
Recognition of Dimethylarginine Analogues by Tandem Tudor Domain Protein Spindlin1



Abstract
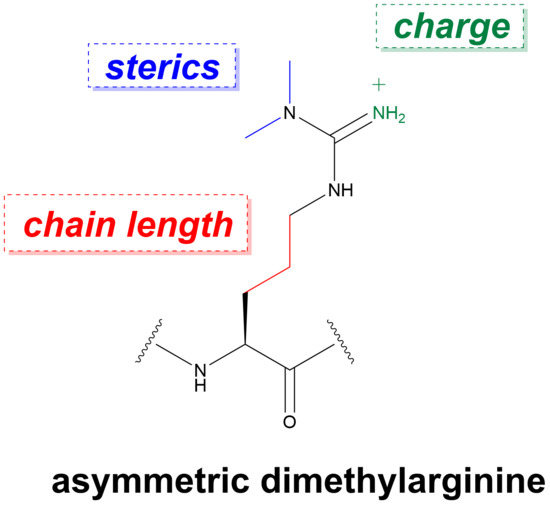
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Synthesis of H3K4me3Cit8me2
4.2. Synthesis of H3K4me3X8
4.3. Expression and Purification of Human Spindlin1
4.4. Isothermal Titration Calorimetry
4.5. Molecular Dynamics Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, J.C.; Rechem, C.V.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedford, M.T.; Clarke, S.G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Morales, Y.; Cáceres, T.; May, K.; Hevel, J.M. Biochemistry and regulation of the protein arginine methyltransferases (PRMTs). Arch. Biochem. Biophys. 2016, 590, 138–152. [Google Scholar] [CrossRef]
- Wesche, J.; Kühn, S.; Kessler, B.M.; Salton, M.; Wolf, A.; Wolf, A. Protein arginine methylation: A prominent modification and its demethylation. Cell. Mol. Life Sci. 2017, 74, 3305–3315. [Google Scholar] [CrossRef]
- Walport, L.J.; Hopkinson, R.J.; Chowdhury, R.; Schiller, R.; Ge, W.; Kawamura, A.; Schofield, C.J. Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat. Commun. 2016, 7, 11974. [Google Scholar] [CrossRef]
- Li, S.; Ali, S.; Duan, X.; Liu, S.; Du, J.; Liu, C.; Dai, H.; Zhou, M.; Zhou, L.; Yang, L.; et al. JMJD1B Demethylates H4R3me2s and H3K9me2 to Facilitate Gene Expression for Development of Hematopoietic Stem and Progenitor Cells. Cell Rep. 2018, 23, 389–403. [Google Scholar] [CrossRef] [Green Version]
- Andrews, F.H.; Strahl, B.D.; Kutateladze, T.G. Insights into newly discovered marks and readers of epigenetic information. Nat. Chem. Biol. 2016, 12, 662–668. [Google Scholar] [CrossRef]
- Gayatri, S.; Bedford, M.T. Readers of histone methylarginine marks. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2014, 1839, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Wang, G.G. Tudor: A versatile family of histone methylation “readers”. Trends Biochem. Sci. 2013, 38, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Sikorsky, T.; Hobor, F.; Krizanova, E.; Pasulka, J.; Kubicek, K.; Stefl, R. Recognition of asymmetrically dimethylated arginine by TDRD3. Nucleic Acids Res. 2012, 40, 11748–11755. [Google Scholar] [CrossRef] [Green Version]
- Supekar, S.; Papageorgiou, A.C.; Gemmecker, G.; Peltzer, R.; Johansson, M.P.; Tripsianes, K.; Sattler, M.; Kaila, V.R.I. Conformational Selection of Dimethylarginine Recognition by the Survival Motor Neuron Tudor Domain. Angew. Chem. Int. Ed. 2018, 57, 486–490. [Google Scholar] [CrossRef]
- Su, X.; Zhu, G.; Ding, X.; Lee, S.Y.; Dou, Y.; Zhu, B.; Wu, W. Molecular basis underlying histone H3 lysine—Arginine methylation pattern readout by Spin/Ssty repeats of Spindlin1. Genes Dev. 2014, 28, 622–636. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.X.; Zeng, Q.; Chen, L.; Du, J.C.; Yan, X.L.; Yuan, H.F.; Zhai, C.; Zhou, J.N.; Jia, Y.L.; Yue, W.; et al. Spindlin1 promotes cancer cell proliferation through activation of WNT/TCF-4 signaling. Mol. Cancer Res. 2012, 10, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, Y.W.; Gao, P. SPIN1, negatively regulated by miR-148/152, enhances Adriamycin resistance via upregulating drug metabolizing enzymes and transporter in breast cancer. J. Exp. Clin. Cancer Res. 2018, 37, 100. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, G.; Su, X.; Li, H.; Wu, W. Nucleolar localization signal and histone methylation reader function is required for SPIN1 to promote rRNA gene expression. Biochem. Biophys. Res. Commun. 2018, 505, 325–332. [Google Scholar] [CrossRef]
