
Kirenol: A Potential Natural Lead Molecule for a New Drug Design, Development, and Therapy for Inflammation


 , , ,
, , ,  ,
,  ,
,  , ,
, ,
Abstract
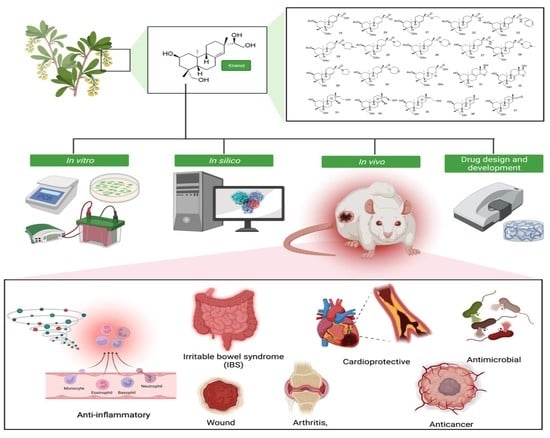
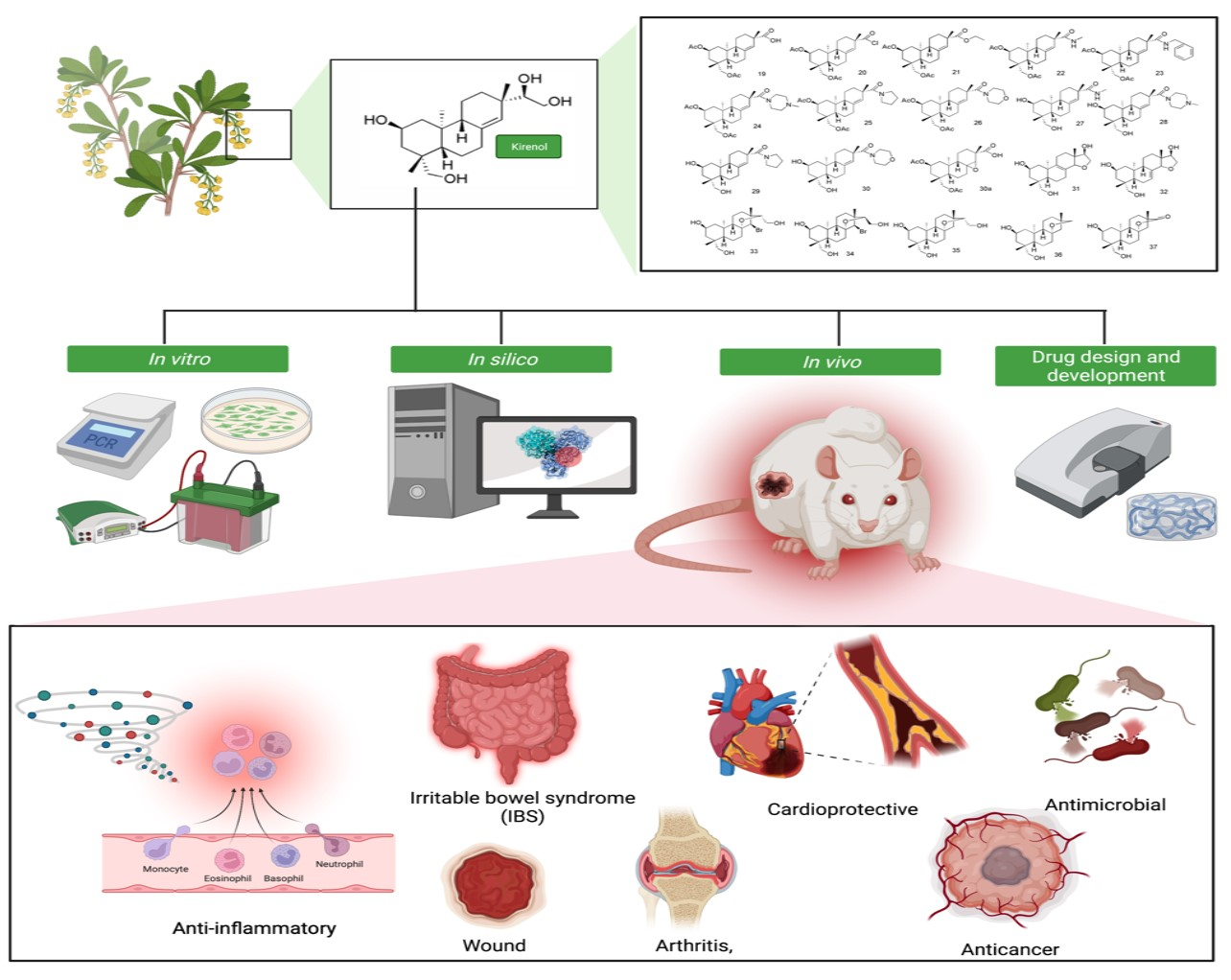
:
1. Introduction
2. The Role of Kirenol against Inflammation
2.1. The Use of Kirenol against Complete Freund’s Adjuvant (CFA)-Induced Chronic Inflammation
2.2. The Use of Kirenol against Neuro-Inflammation
2.3. The Use of Kirenol against Cardiovascular Disease-Related Inflammation
2.4. The Use of Kirenol against Lung Injury-Related Inflammatory Disease
2.5. The Use of Kirenol against Colon Injury
2.6. The Use of Kirenol against Diabetic Wounds
2.7. The Use of Kirenol against Bone Damage
2.8. The Use of Kirenol against Arthritis
3. Overview of Mechanism of Action of Kirenol against Inflammation
4. In Silico Molecular Docking Study
5. Challenges and Opportunities of Kirenol to Be Developed into a Drug Molecule for the Treatment of Inflammation
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dinarello, C.A. Anti-inflammatory agents: Present and future. Cell 2010, 140, 935–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, M. Inflammation and fibrosis. Matrix Biol. 2018, 68, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, J.; Horie, S.; Laffey, J.G. Role of the adaptive immune response in sepsis. Intensive Care Med. Exp. 2020, 8, 1–19. [Google Scholar] [CrossRef]
- Lee, H.; Fessler, M.B.; Qu, P.; Heymann, J.; Kopp, J.B. Macrophage polarization in innate immune responses contributing to pathogenesis of chronic kidney disease. BMC Nephrol. 2020, 21, 1–13. [Google Scholar] [CrossRef]
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like receptors and the control of immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Liu, J.; Cao, X. Cellular and molecular regulation of innate inflammatory responses. Cell. Mol. Immunol. 2016, 13, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Koeberle, A.; Werz, O. Multi-target approach for natural products in inflammation. Drug Discov. Today 2014, 19, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Ci, X.; Li, H.; Yu, Q.; Zhang, X.; Yu, L.; Chen, N.; Song, Y.; Deng, X. Avermectin exerts anti-inflammatory effect by downregulating the nuclear transcription factor kappa-B and mitogen-activated protein kinase activation pathway. Fundam. Clin. Pharmacol. 2009, 23, 449–455. [Google Scholar] [CrossRef] [PubMed]
