
1-Aminoalkylphosphonium Derivatives: Smart Synthetic Equivalents of N-Acyliminium-Type Cations, and Maybe Something More: A Review †

 ,
,  , and
, and
Abstract
:
1. Introduction
2. 1-Aminoalkyltriarylphosphonium Derivatives
2.1. 1-(N-acylamino)alkylphosphonium Salts
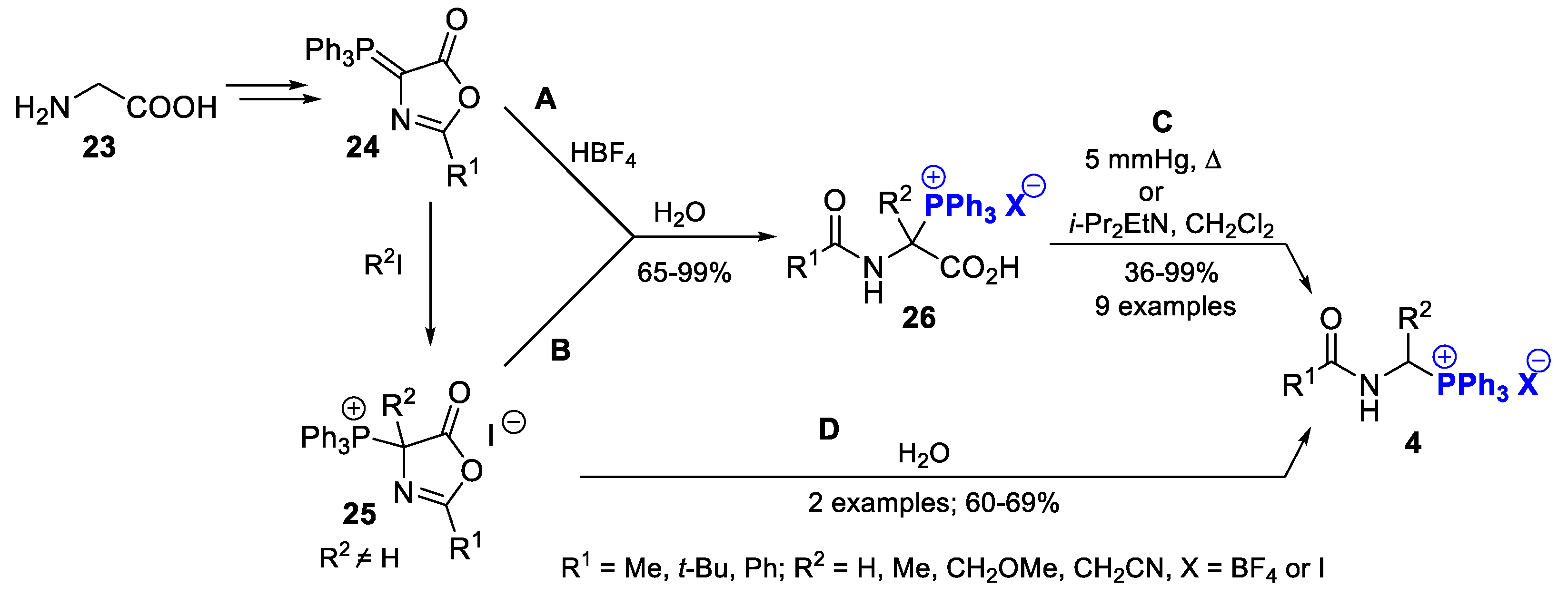
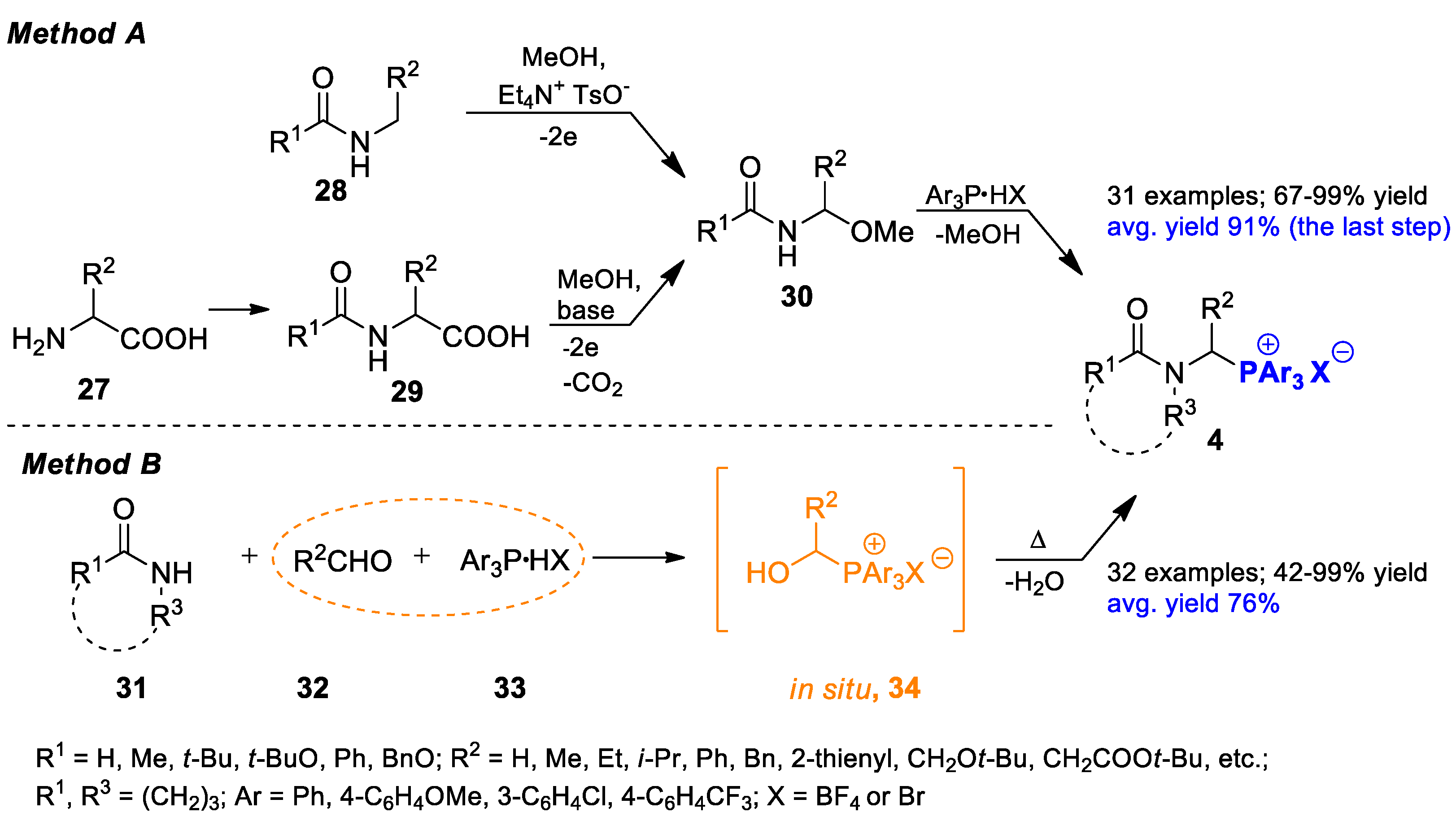
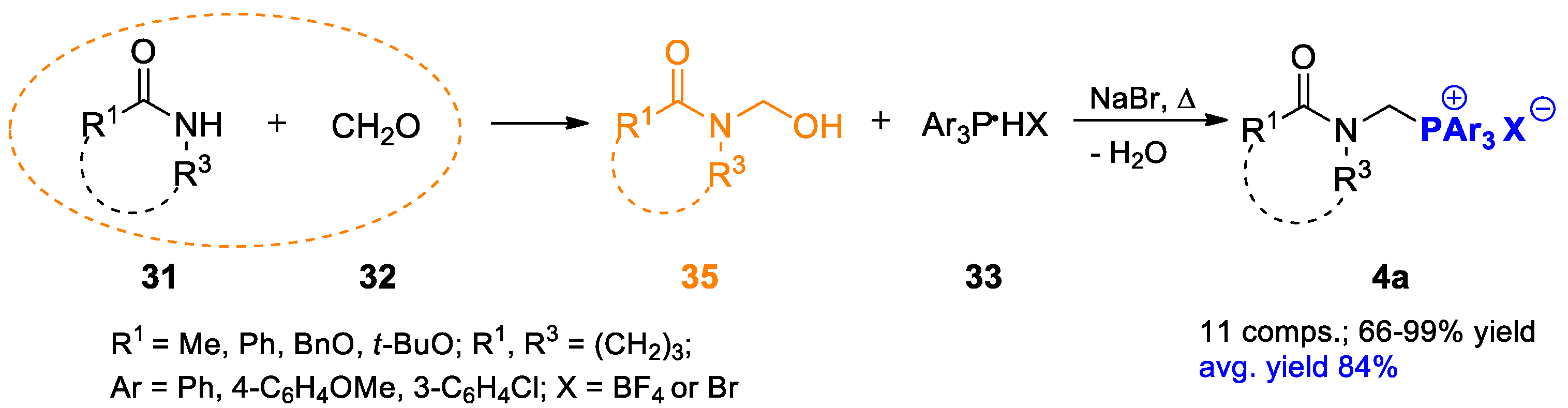
2.1.1. Preparation
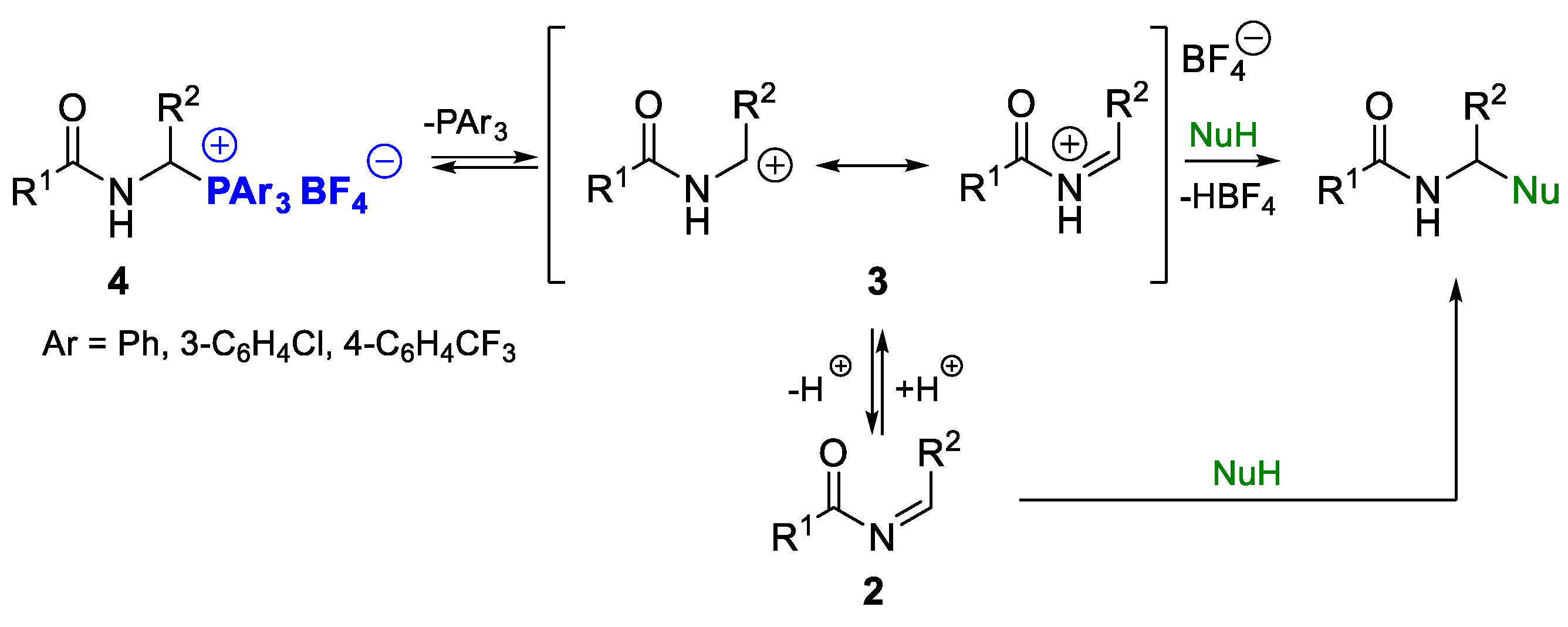
2.1.2. Synthetic Utilization
2.2. 1-Imidoalkyltriarylphosphonium Salts
2.2.1. Preparation
2.2.2. Synthetic Utilization
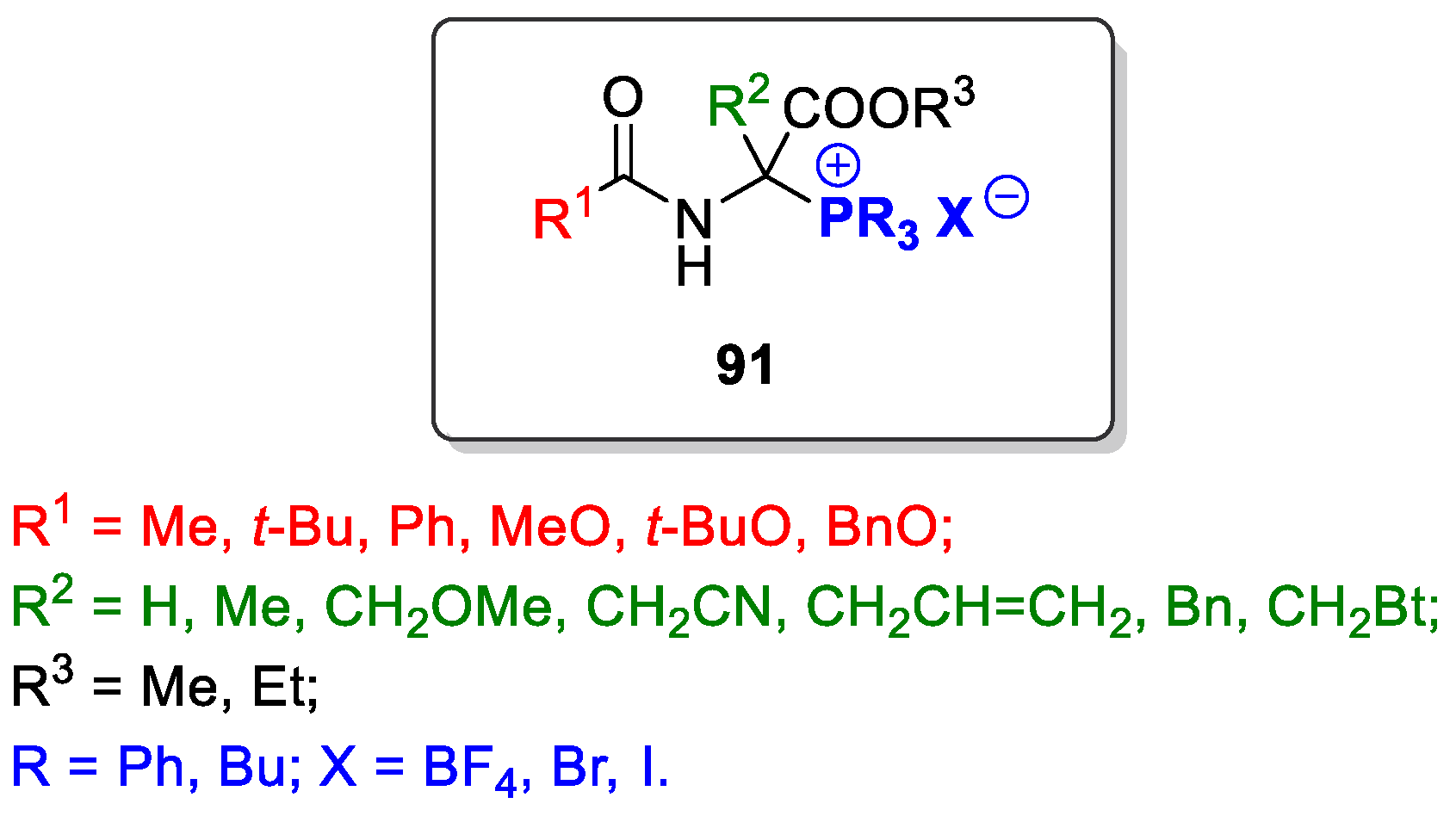
2.3. N-acyl-1-phosphonio-α-amino Acid Esters
2.3.1. Preparation
2.3.2. Synthetic Utilization
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neto, B.A.D.; Rocha, R.O.; Rodrigues, M.O. Catalytic Approaches to Multicomponent Reactions: A Critical Review and Perspectives on the Roles of Catalysis. Molecules 2022, 27, 132. [Google Scholar] [CrossRef]
- Kokkala, P.; Rajeshkumar, T.; Mpakali, A.; Stratikos, E.; Vogiatzis, K.D.; Georgiadis, D. A Carbodiimide-Mediated P–C Bond-Forming Reaction: Mild Amidoalkylation of P-Nucleophiles by Boc-Aminals. Org. Lett. 2021, 23, 1726–1730. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V.; Heydari, M.; Masoumi, B. Organocatalyzed Asymmetric Friedel-Crafts Reactions: An Update. Chem. Rec. 2019, 19, 2236–2340. [Google Scholar] [CrossRef]
