorigenicity of Fibroblasts Expressing a Yeast Proton Pump. Nature 1988, 334, 438–440. [Google Scholar] [CrossRef]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic Extracellular Microenvironment and Cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Astekar, M.; Soi, S.; Manjunatha, B.S.; Shetty, D.C.; Radhakrishnan, R. pH Gradient Reversal: An Emerging Hallmark of Cancers. Recent Pat. Anticancer Drug Discov. 2015, 10, 244–258. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. The Reversed Intra- and Extracellular pH in Tumors as a Unified Strategy to Chemotherapeutic Delivery Using Targeted Nanocarriers. Acta Pharm Sin. B 2021, 11, 2243–2264. [Google Scholar] [CrossRef]
- Chen, M.; Chen, C.; Shen, Z.; Zhang, X.; Chen, Y.; Lin, F.; Ma, X.; Zhuang, C.; Mao, Y.; Gan, H.; et al. Extracellular pH Is a Biomarker Enabling Detection of Breast Cancer and Liver Cancer Using CEST MRI. Oncotarget 2017, 8, 45759–45767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirmanova, M.V.; Druzhkova, I.N.; Lukina, M.M.; Matlashov, M.E.; Belousov, V.V.; Snopova, L.B.; Prodanetz, N.N.; Dudenkova, V.V.; Lukyanov, S.A.; Zagaynova, E.V. Intracellular pH Imaging in Cancer Cells in Vitro and Tumors in Vivo Using the New Genetically Encoded Sensor SypHer2. Biochim. Biophys. Acta 2015, 1850, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. pH Sensing and Regulation in Cancer. Front. Physiol. 2013, 4, 370. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A Perfect Storm for Cancer Progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-Mediated Tumor Invasion: A Multidisciplinary Study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef] [Green Version]
- Gillies, R.J.; Raghunand, N.; Karczmar, G.S.; Bhujwalla, Z.M. MRI of the Tumor Microenvironment. J. Magn. Reson. Imaging 2002, 16, 430–450. [Google Scholar] [CrossRef]
- Martin, G.R.; Jain, R.K. Noninvasive Measurement of Interstitial pH Profiles in Normal and Neoplastic Tissue Using Fluorescence Ratio Imaging Microscopy. Cancer Res. 1994, 54, 5670–5674. [Google Scholar]
- Gillies, R.J.; Liu, Z.; Bhujwalla, Z. 31P-MRS Measurements of Extracellular pH of Tumors Using 3-Aminopropylphosphonate. Am. J. Physiol. 1994, 267 Pt 1, C195–C203. [Google Scholar] [CrossRef]
- Busa, W.B.; Nuccitelli, R. Metabolic Regulation via Intracellular pH. Am. J. Physiol. 1984, 246 Pt 2, R409–R438. [Google Scholar] [CrossRef] [Green Version]
- A Kurkdjian, A.; Guern, J. Intracellular pH: Measurement and Importance in Cell Activity. Annu. Rev. 1989, 40, 271–303. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and Regulators of Intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems Analysis of Intracellular pH Vulnerabilities for Cancer Therapy. Nat. Commun. 2018, 9, 2997. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour Hypoxia Induces a Metabolic Shift Causing Acidosis: A Common Feature in Cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [Green Version]
- Zheng, T.; Jäättelä, M.; Liu, B. pH Gradient Reversal Fuels Cancer Progression. Int. J. Biochem. Cell Biol. 2020, 125, 105796. [Google Scholar] [CrossRef]
- Reshkin, S.J.; Greco, M.R.; Cardone, R.A. Role of pHi, and Proton Transporters in Oncogene-Driven Neoplastic Transformation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, H.M.; Deitmer, J.W. Proton Transport in Cancer Cells: The Role of Carbonic Anhydrases. Int. J. Mol. Sci. 2021, 22, 3171. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Amaravadi, R.K. Targeting the Lysosome in Cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Galenkamp, K.M.O.; Commisso, C. The Golgi as a “Proton Sink” in Cancer. Front. Cell Dev. Biol. 2021, 9, 664295. [Google Scholar] [CrossRef] [PubMed]
- Raghunand, N.; Mahoney, B.P.; Gillies, R.J. Tumor Acidity, Ion Trapping and Chemotherapeutics. II. pH-Dependent Partition Coefficients Predict Importance of Ion Trapping on Pharmacokinetics of Weakly Basic Chemotherapeutic Agents. Biochem. Pharmacol. 2003, 66, 1219–1229. [Google Scholar] [CrossRef]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Omran, Z.; Scaife, P.; Stewart, S.; Rauch, C. Physical and Biological Characteristics of Multi Drug Resistance (MDR): An Integral Approach Considering pH and Drug Resistance in Cancer. Semin. Cancer Biol. 2017, 43, 42–48. [Google Scholar] [CrossRef]
- Hao, G.; Xu, Z.P.; Li, L. Manipulating Extracellular Tumour pH: An Effective Target for Cancer Therapy. RSC Adv. 2018, 8, 22182–22192. [Google Scholar] [CrossRef] [Green Version]
- Theparambil, S.M.; Weber, T.; Schmälzle, J.; Ruminot, I.; Deitmer, J.W. Proton Fall or Bicarbonate Rise: GLYCOLYTIC RATE IN MOUSE ASTROCYTES IS PAVED BY INTRACELLULAR ALKALINIZATION. J. Biol. Chem. 2016, 291, 19108–19117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Cao, M.; Liu, Y.; He, Y.; Wang, Y.; Yang, C.; Wang, W.; Du, Y.; Zhou, M.; Gao, F. Mitochondrial F1Fo-ATP Synthase Translocates to Cell Surface in Hepatocytes and Has High Activity in Tumor-like Acidic and Hypoxic Environment. Acta Biochim. Biophys. Sin. 2010, 42, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.-W.; Choo, H.-J.; Lee, J.-W.; Kim, J.-H.; Ko, Y.-G. Extracellular ATP Is Generated by ATP Synthase Complex in Adipocyte Lipid Rafts. Exp. Mol. Med. 2004, 36, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Bae, T.-J.; Kim, M.-S.; Kim, J.-W.; Kim, B.-W.; Choo, H.-J.; Lee, J.-W.; Kim, K.-B.; Lee, C.S.; Kim, J.-H.; Chang, S.Y.; et al. Lipid Raft Proteome Reveals ATP Synthase Complex in the Cell Surface. Proteomics 2004, 4, 3536–3548. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Qian, Y.; Cao, Y.; Shriwas, P.; Zhang, H.; Chen, X. Extracellular ATP, as an Energy and Phosphorylating Molecule, Induces Different Types of Drug Resistances in Cancer Cells through ATP Internalization and Intracellular ATP Level Increase. Oncotarget 2017, 8, 87860–87877. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Wang, X.; Liu, Y.; Li, Y.; Colvin, R.A.; Tong, L.; Wu, S.; Chen, X. Extracellular ATP Is Internalized by Macropinocytosis and Induces Intracellular ATP Increase and Drug Resistance in Cancer Cells. Cancer Lett. 2014, 351, 242–251. [Google Scholar] [CrossRef]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-Binding Cassette (ABC) Transporter Family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef]
- Sharma, S.; Kalra, H.; Akundi, R.S. Extracellular ATP Mediates Cancer Cell Migration and Invasion Through Increased Expression of Cyclooxygenase 2. Front. Pharmacol. 2020, 11, 617211. [Google Scholar] [CrossRef]
- Alvarez, C.L.; Troncoso, M.F.; Espelt, M.V. Extracellular ATP and Adenosine in Tumor Microenvironment: Roles in Epithelial-Mesenchymal Transition, Cell Migration, and Invasion. J. Cell. Physiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, X.; Li, Y.; Evers, M.; Zhang, H.; Chen, X. Extracellular and Macropinocytosis Internalized ATP Work Together to Induce Epithelial-Mesenchymal Transition and Other Early Metastatic Activities in Lung Cancer. Cancer Cell Int. 2019, 19, 254. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Chen, X. Abstract 3106: Roles of Extracellular ATP in Inducing Cancer Stem Cell (CSC)-like Changes in Non-Small Lung Cancer Cell Line. Cancer Res. 2021, 81 (Suppl. S36), 3106. [Google Scholar] [CrossRef]
- Dratkiewicz, E.; Simiczyjew, A.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. Hypoxia and Extracellular Acidification as Drivers of Melanoma Progression and Drug Resistance. Cells 2021, 10, 862. [Google Scholar] [CrossRef]
- Han, Y.-S.; Lee, J.H.; Yoon, Y.M.; Yun, C.W.; Noh, H.; Lee, S.H. Hypoxia-Induced Expression of Cellular Prion Protein Improves the Therapeutic Potential of Mesenchymal Stem Cells. Cell Death Dis. 2016, 7, e2395. [Google Scholar] [CrossRef]
- Sauer, H.; Dagdanova, A.; Hescheler, J.; Wartenberg, M. Redox-Regulation of Intrinsic Prion Expression in Multicellular Prostate Tumor Spheroids. Free Radic. Biol. Med. 1999, 27, 1276–1283. [Google Scholar] [CrossRef]
- Matafora, V.; Farris, F.; Restuccia, U.; Tamburri, S.; Martano, G.; Bernardelli, C.; Sofia, A.; Pisati, F.; Casagrande, F.; Lazzari, L.; et al. Amyloid Aggregates Accumulate in Melanoma Metastasis Modulating YAP Activity. EMBO Rep. 2020, 21, e50446. [Google Scholar] [CrossRef]
- Dec, R.; Puławski, W.; Dzwolak, W. Selective and Stoichiometric Incorporation of ATP by Self-Assembling Amyloid Fibrils. J. Mater. Chem. B Mater. Biol. Med. 2021, 9, 8626–8630. [Google Scholar] [CrossRef]
- Brown, D.R. ORGINAL ARTICLE Neurodegeneration and Oxidative Stress: Prion Disease Results from Loss of Antioxidant Defence. Folia Neuropathol. 2006, 43, 229–243. [Google Scholar]
- Kim, J.I.; Choi, S.I.; Kim, N.H.; Jin, J.K.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Oxidative Stress and Neurodegeneration in Prion Diseases. Ann. N. Y. Acad. Sci. 2001, 928, 182–186. [Google Scholar] [CrossRef]
- Guentchev, M.; Voigtländer, T.; Haberler, C.; Groschup, M.H.; Budka, H. Evidence for Oxidative Stress in Experimental Prion Disease. Neurobiol. Dis. 2000, 7, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Prasad, K.N.; Bondy, S.C. Oxidative and Inflammatory Events in Prion Diseases: Can They Be Therapeutic Targets? Curr. Aging Sci. 2019, 11, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Boussadia, Z.; Lamberti, J.; Mattei, F.; Pizzi, E.; Puglisi, R.; Zanetti, C.; Pasquini, L.; Fratini, F.; Fantozzi, L.; Felicetti, F.; et al. Acidic Microenvironment Plays a Key Role in Human Melanoma Progression through a Sustained Exosome Mediated Transfer of Clinically Relevant Metastatic Molecules. J. Exp. Clin. Cancer Res. 2018, 37, 245. [Google Scholar] [CrossRef] [PubMed]
- Stüwe, L.; Müller, M.; Fabian, A.; Waning, J.; Mally, S.; Noël, J.; Schwab, A.; Stock, C. pH Dependence of Melanoma Cell Migration: Protons Extruded by NHE1 Dominate Protons of the Bulk Solution. J. Physiol. 2007, 585 Pt 2, 351–360. [Google Scholar] [CrossRef]
- Moreno, A.C.R.; de Freitas Saito, R.; Tiago, M.; Massaro, R.R.; Pagni, R.L.; Pegoraro, R.; da Cruz Souza, P.; Reiter, R.J.; Campa, A.; Soengas, M.S.; et al. Melatonin Inhibits Human Melanoma Cells Proliferation and Invasion via Cell Cycle Arrest and Cytoskeleton Remodeling. Melatonin Res. 2020, 3, 194–209. [Google Scholar] [CrossRef]
- Gonzalez, R.; Sanchez, A.; Ferguson, J.A.; Balmer, C.; Daniel, C.; Cohn, A.; Robinson, W.A. Melatonin Therapy of Advanced Human Malignant Melanoma. Melanoma Res. 1991, 1, 237–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonehara, N.M.; Lacerda, J.Z.; Jardim-Perassi, B.V.; de Paula, R., Jr.; Moschetta-Pinheiro, M.G.; Souza, Y.S.T.; de Andrade, J.C.J.; De Campos Zuccari, D.A.P. Melatonin Regulates Tumor Aggressiveness under Acidosis Condition in Breast Cancer Cell Lines. Oncol. Lett. 2019, 17, 1635–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aust, S.; Brucker, B.; Graf, J.; Klimpfinger, M.; Thalhammer, T. Melatonin Modulates Acid/base Transport in Human Pancreatic Carcinoma Cells. Cell. Physiol. Biochem. 2006, 18, 91–102. [Google Scholar] [CrossRef]
- Henry, R.P. Multiple Roles of Carbonic Anhydrase in Cellular Transport and Metabolism. Annu. Rev. Physiol. 1996, 58, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Breton, S. The Cellular Physiology of Carbonic Anhydrases. JOP 2001, 2 (Suppl. S4), 159–164. [Google Scholar]