- Yang, N.; Wang, W.; Wang, Y.; Wang, M.; Zhao, Q.; Rao, Z.; Zhu, B.; Xu, R.M. Distinct mode of methylated lysine-4 of histone H3 recognition by tandem tudor-like domains of Spindlin1. Proc. Natl. Acad. Sci. USA 2012, 109, 17954–17959. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhan, L.; Wu, M.; Ma, R.; Yao, J.; Xiong, Y.; Pan, Y.; Guan, S.; Zhang, X.; Zang, J. Spindlin-1 recognizes methylations of K20 and R23 of histone H4 tail. FEBS Lett. 2018, 592, 4098–4110. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Liu, Y.; Su, X.; Lee, J.E.J.E.; Song, Y.; Wang, D.; Ge, K.; Gao, J.; Zhang, M.Q.; Li, H. Molecular basis for histone H3 “K4me3-K9me3/2” methylation pattern readout by Spindlin1. J. Biol. Chem. 2020, 295, 16877–16887. [Google Scholar] [CrossRef]
- Ramsey, S.; Nguyen, C.; Salomon-Ferrer, R.; Walker, R.C.; Gilson, M.K.; Kurtzman, T. Solvation thermodynamic mapping of molecular surfaces in ambertools: GIST. J. Comput. Chem. 2016, 37, 2029–2037. [Google Scholar] [CrossRef] [Green Version]
- Kamps, J.J.A.G.; Huang, J.; Poater, J.; Xu, C.; Pieters, B.J.G.E.; Dong, A.; Min, J.; Sherman, W.; Beuming, T.; Matthias Bickelhaupt, F.; et al. Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat. Commun. 2015, 6, 8911. [Google Scholar] [CrossRef]
- Pieters, B.J.G.E.; Wuts, M.H.M.; Poater, J.; Kumar, K.; White, P.B.; Kamps, J.J.A.G.; Sherman, W.; Pruijn, G.J.M.; Paton, R.S.; Beuming, T.; et al. Mechanism of biomolecular recognition of trimethyllysine by the fluorinated aromatic cage of KDM5A PHD3 finger. Commun. Chem. 2020, 3, 69. [Google Scholar] [CrossRef]
- Grant, S.K.; Green, B.G.; Stiffey-Wilusz, J.; Durette, P.L.; Shah, S.K.; Kozarich, J.W. Structural Requirements for Human Inducible Nitric Oxide Synthase Substrates and Substrate Analogue Inhibitors. Biochemistry 1998, 37, 4174–4180. [Google Scholar] [CrossRef]
- Ulhaq, S.; Chinje, E.C.; Naylor, M.A.; Jaffar, M.; Stratford, I.J.; Threadgill, M.D. Heterocyclic analogues of L-citrulline as inhibitors of the isoforms of nitric oxide synthase (NOS) and identification of N(δ)-(4,5-dihydrothiazol-2-yl)ornithine as a potent inhibitor. Bioorg. Med. Chem. 1999, 7, 1787–1796. [Google Scholar] [CrossRef]
- Martin, N.I.; Woodward, J.J.; Winter, M.B.; Beeson, W.T.; Marletta, M.A. Design and synthesis of C5 methylated L-arginine analogues as active site probes for nitric oxide synthase. J. Am. Chem. Soc. 2007, 129, 12563–12570. [Google Scholar] [CrossRef]
- Le, D.D.; Cortesi, A.T.; Myers, S.A.; Burlingame, A.L.; Fujimori, D.G. Site-specific and regiospecific installation of methylarginine analogues into recombinant histones and insights into effector protein binding. J. Am. Chem. Soc. 2013, 135, 2879–2882. [Google Scholar] [CrossRef] [Green Version]
