
Halogen Bonding in Haspin-Halogenated Tubercidin Complexes: Molecular Dynamics and Quantum Chemical Calculations



Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
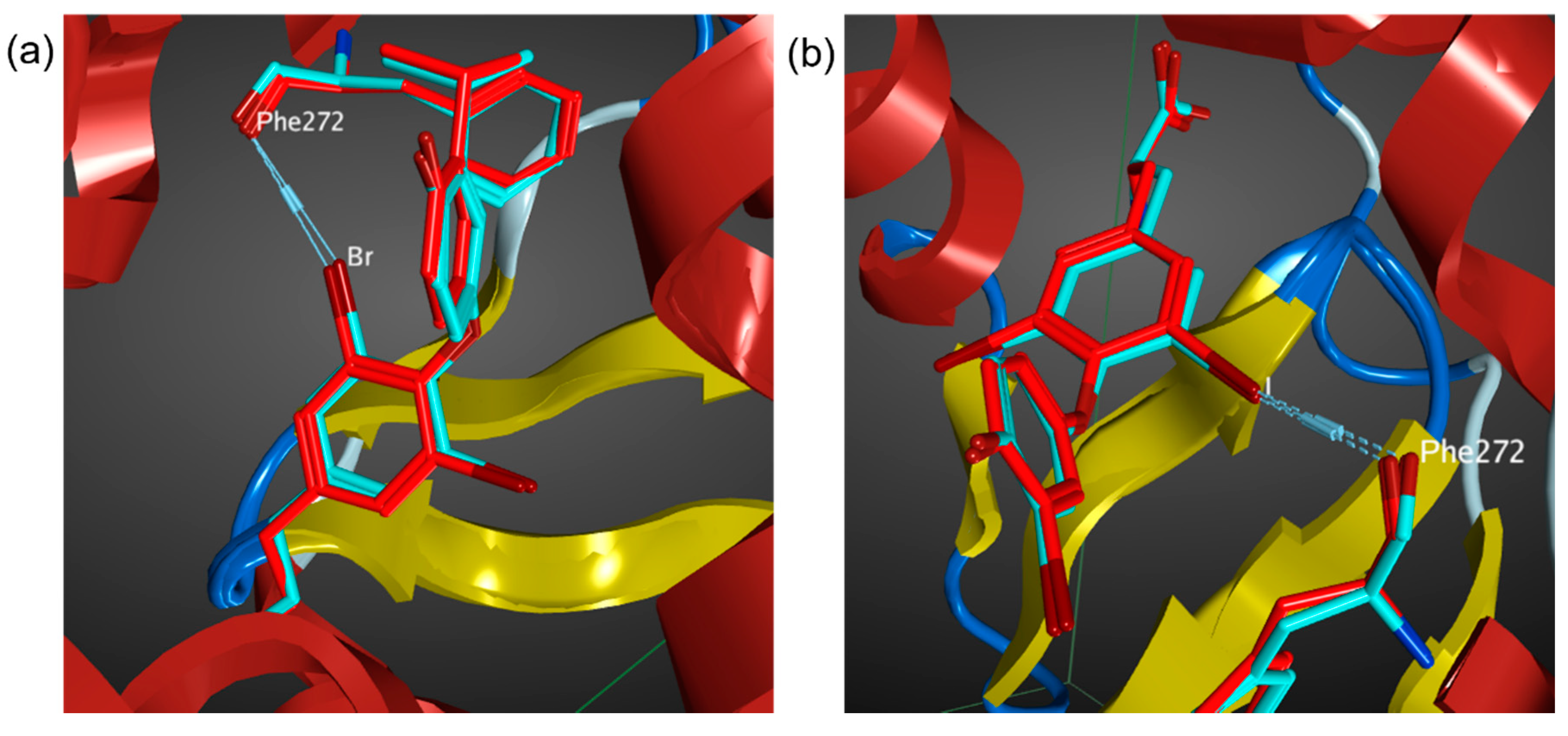
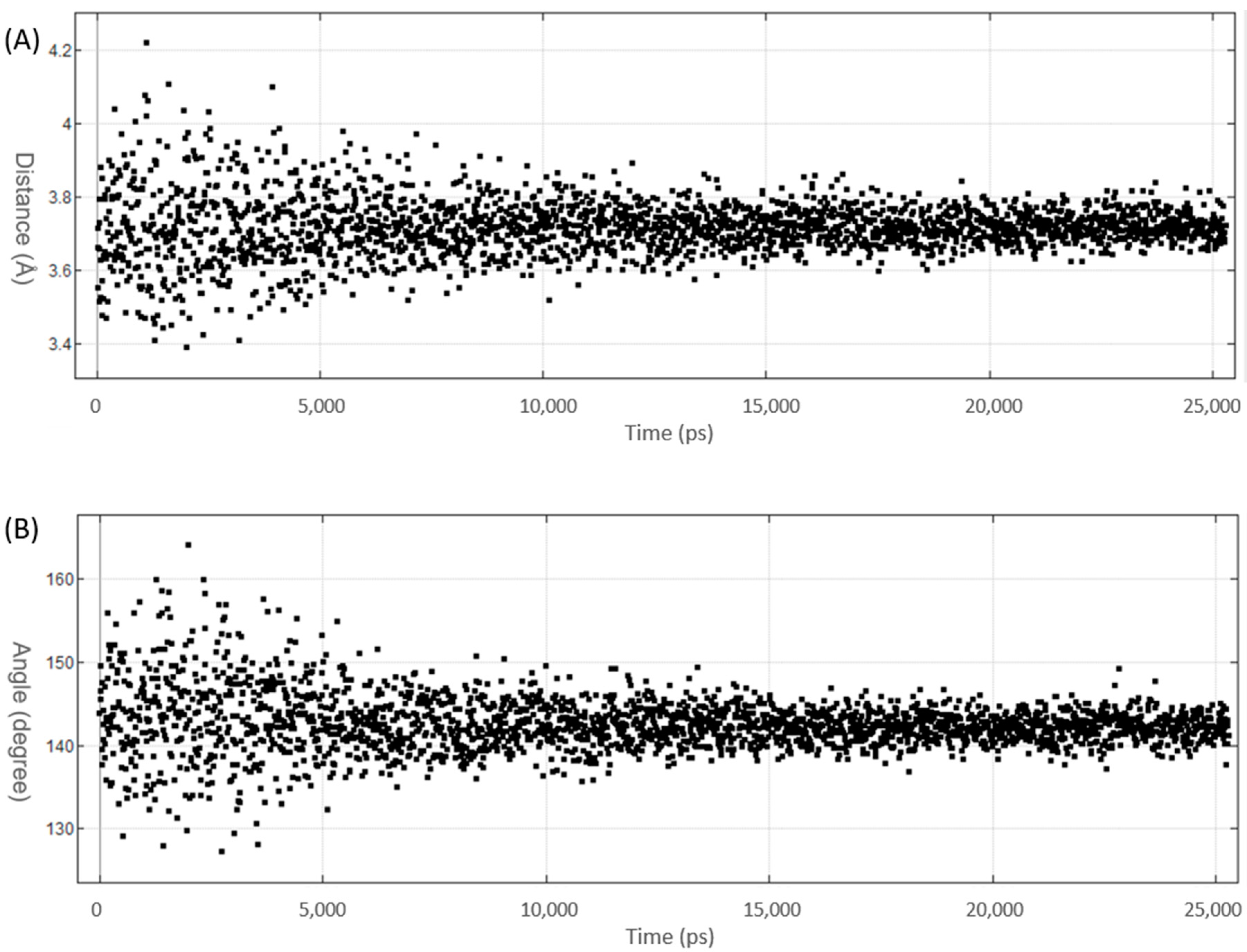
3.1. Benchmark of AMBER14EHT Force Field on XB Ligand–Protein Complexes
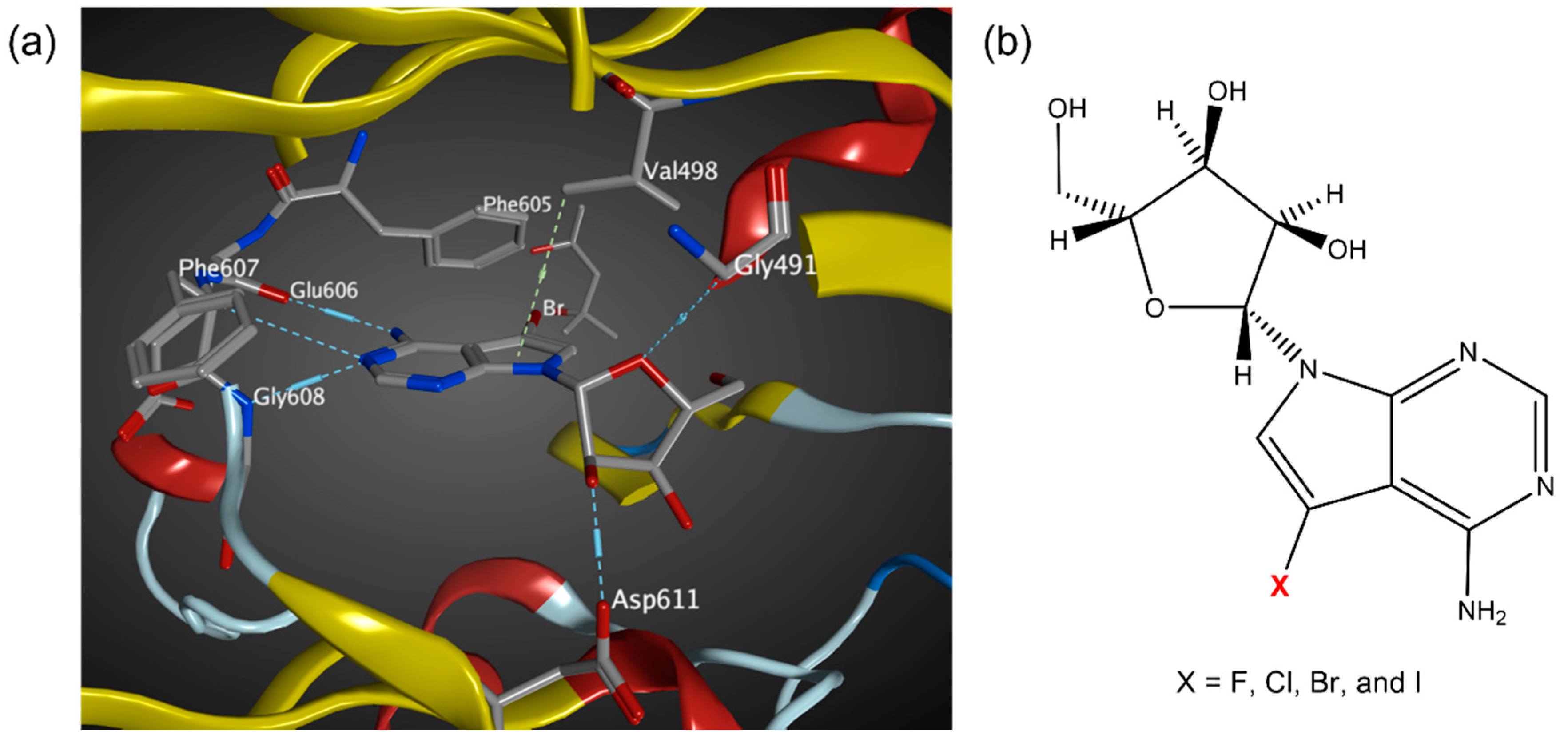
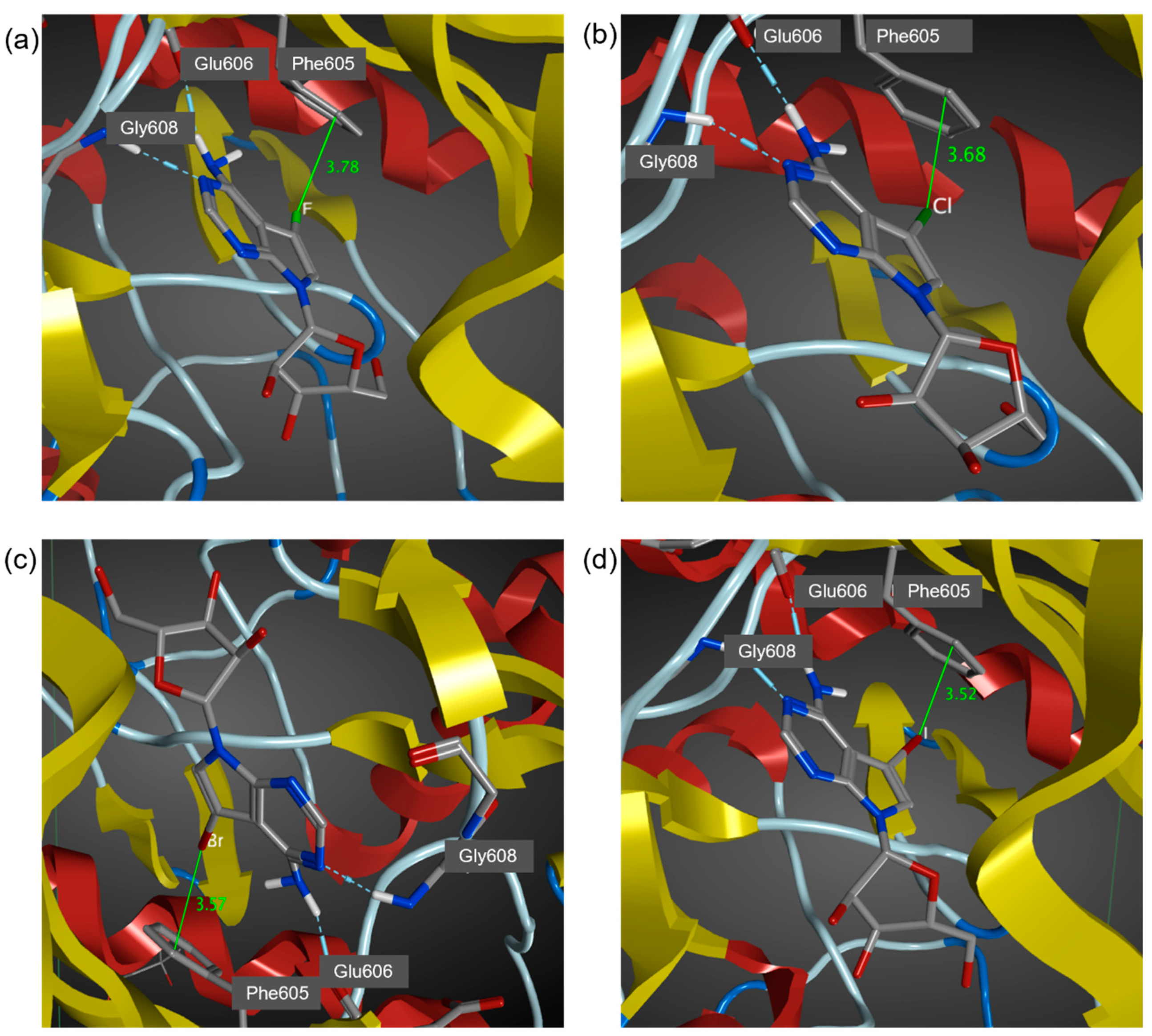
3.2. MD Simulations of Halogenated Tubercidin Ligands with Haspin
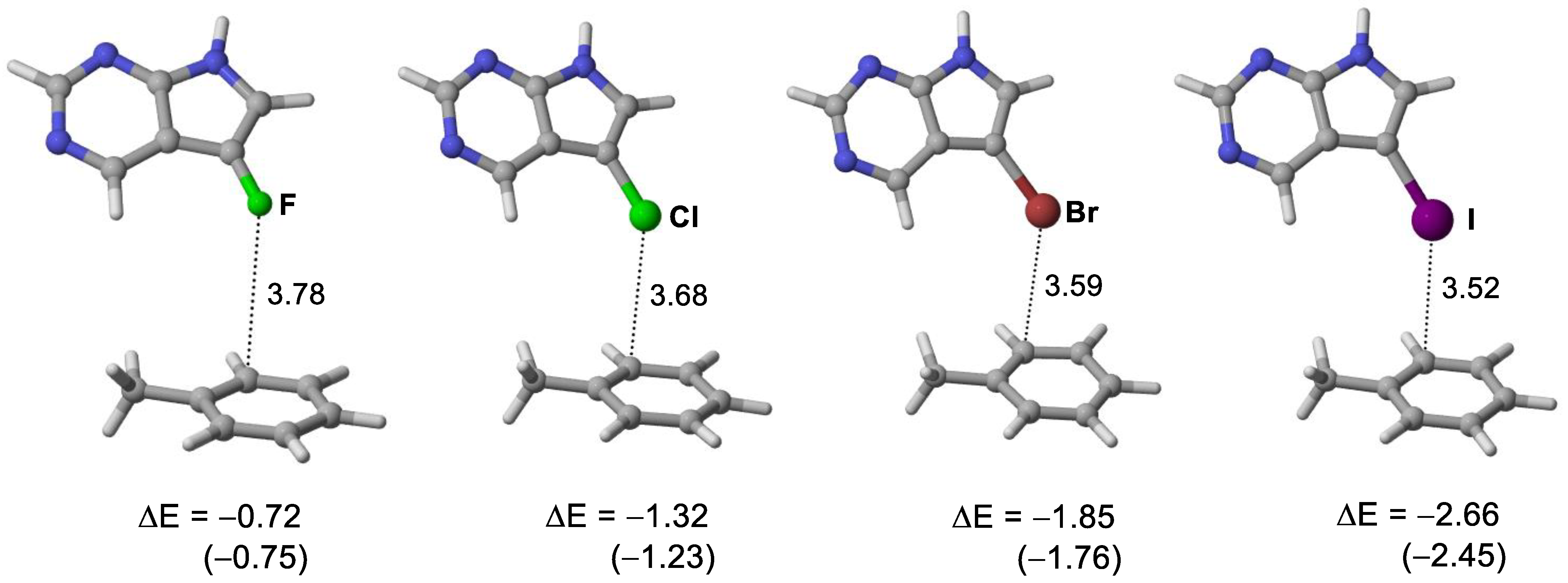
3.3. Quantum Chemical Calculations of Halogen Bond Interaction Energies
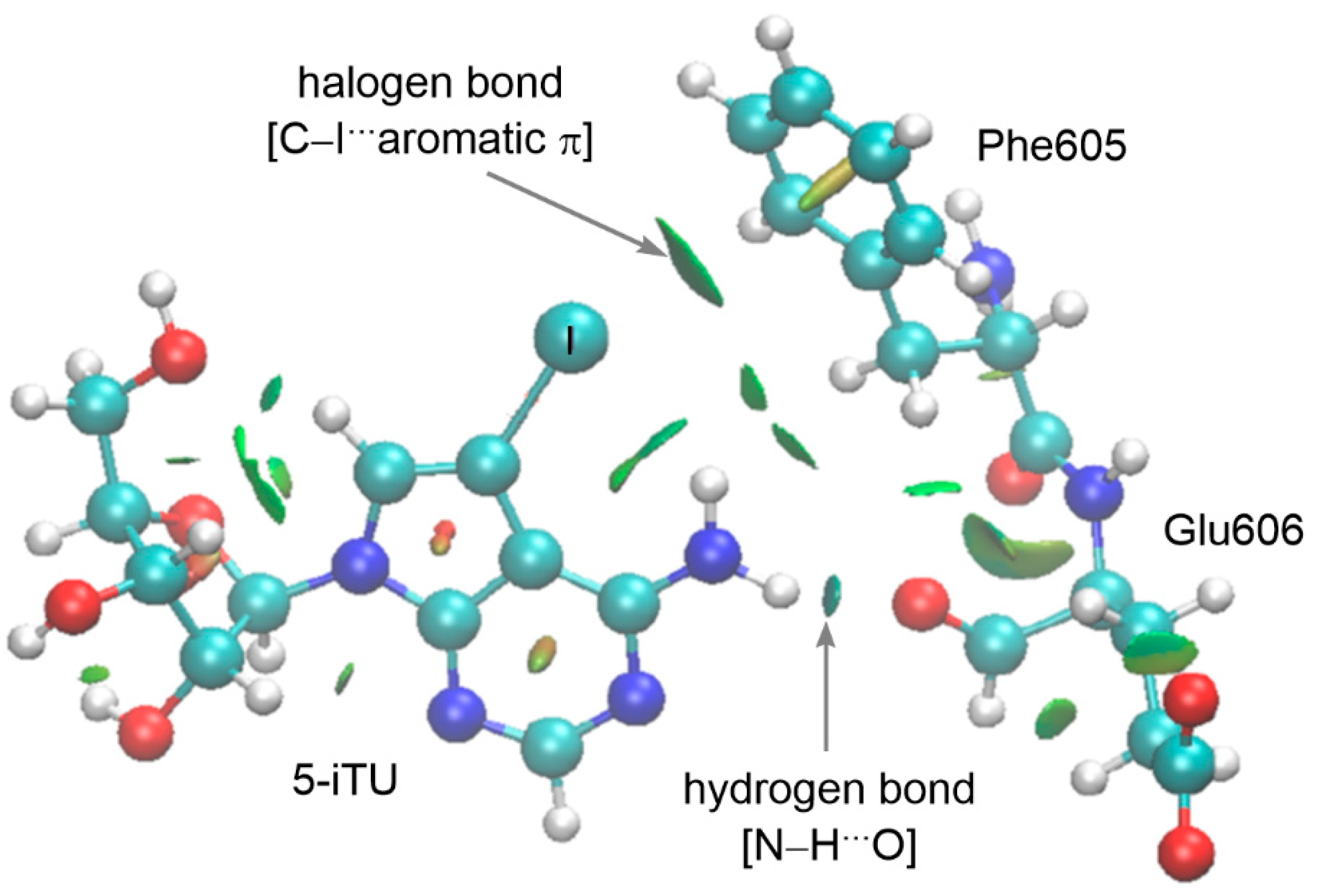
3.4. Analysis of Noncovalent Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Tanaka, H.; Yoshimura, Y.; Nishina, Y.; Nozaki, M.; Nojima, H.; Nishimune, Y. Isolation and characterization of cDNA clones specifically expressed in testicular germ cells. FEBS Lett. 1994, 355, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Yoshimura, Y.; Nozaki, M.; Yomogida, K.; Tsuchida, J.; Tosaka, Y.; Habu, T.; Nakanishi, T.; Okada, M.; Nojima, H.; et al. Identification and characterization of a haploid germ cell-specific nuclear protein kinase (Haspin) in spermatid nuclei and its effects on somatic cells. J. Biol.Chem. 1999, 274, 17049–17057. [Google Scholar] [CrossRef] [Green Version]