- Tafreshi, N.K.; Lloyd, M.C.; Proemsey, J.B.; Bui, M.M.; Kim, J.; Gillies, R.J.; Morse, D.L. Evaluation of CAIX and CAXII Expression in Breast Cancer at Varied O2 Levels: CAIX Is the Superior Surrogate Imaging Biomarker of Tumor Hypoxia. Mol. Imaging Biol. 2016, 18, 219–231. [Google Scholar] [CrossRef]
- Potter, C.; Harris, A.L. Hypoxia Inducible Carbonic Anhydrase IX, Marker of Tumour: Hypoxia, Survival Pathway and Therapy Target. Cell Cycle 2004, 3, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-Inducible Carbonic Anhydrase IX and XII Promote Tumor Cell Growth by Counteracting Acidosis through the Regulation of the Intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [Green Version]
- de Lima Mota, A.; Jardim-Perassi, B.V.; de Castro, T.B.; Colombo, J.; Sonehara, N.M.; Nishiyama, V.K.G.; Pierri, V.A.G.; de Campos Zuccari, D.A.P. Melatonin Modifies Tumor Hypoxia and Metabolism by Inhibiting HIF-1α and Energy Metabolic Pathway in the in Vitro and in Vivo Models of Breast Cancer. Melatonin Res. 2019, 2, 83–98. [Google Scholar] [CrossRef]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and Developmental Control of O2 Homeostasis by Hypoxia-Inducible Factor 1 Alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swenson, E.R. Hypoxia and Its Acid-Base Consequences: From Mountains to Malignancy. Adv. Exp. Med. Biol. 2016, 903, 301–323. [Google Scholar] [CrossRef]
- Magovern, G.J.; Cartwright, R.S.; Neville, J.F., Jr.; Kent, E.M. Metabolic Acidosis and the Dissociation Curve of Hemoglobin during Extracorporeal Circulation. J. Thorac. Cardiovasc. Surg. 1959, 38, 561–572. [Google Scholar] [CrossRef]
- Liu, C.; Lin, Q.; Yun, Z. Cellular and Molecular Mechanisms Underlying Oxygen-Dependent Radiosensitivity. Radiat. Res. 2015, 183, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, D.P.; Kandle, P.F.; Murray, I.; Dhamoon, A.S. Physiology, Oxyhemoglobin Dissociation Curve. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Darling, R.C.; Roughton, F.J.W. The Effect Of Methemoglobin on the Equilibrium Between Oxygen and Hemoglobin. Am. J. Physiol.-Leg. Content 1942, 137, 56–68. [Google Scholar] [CrossRef]
- Hirst, D.G.; Wood, P.J. The Influence of Haemoglobin Affinity for Oxygen on Tumour Radiosensitivity. Br. J. Cancer 1987, 55, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkpatrick, J.P.; Cárdenas-Navia, L.I.; Dewhirst, M.W. Predicting the Effect of Temporal Variations in pO2 on Tumor Radiosensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.D.; Chavaudra, N.; Martin, L.; Guichard, M. Correlation between Radiosensitivity, Percentage Hypoxic Cells and pO2 Measurements in One Rodent and Two Human Tumor Xenografts. Radiat. Res. 1994, 139, 1–8. [Google Scholar] [CrossRef]
- Lamonte, G.; Tang, X.; Chen, J.L.-Y.; Wu, J.; Ding, C.-K.C.; Keenan, M.M.; Sangokoya, C.; Kung, H.-N.; Ilkayeva, O.; Boros, L.G.; et al. Acidosis Induces Reprogramming of Cellular Metabolism to Mitigate Oxidative Stress. Cancer Metab. 2013, 1, 23. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, D.; Simon, M.C. Hypoxia, Lipids, and Cancer: Surviving the Harsh Tumor Microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybner, C.; Finel-Szermanski, S.; Felin, M.; Sahraoui, T.; Rousseau, C.; Fournier, J.G.; Sève, A.P.; Botti, J. The Cellular Prion Protein: A New Partner of the Lectin CBP70 in the Nucleus of NB4 Human Promyelocytic Leukemia Cells. J. Cell. Biochem. 2002, 84, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, T.; Tsuchiya, K.; Sato, I.; Takeyama, N.; Ueda, S.; Tagawa, Y.; Kimura, K.M.; Nakamura, I.; Wu, G.; Sakudo, A.; et al. A Monoclonal Antibody (1D12) Defines Novel Distribution Patterns of Prion Protein (PrP) as Granules in Nucleus. Biochem. Biophys. Res. Commun. 2008, 366, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Strom, A.; Wang, G.-S.; Picketts, D.J.; Reimer, R.; Stuke, A.W.; Scott, F.W. Cellular Prion Protein Localizes to the Nucleus of Endocrine and Neuronal Cells and Interacts with Structural Chromatin Components. Eur. J. Cell Biol. 2011, 90, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Fouquet, S.; Strup-Perrot, C.; Pichol Thievend, C.; Petit, C.; Loew, D.; Faussat, A.-M.; Yvernault, L.; Pinçon-Raymond, M.; Chambaz, J.; et al. The Cellular Prion Protein PrP(c) Is Involved in the Proliferation of Epithelial Cells and in the Distribution of Junction-Associated Proteins. PLoS ONE 2008, 3, e3000. [Google Scholar] [CrossRef]
- Yao, Y.; Dai, W. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 1000165. [Google Scholar] [CrossRef] [PubMed]
- Hornshaw, M.P.; McDermott, J.R.; Candy, J.M.; Lakey, J.H. Copper Binding to the N-Terminal Tandem Repeat Region of Mammalian and Avian Prion Protein: Structural Studies Using Synthetic Peptides. Biochem. Biophys. Res. Commun. 1995, 214, 993–999. [Google Scholar] [CrossRef]
- Caputo, A.; Sarnataro, D.; Campana, V.; Costanzo, M.; Negro, A.; Sorgato, M.C.; Zurzolo, C. Doppel and PrPC Co-Immunoprecipitate in Detergent-Resistant Membrane Domains of Epithelial FRT Cells. Biochem. J. 2009, 425, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martellucci, S.; Santacroce, C.; Santilli, F.; Manganelli, V.; Sorice, M.; Mattei, V. Prion Protein in Stem Cells: A Lipid Raft Component Involved in the Cellular Differentiation Process. Int. J. Mol. Sci. 2020, 21, 4168. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Baskakov, I.V. The Cellular Form of the Prion Protein Is Involved in Controlling Cell Cycle Dynamics, Self-Renewal, and the Fate of Human Embryonic Stem Cell Differentiation. J. Neurochem. 2013, 124, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.C.; Steele, A.D.; Lindquist, S.; Lodish, H.F. Prion Protein Is Expressed on Long-Term Repopulating Hematopoietic Stem Cells and Is Important for Their Self-Renewal. Proc. Natl. Acad. Sci. USA 2006, 103, 2184–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodelet, V.C.; Cashman, N.R. Prion Protein Expression in Human Leukocyte Differentiation. Blood 1998, 91, 1556–1561. [Google Scholar] [CrossRef]
- Barba-Bon, A.; Nilam, M.; Hennig, A. Supramolecular Chemistry in the Biomembrane. ChemBioChem 2020, 21, 886–910. [Google Scholar] [CrossRef]
- Martellucci, S.; Santacroce, C.; Manganelli, V.; Santilli, F.; Piccoli, L.; Cassetta, M.; Misasi, R.; Sorice, M.; Mattei, V. Isolation, Propagation, and Prion Protein Expression During Neuronal Differentiation of Human Dental Pulp Stem Cells. J. Vis. Exp. 2019, 18, e59282. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Ge, F.; Guo, C.; Luo, G.; Wang, X.; Han, G.; Zhang, D.; Wang, J.; Li, K.; Pan, Y.; et al. Inhibition of PI3K/Akt Partially Leads to the Inhibition of PrP(C)-Induced Drug Resistance in Gastric Cancer Cells. FEBS J. 2009, 276, 685–694. [Google Scholar] [CrossRef]
- Pushie, M.J.; Pickering, I.J.; Martin, G.R.; Tsutsui, S.; Jirik, F.R.; George, G.N. Prion Protein Expression Level Alters Regional Copper, Iron and Zinc Content in the Mouse Brain. Metallomics 2011, 3, 206–214. [Google Scholar] [CrossRef]
- Gasperini, L.; Meneghetti, E.; Legname, G.; Benetti, F. In Absence of the Cellular Prion Protein, Alterations in Copper Metabolism and Copper-Dependent Oxidase Activity Affect Iron Distribution. Front. Neurosci. 2016, 10, 437. [Google Scholar] [CrossRef] [Green Version]
- Osaki, S.; Johnson, D.A.; Frieden, E. The Mobilization of Iron from the Perfused Mammalian Liver by a Serum Copper Enzyme, Ferroxidase, I.J. Biol. Chem. 1971, 246, 3018–3023. [Google Scholar] [CrossRef]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on Iron and Its Importance for Human Health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar]
- Singh, A.; Mohan, M.L.; Isaac, A.O.; Luo, X.; Petrak, J.; Vyoral, D.; Singh, N. Prion Protein Modulates Cellular Iron Uptake: A Novel Function with Implications for Prion Disease Pathogenesis. PLoS ONE 2009, 4, e4468. [Google Scholar] [CrossRef]
- Singh, A.; Isaac, A.O.; Luo, X.; Mohan, M.L.; Cohen, M.L.; Chen, F.; Kong, Q.; Bartz, J.; Singh, N. Abnormal Brain Iron Homeostasis in Human and Animal Prion Disorders. PLoS Pathog. 2009, 5, e1000336. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Marín-Hernández, A.; Gallardo-Pérez, J.C.; Rodríguez-Enríquez, S.; Encalada, R.; Moreno-Sánchez, R.; Saavedra, E. Modeling Cancer Glycolysis. Biochim. Biophys. Acta 2011, 1807, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging Roles and the Regulation of Aerobic Glycolysis in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef]
- Lorito, N.; Bacci, M.; Smiriglia, A.; Mannelli, M.; Parri, M.; Comito, G.; Ippolito, L.; Giannoni, E.; Bonechi, M.; Benelli, M.; et al. Glucose Metabolic Reprogramming of ER Breast Cancer in Acquired Resistance to the CDK4/6 Inhibitor Palbociclib. Cells 2020, 9, 668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, H.; Yang, H.; Bai, M.; Ning, T.; Deng, T.; Liu, R.; Fan, Q.; Zhu, K.; Li, J.; et al. Exosome-Delivered circRNA Promotes Glycolysis to Induce Chemoresistance through the miR-122-PKM2 Axis in Colorectal Cancer. Mol. Oncol. 2020, 14, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Kania, K.D.; Blauz, A.; Ciszewski, W.M. The Lactate Receptor (HCAR1/GPR81) Contributes to Doxorubicin Chemoresistance via ABCB1 Transporter up-Regulation in Human Cervical Cancer HeLa Cells. J. Physiol. Pharmacol. 2017, 68, 555–564. [Google Scholar]
- Ruprecht, B.; Zaal, E.A.; Zecha, J.; Wu, W.; Berkers, C.R.; Kuster, B.; Lemeer, S. Lapatinib Resistance in Breast Cancer Cells Is Accompanied by Phosphorylation-Mediated Reprogramming of Glycolysis. Cancer Res. 2017, 77, 1842–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldonza, M.B.D.; Hong, J.-Y.; Lee, S.K. Paclitaxel-Resistant Cancer Cell-Derived Secretomes Elicit ABCB1-Associated Docetaxel Cross-Resistance and Escape from Apoptosis through FOXO3a-Driven Glycolytic Regulation. Exp. Mol. Med. 2017, 49, e286. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, J.A.; Wanka, C.; Burger, M.C.; Urban, H.; Hartel, I.; von Renesse, J.; Harter, P.N.; Mittelbronn, M.; Steinbach, J.P.; Rieger, J. Suppression of Oxidative Phosphorylation Confers Resistance against Bevacizumab in Experimental Glioma. J. Neurochem. 2018, 144, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Jiang, H.; Wu, C.; Jiang, Y.; Xiao, L.; Tian, Y. Overcoming Cetuximab Resistance in Ewing’s Sarcoma by Inhibiting Lactate Dehydrogenase-A. Mol. Med. Rep. 2016, 14, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Beauchamp, L.; Himonas, E.; Helgason, G.V. Mitochondrial Metabolism as a Potential Therapeutic Target in Myeloid Leukaemia. Leukemia 2021, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.-E.; Chiche, J. Metabolic Reprogramming of Non-Hodgkin’s B-Cell Lymphomas and Potential Therapeutic Strategies. Front. Oncol. 2018, 8, 556. [Google Scholar] [CrossRef]
- Reyes-Castellanos, G.; Masoud, R.; Carrier, A. Mitochondrial Metabolism in PDAC: From Better Knowledge to New Targeting Strategies. Biomedicines 2020, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Salhi, A.; Jordan, A.C.; Bochaca, I.I.; Izsak, A.; Darvishian, F.; Houvras, Y.; Giles, K.M.; Osman, I. Oxidative Phosphorylation Promotes Primary Melanoma Invasion. Am. J. Pathol. 2020, 190, 1108–1117. [Google Scholar] [CrossRef]