- Al Temimi, A.H.K.; Belle, R.; Kumar, K.; Poater, J.; Betlem, P.; Pieters, B.J.G.E.; Paton, R.S.; Bickelhaupt, F.M.; Mecinović, J. Recognition of shorter and longer trimethyllysine analogues by epigenetic reader proteins. Chem. Commun. 2018, 54, 2409–2412. [Google Scholar] [CrossRef] [Green Version]
- Belle, R.; Al Temimi, A.H.K.; Kumar, K.; Pieters, B.J.G.E.; Tumber, A.; Dunford, J.E.; Johansson, C.; Oppermann, U.; Brown, T.; Schofield, C.J.; et al. Investigating d-lysine stereochemistry for epigenetic methylation, demethylation and recognition. Chem. Commun. 2017, 53, 13264–13267. [Google Scholar] [CrossRef]
- Maas, M.N.; Hintzen, J.C.J.; Porzberg, M.R.B.; Mecinović, J. Trimethyllysine: From carnitine biosynthesis to epigenetics. Int. J. Mol. Sci. 2020, 21, 9451. [Google Scholar] [CrossRef]
- Hughes, R.M.; Wiggins, K.R.; Khorasanizadeh, S.; Waters, M.L. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect. Proc. Natl. Acad. Sci. USA 2007, 104, 11184–11188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieters, B.J.G.E.; Hintzen, J.C.J.; Grobben, Y.; Al Temimi, A.H.K.; Kamps, J.J.A.G.; Mecinović, J. Installation of Trimethyllysine Analogs on Intact Histones via Cysteine Alkylation. Bioconjug. Chem. 2019, 30, 952–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hintzen, J.C.J.; Poater, J.; Kumar, K.; Al Temimi, A.H.K.; Pieters, B.J.G.E.; Paton, R.S.; Bickelhaupt, F.M.; Mecinović, J. Comparison of molecular recognition of trimethyllysine and trimethylthialysine by epigenetic reader proteins. Molecules 2020, 25, 1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luise, C.; Robaa, D.; Regenass, P.; Maurer, D.; Ostrovskyi, D.; Seifert, L.; Bacher, J.; Burgahn, T.; Wagner, T.; Seitz, J.; et al. Structure-based design, docking and binding free energy calculations of A366 derivatives as Spindlin1 inhibitors. Int. J. Mol. Sci. 2021, 22, 5910. [Google Scholar] [CrossRef]
- Fagan, V.; Johansson, C.; Gileadi, C.; Monteiro, O.; Dunford, J.E.; Nibhani, R.; Philpott, M.; Malzahn, J.; Wells, G.; Faram, R.; et al. A chemical probe for tudor domain protein Spindlin1 to investigate chromatin function. J. Med. Chem. 2019, 62, 9008–9025. [Google Scholar] [CrossRef]
- Xiong, Y.; Greschik, H.; Johansson, C.; Seifert, L.; Bacher, J.; Park, K.S.; Babault, N.; Martini, M.; Fagan, V.; Li, F.; et al. Discovery of a potent and selective fragment-like inhibitor of methyllysine reader protein Spindlin 1 (SPIN1). J. Med. Chem. 2019, 62, 8996–9007. [Google Scholar] [CrossRef]
- Robaa, D.; Wagner, T.; Luise, C.; Carlino, L.; McMillan, J.; Flaig, R.; Schüle, R.; Jung, M.; Sippl, W. Identification and structure–activity relationship studies of small-molecule inhibitors of the methyllysine reader protein Spindlin1. ChemMedChem 2016, 11, 2327–2338. [Google Scholar] [CrossRef]
- Wagner, T.; Greschik, H.; Burgahn, T.; Schmidtkunz, K.; Schott, A.K.; McMillan, J.; Baranauskiene, L.; Xiong, Y.; Fedorov, O.; Jin, J.; et al. Identification of a small-molecule ligand of the epigenetic reader protein Spindlin1 via a versatile screening platform. Nucleic Acids Res. 2016, 44, e88. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-1: Maestro, Schrödinger; Limited Liability Company: New York, NY, USA, 2019.