- Aranzamendi, E.; Sotomayor, N.; Lete, E. Phenolic Activation in Chiral Brønsted Acid-Catalyzed Intramolecular α-Amidoalkylation Reactions for the Synthesis of Fused Isoquinolines. ACS Omega 2017, 2, 2706–2718. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-Y.; Cai, C.; Yang, X.; Lv, Z.-C.; Schneider, U. Catalytic Asymmetric Reactions with N,O-Aminals. ACS Catal. 2016, 6, 5747–5763. [Google Scholar] [CrossRef]
- Kataja, A.O.; Masson, G. Imine and iminium precursors as versatile intermediates in enantioselective organocatalysis. Tetrahedron 2014, 70, 8783–8815. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Zhang, H.C.; Cohen, J.H.; Turchi, I.J.; Maryanoff, C.A. Cyclizations of N-acyliminium ions. Chem. Rev. 2004, 104, 1431–1628. [Google Scholar] [CrossRef]
- Yazici, A.; Pyne, S.G. Intermolecular addition reactions of N-acyliminium ions (Part I). Synthesis 2009, 339–368. [Google Scholar] [CrossRef] [Green Version]
- Yazici, A.; Pyne, S.G. Intermolecular addition reactions of N-acyliminium ions (Part II). Synthesis 2009, 513–541. [Google Scholar] [CrossRef] [Green Version]
- Aranzamendi, E.; Arrasate, S.; Sotomayor, N.; González-Díaz, H.; Lete, E. Chiral Brønsted Acid Catalyzed Enantioselective α-Amidoalkylation Reactions: A Joint Experimental and Predictive Study. ChemistryOpen 2016, 5, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Shi, X.; Li, J.; Hou, Z.; Song, Z.; Su, X.; Peng, D.; Wang, F.; Yu, Y.; Zhao, G. Nickel-Catalyzed Amidoalkylation Reaction of γ-Hydroxy Lactams: An Access to 3-Substituted Isoindolinones. ACS Omega 2019, 4, 19420–19436. [Google Scholar] [CrossRef] [Green Version]