- Schöpf, B.; Weissensteiner, H.; Schäfer, G.; Fazzini, F.; Charoentong, P.; Naschberger, A.; Rupp, B.; Fendt, L.; Bukur, V.; Giese, I.; et al. OXPHOS Remodeling in High-Grade Prostate Cancer Involves mtDNA Mutations and Increased Succinate Oxidation. Nat. Commun. 2020, 11, 1487. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [Green Version]
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.-V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C.; et al. Metabolic Reprogramming of Oncogene-Addicted Cancer Cells to OXPHOS as a Mechanism of Drug Resistance. Redox Biol. 2019, 25, 101076. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.-S.; Seo, J.; Lee, S.-H.; Kang, J.H.; Song, J.; Kim, S.-Y. Targeting Mitochondrial Oxidative Phosphorylation Abrogated Irinotecan Resistance in NSCLC. Sci. Rep. 2018, 8, 15707. [Google Scholar] [CrossRef] [PubMed]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.J.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1α-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-S.; Lee, H.; Jang, H.; Woo, S.M.; Park, J.B.; Lee, S.-H.; Kang, J.H.; Kim, H.Y.; Song, J.; Kim, S.-Y. Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling. Cells 2020, 9, 2013. [Google Scholar] [CrossRef] [PubMed]
- Denise, C.; Paoli, P.; Calvani, M.; Taddei, M.L.; Giannoni, E.; Kopetz, S.; Kazmi, S.M.A.; Pia, M.M.; Pettazzoni, P.; Sacco, E.; et al. 5-Fluorouracil Resistant Colon Cancer Cells Are Addicted to OXPHOS to Survive and Enhance Stem-like Traits. Oncotarget 2015, 6, 41706–41721. [Google Scholar] [CrossRef] [Green Version]
- Salunkhe, S.; Mishra, S.V.; Ghorai, A.; Hole, A.; Chandrani, P.; Dutt, A.; Chilakapati, M.; Dutt, S. Metabolic Rewiring in Drug Resistant Cells Exhibit Higher OXPHOS and Fatty Acids as Preferred Major Source to Cellular Energetics. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148300. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Arismendi-Morillo, G.; Mukherjee, P.; Chinopoulos, C. On the Origin of ATP Synthesis in Cancer. iScience 2020, 23, 101761. [Google Scholar] [CrossRef]
- Veech, R.L.; Lawson, J.W.; Cornell, N.W.; Krebs, H.A. Cytosolic Phosphorylation Potential. J. Biol. Chem. 1979, 254, 6538–6547. [Google Scholar] [CrossRef]
- Veech, R.L.; King, M.T.; Pawlosky, R.; Bradshaw, P.C.; Curtis, W. Relationship between Inorganic Ion Distribution, Resting Membrane Potential, and the ΔG’ of ATP Hydrolysis: A New Paradigm. FASEB J. 2019, 33, 13126–13130. [Google Scholar] [CrossRef] [Green Version]
- Poljsak, B.; Kovac, V.; Dahmane, R.; Levec, T.; Starc, A. Cancer Etiology: A Metabolic Disease Originating from Life’s Major Evolutionary Transition? Oxid. Med. Cell. Longev. 2019, 2019, 7831952. [Google Scholar] [CrossRef] [Green Version]
- Fiorito, V.; Chiabrando, D.; Petrillo, S.; Bertino, F.; Tolosano, E. The Multifaceted Role of Heme in Cancer. Front. Oncol. 2019, 9, 1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yu, L.; Ding, J.; Chen, Y. Iron Metabolism in Cancer. Int. J. Mol. Sci. 2018, 20, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazan, H.H.; Urfali-Mamatoglu, C.; Gunduz, U. Iron Metabolism and Drug Resistance in Cancer. Biometals 2017, 30, 629–641. [Google Scholar] [CrossRef]
- Forciniti, S.; Greco, L.; Grizzi, F.; Malesci, A.; Laghi, L. Iron Metabolism in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Fan, Z.; Yang, Y.; Gu, C. Iron Metabolism and Its Contribution to Cancer (Review). Int. J. Oncol. 2019, 54, 1143–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, M.; Xue, X. Targeting Iron Metabolism in Cancer Therapy. Theranostics 2021, 11, 8412–8429. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered Iron Metabolism and Impact in Cancer Biology, Metastasis, and Immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Bastide, N.; Morois, S.; Cadeau, C.; Kangas, S.; Serafini, M.; Gusto, G.; Dossus, L.; Pierre, F.H.; Clavel-Chapelon, F.; Boutron-Ruault, M.-C. Heme Iron Intake, Dietary Antioxidant Capacity, and Risk of Colorectal Adenomas in a Large Cohort Study of French Women. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 640–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastide, N.M.; Chenni, F.; Audebert, M.; Santarelli, R.L.; Taché, S.; Naud, N.; Baradat, M.; Jouanin, I.; Surya, R.; Hobbs, D.A.; et al. A Central Role for Heme Iron in Colon Carcinogenesis Associated with Red Meat Intake. Cancer Res. 2015, 75, 870–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Mackey, M.M.; Diaz, A.A.; Cox, D.P. Hydroxyl Radical Is Produced via the Fenton Reaction in Submitochondrial Particles under Oxidative Stress: Implications for Diseases Associated with Iron Accumulation. Redox Rep. 2009, 14, 102–108. [Google Scholar] [CrossRef]
- Oliver, T.; Sánchez-Baracaldo, P.; Larkum, A.W.; Rutherford, A.W.; Cardona, T. Time-Resolved Comparative Molecular Evolution of Oxygenic Photosynthesis. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148400. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Chrysina, M.; Cox, N. Water Oxidation in Photosystem II. Photosynth. Res. 2019, 142, 105–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rantamäki, S.; Meriluoto, J.; Spoof, L.; Puputti, E.-M.; Tyystjärvi, T.; Tyystjärvi, E. Oxygen Produced by Cyanobacteria in Simulated Archaean Conditions Partly Oxidizes Ferrous Iron but Mostly Escapes-Conclusions about Early Evolution. Photosynth. Res. 2016, 130, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Benger, G.; Govindjee. The Mechanism of Photosynthetic Water Oxidation. Photosynth. Res. 1985, 6, 33–55. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Mahad, D.J. A Method to Detect Cytochrome c Oxidase Activity and Mitochondrial Proteins in Oligodendrocytes. Methods Mol. Biol. 2019, 1936, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Tsukihara, T.; Shimokata, K.; Katayama, Y.; Shimada, H.; Muramoto, K.; Aoyama, H.; Mochizuki, M.; Shinzawa-Itoh, K.; Yamashita, E.; Yao, M.; et al. The Low-Spin Heme of Cytochrome c Oxidase as the Driving Element of the Proton-Pumping Process. Proc. Natl. Acad. Sci. USA 2003, 100, 15304–15309. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, P. Observations on the Oxidation of Cytochrome C. Arch. Biochem. Biophys. 1964, 106, 25–48. [Google Scholar] [CrossRef]
- Hooda, J.; Shah, A.; Zhang, L. Heme, an Essential Nutrient from Dietary Proteins, Critically Impacts Diverse Physiological and Pathological Processes. Nutrients 2014, 6, 1080–1102. [Google Scholar] [CrossRef] [Green Version]
- Reedy, C.J.; Gibney, B.R. Heme Protein Assemblies. Chem. Rev. 2004, 104, 617–649. [Google Scholar] [CrossRef]
- Sachar, M.; Anderson, K.E.; Ma, X. Protoporphyrin IX: The Good, the Bad, and the Ugly. J. Pharmacol. Exp. Ther. 2016, 356, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Ajioka, R.S.; Phillips, J.D.; Kushner, J.P. Biosynthesis of Heme in Mammals. Biochim. Biophys. Acta 2006, 1763, 723–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorito, V.; Allocco, A.L.; Petrillo, S.; Gazzano, E.; Torretta, S.; Marchi, S.; Destefanis, F.; Pacelli, C.; Audrito, V.; Provero, P.; et al. The Heme Synthesis-Export System Regulates the Tricarboxylic Acid Cycle Flux and Oxidative Phosphorylation. Cell Rep. 2021, 35, 109252. [Google Scholar] [CrossRef]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a Human Heme Exporter That Is Essential for Erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.; Ding, J.; Yang, Z.; Li, H.; Ding, H.; Chen, Q. LncRNA FLVCR1-AS1 Promotes Proliferation, Migration and Activates Wnt/β-Catenin Pathway through miR-381-3p/CTNNB1 Axis in Breast Cancer. Cancer Cell Int. 2020, 20, 1–12. [Google Scholar] [CrossRef]
- Lin, H.; Shangguan, Z.; Zhu, M.; Bao, L.; Zhang, Q.; Pan, S. lncRNA FLVCR1-AS1 Silencing Inhibits Lung Cancer Cell Proliferation, Migration, and Invasion by Inhibiting the Activity of the Wnt/β-Catenin Signaling Pathway. J. Cell. Biochem. 2019, 120, 10625–10632. [Google Scholar] [CrossRef]
- Peng, C.; Song, Y.; Chen, W.; Wang, X.; Liu, X.; Wang, F.; Wu, D.; Ma, S.; Wang, X.; Gao, C. FLVCR1 Promotes the Proliferation and Tumorigenicity of Synovial Sarcoma through Inhibiting Apoptosis and Autophagy. Int. J. Oncol. 2018, 52, 1559–1568. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.; Kumari, S.; Kalainayakan, S.P.; Campbell, J., 3rd; Ghosh, P.; Zhou, H.; FitzGerald, K.E.; Li, M.; Mason, R.P.; Zhang, L.; et al. The Vascular Disrupting Agent Combretastatin A-4 Phosphate Causes Prolonged Elevation of Proteins Involved in Heme Flux and Function in Resistant Tumor Cells. Oncotarget 2018, 9, 4090–4101. [Google Scholar] [CrossRef] [Green Version]
- Hooda, J.; Cadinu, D.; Alam, M.M.; Shah, A.; Cao, T.M.; Sullivan, L.A.; Brekken, R.; Zhang, L. Enhanced Heme Function and Mitochondrial Respiration Promote the Progression of Lung Cancer Cells. PLoS ONE 2013, 8, e63402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalainayakan, S.P.; FitzGerald, K.E.; Konduri, P.C.; Vidal, C.; Zhang, L. Essential Roles of Mitochondrial and Heme Function in Lung Cancer Bioenergetics and Tumorigenesis. Cell Biosci. 2018, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Panigaj, M.; Brouckova, A.; Glierova, H.; Dvorakova, E.; Simak, J.; Vostal, J.G.; Holada, K. Underestimation of the Expression of Cellular Prion Protein on Human Red Blood Cells. Transfusion 2011, 51, 1012–1021. [Google Scholar] [CrossRef]
- Holada, K.; Vostal, J.G. Different Levels of Prion Protein (PrPc) Expression on Hamster, Mouse and Human Blood Cells. Br. J. Haematol. 2000, 110, 472–480. [Google Scholar] [CrossRef]
- Ogun, A.S.; Joy, N.V.; Valentine, M. Biochemistry, Heme Synthesis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Marengo-Rowe, A.J. Structure-Function Relations of Human Hemoglobins. In Baylor University Medical Center; 2006; Volume 19, pp. 239–245. [Google Scholar] [CrossRef]
- Frey, P.A.; Reed, G.H. The Ubiquity of Iron. ACS Chem. Biol. 2012, 7, 1477–1481. [Google Scholar] [CrossRef]
- Huang, X.; Groves, J.T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soutyrine, A.; Yogasingam, N.; Huang, H.; Mitchell, G. Effects of Heme-PrP Complex on Cell-Free Conversion and Peroxidase-Linked Immunodetection of Prions in Blood-Based Assays. Res. Vet. Sci. 2015, 101, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Raymond, L.D.; Schoen, B.; Raymond, G.J.; Kett, L.; Moore, R.A.; Johnson, L.M.; Taubner, L.; Speare, J.O.; Onwubiko, H.A.; et al. Hemin Interactions and Alterations of the Subcellular Localization of Prion Protein. J. Biol. Chem. 2007, 282, 36525–36533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, B.S.; Liu, T.; Li, R.; Pan, T.; Petersen, R.B.; Smith, M.A.; Gambetti, P.; Perry, G.; Manson, J.C.; Brown, D.R.; et al. Increased Levels of Oxidative Stress Markers Detected in the Brains of Mice Devoid of Prion Protein. J. Neurochem. 2001, 76, 565–572. [Google Scholar] [CrossRef] [Green Version]