- Polioudaki, H.; Markaki, Y.; Kourmouli, N.; Dialynas, G.; Theodoropoulos, P.A.; Singh, P.B.; Georgatos, S.D. Mitotic phosphorylation of histone H3 at threonine 3. FEBS Lett. 2004, 560, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.M.G. Haspin-like proteins: A new family of evolutionarily conserved putative eukaryotic protein kinases. Protein Sci. 2001, 10, 1677–1684. [Google Scholar] [CrossRef]
- Hindriksen, S.; Bramer, A.J.; Truong, M.A.; Vromans, M.J.M.; Post, J.B.; Verlaan-Klink, I.; Snippert, H.J.; Lens, S.M.A.; Hadders, M.A. Baculoviral delivery of CRISPR/Cas9 facilitates efficient genome editing in human cells. PLoS ONE 2017, 12, e0179514. [Google Scholar] [CrossRef]
- Higgins, J.M.G. Structure, function and evolution of haspin and haspin-related proteins, a distinctive group of eukaryotic protein kinases. Cell Mol. Life Sci. 2003, 60, 446–462. [Google Scholar] [CrossRef]
- Dai, J.; Sultan, S.; Taylor, S.S.; Higgins, J.M.G. The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2005, 19, 472–488. [Google Scholar] [CrossRef] [Green Version]
- Markaki, Y.; Christogianni, A.; Politou, A.S.; Georgatos, S.D. Phosphorylation of histone H3 at Thr3 is part of a combinatorial pattern that marks and configures mitotic chromatin. J. Cell Sci. 2009, 122, 2809–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, A.E.; Ghenoiu, C.; Xue, J.Z.; Zierhut, C.; Kimura, H.; Funabiki, H. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science 2010, 330, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Dai, J.; Daum, J.R.; Niedzialkowska, E.; Banerjee, B.; Stukenberg, P.T.; Gorbsky, G.J.; Higgins, J.M.G. Histone H3 Thr-3 phosphorylation by haspin positions Aurora B at centromeres in mitosis. Science 2010, 330, 231–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Kelly, A.E.; Funabiki, H.; Patel, D.J. Structural basis for recognition of H3T3ph and Smac/DIABLO N-terminal peptides by human survivin. Structure 2012, 20, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Biggins, S.; Murray, A.W. The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev. 2001, 15, 3118–3129. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Iguchi, N.; Nakamura, Y.; Kohroki, J.; de Carvalho, C.E.; Nishimune, Y. Cloning and characterization of human haspin gene encoding haploid germ cell-specific nuclear protein kinase. Mol. Hum. Reprod. 2001, 7, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Hauf, S.; Cole, R.W.; LaTerra, S.; Zimmer, C.; Schnapp, G.; Walter, R.; Heckel, A.; van Meel, J.; Rieder, C.L.; Peters, J.-M. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 2003, 161, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, B.A.; Kung, C.; Shokat, K.M.; Biggins, S. The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat. Cell Biol. 2006, 8, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.; Capasso, P.; Tortorici, M.; Forneris, F.; de