- Mazurkiewicz, R.; Październiok-Holewa, A.; Adamek, J.; Zielińska, K. α-Amidoalkylating agents: Structure, synthesis, reactivity and application. Adv. Heterocycl. Chem. 2014, 111, 43–94. [Google Scholar] [CrossRef]
- Touati, B.; El Bouakher, A.; Taillier, C.; Othman, R.B.; Trabelsi-Ayadi, M.; Antoniotti, S.; DuÇach, E.; Dalla, V. Enolizable Carbonyls and N,O-Acetals: A Rational Approach for Room-Temperature Lewis Superacid-Catalyzed Directa-Amidoalkylation of Ketones and Aldehydes. Chem. Eur. J. 2016, 22, 6012–6022. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.E.; Manolikakes, G. Bi(OTf)3-Catalyzed Multicomponent α-Amidoalkylation Reactions. J. Org. Chem. 2015, 80, 6193–6212. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, M.G.; Olga, V.; Turova, O.V.; Zlotin, S.G. The progress in the chemistry of N-acyliminium ions and their use in stereoselective organic synthesis. Russ. Chem. Rev. 2017, 86, 1–17. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Abdel-Fattah, A.A.A.; Celik, I. Benzotriazole-mediated amidoalkylations of nitroalkanes, nitriles, alkynes and esters. ARKIVOC 2007, 11, 96–113. [Google Scholar] [CrossRef] [Green Version]
- Katritzky, A.R.; Manju, K.; Singh, S.K.; Meher, N.K. Benzotriazole mediated amino-, amido-, alkoxy- and alkylthioalkylation. Tetrahedron 2005, 61, 2555–2581. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Mehta, S.; He, H.Y. Syntheses of Pyrrolo- and Indoloisoquinolinones by Intramolecular Cyclizations of 1-(2-Arylethyl)-5-benzotriazolylpyrrolidin-2-ones and 3-Benzotriazolyl-2-(2-arylethyl)-1-isoindolinones. J. Org. Chem. 2001, 66, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Kirichenko, K.; Elsayed, A.M.; Ji, Y.; Fang, Y. Convenient Preparation of tert-Butyl β-(Protected amino)esters. J. Org. Chem. 2002, 67, 4957–4959. [Google Scholar] [CrossRef]
- Petrini, M. α-Amido Sulfones as Stable Precursors of Reactive N-Acylimino Derivatives. Chem. Rev. 2005, 105, 3949–3977. [Google Scholar] [CrossRef]
- Ballini, R.; Palmieri, A.; Petrini, M.; Torregiani, E. Solventless Clay-Promoted Friedel−Crafts Reaction of Indoles with α-Amido Sulfones: Unexpected Synthesis of 3-(1-Arylsulfonylalkyl) Indoles. Org. Lett. 2006, 8, 4093–4096. [Google Scholar] [CrossRef]
- Das, B.; Damodar, K.; Bhunia, N. A Simple and Efficient Access to α-Amino Phosphonates from N-Benzyloxycarbonylamino Sulfones Using Indium(III) Chloride. J. Org. Chem. 2009, 74, 5607–5609. [Google Scholar] [CrossRef] [PubMed]
- Marcantoni, E.; Palmieri, A.; Petrini, M. Recent synthetic applications of α-amido sulfones as precursors of N-acylimino derivatives. Org. Chem. Front. 2019, 6, 2142–2182. [Google Scholar] [CrossRef]
- Drach, B.; Kirsanov, A.; Sviridov, E. Reaction of N-chloromethyl amides of acids with triphenylphosphine. Zh. Obshch. Khim. 1972, 42, 953–954. [Google Scholar]
- Smolii, O.B.; Brovarets, V.S.; Drach, B.S. Substituted Methylphosphonium Salts with an Imidoyl Chloride Group. Zh. Obshch. Khim. 1986, 56, 2802–2803. [Google Scholar]