- White, A.R.; Collins, S.J.; Maher, F.; Jobling, M.F.; Stewart, L.R.; Thyer, J.M.; Beyreuther, K.; Masters, C.L.; Cappai, R. Prion Protein-Deficient Neurons Reveal Lower Glutathione Reductase Activity and Increased Susceptibility to Hydrogen Peroxide Toxicity. Am. J. Pathol. 1999, 155, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Tamada, M.; Nagano, O.; Tateyama, S.; Ohmura, M.; Yae, T.; Ishimoto, T.; Sugihara, E.; Onishi, N.; Yamamoto, T.; Yanagawa, H.; et al. Modulation of Glucose Metabolism by CD44 Contributes to Antioxidant Status and Drug Resistance in Cancer Cells. Cancer Res. 2012, 72, 1438–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossu, V.; Bonanomi, M.; Bauckneht, M.; Ravera, S.; Righi, N.; Miceli, A.; Morbelli, S.; Orengo, A.M.; Piccioli, P.; Bruno, S.; et al. Two High-Rate Pentose-Phosphate Pathways in Cancer Cells. Sci. Rep. 2020, 10, 22111. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zheng, Q.; Meng, X. Hyperglycemia and Chemoresistance in Breast Cancer: From Cellular Mechanisms to Treatment Response. Front. Oncol. 2021, 11, 628359. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Meng, Y.-Q.; Xu, X.-F.; Gu, J. Blockade of GLUT1 by WZB117 Resensitizes Breast Cancer Cells to Adriamycin. Anticancer Drugs 2017, 28, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient Transporters in Cancer: Relevance to Warburg Hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef]
- Simcox, J.A.; Mitchell, T.C.; Gao, Y.; Just, S.F.; Cooksey, R.; Cox, J.; Ajioka, R.; Jones, D.; Lee, S.-H.; King, D.; et al. Dietary Iron Controls Circadian Hepatic Glucose Metabolism through Heme Synthesis. Diabetes 2015, 64, 1108–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.-Q.; Sun, Y.-P.; Ruan, C.-P.; Xu, X.-Y.; Ge, J.-H.; He, J.; Xu, Z.-D.; Wang, Q.; Gao, W.-C. Cellular Prion Protein Promotes Glucose Uptake through the Fyn-HIF-2α-Glut1 Pathway to Support Colorectal Cancer Cell Survival. Cancer Sci. 2011, 102, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Soto, C. Prion-Like Protein Aggregates and Type 2 Diabetes. Cold Spring Harb. Perspect. Med. 2017, 7, a024315. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Dey, S.G. Heme Bound Amylin: Spectroscopic Characterization, Reactivity, and Relevance to Type 2 Diabetes. Inorg. Chem. 2013, 52, 5226–5235. [Google Scholar] [CrossRef]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kisilevsky, R.; Szarek, W.A.; Kahn, S.E. Oxidative Stress Is Induced by Islet Amyloid Formation and Time-Dependently Mediates Amyloid-Induced Beta Cell Apoptosis. Diabetologia 2009, 52, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppenol, W.H.; Stanbury, D.M.; Bounds, P.L. Electrode Potentials of Partially Reduced Oxygen Species, from Dioxygen to Water. Free Radic. Biol. Med. 2010, 49, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, Z.; Xu, H.; Zhang, P.; Gao, Z.; Li, H. Insulin Enhances the Peroxidase Activity of Heme by Forming Heme-Insulin Complex: Relevance to Type 2 Diabetes Mellitus. Int. J. Biol. Macromol. 2017, 102, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Navarrete, J.M.; Rodríguez, A.; Ortega, F.; Becerril, S.; Sabater-Masdeu, M.; Latorre, J.; Ricart, W.; Frühbeck, G.; Fernández-Real, J.M. Increased Adipose Tissue Heme Levels and Exportation Are Associated with Altered Systemic Glucose Metabolism. Sci. Rep. 2017, 7, 5305. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, K.T.; Chang, H.-C.; Ardehali, H. Role of Heme in Cardiovascular Physiology and Disease. J. Am. Heart Assoc. 2015, 4, e001138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A Possible Physiological Mediator of Low Density Lipoprotein Oxidation and Endothelial Injury. Arterioscler. Thromb. 1991, 11, 1700–1711. [Google Scholar] [CrossRef] [Green Version]
- Laird, M.D.; Wakade, C.; Alleyne, C.H., Jr.; Dhandapani, K.M. Hemin-Induced Necroptosis Involves Glutathione Depletion in Mouse Astrocytes. Free Radic. Biol. Med. 2008, 45, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.R.; Dang, T.N.; Dringen, R.; Bishop, G.M. Hemin Toxicity: A Preventable Source of Brain Damage Following Hemorrhagic Stroke. Redox Rep. 2009, 14, 228–235. [Google Scholar] [CrossRef]
- Harvey, J.W.; Beutler, E. Binding of Heme by Glutathione S-Transferase: A Possible Role of the Erythrocyte Enzyme. Blood 1982, 60, 1227–1230. [Google Scholar] [CrossRef]
- Aft, R.L.; Mueller, G.C. Degradation and Covalent Cross-Linking of Glutathione Reductase by Hemin. Life Sci. 1985, 36, 2153–2161. [Google Scholar] [CrossRef]
- Ye, W.; Zhang, L. Heme Controls the Expression of Cell Cycle Regulators and Cell Growth in HeLa Cells. Biochem. Biophys. Res. Commun. 2004, 315, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Aboelella, N.S.; Brandle, C.; Kim, T.; Ding, Z.-C.; Zhou, G. Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy. Cancers 2021, 13, 986. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Singh, N. Prion Protein-Hemin Interaction Upregulates Hemoglobin Synthesis: Implications for Cerebral Hemorrhage and Sporadic Creutzfeldt-Jakob Disease. J. Alzheimers Dis. 2016, 51, 107–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mense, S.M.; Zhang, L. Heme: A Versatile Signaling Molecule Controlling the Activities of Diverse Regulators Ranging from Transcription Factors to MAP Kinases. Cell Res. 2006, 16, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Padmanaban, G.; Venkateswar, V.; Rangarajan, P.N. Haem as a Multifunctional Regulator. Trends Biochem. Sci. 1989, 14, 492–496. [Google Scholar] [CrossRef]
- Zhu, Y.; Hon, T.; Zhang, L. Heme Initiates Changes in the Expression of a Wide Array of Genes during the Early Erythroid Differentiation Stage. Biochem. Biophys. Res. Commun. 1999, 258, 87–93. [Google Scholar] [CrossRef]
- Ulianov, S.V.; Galitsyna, A.A.; Flyamer, I.M.; Golov, A.K.; Khrameeva, E.E.; Imakaev, M.V.; Abdennur, N.A.; Gelfand, M.S.; Gavrilov, A.A.; Razin, S.V. Activation of the Alpha-Globin Gene Expression Correlates with Dramatic Upregulation of Nearby Non-Globin Genes and Changes in Local and Large-Scale Chromatin Spatial Structure. Epigenetics Chromatin 2017, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Liao, R.; Zheng, Y.; Liu, X.; Zhang, Y.; Seim, G.; Tanimura, N.; Wilson, G.M.; Hematti, P.; Coon, J.J.; Fan, J.; et al. Discovering How Heme Controls Genome Function Through Heme-Omics. Cell Rep. 2020, 31, 107832. [Google Scholar] [CrossRef]
- Caughey, W.S.; Raymond, L.D.; Horiuchi, M.; Caughey, B. Inhibition of Protease-Resistant Prion Protein Formation by Porphyrins and Phthalocyanines. Proc. Natl. Acad. Sci. USA 1998, 95, 12117–12122. [Google Scholar] [CrossRef] [Green Version]
- Sadrzadeh, S.M.; Graf, E.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin. A Biologic Fenton Reagent. J. Biol. Chem. 1984, 259, 14354–14356. [Google Scholar] [CrossRef]
- Richards, R.S.; Roberts, T.K.; Dunstan, R.H.; McGregor, N.R.; Butt, H.L. Erythrocyte Antioxidant Systems Protect Cultured Endothelial Cells against Oxidant Damage. Biochem. Mol. Biol. Int. 1998, 46, 857–865. [Google Scholar] [CrossRef]
- Rifkind, J.M.; Nagababu, E. Hemoglobin Redox Reactions and Red Blood Cell Aging. Antioxid. Redox Signal. 2013, 18, 2274–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Navarro, G.; Martínez-Pinilla, E. Antioxidant Defense Mechanisms in Erythrocytes and in the Central Nervous System. Antioxidants 2019, 8, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansouri, A.; Perry, C.A. Hemoglobin Autoxidation at Physiological Concentrations. Hemoglobin 1987, 11, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Kanias, T.; Acker, J.P. Biopreservation of Red Blood Cells—The Struggle with Hemoglobin Oxidation. FEBS J. 2010, 277, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Free-Radical Production and Oxidative Reactions of Hemoglobin. Environ. Health Perspect. 1985, 64, 321–330. [Google Scholar] [CrossRef]
- Kassa, T.; Jana, S.; Meng, F.; Alayash, A.I. Differential Heme Release from Various Hemoglobin Redox States and the Upregulation of Cellular Heme Oxygenase-1. FEBS Open Bio 2016, 6, 876–884. [Google Scholar] [CrossRef]
- Hultquist, D.E.; Passon, P.G. Catalysis of Methaemoglobin Reduction by Erythrocyte Cytochrome B5 and Cytochrome B5 Reductase. Nat. New Biol. 1971, 229, 252–254. [Google Scholar] [CrossRef]
- Siendones, E.; SantaCruz-Calvo, S.; Martín-Montalvo, A.; Cascajo, M.V.; Ariza, J.; López-Lluch, G.; Villalba, J.M.; Acquaviva-Bourdain, C.; Roze, E.; Bernier, M.; et al. Membrane-Bound CYB5R3 Is a Common Effector of Nutritional and Oxidative Stress Response through FOXO3a and Nrf2. Antioxid. Redox Signal. 2014, 21, 1708–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulbarelli, A.; Valentini, A.; DeSilvestris, M.; Cappellini, M.D.; Borgese, N. An Erythroid-Specific Transcript Generates the Soluble Form of NADH-Cytochrome b5 Reductase in Humans. Blood 1998, 92, 310–319. [Google Scholar] [CrossRef]
- Passon, P.G.; Hultquist, D.E. Soluble Cytochrome B 5 Reductase from Human Erythrocytes. Biochim. Biophys. Acta 1972, 275, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Choury, D.; Leroux, A.; Kaplan, J.C. Membrane-Bound Cytochrome b5 Reductase (methemoglobin Reductase) in Human Erythrocytes. Study in Normal and Methemoglobinemic Subjects. J. Clin. Investig. 1981, 67, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Borgese, N.; Pietrini, G.; Gaetani, S. Concentration of NADH-Cytochrome b5 Reductase in Erythrocytes of Normal and Methemoglobinemic Individuals Measured with a Quantitative Radioimmunoblotting Assay. J. Clin. Investig. 1987, 80, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Samhan-Arias, A.K.; Garcia-Bereguiain, M.A.; Martin-Romero, F.J.; Gutierrez-Merino, C. Clustering of Plasma Membrane-Bound Cytochrome b5 Reductase within “Lipid Raft” Microdomains of the Neuronal Plasma Membrane. Mol. Cell. Neurosci. 2009, 40, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Hayashi, S.; Yoshida, T. Participation of a Cytochrome b5-like Hemoprotein of Outer Mitochondrial Membrane (OM Cytochrome B) in NADH-Semidehydroascorbic Acid Reductase Activity of Rat Liver. Biochem. Biophys. Res. Commun. 1981, 101, 591–598. [Google Scholar] [CrossRef]
- Fiaschi, T.; Chiarugi, P. Oxidative Stress, Tumor Microenvironment, and Metabolic Reprogramming: A Diabolic Liaison. Int. J. Cell Biol. 2012, 2012, 762825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogasawara, Y.; Funakoshi, M.; Ishii, K. Glucose Metabolism Is Accelerated by Exposure to T-Butylhydroperoxide during NADH Consumption in Human Erythrocytes. Blood Cells Mol. Dis. 2008, 41, 237–243. [Google Scholar] [CrossRef]
- Crespi, F.; Ratti, E.; Trist, D.G. Melatonin, a Hormone Monitorable in Vivo by Voltammetry? Analyst 1994, 119, 2193–2197. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Liebman, J.F. The Oxidizing Nature of the Hydroxyl Radical. A Comparison with the Ferryl Ion (FeO2+). J. Phys. Chem. 1984, 88, 99–101. [Google Scholar] [CrossRef]
- Tesoriere, L.; D’Arpa, D.; Conti, S.; Giaccone, V.; Pintaudi, A.M.; Livrea, M.A. Melatonin Protects Human Red Blood Cells from Oxidative Hemolysis: New Insights into the Radical-Scavenging Activity. J. Pineal Res. 1999, 27, 95–105. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Sainz, R.M.; Mayo, J.C.; Leon, J.; Hardeland, R.; Poeggeler, B.; Reiter, R.J. Interactions between Melatonin and Nicotinamide Nucleotide: NADH Preservation in Cells and in Cell-Free Systems by Melatonin. J. Pineal Res. 2005, 39, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Reiter, R.J.; Poeggeler, B.; Tan, D.X. The Significance of the Metabolism of the Neurohormone Melatonin: Antioxidative Protection and Formation of Bioactive Substances. Neurosci. Biobehav. Rev. 1993, 17, 347–357. [Google Scholar] [CrossRef]