- Schrödinger Release 2019-1: Prime, Schrödinger; Limited Liability Company: New York, NY, USA, 2019.
- Rostkowski, M.; Olsson, M.H.; Søndergaard, C.R.; Jensen, J.H. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 2011, 11, 6. [Google Scholar] [CrossRef] [Green Version]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins Struct. Funct. Genet. 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Sindhikara, D.J.; Yoshida, N.; Hirata, F. Placevent: An algorithm for prediction of explicit solvent atom distribution-Application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 2012, 33, 1536–1543. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kd (nM) | ΔG° | ΔH° | −TΔS° | |
|---|---|---|---|---|
| H3K4me3R8me2a | 28 ± 0.5 | −10.3 ± 0.02 | −17.3 ± 1.9 | 7.0 ± 1.9 |
| H3K4me3Cit8me2 | 81 ± 6 | −9.7 ± 0.04 | −15.6 ± 0.3 | 5.9 ± 0.2 |
| H3K4me3hR8me2a | 56 ± 15 | −9.9 ± 0.2 | −20.9 ± 0.7 | 11.0 ± 0.7 |
| H3K4me3nR8me2a | 42 ± 8 | −10.1 ± 0.1 | −16.9 ± 1.3 | 6.8 ± 1.2 |
| H3K4me3R8etme | 88 ± 6 | −9.6 ± 0.1 | −20.8 ± 0.6 | 11.2 ± 0.6 |
| H3K4me3R8et2a | 46 ± 4 | −10.0 ± 0.1 | −19.3 ± 0.6 | 9.3 ± 0.6 |
| H3K4me3R8pip | 81 ± 19 | −9.7 ± 0.2 | −20.9 ± 0.3 | 11.2 ± 0.1 |
| H3K4me3R8pyr | 102 ± 12 | −9.5 ± 0.1 | −22.5 ± 0.4 | 13.0 ± 0.5 |
| H3K4me3G8 | 80 ± 15 | −9.7 ± 0.1 | −15.5 ± 0.5 | 5.8 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porzberg, M.R.B.; Moesgaard, L.; Johansson, C.; Oppermann, U.; Kongsted, J.; Mecinović, J. Recognition of Dimethylarginine Analogues by Tandem Tudor Domain Protein Spindlin1. Molecules 2022, 27, 983. https://doi.org/10.3390/molecules27030983
Porzberg MRB, Moesgaard L, Johansson C, Oppermann U, Kongsted J, Mecinović J. Recognition of Dimethylarginine Analogues by Tandem Tudor Domain Protein Spindlin1. Molecules. 2022; 27(3):983. https://doi.org/10.3390/molecules27030983
Chicago/Turabian StylePorzberg, Miriam R. B., Laust Moesgaard, Catrine Johansson, Udo Oppermann, Jacob Kongsted, and Jasmin Mecinović. 2022. "Recognition of Dimethylarginine Analogues by Tandem Tudor Domain Protein Spindlin1" Molecules 27, no. 3: 983. https://doi.org/10.3390/molecules27030983
APA StylePorzberg, M. R. B., Moesgaard, L., Johansson, C., Oppermann, U., Kongsted, J., & Mecinović, J. (2022). Recognition of Dimethylarginine Analogues by Tandem Tudor Domain Protein Spindlin1. Molecules, 27(3), 983. https://doi.org/10.3390/molecules27030983