- Smolii, O.B.; Brovarets, V.S.; Pirozhenko, V.V.; Drach, B.S. Cyclocondensation of N-Substituted Imidoyl Chlorides Containing a Phosphonium Group. Zh. Obshch. Khim. 1988, 58, 2465–2471. [Google Scholar]
- Kasukhin, L.F.; Brovarets, V.S.; Smolii, O.B.; Kurg, V.V.; Budnik, L.V.; Drach, B.S. N-Acylaminomethyl and Substituted 1-Acylaminoethenylphosphonium Salts As Inhibitors of Acetylcholinesterase. Zh. Obshch. Khim. 1991, 61, 2679–2684. [Google Scholar]
- Devlin, C.J.; Walker, B.J. Reactions of bromonitroalkenes with tervalent phosphorus. Part II. Reaction in methanol. J. Chem. Soc. Perkin Trans. 1974, 1, 453–460. [Google Scholar] [CrossRef]
- Petersen, H.; Reuther, W. α-Ureidoalkylierung von Phosphor (III)-Verbindungen. Justus Liebigs Ann. Chem. 1972, 766, 58–72. [Google Scholar] [CrossRef]
- Kozhushko, B.N.; Gumenyuk, A.V.; Palichuk, Y.A.; Shokol, V.A. Trialkyl- et Triaryl-(Isocyanatomethyl) Chlorophosphoranes. Zh. Obshch. Khim. 1977, 47, 333–339. [Google Scholar]
- Shokol, V.A.; Silina, E.B.; Kozushko, B.N.; Golik, G.A. Bromomethyl Isocyanate and Its Phosphorylated Derivatives. Zh. Obshch. Khim. 1979, 49, 312–316. [Google Scholar]
- Kozhushko, B.N.; Silina, E.B.; Gumenyuk, A.V.; Turov, A.V.; Shokol, V.A. Triaryl(Isocyanatomethyl)Phosphonium Iodides. Zh. Obshch. Khim. 1980, 50, 2210–2215. [Google Scholar]
- Shokol, V.A.; Kozushko, B.N.; Gumenyuk, A.V. Trialkyl- And Aryldialkyl(Isocyanatomethyl)Ammonium Chlorides. Zh. Obshch. Khim. 1977, 47, 1110–1118. [Google Scholar]