- Martinez, G.R.; Almeida, E.A.; Klitzke, C.F.; Onuki, J.; Prado, F.M.; Medeiros, M.H.G.; Di Mascio, P. Measurement of Melatonin and Its Metabolites: Importance for the Evaluation of Their Biological Roles. Endocrine 2005, 27, 111–118. [Google Scholar] [CrossRef]
- Morabito, R.; Remigante, A.; Marino, A. Melatonin Protects Band 3 Protein in Human Erythrocytes against H2O2-Induced Oxidative Stress. Molecules 2019, 24, 2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morabito, R.; Romano, O.; La Spada, G.; Marino, A. H2O2-Induced Oxidative Stress Affects SO4= Transport in Human Erythrocytes. PLoS ONE 2016, 11, e0146485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.S.; Kim, S.M.; Lee, J.H.; Lee, S.H. Co-Administration of Melatonin Effectively Enhances the Therapeutic Effects of Pioglitazone on Mesenchymal Stem Cells Undergoing Indoxyl Sulfate-Induced Senescence through Modulation of Cellular Prion Protein Expression. Int. J. Mol. Sci. 2018, 19, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.-S.; Yoon, Y.M.; Go, G.; Lee, J.H.; Lee, S.H. Melatonin Protects Human Renal Proximal Tubule Epithelial Cells Against High Glucose-Mediated Fibrosis via the Cellular Prion Protein-TGF-β-Smad Signaling Axis. Int. J. Med. Sci. 2020, 17, 1235–1245. [Google Scholar] [CrossRef]
- Alvarez-Artime, A.; Cernuda-Cernuda, R.; Artime-Naveda, F.; Cepas, V.; Gonzalez-Menendez, P.; Fernadez-Vega, S.; Quiros-Gonzalez, I.; Sainz, R.M.; Mayo, J.C. Melatonin-Induced Cytoskeleton Reorganization Leads to Inhibition of Melanoma Cancer Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 548. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of Hypoxia in Cancer Therapy by Regulating the Tumor Microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [Green Version]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milane, L.; Duan, Z.; Amiji, M. Role of Hypoxia and Glycolysis in the Development of Multi-Drug Resistance in Human Tumor Cells and the Establishment of an Orthotopic Multi-Drug Resistant Tumor Model in Nude Mice Using Hypoxic Pre-Conditioning. Cancer Cell Int. 2011, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Pourhasanzade, F.; Sabzpoushan, S.H. A New Mathematical Model for Controlling Tumor Growth Based on Microenvironment Acidity and Oxygen Concentration. Biomed. Res. Int. 2021, 2021, 8886050. [Google Scholar] [CrossRef]
- Mu, Q.; Najafi, M. Modulation of the Tumor Microenvironment (TME) by Melatonin. Eur. J. Pharmacol. 2021, 907, 174365. [Google Scholar] [CrossRef]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The Role of Hypoxia in the Tumor Microenvironment and Development of Cancer Stem Cell: A Novel Approach to Developing Treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Bastani, S.; Akbarzadeh, M.; Rastgar Rezaei, Y.; Farzane, A.; Nouri, M.; Mollapour Sisakht, M.; Fattahi, A.; Akbarzadeh, M.; Reiter, R.J. Melatonin as a Therapeutic Agent for the Inhibition of Hypoxia-Induced Tumor Progression: A Description of Possible Mechanisms Involved. Int. J. Mol. Sci. 2021, 22, 874. [Google Scholar] [CrossRef] [PubMed]
- Finger, E.C.; Giaccia, A.J. Hypoxia, Inflammation, and the Tumor Microenvironment in Metastatic Disease. Cancer Metastasis Rev. 2010, 29, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Bristow, R.G.; Hill, R.P. Hypoxia and Metabolism. Hypoxia, DNA Repair and Genetic Instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. The Role of Hypoxia-Induced Factors in Tumor Progression. Oncologist 2004, 9 (Suppl. S5), 10–17. [Google Scholar] [CrossRef]
- Fuller, G.G.; Han, T.; Freeberg, M.A.; Moresco, J.J.; Ghanbari Niaki, A.; Roach, N.P.; Yates, J.R., 3rd; Myong, S.; Kim, J.K. RNA Promotes Phase Separation of Glycolysis Enzymes into Yeast G Bodies in Hypoxia. Elife 2020, 9, e48480. [Google Scholar] [CrossRef]
- Jin, M.; Fuller, G.G.; Han, T.; Yao, Y.; Alessi, A.F.; Freeberg, M.A.; Roach, N.P.; Moresco, J.J.; Karnovsky, A.; Baba, M.; et al. Glycolytic Enzymes Coalesce in G Bodies under Hypoxic Stress. Cell Rep. 2017, 20, 895–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garai, S.; Citu; Singla-Pareek, S.L.; Sopory, S.K.; Kaur, C.; Yadav, G. Complex Networks of Prion-Like Proteins Reveal Cross Talk Between Stress and Memory Pathways in Plants. Front. Plant. Sci. 2021, 12, 707286. [Google Scholar] [CrossRef]
- Godet, I.; Mamo, M.; Thurnheer, A.; Rosen, D.M.; Gilkes, D.M. Post-Hypoxic Cells Promote Metastatic Recurrence after Chemotherapy Treatment in TNBC. Cancers 2021, 13, 5509. [Google Scholar] [CrossRef] [PubMed]
- Gomatou, G.; Syrigos, N.; Vathiotis, I.A.; Kotteas, E.A. Tumor Dormancy: Implications for Invasion and Metastasis. Int. J. Mol. Sci. 2021, 22, 4862. [Google Scholar] [CrossRef]
- Perego, M.; Tyurin, V.A.; Tyurina, Y.Y.; Yellets, J.; Nacarelli, T.; Lin, C.; Nefedova, Y.; Kossenkov, A.; Liu, Q.; Sreedhar, S.; et al. Reactivation of Dormant Tumor Cells by Modified Lipids Derived from Stress-Activated Neutrophils. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- van der Kamp, M.W.; Daggett, V. Influence of pH on the Human Prion Protein: Insights into the Early Steps of Misfolding. Biophys. J. 2010, 99, 2289–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reithmeier, R.A.F.; Casey, J.R.; Kalli, A.C.; Sansom, M.S.P.; Alguel, Y.; Iwata, S. Band 3, the Human Red Cell Chloride/bicarbonate Anion Exchanger (AE1, SLC4A1), in a Structural Context. Biochim. Biophys. Acta 2016, 1858 Pt A, 1507–1532. [Google Scholar] [CrossRef]
- Pushkin, A.; Kurtz, I. SLC4 Base (HCO3−, CO32−) Transporters: Classification, Function, Structure, Genetic Diseases, and Knockout Models. Am. J. Physiol -Ren. Physiol. 2006, 290, F580–F599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passow, H. Molecular Aspects of Band 3 Protein-Mediated Anion Transport across the Red Blood Cell Membrane. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 1986; Volume 103, pp. 61–203. [Google Scholar] [CrossRef]
- Rivera-Santiago, R.; Harper, S.L.; Sriswasdi, S.; Hembach, P.; Speicher, D.W. Full-Length Anion Exchanger 1 Structure and Interactions with Ankyrin-1 Determined by Zero Length Crosslinking of Erythrocyte Membranes. Structure 2017, 25, 132–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Lykotrafitis, G. Erythrocyte Membrane Model with Explicit Description of the Lipid Bilayer and the Spectrin Network. Biophys. J. 2014, 107, 642–653. [Google Scholar] [CrossRef] [Green Version]
- Grey, J.L.; Kodippili, G.C.; Simon, K.; Low, P.S. Identification of Contact Sites between Ankyrin and Band 3 in the Human Erythrocyte Membrane. Biochemistry 2012, 51, 6838–6846. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, N. The Role of Band 3 Protein in Oxygen Delivery by Red Blood Cells. Indian, J. Clin. Biochem. 1999, 14, 49–58. [Google Scholar] [CrossRef] [Green Version]
- van den Akker, E.; Satchwell, T.J.; Williamson, R.C.; Toye, A.M. Band 3 Multiprotein Complexes in the Red Cell Membrane; of Mice and Men. Blood Cells Mol. Dis. 2010, 45, 1–8. [Google Scholar] [CrossRef]
- Sleep, J.; Wilson, D.; Simmons, R.; Gratzer, W. Elasticity of the Red Cell Membrane and Its Relation to Hemolytic Disorders: An Optical Tweezers Study. Biophys. J. 1999, 77, 3085–3095. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Dou, W.; Wang, C.; Sun, Y. Stiffness and ATP Recovery of Stored Red Blood Cells in Serum. Microsyst Nanoeng 2019, 5, 51. [Google Scholar] [CrossRef]
- Huisjes, R.; Bogdanova, A.; van Solinge, W.W.; Schiffelers, R.M.; Kaestner, L.; van Wijk, R. Squeezing for Life-Properties of Red Blood Cell Deformability. Front. Physiol. 2018, 9, 656. [Google Scholar] [CrossRef] [PubMed]
- Baier, D.; Müller, T.; Mohr, T.; Windberger, U. Red Blood Cell Stiffness and Adhesion Are Species-Specific Properties Strongly Affected by Temperature and Medium Changes in Single Cell Force Spectroscopy. Molecules 2021, 26, 2771. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, S.; Saldanha, C. An Overview about Erythrocyte Membrane. Clin. Hemorheol. Microcirc. 2010, 44, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrales, P. Effects of Erythrocyte Flexibility on Microvascular Perfusion and Oxygenation during Acute Anemia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1206–H1215. [Google Scholar] [CrossRef] [PubMed]
- Chien, S. Red Cell Deformability and Its Relevance to Blood Flow. Annu. Rev. Physiol. 1987, 49, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Fraser, G.M.; Dias, G.M.; Goldman, D.; Ellis, C. Red Blood Cell Flux in Isolated Capillary Bifurcations. FASEB J. 2009, 23, 949.5. [Google Scholar] [CrossRef]
- Kimura, H.; Braun, R.D.; Ong, E.T.; Hsu, R.; Secomb, T.W.; Papahadjopoulos, D.; Hong, K.; Dewhirst, M.W. Fluctuations in Red Cell Flux in Tumor Microvessels Can Lead to Transient Hypoxia and Reoxygenation in Tumor Parenchyma. Cancer Res. 1996, 56, 5522–5528. [Google Scholar]
- Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Red Blood Cell Oxidative Stress Impairs Oxygen Delivery and Induces Red Blood Cell Aging. Front. Physiol. 2014, 5, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çimen, M.Y.B. Free Radical Metabolism in Human Erythrocytes. Clin. Chim. Acta 2008, 390, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Barodka, V.M.; Nagababu, E.; Mohanty, J.G.; Nyhan, D.; Berkowitz, D.E.; Rifkind, J.M.; Strouse, J.J. New Insights Provided by a Comparison of Impaired Deformability with Erythrocyte Oxidative Stress for Sickle Cell Disease. Blood Cells Mol. Dis. 2014, 52, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zennadi, R. The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants 2021, 10, 1608. [Google Scholar] [CrossRef] [PubMed]
- Fertrin, K.Y.; Costa, F.F. Genomic Polymorphisms in Sickle Cell Disease: Implications for Clinical Diversity and Treatment. Expert Rev. Hematol. 2010, 3, 443–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunn, H.F. Pathogenesis and Treatment of Sickle Cell Disease. N. Engl. J. Med. 1997, 337, 762–769. [Google Scholar] [CrossRef]
- Spector, J.; Kodippili, G.C.; Ritchie, K.; Low, P.S. Single Molecule Studies of the Diffusion of Band 3 in Sickle Cell Erythrocytes. PLoS ONE 2016, 11, e0162514. [Google Scholar] [CrossRef]
- Efferth, T.; Schwarzl, S.M.; Smith, J.; Osieka, R. Role of Glucose-6-Phosphate Dehydrogenase for Oxidative Stress and Apoptosis. Cell Death Differ. 2006, 13, 527–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, H.; Mohiuddin, S.S. Biochemistry, Hexose Monophosphate Pathway. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- van Zwieten, R.; Verhoeven, A.J.; Roos, D. Inborn Defects in the Antioxidant Systems of Human Red Blood Cells. Free Radic. Biol. Med. 2014, 67, 377–386. [Google Scholar] [CrossRef]
- Davidson, W.D.; Tanaka, K.R. Factors Affecting Pentose Phosphate Pathway Activity in Human Red Cells. Br. J. Haematol. 1972, 23, 371–385. [Google Scholar] [CrossRef]
- Omachi, A.; Scott, C.B.; Parry, T.E. Influence of Glycolysis on NADH Content in Human Erythrocytes. Am. J. Physiol. 1969, 216, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Rapoprot, I.; Berger, H.; Rapoport, S.M.; Elsner, R.; Gerber, G. Response of the Glycolysis of Human Erythrocytes to the Transition from the Oxygenated to the Deoxygenated State at Constant Intracellular pH. Biochim. Biophys. Acta 1976, 428, 193–204. [Google Scholar] [CrossRef]