- French, J.A.; Koepp, M.; Naegelin, Y.; Vigevano, F.; Auvin, S.; Rho, J.M.; Rosenberg, E.; Devinsky, O.; Olofsson, P.S.; Dichter, M.A. Clinical studies and anti-inflammatory mechanisms of treatments. Epilepsia 2017, 58, 69–82. [Google Scholar] [CrossRef]
- Ponder, A.; Long, M.D. A clinical review of recent findings in the epidemiology of inflammatory bowel disease. Clin. Epidemiol. 2013, 5, 237. [Google Scholar]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Calixto, J.B.; Otuki, M.F.; Santos, A.R. Anti-inflammatory compounds of plant origin. Part I. Action on arachidonic acid pathway, nitric oxide and nuclear factor κ B (NF-κB). Planta Med. 2003, 69, 973–983. [Google Scholar]
- Zhu, F.; Du, B.; Xu, B. Anti-inflammatory effects of phytochemicals from fruits, vegetables, and food legumes: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1260–1270. [Google Scholar] [CrossRef]
- Li, H.; Li, P.Y.; Yeung, P. A simple HPLC assay for ginsenoside-Rh2 in plasma and its application for pharmacokinetic study in rats. Nat. Prod. Chem. Res. 2013. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.R.; Altyar, A.E.; Sindi, I.A.; El-Agamy, D.S.; Abdallah, H.M.; Mohamed, S.G.; Mohamed, G.A. Kirenol: A promising bioactive metabolite from siegesbeckia species: A detailed review. J. Ethnopharmacol. 2021, 281, 114552. [Google Scholar] [CrossRef] [PubMed]
- Stils Jr, H.F. Adjuvants and antibody production: Dispelling the myths associated with Freund’s complete and other adjuvants. ILAR J. 2005, 46, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billiau, A.; Matthys, P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J. Leukoc. Biol. 2001, 70, 849–860. [Google Scholar] [PubMed]
- Wang, J.-P.; Zhou, Y.-M.; Ye, Y.-J.; Shang, X.-M.; Cai, Y.-L.; Xiong, C.-M.; Wu, Y.-X.; Xu, H.-X. Topical anti-inflammatory and analgesic activity of kirenol isolated from Siegesbeckia orientalis. J. Ethnopharmacol. 2011, 137, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Yang, R.; Yang, L.; Fan, X.; Liu, W.; Deng, W. Kirenol attenuates experimental autoimmune encephalomyelitis by inhibiting differentiation of Th1 and th17 cells and inducing apoptosis of effector T cells. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Kim, S.H.; Lee, H.K.; Cho, Y.; Kang, J.; Sung, S.H. ent-kaurane and ent-pimarane diterpenes from Siegesbeckia pubescens inhibit lipopolysaccharide-induced nitric oxide production in BV2 microglia. Biol. Pharm. Bull. 2014, 37, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Alzahrani, A.M.; Ahmed, E.A.; Veeraraghavan, V.P. Kirenol inhibits B[a]P-induced oxidative stress and apoptosis in endothelial cells via modulation of the Nrf2 signaling pathway. Oxidative Med. Cell. Longev. 2021, 2021, 5585303. [Google Scholar] [CrossRef]
- Lin, F.C.-F.; Lee, S.-S.; Li, Y.-C.; Ho, Y.-C.; Chen, W.-Y.; Chen, C.-J.; Lee, M.-W.; Yeh, K.-L.; Tsai, S.C.-S.; Kuan, Y.-H. Protective Effects of Kirenol against Lipopolysaccharide-Induced Acute Lung Injury through the Modulation of the Proinflammatory NFκB Pathway and the AMPK2-/Nrf2-Mediated HO-1/AOE Pathway. Antioxidants 2021, 10, 204. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef]
- Liu, X.H.; Du, Y.J.; Liu, G.X.; Dan, G.M.; Tong, X.; Xiao, J. Kirenol relieves dextran sulfate sodium-induced ulcerative colitis in mice by inhibiting inflammatory cytokines and inducing CD4+ T lymphocyte apoptosis. J. South. Med. Univ. 2019, 39, 1387–1392. [Google Scholar]
- Gupta, A.; Upadhyay, N.K.; Sawhney, R.; Kumar, R. A poly-herbal formulation accelerates normal and impaired diabetic wound healing. Wound Repair Regen. 2008, 16, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, S.F.; Caruso, G.I.; De Pasquale, R.; Sortino, M.A.; Merlo, S. The treatment of impaired wound healing in diabetes: Looking among old drugs. Pharmaceuticals 2020, 13, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, J.L.; Wyant, W.A.; Abdo Abujamra, B.; Kirsner, R.S.; Jozic, I. Diabetic Wound-Healing Science. Medicina 2021, 57, 1072. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Yang, M.; Chen, J.; Ma, S.; Wang, N. Anti-inflammatory and wound healing potential of kirenol in diabetic rats through the suppression of inflammatory markers and matrix metalloproteinase expressions. Biomed. Pharmacother. 2020, 129, 110475. [Google Scholar] [CrossRef]
- Lewiecki, E.M. New targets for intervention in the treatment of postmenopausal osteoporosis. Nat. Rev. Rheumatol. 2011, 7, 631–638. [Google Scholar] [CrossRef]