Marco, A.; Mattevi, A.; Musacchio. Crystal structure of the catalytic domain of Haspin, an atypical kinase implicated in chromatin organization. Proc. Natl. Acad. Sci. USA 2009, 106, 20204–20209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswaran, J.; Patnaik, D.; Filippakopoulos, P.; Wang, F.; Stein, R.L.; Murray, J.W.; Higgins, J.M.G.; Knapp, S. Structure and functional characterization of the atypical human kinase haspin. Proc. Natl. Acad. Sci. USA 2009, 106, 20198–20203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heroven, C.; Georgi, V.; Ganotra, G.K.; Brennan, P.; Wolfreys, F.; Wade, R.C.; Fernandez-Montalvan, A.E.; Chaikuad, A.; Knapp, S. Halogen-aromatic π interactions modulate inhibitor residence times. Angew. Chem. Int. Ed. Engl. 2018, 57, 7220–7224. [Google Scholar] [CrossRef]
- Newby, A.C. The role of adenosine kinase in regulating adenosine concentration. Biochem. J. 1985, 226, 343–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorov, O.; Marsden, B.; Pogacic, V.; Rellos, P.; Muller, S.; Bullock, A.N. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 20523–20528. [Google Scholar] [CrossRef] [Green Version]
- Balzano, D.; Santaguida, S.; Musacchio, A.; Villa, F. A general framework for inhibitor resistance in protein kinases. Chem. Biol. 2011, 18, 966–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Antoni, A.; Maffini, S.; Knapp, S.; Musacchio, A.; Santaguida, S. A