- Stefanovic, M.; Puchulu-Campanella, E.; Kodippili, G.; Low, P.S. Oxygen Regulates the Band 3-Ankyrin Bridge in the Human Erythrocyte Membrane. Biochem. J. 2013, 449, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grygorczyk, R.; Orlov, S.N. Effects of Hypoxia on Erythrocyte Membrane Properties-Implications for Intravascular Hemolysis and Purinergic Control of Blood Flow. Front. Physiol. 2017, 8, 1110. [Google Scholar] [CrossRef] [Green Version]
- Anong, W.A.; Weis, T.L.; Low, P.S. Rate of Rupture and Reattachment of the Band 3-Ankyrin Bridge on the Human Erythrocyte Membrane. J. Biol. Chem. 2006, 281, 22360–22366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, P.S.; Allen, D.P.; Zioncheck, T.F.; Chari, P.; Willardson, B.M.; Geahlen, R.L.; Harrison, M.L. Tyrosine Phosphorylation of Band 3 Inhibits Peripheral Protein Binding. J. Biol. Chem. 1987, 262, 4592–4596. [Google Scholar] [CrossRef]
- Lewis, I.A.; Campanella, M.E.; Markley, J.L.; Low, P.S. Role of Band 3 in Regulating Metabolic Flux of Red Blood Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 18515–18520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanella, M.E.; Chu, H.; Wandersee, N.J.; Peters, L.L.; Mohandas, N.; Gilligan, D.M.; Low, P.S. Characterization of Glycolytic Enzyme Interactions with Murine Erythrocyte Membranes in Wild-Type and Membrane Protein Knockout Mice. Blood 2008, 112, 3900–3906. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, A.; Ferru, E.; Pau, M.C.; Khadjavi, A.; Mandili, G.; Mattè, A.; Spano, A.; De Franceschi, L.; Pippia, P.; Turrini, F. Band 3 Erythrocyte Membrane Protein Acts as Redox Stress Sensor Leading to Its Phosphorylation by P (72) Syk. Oxid. Med. Cell. Longev. 2016, 2016, 6051093. [Google Scholar] [CrossRef] [Green Version]
- Ferru, E.; Giger, K.; Pantaleo, A.; Campanella, E.; Grey, J.; Ritchie, K.; Vono, R.; Turrini, F.; Low, P.S. Regulation of Membrane-Cytoskeletal Interactions by Tyrosine Phosphorylation of Erythrocyte Band 3. Blood 2011, 117, 5998–6006. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, A.; Ferru, E.; Giribaldi, G.; Mannu, F.; Carta, F.; Matte, A.; de Franceschi, L.; Turrini, F. Oxidized and Poorly Glycosylated Band 3 Is Selectively Phosphorylated by Syk Kinase to Form Large Membrane Clusters in Normal and G6PD-Deficient Red Blood Cells. Biochem. J. 2009, 418, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Jarolim, P.; Lahav, M.; Liu, S.C.; Palek, J. Effect of Hemoglobin Oxidation Products on the Stability of Red Cell Membrane Skeletons and the Associations of Skeletal Proteins: Correlation with a Release of Hemin. Blood 1990, 76, 2125–2131. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.C.; Zhai, S.; Lawler, J.; Palek, J. Hemin-Mediated Dissociation of Erythrocyte Membrane Skeletal Proteins. J. Biol. Chem. 1985, 260, 12234–12239. [Google Scholar] [CrossRef]
- An, X.L.; Takakuwa, Y.; Nunomura, W.; Manno, S.; Mohandas, N. Modulation of Band 3-Ankyrin Interaction by Protein 4.1. Functional Implications in Regulation of Erythrocyte Membrane Mechanical Properties. J. Biol. Chem. 1996, 271, 33187–33191. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, T.H.; Frezzatti, W.A., Jr.; Schreier, S. Hemin-Induced Lipid Membrane Disorder and Increased Permeability: A Molecular Model for the Mechanism of Cell Lysis. Arch. Biochem. Biophys. 1993, 307, 96–103. [Google Scholar] [CrossRef]
- Ludlow, J.T.; Wilkerson, R.G.; Nappe, T.M. Methemoglobinemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Wright, R.O.; Lewander, W.J.; Woolf, A.D. Methemoglobinemia: Etiology, Pharmacology, and Clinical Management. Ann. Emerg. Med. 1999, 34, 646–656. [Google Scholar] [CrossRef]
- Zhang, Y.; Manning, L.R.; Falcone, J.; Platt, O.; Manning, J.M. Human Erythrocyte Membrane Band 3 Protein Influences Hemoglobin Cooperativity: POSSIBLE EFFECT ON OXYGEN TRANSPORT. J. Biol. Chem. 2003, 278, 39565–39571. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; McKenna, M.M.; Krump, N.A.; Zheng, S.; Mendelsohn, L.; Thein, S.L.; Garrett, L.J.; Bodine, D.M.; Low, P.S. Reversible Binding of Hemoglobin to Band 3 Constitutes the Molecular Switch That Mediates O2 Regulation of Erythrocyte Properties. Blood 2016, 128, 2708–2716. [Google Scholar] [CrossRef]
- Walder, J.A.; Chatterjee, R.; Steck, T.L.; Low, P.S.; Musso, G.F.; Kaiser, E.T.; Rogers, P.H.; Arnone, A. The Interaction of Hemoglobin with the Cytoplasmic Domain of Band 3 of the Human Erythrocyte Membrane. J. Biol. Chem. 1984, 259, 10238–10246. [Google Scholar] [CrossRef]
- Chu, H.; Breite, A.; Ciraolo, P.; Franco, R.S.; Low, P.S. Characterization of the Deoxyhemoglobin Binding Site on Human Erythrocyte Band 3: Implications for O2 Regulation of Erythrocyte Properties. Blood 2008, 111, 932–938. [Google Scholar] [CrossRef] [Green Version]
- Stromme, J.H.; Eldjarn, L. The Role of the Pentose Phosphate Pathway in the Reduction of Methaemoglobin in Human Erythrocytes. Biochem. J. 1962, 84, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Hlutkin, S.; Zinchuk, V. Effect of Melatonin on the Blood Oxygen Transport during Hypothermia and Rewarming in Rats. Adv. Med. Sci. 2008, 53, 234–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditlev, J.A. Membrane-Associated Phase Separation: Organization and Function Emerge from a Two-Dimensional Milieu. J. Mol. Cell Biol. 2021, 13, 319–324. [Google Scholar] [CrossRef]
- Zhang, C.; Rabouille, C. Membrane-Bound Meet Membraneless in Health and Disease. Cells 2019, 8, 1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernon, R.M.; Chong, P.A.; Tsang, B.; Kim, T.H.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J.D. Pi-Pi Contacts Are an Overlooked Protein Feature Relevant to Phase Separation. Elife 2018, 7, e31486. [Google Scholar] [CrossRef]
- Lee, I.-H.; Imanaka, M.Y.; Modahl, E.H.; Torres-Ocampo, A.P. Lipid Raft Phase Modulation by Membrane-Anchored Proteins with Inherent Phase Separation Properties. ACS Omega 2019, 4, 6551–6559. [Google Scholar] [CrossRef] [PubMed]
- Hicks, M.R.; Gill, A.C.; Bath, I.K.; Rullay, A.K.; Sylvester, I.D.; Crout, D.H.; Pinheiro, T.J.T. Synthesis and Structural Characterization of a Mimetic Membrane-Anchored Prion Protein. FEBS J. 2006, 273, 1285–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, F.; Watt, N.T.; Walmsley, A.R.; Hooper, N.M. Tethering the N-Terminus of the Prion Protein Compromises the Cellular Response to Oxidative Stress. J. Neurochem. 2003, 84, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Baron, G.S.; Caughey, B. Effect of Glycosylphosphatidylinositol Anchor-Dependent and -Independent Prion Protein Association with Model Raft Membranes on Conversion to the Protease-Resistant Isoform. J. Biol. Chem. 2003, 278, 14883–14892. [Google Scholar] [CrossRef] [Green Version]
- Sanghera, N.; Pinheiro, T.J.T. Binding of Prion Protein to Lipid Membranes and Implications for Prion Conversion. J. Mol. Biol. 2002, 315, 1241–1256. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, F.; Hu, Y.; Wang, X.; Wang, X.; Jin, C.; Ma, J. Lipid Interaction Converts Prion Protein to a PrPSc-like Proteinase K-Resistant Conformation under Physiological Conditions. Biochemistry 2007, 46, 7045–7053. [Google Scholar] [CrossRef]
- Chen, W.; van der Kamp, M.W.; Daggett, V. Structural and Dynamic Properties of the Human Prion Protein. Biophys. J. 2014, 106, 1152–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, J.; Sreeramulu, S.; Schmidt, T.L.; Richter, C.; Vonck, J.; Heckel, A.; Glaubitz, C.; Schwalbe, H. Prion Protein Amyloid Formation Involves Structural Rearrangements in the C-Terminal Domain. ChemBioChem 2010, 11, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.K.; Srivastava, A.K.; Srinivas, V.; Chary, K.V.R.; Rao, C.M. Copper Alters Aggregation Behavior of Prion Protein and Induces Novel Interactions between Its N- and C-Terminal Regions. J. Biol. Chem. 2011, 286, 38533–38545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spagnolli, G.; Rigoli, M.; Orioli, S.; Sevillano, A.M.; Faccioli, P.; Wille, H.; Biasini, E.; Requena, J.R. Full Atomistic Model of Prion Structure and Conversion. PLoS Pathog. 2019, 15, e1007864. [Google Scholar] [CrossRef] [Green Version]
- Kabani, M.; Cosnier, B.; Bousset, L.; Rousset, J.-P.; Melki, R.; Fabret, C. A Mutation within the C-Terminal Domain of Sup35p That Affects [PSI+] Prion Propagation. Mol. Microbiol. 2011, 81, 640–658. [Google Scholar] [CrossRef] [PubMed]
- Zanusso, G.; Petersen, R.B.; Jin, T.; Jing, Y.; Kanoush, R.; Ferrari, S.; Gambetti, P.; Singh, N. Proteasomal Degradation and N-Terminal Protease Resistance of the Codon 145 Mutant Prion Protein. J. Biol. Chem. 1999, 274, 23396–23404. [Google Scholar] [CrossRef] [Green Version]
- Westergard, L.; Turnbaugh, J.A.; Harris, D.A. A Naturally Occurring C-Terminal Fragment of the Prion Protein (PrP) Delays Disease and Acts as a Dominant-Negative Inhibitor of PrPSc Formation. J. Biol. Chem. 2011, 286, 44234–44242. [Google Scholar] [CrossRef] [Green Version]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W.; et al. Prion Protein Accumulation and Neuroprotection in Hypoxic Brain Damage. Am. J. Pathol. 2004, 165, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Shyu, W.-C.; Lin, S.-Z.; Chiang, M.-F.; Ding, D.-C.; Li, K.-W.; Chen, S.-F.; Yang, H.-I.; Li, H. Overexpression of PrPC by Adenovirus-Mediated Gene Targeting Reduces Ischemic Injury in a Stroke Rat Model. J. Neurosci. 2005, 25, 8967–8977. [Google Scholar] [CrossRef] [Green Version]
- Spudich, A.; Frigg, R.; Kilic, E.; Kilic, U.; Oesch, B.; Raeber, A.; Bassetti, C.L.; Hermann, D.M. Aggravation of Ischemic Brain Injury by Prion Protein Deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol. Dis. 2005, 20, 442–449. [Google Scholar] [CrossRef]
- Weise, J.; Sandau, R.; Schwarting, S.; Crome, O.; Wrede, A.; Schulz-Schaeffer, W.; Zerr, I.; Bähr, M. Deletion of Cellular Prion Protein Results in Reduced Akt Activation, Enhanced Postischemic Caspase-3 Activation, and Exacerbation of Ischemic Brain Injury. Stroke 2006, 37, 1296–1300. [Google Scholar] [CrossRef] [Green Version]
- Vriend, J.; Reiter, R.J. Breast Cancer Cells: Modulation by Melatonin and the Ubiquitin-Proteasome System—A Review. Mol. Cell. Endocrinol. 2015, 417, 1–9. [Google Scholar] [CrossRef]
- Song, J.; Ma, S.-J.; Luo, J.-H.; Zhang, H.; Wang, R.-X.; Liu, H.; Li, L.; Zhang, Z.-G.; Zhou, R.-X. Melatonin Induces the Apoptosis and Inhibits the Proliferation of Human Gastric Cancer Cells via Blockade of the AKT/MDM2 Pathway. Oncol. Rep. 2018, 39, 1975–1983. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zeng, S. Melatonin Promotes Ubiquitination of Phosphorylated Pro-Apoptotic Protein Bcl-2-Interacting Mediator of Cell Death-Extra Long (BimEL) in Porcine Granulosa Cells. Int. J. Mol. Sci. 2018, 19, 3431. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, C.L.; Marquardt, D.; Dies, H.; Kučerka, N.; Yamani, Z.; Harroun, T.A.; Katsaras, J.; Shi, A.-C.; Rheinstädter, M.C. The Observation of Highly Ordered Domains in Membranes with Cholesterol. PLoS ONE 2013, 8, e66162. [Google Scholar] [CrossRef] [Green Version]