- Niu, Y.; Li, Y.; Kong, X.; Zhang, R.; Sun, Y.; Li, Q.; Li, C.; Liu, L.; Wang, J.; Mei, Q. The beneficial effect of Radix Dipsaci total saponins on bone metabolism in vitro and in vivo and the possible mechanisms of action. Osteoporos. Int. 2012, 23, 2649–2660. [Google Scholar] [CrossRef]
- Zou, B.; Zheng, J.; Deng, W.; Tan, Y.; Jie, L.; Qu, Y.; Yang, Q.; Ke, M.; Ding, Z.; Chen, Y. Kirenol inhibits RANKL-induced osteoclastogenesis and prevents ovariectomized-induced osteoporosis via suppressing the Ca2+-NFATc1 and Cav-1 signaling pathways. Phytomedicine 2021, 80, 153377. [Google Scholar] [CrossRef]
- Kim, M.-B.; Song, Y.; Hwang, J.-K. Kirenol stimulates osteoblast differentiation through activation of the BMP and Wnt/β-catenin signaling pathways in MC3T3-E1 cells. Fitoterapia 2014, 98, 59–65. [Google Scholar] [CrossRef]
- Karaman, İ.; Günay, A.E.; Yerer, M.B.; Demirpolat, E.; Doğan, S.; Yay, A.H.; Kafadar, İ.H. Effect of kirenol on the interaction between the WNT/β-Catenin and RUNX2/TCF/LEF1 pathways in fracture healing in vivo. Acta Orthop. Et Traumatol. Turc. 2020, 54, 320–329. [Google Scholar] [CrossRef]
- Wu, J.; Li, Q.; Jin, L.; Qu, Y.; Liang, B.-B.; Zhu, X.-T.; Du, H.-Y.; Jie, L.-G.; Yu, Q.-H. Kirenol inhibits the function and inflammation of fibroblast-like synoviocytes in rheumatoid arthritis in vitro and in vivo. Front. Immunol. 2019, 10, 1304. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-M.; Zhu, S.-G.; Wu, Z.-W.; Lu, Y.; Fu, H.-Z.; Qian, R.-Q. Kirenol upregulates nuclear Annexin-1 which interacts with NF-κB to attenuate synovial inflammation of collagen-induced arthritis in rats. J. Ethnopharmacol. 2011, 137, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xiao, J.; Wu, Z.; Wang, Z.; Fu, H.; Chen, Y.; Qian, R. Effects of kirenol on bovine type II collagen-induced rat lymphocytes in vivo and in vitro. Nan Fang Yi Ke Da Xue Xue Bao J. South. Med. Univ. 2012, 32, 1–6. [Google Scholar]