small-molecule inhibitor of Haspin alters the kinetochore functions of Aurora B. J. Cell Biol. 2012, 199, 269–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Ulyanova, N.P.; Daum, J.R.; Patnaik, D.; Kateneva, A.V.; Gorbsky, G.J.; Higgins, J.M.G. Haspin inhibitors reveal centromeric functions of Aurora B in chromosome segregation. J. Cell Biol. 2012, 199, 251–268. [Google Scholar] [CrossRef] [Green Version]
- Kozgunova, E.; Suzuki, T.; Ito, M.; Higashiyama, T.; Kurihara, D. Haspin has multiple functions in the plant cell division regulatory network. Plant Cell Physiol. 2016, 57, 848–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. Proceedings of “Modeling interactions in biomolecules II”, Prague, September 5th–9th, 2005. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y. Chen, K.; Zhu, W. Halogen Bond: Its Role beyond Drug−Target Binding Affinity for Drug Discovery and Development. J. Chem. Inf. Model. 2014, 54, 69–78. [Google Scholar] [CrossRef]
- Carter, M.; Rappé, A.K.; Ho, P.S. Scalable Anisotropic Shape and Electrostatic Models for Biological Bromine Halogen Bond. J. Chem. Theory Comput. 2012, 8, 2461–2473. [Google Scholar] [CrossRef]
- Scholfield, M.R.; Vander Zanden, C.M.; Carter, M.; Ho, P.S. Halogen bonding (X-bonding): A biological perspective. Prot. Sci. 2013, 22, 139–152. [Google Scholar] [CrossRef]