- Almeida, P.F.F.; Pokorny, A.; Hinderliter, A. Thermodynamics of Membrane Domains. Biochim. Biophys. Acta 2005, 1720, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Toomre, D. Lipid Rafts and Signal Transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Bagnat, M.; Keränen, S.; Shevchenko, A.; Shevchenko, A.; Simons, K. Lipid Rafts Function in Biosynthetic Delivery of Proteins to the Cell Surface in Yeast. Proc. Natl. Acad. Sci. USA 2000, 97, 3254–3259. [Google Scholar] [CrossRef]
- Ikonen, E. Roles of Lipid Rafts in Membrane Transport. Curr. Opin. Cell Biol. 2001, 13, 470–477. [Google Scholar] [CrossRef]
- Preta, G. New Insights Into Targeting Membrane Lipids for Cancer Therapy. Front. Cell Dev. Biol 2020, 8, 571237. [Google Scholar] [CrossRef]
- Greenlee, J.D.; Subramanian, T.; Liu, K.; King, M.R. Rafting Down the Metastatic Cascade: The Role of Lipid Rafts in Cancer Metastasis, Cell Death, and Clinical Outcomes. Cancer Res. 2021, 81, 5–17. [Google Scholar] [CrossRef]
- Codini, M.; Garcia-Gil, M.; Albi, E. Cholesterol and Sphingolipid Enriched Lipid Rafts as Therapeutic Targets in Cancer. Int. J. Mol. Sci. 2021, 22, 726. [Google Scholar] [CrossRef]
- Gomà, A.; Mir, R.; Martínez-Soler, F.; Tortosa, A.; Vidal, A.; Condom, E.; Pérez-Tomás, R.; Giménez-Bonafé, P. Multidrug Resistance Protein 1 Localization in Lipid Raft Domains and Prostasomes in Prostate Cancer Cell Lines. Onco. Targets. Ther. 2014, 7, 2215–2225. [Google Scholar] [CrossRef] [Green Version]
- Ye, D.M.; Ye, S.C.; Yu, S.Q.; Shu, F.F.; Xu, S.S.; Chen, Q.Q.; Wang, Y.L.; Tang, Z.T.; Pan, C. Drug-Resistance Reversal in Colorectal Cancer Cells by Destruction of Flotillins, the Key Lipid Rafts Proteins. Neoplasma 2019, 66, 576–583. [Google Scholar] [CrossRef]
- Taylor, D.R.; Whitehouse, I.J.; Hooper, N.M. Glypican-1 Mediates Both Prion Protein Lipid Raft Association and Disease Isoform Formation. PLoS Pathog. 2009, 5, e1000666. [Google Scholar] [CrossRef] [Green Version]
- Fremuntova, Z.; Mosko, T.; Soukup, J.; Kucerova, J.; Kostelanska, M.; Hanusova, Z.B.; Filipova, M.; Cervenakova, L.; Holada, K. Changes in Cellular Prion Protein Expression, Processing and Localisation during Differentiation of the Neuronal Cell Line CAD 5. Biol. Cell 2020, 112, 1–21. [Google Scholar] [CrossRef]
- Martellucci, S.; Santacroce, C.; Santilli, F.; Piccoli, L.; Delle Monache, S.; Angelucci, A.; Misasi, R.; Sorice, M.; Mattei, V. Cellular and Molecular Mechanisms Mediated by recPrPC Involved in the Neuronal Differentiation Process of Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 345. [Google Scholar] [CrossRef] [Green Version]
- Nieznanski, K. Interactions of Prion Protein with Intracellular Proteins: So Many Partners and No Consequences? Cell. Mol. Neurobiol. 2010, 30, 653–666. [Google Scholar] [CrossRef]
- Kourie, J.I. Mechanisms of Prion-Induced Modifications in Membrane Transport Properties: Implications for Signal Transduction and Neurotoxicity. Chem. Biol. Interact. 2001, 138, 1–26. [Google Scholar] [CrossRef]
- Singh, N.; Gu, Y.; Bose, S.; Kalepu, S.; Mishra, R.S.; Verghese, S. Prion Peptide 106-126 as a Model for Prion Replication and Neurotoxicity. Front. Biosci. 2002, 7, a60–a71. [Google Scholar] [CrossRef]
- Forloni, G.; Chiesa, R.; Bugiani, O.; Salmona, M.; Tagliavini, F. Review: PrP 106-126-25 Years after. Neuropathol. Appl. Neurobiol. 2019, 45, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Salmona, M.; Forloni, G.; Diomede, L.; Algeri, M.; De Gioia, L.; Angeretti, N.; Giaccone, G.; Tagliavini, F.; Bugiani, O. A Neurotoxic and Gliotrophic Fragment of the Prion Protein Increases Plasma Membrane Microviscosity. Neurobiol. Dis. 1997, 4, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.C.; Mirzabekov, T.; Kagan, B.L. Channel Formation by a Neurotoxic Prion Protein Fragment. J. Biol. Chem. 1997, 272, 44–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourie, J.I.; Culverson, A. Prion Peptide Fragment PrP[106–126] Forms Distinct Cation Channel Types. J. Neurosci. Res. 2000, 62, 120–133. [Google Scholar] [CrossRef]
- Kourie, J.I.; Shorthouse, A.A. Properties of Cytotoxic Peptide-Formed Ion Channels. Am. J. Physiol. Cell Physiol. 2000, 278, C1063–C1087. [Google Scholar] [CrossRef] [PubMed]
- DeArmond, S.J.; Qiu, Y.; Wong, K.; Nixon, R.; Hyun, W.; Prusiner, S.B.; Mobley, W.C. Abnormal Plasma Membrane Properties and Functions in Prion-Infected Cell Lines. Cold Spring Harb. Symp. Quant. Biol. 1996, 61, 531–540. [Google Scholar]
- Wong, K.; Qiu, Y.; Hyun, W.; Nixon, R.; VanCleff, J.; Sanchez-Salazar, J.; Prusiner, S.B.; DeArmond, S.J. Decreased Receptor-Mediated Calcium Response in Prion-Infected Cells Correlates with Decreased Membrane Fluidity and IP3 Release. Neurology 1996, 47, 741–750. [Google Scholar] [CrossRef]
- Pan, J.; Sahoo, P.K.; Dalzini, A.; Hayati, Z.; Aryal, C.M.; Teng, P.; Cai, J.; Rodriguez Gutierrez, H.; Song, L. Membrane Disruption Mechanism of a Prion Peptide (106-126) Investigated by Atomic Force Microscopy, Raman and Electron Paramagnetic Resonance Spectroscopy. J. Phys. Chem. B 2017, 121, 5058–5071. [Google Scholar] [CrossRef] [Green Version]
- Mahal, S.P.; Jablonski, J.; Suponitsky-Kroyter, I.; Oelschlegel, A.M.; Herva, M.E.; Oldstone, M.; Weissmann, C. Propagation of RML Prions in Mice Expressing PrP Devoid of GPI Anchor Leads to Formation of a Novel, Stable Prion Strain. PLoS Pathog. 2012, 8, e1002746. [Google Scholar] [CrossRef] [Green Version]
- Roseboom, P.H.; Namboodiri, M.A.; Zimonjic, D.B.; Popescu, N.C.; Rodriguez, I.R.; Gastel, J.A.; Klein, D.C. Natural Melatonin “Knockdown” in C57BL/6J Mice: Rare Mechanism Truncates Serotonin N-Acetyltransferase. Brain Res. Mol. Brain Res. 1998, 63, 189–197. [Google Scholar] [CrossRef]
- Vivien-Roels, B.; Malan, A.; Rettori, M.C.; Delagrange, P.; Jeanniot, J.P.; Pévet, P. Daily Variations in Pineal Melatonin Concentrations in Inbred and Outbred Mice. J. Biol. Rhythms 1998, 13, 403–409. [Google Scholar] [CrossRef]
- Dupiereux, I.; Zorzi, W.; Lins, L.; Brasseur, R.; Colson, P.; Heinen, E.; Elmoualij, B. Interaction of the 106-126 Prion Peptide with Lipid Membranes and Potential Implication for Neurotoxicity. Biochem. Biophys. Res. Commun. 2005, 331, 894–901. [Google Scholar] [CrossRef]
- Oku, N.; Shibamoto, S.; Ito, F.; Gondo, H.; Nango, M. Low pH Induced Membrane Fusion of Lipid Vesicles Containing Proton-Sensitive Polymer. Biochemistry 1987, 26, 8145–8150. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.; Yatvin, M.B.; Huang, L. pH-Sensitive Liposomes: Acid-Induced Liposome Fusion. Proc. Natl. Acad. Sci. USA 1984, 81, 1715–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.-T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The Role of Cholesterol in Membrane Fusion. Chem. Phys. Lipids 2016, 199, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Laszlo, L.; Prusiner, S.B. Cholesterol Depletion and Modification of COOH-Terminal Targeting Sequence of the Prion Protein Inhibit Formation of the Scrapie Isoform. J. Cell Biol. 1995, 129, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Tobert, J.A. Lovastatin and beyond: The History of the HMG-CoA Reductase Inhibitors. Nat. Rev. Drug Discov. 2003, 2, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, P.I.; Akimov, S.A.; Chizmadzhev, Y.A.; Zimmerberg, J.; Cohen, F.S. Line Tension and Interaction Energies of Membrane Rafts Calculated from Lipid Splay and Tilt. Biophys. J. 2005, 88, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.-C.; Feigenson, G.W. Lowering Line Tension with High Cholesterol Content Induces a Transition from Macroscopic to Nanoscopic Phase Domains in Model Biomembranes. Biochim. Biophys. Acta (BBA)-Biomembr. 2019, 1861, 478–485. [Google Scholar] [CrossRef]
- Ayuyan, A.G.; Cohen, F.S. Lipid Peroxides Promote Large Rafts: Effects of Excitation of Probes in Fluorescence Microscopy and Electrochemical Reactions during Vesicle Formation. Biophys. J. 2006, 91, 2172–2183. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; Gajate, C. Lipid Rafts and Clusters of Apoptotic Signaling Molecule-Enriched Rafts in Cancer Therapy. Future Oncol. 2010, 6, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Cedó, L.; Reddy, S.T.; Mato, E.; Blanco-Vaca, F.; Escolà-Gil, J.C. HDL and LDL: Potential New Players in Breast Cancer Development. J. Clin. Med. Res. 2019, 8, 853. [Google Scholar] [CrossRef] [Green Version]
- Jamnagerwalla, J.; Howard, L.E.; Allott, E.H.; Vidal, A.C.; Moreira, D.M.; Castro-Santamaria, R.; Andriole, G.L.; Freeman, M.R.; Freedland, S.J. Serum Cholesterol and Risk of High-Grade Prostate Cancer: Results from the REDUCE Study. Prostate Cancer Prostatic Dis. 2018, 21, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Park, M.J.; Ye, S.-K.; Kim, C.-W.; Kim, Y.-N. Elevated Levels of Cholesterol-Rich Lipid Rafts in Cancer Cells Are Correlated with Apoptosis Sensitivity Induced by Cholesterol-Depleting Agents. Am. J. Pathol. 2006, 168, 1107–1118; quiz 1404–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopecka, J.; Trouillas, P.; Gašparović, A.Č.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and Cholesterol: Inducers of Cancer Multidrug Resistance and Therapeutic Targets. Drug Resist. Updat. 2020, 49, 100670. [Google Scholar] [CrossRef]
- Yan, A.; Jia, Z.; Qiao, C.; Wang, M.; Ding, X. Cholesterol Metabolism in Drug-resistant Cancer (Review). Int. J. Oncol. 2020, 57, 1103–1115. [Google Scholar] [CrossRef]
- Brindisi, M.; Fiorillo, M.; Frattaruolo, L.; Sotgia, F.; Lisanti, M.P.; Cappello, A.R. Cholesterol and Mevalonate: Two Metabolites Involved in Breast Cancer Progression and Drug Resistance through the ERRα Pathway. Cells 2020, 9, 1819. [Google Scholar] [CrossRef] [PubMed]
- Yun, U.-J.; Lee, J.-H.; Koo, K.H.; Ye, S.-K.; Kim, S.-Y.; Lee, C.-H.; Kim, Y.-N. Lipid Raft Modulation by Rp1 Reverses Multidrug Resistance via Inactivating MDR-1 and Src Inhibition. Biochem. Pharmacol. 2013, 85, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol Level Affects Surface Charge of Lipid Membranes in Saline Solution. Sci. Rep. 2014, 4, 5005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finot, E.; Leonenko, Y.; Moores, B.; Eng, L.; Amrein, M.; Leonenko, Z. Effect of Cholesterol on Electrostatics in Lipid-Protein Films of a Pulmonary Surfactant. Langmuir 2010, 26, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Eckert, G.P.; Kirsch, C.; Leutz, S.; Wood, W.G.; Müller, W.E. Cholesterol Modulates Amyloid Beta-Peptide’s Membrane Interactions. Pharmacopsychiatry 2003, 36 (Suppl. S2), 136–143. [Google Scholar] [CrossRef]
- Choi, Y.; Attwood, S.J.; Hoopes, M.I.; Drolle, E.; Karttunen, M.; Leonenko, Z. Melatonin Directly Interacts with Cholesterol and Alleviates Cholesterol Effects in Dipalmitoylphosphatidylcholine Monolayers. Soft Matter 2014, 10, 206–213. [Google Scholar] [CrossRef]
- Bongiorno, D.; Ceraulo, L.; Ferrugia, M.; Filizzola, F.; Giordano, C.; Ruggirello, A.; Liveri, V.T. H-NMR and FT-IR Study of the State of Melatonin Confined in Membrane Models: Location and Interactions of Melatonin in Water Free Lecithin and AOT Reversed Micelles. Arkivoc 2004, 2004, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Rudzite, V.; Jurika, E.; Jirgensons, J. Changes in Membrane Fluidity Induced by Tryptophan and Its Metabolites. In Tryptophan, Serotonin, and Melatonin: Basic Aspects and Applications; Huether, G., Kochen, W., Simat, T.J., Steinhart, H., Eds.; Springer US: Boston, MA, USA, 1999; pp. 353–367. [Google Scholar] [CrossRef]