- Lu, Y.; Xiao, J.; Wu, Z.-W.; Wang, Z.-M.; Hu, J.; Fu, H.-Z.; Chen, Y.-Y.; Qian, R.-Q. Kirenol exerts a potent anti-arthritic effect in collagen-induced arthritis by modifying the T cells balance. Phytomedicine 2012, 19, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Qu, Y.; Deng, J.-X.; Liang, W.-Y.; Jiang, Z.-L.; Lai, R.; Yu, Q.-H. Molecular docking studies of kirenol a traditional Chinese medicinal compound against rheumatoid arthritis cytokine drug targets (TNF-α, IL-1 and IL-6). Biomed. Res. 2017, 28, 1992–1995. [Google Scholar]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in inflammatory disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.; West, A.; Ghosh, S. NF-κ B and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [Green Version]
- Cross, M.L.; Ganner, A.; Teilab, D.; Fray, L.M. Patterns of cytokine induction by gram-positive and gram-negative probiotic bacteria. FEMS Immunol. Med. Microbiol. 2004, 42, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Prescott, J.A.; Mitchell, J.P.; Cook, S.J. Inhibitory feedback control of NF-κB signalling in health and disease. Biochem. J. 2021, 478, 2619–2664. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Malkowski, M.G. Substrate-selective inhibition of cyclooxygeanse-2 by fenamic acid derivatives is dependent on peroxide tone. J. Biol. Chem. 2016, 291, 15069–15081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, N.C.; Gerstmeier, J.; Schexnaydre, E.E.; Börner, F.; Garscha, U.; Neau, D.B.; Werz, O.; Newcomer, M.E. Structural and mechanistic insights into 5-lipoxygenase inhibition by natural products. Nat. Chem. Biol. 2020, 16, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.; Bernardi, D.I.; Fill, T.; Fernandes, A.A.; Jurberg, I.D. The chemistry and biology of guanidine secondary metabolites. Nat. Prod. Rep. 2021, 38, 586–667. [Google Scholar] [CrossRef]
- Kircher, M.; Herhaus, P.; Schottelius, M.; Buck, A.K.; Werner, R.A.; Wester, H.-J.; Keller, U.; Lapa, C. CXCR4-directed theranostics in oncology and inflammation. Ann. Nucl. Med. 2018, 32, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, K.; Suno, R.; Hotta, Y.; Yamashita, K.; Hirata, K.; Yamamoto, M.; Narumiya, S.; Iwata, S.; Kobayashi, T. Crystal structure of the endogenous agonist-bound prostanoid receptor EP3. Nat. Chem. Biol. 2019, 15, 8–10. [Google Scholar] [CrossRef]
- Song, X.L.; Zhang, Q.Y.; Wang, Z.M.; Fu, H.Z.; Qian, R.Q. A rapid and simple RP-HPLC method for quantification of kirenol in rat plasma after oral administration and its application to pharmacokinetic study. Biomed. Chromatogr. 2011, 25, 542–546. [Google Scholar] [CrossRef]