- Parisini, E.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding in halocarbon-protein complexes: A structural survey. Chem. Soc. Rev. 2011, 40, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.J.; Mak, A.M.; Sullivan, M.B.; Wong, M.W. Site specificity of halogen bonding involving aromatic acceptors. Phys. Chem. Chem. Phys. 2018, 20, 8685–8694. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.J.; Mak, A.M.; Wong, M.W. Nature of halogen bonding involving π-systems, nitroxide radicals and carbenes: A highlight of the importance of charge transfer. Phys. Chem. Chem. Phys. 2018, 20, 26463–26478. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2020. [Google Scholar]
- Labute, P.; Williams, C. Application of Hückel Theory to Pharmacophore Discovery. CICSJ Bull. 2015, 33, 33–40. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, D. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 128, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: Benchmarks and Theoretical Analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Zhang, Z.; Alexov, E. On the Dielectric “Constant” of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theory Comput. 2013, 9, 2126–2136. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Himo, F. The quantum chemical cluster approach for modeling enzyme reactions. WIREs Comput. Mol. Sci. 2011, 1, 323–336. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kee, C.W.; Wong, M.W. In silico design of halogen-bonding-based organocatalyst for Diels–Alder reaction, Claisen rearrangement, and Cope-type hydroamination. J. Org. Chem. 2016, 81, 7459–7470. [Google Scholar] [CrossRef]
- Narth, C.; Maroun, Z.; Boto, R.; Chaudret, C.; Bonnet, M.L.; Piquemal, J.-P.; Contreras-Garcia, J. A Complete NCI Perspective: From New Bonds to Reactivity. In Challenges and Advances in Computational Chemistry and Physics; Springer Nature: Basingstoke, UK, 2016; Chapter 18. [Google Scholar]
- Song, S.; Wang, L.; Su, J.; Xu, Z.; Hsu, C.-H.; Lyu, P.; Li, J.; Peng, X.; Takahiro, K.; Telychko, M.; et al. Manifold Dynamic Non-Covalent Interactions for Steering Molecular Assembly and Cyclization. Chem. Sci. 2021, 12, 11659–11667. [Google Scholar] [CrossRef]
- Masaryk, L.; Moncol, J.; Herchel, R.; Nemec, I. Halogen Bonding in New Dichloride-Cobalt(II) Complex with Iodo Substituted Chalcone Ligands. Crystals 2020, 10, 354. [Google Scholar] [CrossRef]
- Koehler, K.; Gordon, S.; Brandt, P.; Carlsson, B.; Backsbro-Saeidi, A.; Apelqvist, T.; Agback, P.; Grover, G.J.; Nelson, W.; Grynfarb, M.; et al. Thyroid Receptor Ligands. 6. A High Affinity “Direct Antagonist” Selective for the Thyroid Hormone Receptor. J. Med. Chem. 2006, 49, 6635–6637. [Google Scholar] [CrossRef]