- de Lima, V.R.; Caro, M.S.B.; Munford, M.L.; Desbat, B.; Dufourc, E.; Pasa, A.A.; Creczynski-Pasa, T.B. Influence of Melatonin on the Order of Phosphatidylcholine-Based Membranes. J. Pineal Res. 2010, 49, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Drolle, E.; Kučerka, N.; Hoopes, M.I.; Choi, Y.; Katsaras, J.; Karttunen, M.; Leonenko, Z. Effect of Melatonin and Cholesterol on the Structure of DOPC and DPPC Membranes. Biochim. Biophys. Acta 2013, 1828, 2247–2254. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Pérez, E.J.; Sepúlveda, F.J.; Peters, C.; Bascuñán, D.; Riffo-Lepe, N.O.; González-Sanmiguel, J.; Sánchez, S.A.; Peoples, R.W.; Vicente, B.; Aguayo, L.G. Effect of Cholesterol on Membrane Fluidity and Association of Aβ Oligomers and Subsequent Neuronal Damage: A Double-Edged Sword. Front. Aging Neurosci. 2018, 10, 226. [Google Scholar] [CrossRef] [PubMed]
- Dies, H.; Cheung, B.; Tang, J.; Rheinstädter, M.C. The Organization of Melatonin in Lipid Membranes. Biochim. Biophys. Acta 2015, 1848, 1032–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venegas, C.; García, J.A.; Escames, G.; Ortiz, F.; López, A.; Doerrier, C.; García-Corzo, L.; López, L.C.; Reiter, R.J.; Acuña-Castroviejo, D. Extrapineal Melatonin: Analysis of Its Subcellular Distribution and Daily Fluctuations. J. Pineal Res. 2012, 52, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Liang, Q.; Tieleman, D.P. Interactions between Band 3 Anion Exchanger and Lipid Nanodomains in Ternary Lipid Bilayers: Atomistic Simulations. J. Phys. Chem. B 2020, 124, 3054–3064. [Google Scholar] [CrossRef]
- Ciana, A.; Achilli, C.; Minetti, G. Membrane Rafts of the Human Red Blood Cell. Mol. Membr. Biol. 2014, 31, 47–57. [Google Scholar] [CrossRef]
- Murphy, S.C.; Samuel, B.U.; Harrison, T.; Speicher, K.D.; Speicher, D.W.; Reid, M.E.; Prohaska, R.; Low, P.S.; Tanner, M.J.; Mohandas, N.; et al. Erythrocyte Detergent-Resistant Membrane Proteins: Their Characterization and Selective Uptake during Malarial Infection. Blood 2004, 103, 1920–1928. [Google Scholar] [CrossRef] [Green Version]
- Yeagle, P.L. Cholesterol and the Cell Membrane. Biochim. Biophys. Acta 1985, 822, 267–287. [Google Scholar] [CrossRef]
- Cooper, R.A. Influence of Increased Membrane Cholesterol on Membrane Fluidity and Cell Function in Human Red Blood Cells. J. Supramol. Struct. 1978, 8, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Maneri, L.R.; Low, P.S. Structural Stability of the Erythrocyte Anion Transporter, Band 3, in Different Lipid Environments. A Differential Scanning Calorimetric Study. J. Biol. Chem. 1988, 263, 16170–16178. [Google Scholar] [CrossRef]
- Groves, J.D.; Wang, L.; Tanner, M.J. Complementation Studies with Co-Expressed Fragments of Human Red Cell Band 3 (AE1): The Assembly of the Anion-Transport Domain in Xenopus Oocytes and a Cell-Free Translation System. Biochem. J. 1998, 332 Pt 1, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargreaves, W.R.; Giedd, K.N.; Verkleij, A.; Branton, D. Reassociation of Ankyrin with Band 3 in Erythrocyte Membranes and in Lipid Vesicles. J. Biol. Chem. 1980, 255, 11965–11972. [Google Scholar] [CrossRef]
- Cordeiro, R.M. Reactive Oxygen Species at Phospholipid Bilayers: Distribution, Mobility and Permeation. Biochim. Biophys. Acta 2014, 1838 Pt B, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, M.C.; Yusupov, M.; Bogaerts, A.; Cordeiro, R.M. Lipid Oxidation: Role of Membrane Phase-Separated Domains. J. Chem. Inf. Model. 2021, 61, 2857–2868. [Google Scholar] [CrossRef] [PubMed]
- Wong-Ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.-M.; Tieleman, D.P.; Monticelli, L. Effect of Lipid Peroxidation on the Properties of Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef] [Green Version]
- Kaplán, P.; Racay, P.; Lehotský, J.; Mézesová, V. Change in Fluidity of Brain Endoplasmic Reticulum Membranes by Oxygen Free Radicals: A Protective Effect of Stobadine, Alpha-Tocopherol Acetate, and Butylated Hydroxytoluene. Neurochem. Res. 1995, 20, 815–820. [Google Scholar] [CrossRef]
- Miller, Y.I.; Navia-Pelaez, J.M.; Corr, M.; Yaksh, T.L. Lipid Rafts in Glial Cells: Role in Neuroinflammation and Pain Processing: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Sviridov, D.; Mukhamedova, N.; Miller, Y.I. Lipid Rafts as a Therapeutic Target: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, M.; Kenworthy, A.K. Lipid Peroxidation Enhances LO/LD Domain Phase Separation in Giant Plasma Membrane Vesicles. Biophys. J. 2021, 120, 324a. [Google Scholar] [CrossRef]
- Dies, H.; Toppozini, L.; Rheinstädter, M.C. The Interaction between Amyloid-β Peptides and Anionic Lipid Membranes Containing Cholesterol and Melatonin. PLoS ONE 2014, 9, e99124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceraulo, L.; Ferrugia, M.; Tesoriere, L.; Segreto, S.; Livrea, M.A.; Turco Liveri, V. Interactions of Melatonin with Membrane Models: Portioning of Melatonin in AOT and Lecithin Reversed Micelles. J. Pineal Res. 1999, 26, 108–112. [Google Scholar] [CrossRef]
- Aikens, J.; Dix, T.A. Perhydroxyl Radical (HOO.) Initiated Lipid Peroxidation. The Role of Fatty Acid Hydroperoxides. J. Biol. Chem. 1991, 266, 15091–15098. [Google Scholar] [CrossRef]
- Brazier, M.W.; Lewis, V.; Ciccotosto, G.D.; Klug, G.M.; Lawson, V.A.; Cappai, R.; Ironside, J.W.; Masters, C.L.; Hill, A.F.; White, A.R.; et al. Correlative Studies Support Lipid Peroxidation Is Linked to PrP(res) Propagation as an Early Primary Pathogenic Event in Prion Disease. Brain Res. Bull. 2006, 68, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Mathiesen, B.; Kindem, K.; Galappathi, K. Acidic Extracellular pH Promotes Experimental Metastasis of Human Melanoma Cells in Athymic Nude Mice. Cancer Res. 2006, 66, 6699–6707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.Y.; Ryu, J.M.; Lee, S.H.; Park, J.H.; Han, H.J. Lipid Rafts Play an Important Role for Maintenance of Embryonic Stem Cell Self-Renewal. J. Lipid Res. 2010, 51, 2082–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alomari, M.; Almohazey, D.; Almofty, S.A.; Khan, F.A.; Al Hamad, M.; Ababneh, D. Role of Lipid Rafts in Hematopoietic Stem Cells Homing, Mobilization, Hibernation, and Differentiation. Cells 2019, 8, 630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, G.; Jin, Y. Role of Oct4 in Maintaining and Regaining Stem Cell Pluripotency. Stem Cell Res. Ther. 2010, 1, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Orkin, S.H. Embryonic Stem Cell-Specific Signatures in Cancer: Insights into Genomic Regulatory Networks and Implications for Medicine. Genome Med. 2011, 3, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Yu, X.; Liu, S. Pluripotency Transcription Factors and Cancer Stem Cells: Small Genes Make a Big Difference. Chin. J. Cancer 2013, 32, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Go, G.; Lee, S.H. PrPC Regulates the Cancer Stem Cell Properties via Interaction With c-Met in Colorectal Cancer Cells. Anticancer Res. 2021, 41, 3459–3470. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, N.; Zhang, T.; Wang, M.; Ge, W.; Wang, X. Roles of Melatonin in Goat Hair Follicle Stem Cell Proliferation and Pluripotency Through Regulating the Wnt Signaling Pathway. Front. Cell Dev. Biol 2021, 9, 686805. [Google Scholar] [CrossRef]
- Georgiev, G.N.; Marinova, E.; Konakchieva, R.; Todorov, P. Melatonin Selectively Influences the Transcription of Pluripotency and Differentiation Markers in Human Non-Cancer Cells. Biotechnol. Biotechnol. Equip. 2019, 33, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Akbarzadeh, M.; Movassaghpour, A.A.; Ghanbari, H.; Kheirandish, M.; Fathi Maroufi, N.; Rahbarghazi, R.; Nouri, M.; Samadi, N. The Potential Therapeutic Effect of Melatonin on Human Ovarian Cancer by Inhibition of Invasion and Migration of Cancer Stem Cells. Sci. Rep. 2017, 7, 17062. [Google Scholar] [CrossRef]



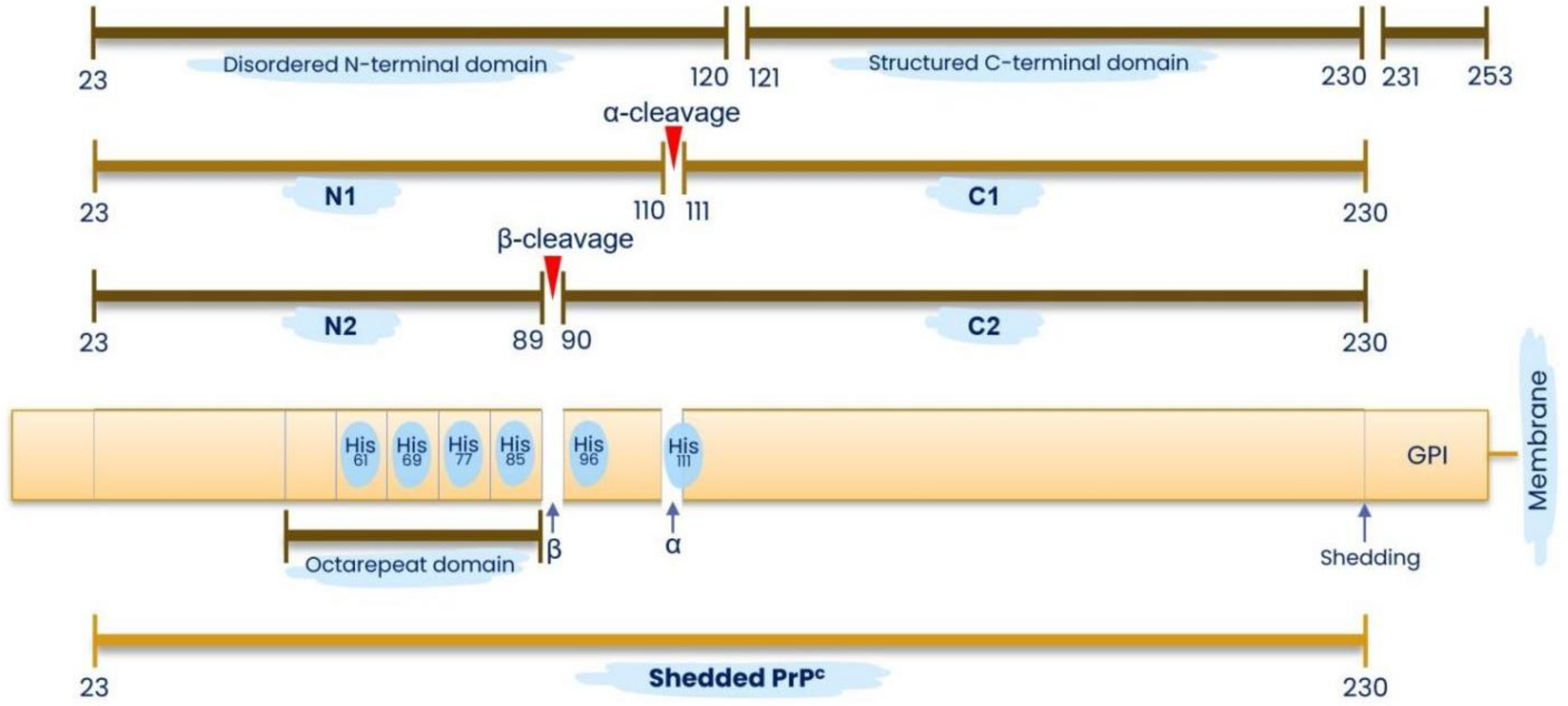{kind=link}
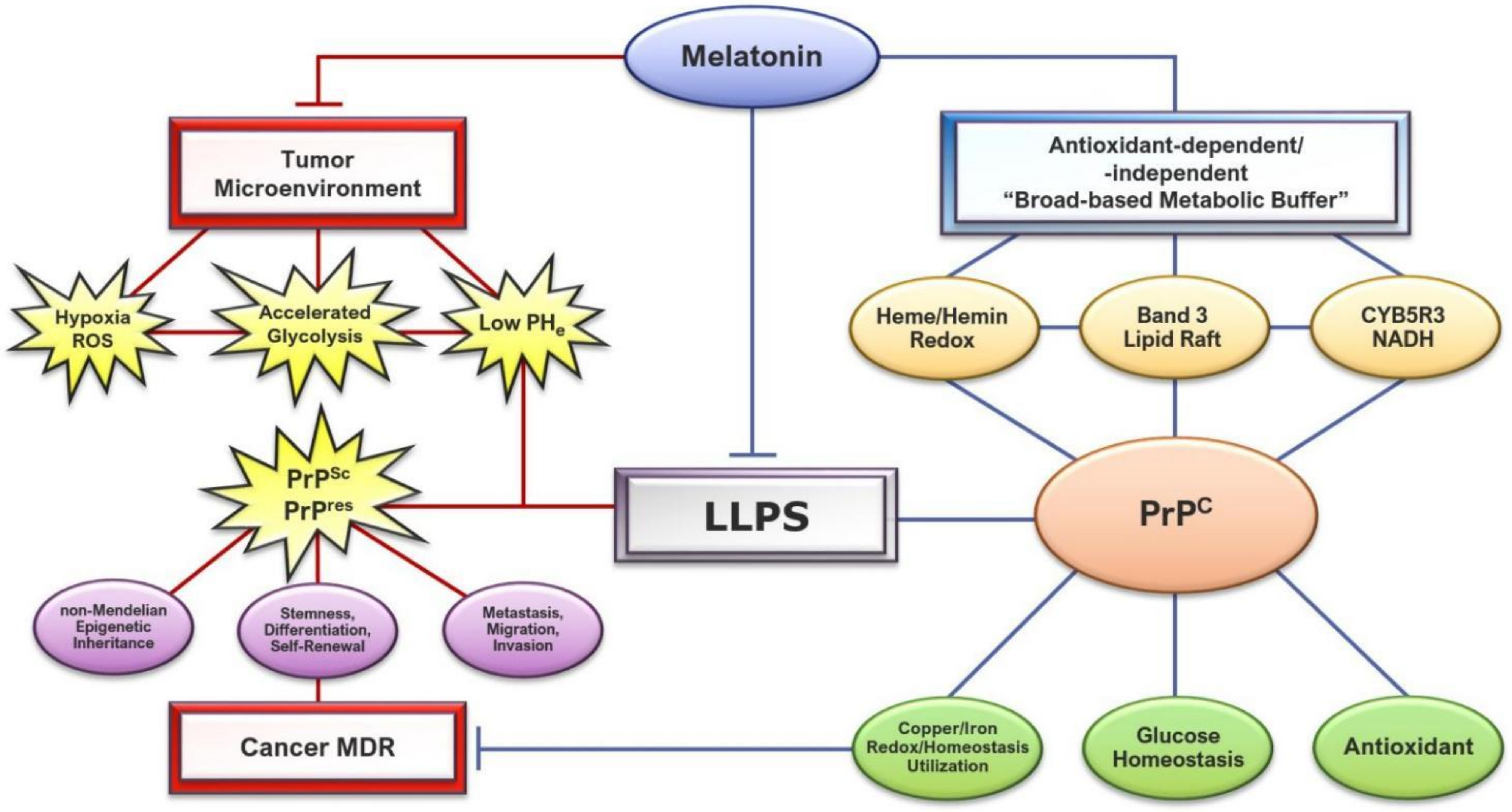{kind=link}
| Model/Description | Melatonin Doses | Melatonin’s Effects | Reference |
|---|---|---|---|
| MSCs/Model of ER stress–induced ischaemic injury. | 1 μM MEL pretreatment 30 min at 37 °C. | Increased expression of PrPC and antioxidant enzymes to reduce oxidative stress. | [113] |
| MSCs/Model of indoxyl sulfate-induced senescence. | 1 µM MEL + 5 µM pioglitazone. | Treatment promoted highest MSC growth rates and inhibited senescence via enhanced PrPC expression. | [647] |
| TH1/Model of high glucose-mediated fibrosis. | 1 µM MEL as pretreatment. | Prevented high glucose-induced fibrosis by recovering PrPC expression to augment antioxidant protection. | [648] |
| SNU-C5/WT cells/Model of colorectal cancer cell apoptosis. | 1 mM MEL treatment 24 h. | Reduced PrPC and PINK1 expression to increase mitochondrial superoxide. | [114] |
| Human colon CSCs (S707)/Model of PRNP overexpression. | 500 μM MEL + 1 μM 5-FU treatment for 72 h. | Treatment suppressed proliferation and increased apoptosis by inhibiting PrPC-OCT4 axis. | [115] |
| Murine/Model of human CSCs (S707) xenograft tumorigenesis. | 500 μM MEL + 1 μM 5-FU treatment for 72 h. | Treatment decreased PrPC expression to reduce tumor volume and suppress cell proliferation. | [115] |
| SNU-C5/Oxal-R/Model of PrPC expression in oxaliplatin-resistant colon cancer cells. | 500 μM MEL + 1 μM oxaliplatin for 24 h. | MEL induced oxaliplatin-mediated apoptosis via blockade of PrPC-mediated antioxidant activities. | [116] |
| PC12/Model of paraquat-induced NADH depletion. | 1 mM MEL incubation at 35 °C for 1 h. | Prevented the loss of NADH/NAD+ caused by paraquat treatment. | [642] |
| Oxyhemoglobin/Model of vanadate-induced NADH oxidation. | 2 mM MEL. | Treatment conferred the highest level of protection against NADH oxidation compared to lower doses. | [642] |
| Murine/Model of B16-F10 melanoma cell proliferation. | 1 mM MEL 24 h I incubation. | Significantly reduced growth rate and migration. | [649] |
| C57BL/6J mice/Model of lung metastasis via B16-F10 cell injection. | 20 mg/kg in drinking water or IP injection for 15 days. | Melatonin did not alter cell migration or proliferation. | [649] |
| Kunming mice/Model of copper-induced liver injury. | 50 mg/kg IP injection once daily, 3 times. | Inhibited copper-induced hepatotoxicity and DNA damage via copper chelation, preventing formation of hydroxyl radical. | [430] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loh, D.; Reiter, R.J. Melatonin: Regulation of Prion Protein Phase Separation in Cancer Multidrug Resistance. Molecules 2022, 27, 705. https://doi.org/10.3390/molecules27030705
Loh D, Reiter RJ. Melatonin: Regulation of Prion Protein Phase Separation in Cancer Multidrug Resistance. Molecules. 2022; 27(3):705. https://doi.org/10.3390/molecules27030705
Chicago/Turabian StyleLoh, Doris, and Russel J. Reiter. 2022. "Melatonin: Regulation of Prion Protein Phase Separation in Cancer Multidrug Resistance" Molecules 27, no. 3: 705. https://doi.org/10.3390/molecules27030705
APA StyleLoh, D., & Reiter, R. J. (2022). Melatonin: Regulation of Prion Protein Phase Separation in Cancer Multidrug Resistance. Molecules, 27(3), 705. https://doi.org/10.3390/molecules27030705