- Huo, L.; Jiang, Z.; Lei, M.; Wang, X.; Guo, X. Simultaneous quantification of Kirenol and ent-16β, 17-dihydroxy-kauran-19-oic acid from Herba siegesbeckiae in rat plasma by liquid chromatography–tandem mass spectrometry and its application to pharmacokinetic studies. J. Chromatogr. B 2013, 937, 18–24. [Google Scholar] [CrossRef]
- Yin, Y.; Sun, Y.; Jiang, Z. Pharmacokinetics study of two active diterpenoids from Herba siegesbeckiae in rat plasma. Yao Xue Xue Bao = Acta Pharm. Sin. 2016, 51, 631–636. [Google Scholar]
- Martinho, N.; Damgé, C.; Reis, C.P. Recent advances in drug delivery systems. J. Biomater. Nanobiotechnol. 2011, 2, 510–526. [Google Scholar] [CrossRef] [Green Version]
- Jahangirian, H.; Lemraski, E.G.; Webster, T.J.; Rafiee-Moghaddam, R.; Abdollahi, Y. A review of drug delivery systems based on nanotechnology and green chemistry: Green nanomedicine. Int. J. Nanomed. 2017, 12, 2957–2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
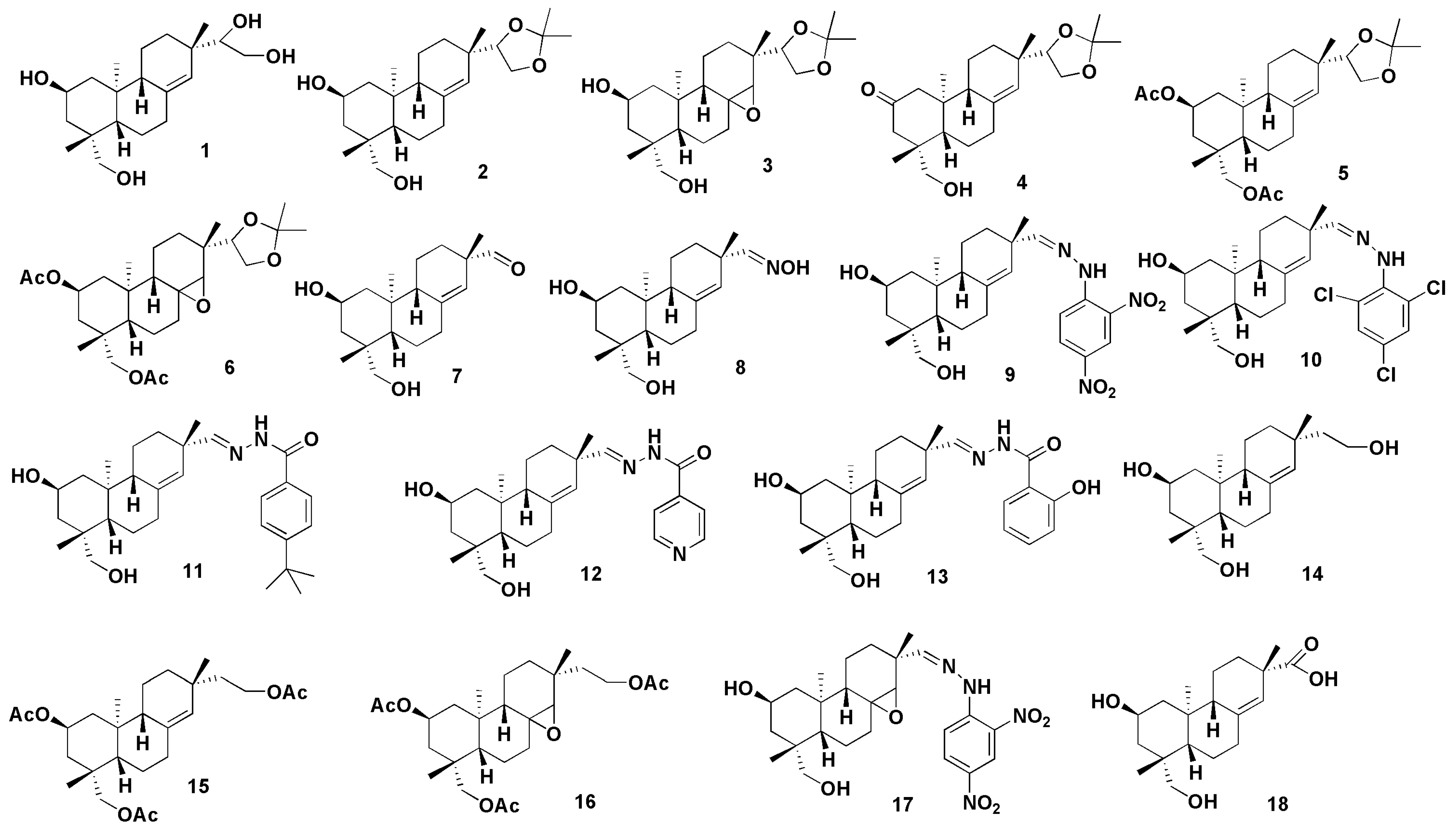
- Wang, J.L.; Sun, H.Y.; Li, Y.F.; Chu, H.P.; Sun, J.Y. Synthesis and preliminary anti-inflammatory activity exploration of novel derivatives of kirenol. N. J. Chem. 2020, 44, 19250–19261. [Google Scholar] [CrossRef]
- Wang, J.; Wu, M.; Gao, C.; Fu, H. Semisynthesis of epoxy-pimarane diterpenoids from kirenol and their FXa inhibition activities. Bioorganic Med. Chem. 2019, 27, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, H.; Fu, H. Semisynthesis of ent-norstrobane diterpenoids as potential inhibitor for factor Xa. Bioorganic Med. Chem. Lett. 2018, 28, 3813–3815. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Protein | Name of the Protein | Ligand | MolDock Score | Rerank Score | HBond | Amino Acid Residue ID |
|---|---|---|---|---|---|---|---|
| 1. | 5IKR | COX-2 | Kirenol | −91.509 | −78.647 | −4.718 | 5IKR (B): Arg 120, Glu 524, Leu 80, Leu 123, Leu 472, Lys 83, Met 471, Phe 470, Pro 84, Pro 86, Ser 119, Thr 85, Tyr 115, HOH 754 (B) (water) 107. |
| 2. | 6NCF | LOX-5 | Kirenol | −82.439 | −72.048 | −9.983 | 6NCF (B): Arg 47, Arg 401, Asn 613, Asp 170, Gln 15, Glu 16, Glu 612, Glu 614, Glu 622, Leu 615, Phe 402, Ser 14. |
| 3. | 4RWS | CXCR4 | Kirenol | −69.913 | −59.367 | −9.542 | 4RWS (A): Arg 188, Asp 262, Cys 187, His 113, Ile 259, Ile 284, Leu 266, Tyr 190, Tyr 255, 4RWS (C): Ala 3, Cys 5, Gly 2, His 6, Leu 1. |