- Sandler, B.; Webb, P.; Apriletti, J.W.; Huber, B.R.; Togashi, M.; Lima, S.T.C.; Juric, S.; Nilsson, S.; Wagner, T.; Fletterick, R.J.; et al. Thyroxine-Thyroid Hormone Receptor Interactions. J. Biol. Chem. 2004, 279, 55801–55808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided. Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | X∙∙∙π Closest Carbon Distance (Å) a | Sum of VDW Radii (Å) b | Binding Energy (kcal/moL) c |
|---|---|---|---|
| 5-iTu | 3.52 (3.38) | 3.68 | −48.9 |
| 5-brTu | 3.57 (3.28) | 3.55 | −48.6 |
| 5-clTu | 3.68 (3.39) | 3.45 | −47.9 |
| 5-fTu | 3.78 (3.48) | 3.17 | −46.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Wong, M.W. Halogen Bonding in Haspin-Halogenated Tubercidin Complexes: Molecular Dynamics and Quantum Chemical Calculations. Molecules 2022, 27, 706. https://doi.org/10.3390/molecules27030706
Zhou Y, Wong MW. Halogen Bonding in Haspin-Halogenated Tubercidin Complexes: Molecular Dynamics and Quantum Chemical Calculations. Molecules. 2022; 27(3):706. https://doi.org/10.3390/molecules27030706
Chicago/Turabian StyleZhou, Yujing, and Ming Wah Wong. 2022. "Halogen Bonding in Haspin-Halogenated Tubercidin Complexes: Molecular Dynamics and Quantum Chemical Calculations" Molecules 27, no. 3: 706. https://doi.org/10.3390/molecules27030706
APA StyleZhou, Y., & Wong, M. W. (2022). Halogen Bonding in Haspin-Halogenated Tubercidin Complexes: Molecular Dynamics and Quantum Chemical Calculations. Molecules, 27(3), 706. https://doi.org/10.3390/molecules27030706