| 4. | 6AK3 | Human prostaglandin E receptor EP3 | Kirenol | −83.287 | −66.223 | −1.442 | 6AK3 (A): Gly 65, Ile 340, Leu 60, Leu 341, Leu 349, Thr 61, Trp 344, Val 64, 6AK3 (B): Gly 65, Leu 60, Leu 341, Thr 61, Trp 344, Val 64. |
| Property/Rule | Result |
|---|---|
| Molecular formula | C20H34O4 |
| Molecular weight | 338.50 |
| Hydrogen bond donors | 4 |
| Hydrogen bond acceptors | 4 |
| Rotatable bonds | 3 |
| Log P (Partition coefficient, Predicted value) | 1.898 |
| Molar refractivity | 95.87 cm3 |
| Topological polar surface area | 80.9 Ų |
| Lipinski’s Rule of Five | Passed |
| Ghose_Filter | Passed |
| Veber’s Rule | Passed |
| BBB Likeness Rule | Passed |
| Unweighted QED | Passed |
| Weighted QED | Passed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasir, N.N.; Sekar, M.; Fuloria, S.; Gan, S.H.; Rani, N.N.I.M.; Ravi, S.; Begum, M.Y.; Chidambaram, K.; Sathasivam, K.V.; Jeyabalan, S.; et al. Kirenol: A Potential Natural Lead Molecule for a New Drug Design, Development, and Therapy for Inflammation. Molecules 2022, 27, 734. https://doi.org/10.3390/molecules27030734
Nasir NN, Sekar M, Fuloria S, Gan SH, Rani NNIM, Ravi S, Begum MY, Chidambaram K, Sathasivam KV, Jeyabalan S, et al. Kirenol: A Potential Natural Lead Molecule for a New Drug Design, Development, and Therapy for Inflammation. Molecules. 2022; 27(3):734. https://doi.org/10.3390/molecules27030734
Chicago/Turabian StyleNasir, Naurah Nabihah, Mahendran Sekar, Shivkanya Fuloria, Siew Hua Gan, Nur Najihah Izzati Mat Rani, Subban Ravi, M. Yasmin Begum, Kumarappan Chidambaram, Kathiresan V. Sathasivam, Srikanth Jeyabalan, and et al. 2022. "Kirenol: A Potential Natural Lead Molecule for a New Drug Design, Development, and Therapy for Inflammation" Molecules 27, no. 3: 734. https://doi.org/10.3390/molecules27030734
APA StyleNasir, N. N., Sekar, M., Fuloria, S., Gan, S. H., Rani, N. N. I. M., Ravi, S., Begum, M. Y., Chidambaram, K., Sathasivam, K. V., Jeyabalan, S., Dhiravidamani, A., Thangavelu, L., Lum, P. T., Subramaniyan, V., Wu, Y. S., Azad, A. K., & Fuloria, N. K. (2022). Kirenol: A Potential Natural Lead Molecule for a New Drug Design, Development, and Therapy for Inflammation. Molecules, 27(3), 734. https://doi.org/10.3390/molecules27030734