- Zinner, G.; Fehlhammer, W.P. Isocyanomethylenetriphenylphosphorane. Angew. Chem. Int. Ed. Engl. 1985, 24, 979–980. [Google Scholar] [CrossRef]
- Frank, A.W.; Drake, G.L. Synthesis and properties of carbamate derivatives of tetrakis(hydroxymethyl)phosphonium chloride. J. Org. Chem. 1977, 42, 4040–4045. [Google Scholar] [CrossRef]
- Devlin, C.J.; Walker, B.J. A possible azirine intermediate in the reaction of bromonitroalkenes with triphenylphosphine. J. Chem. Soc. D Chem. Comm. 1970, 917–918. [Google Scholar] [CrossRef]
- Drach, B.S.; Dolgushina, I.Y.; Sinitsa, A.D. Application of Omega-Chloro-Omega-Acylamidoacetophenones for Synthesis of Phosphorylated Oxazoles. Zh. Obshch. Khim. 1975, 45, 1251–1255. [Google Scholar]
- Brovarets, V.S.; Lobanov, O.P.; Vinogradova, T.K.; Drach, B.S. Preparation and Properties of 2-Chloro-1-Acylaminovinyltriphenylphosphonium Chlorides. Zh. Obshch. Khim. 1984, 54, 288–301. [Google Scholar]
- Drach, B.S.; Brovarets, V.S.; Smolii, O.B. Acylamino-Substituted Vinylphosphonium Salts in Syntheses of Derivatives of Nitrogen Heterocycles. Russ. J. Gen. Chem. 2002, 72, 1661–1687. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Pierwocha, A.W. Phosphoranylidene-5(4H)-oxazolones–A novel synthesis and properties. Monatsh. Chem. 1996, 127, 219–225. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Październiok-Holewa, A.; Grymel, M. Synthesis and decarboxylation of N-acyl-α-triphenylphosphonio-α-amino acids: A new synthesis of α-(N-acylamino)alkyltriphenylphosphonium salts. Tetrahedron Lett. 2008, 49, 1801–1803. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Październiok-Holewa, A.; Grymel, M. N-Acyl-α-triphenylphosphonio-α-amino Acids: Synthesis and Decarboxylation to α-(N-Acylamino)alkyltriphenylphosphonium Salts. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 1017–1027. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Simka, W.; Gajos, A.; Szymura, K. α-Amidoalkylating Agents from N-Acyl-α-amino Acids: 1-(N-Acylamino)alkyltriphenylphosphonium Salts. J. Org. Chem. 2012, 77, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Adamek, J.; Zieleźny, P.; Erfurt, K. N-protected 1-aminoalkylphosphonium salts from amides, carbamates, lactams, or imides. J. Org. Chem. 2021, 86, 5852–5862. [Google Scholar] [CrossRef]
- Walęcka-Kurczyk, A.; Adamek, J.; Walczak, K.; Michalak, M.; Październiok-Holewa, A. Non-Kolbe electrolysis of N-protected-α-amino acids: A standardized method for the synthesis of N-protected (1-methoxyalkyl)amines. RSC Adv. 2022, 12, 2107–2114. [Google Scholar] [CrossRef]
- Adamek, J.; Węgrzyk, A.; Kończewicz, J.; Walczak, K.; Erfurt, K. 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications. Molecules 2018, 23, 2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamek, J.; Mazurkiewicz, R.; Październiok-Holewa, A.; Grymel, M.; Kuźnik, A.; Zielińska, K. 1-(N-Acylamino)alkyl Sulfones from N-Acyl-α-amino Acids or N-Alkylamides. J. Org. Chem. 2014, 79, 2765–2770. [Google Scholar] [CrossRef]
- Kozicka, D.; Zieleźny, P.; Erfurt, K.; Adamek, J. Amide-type substrates in the synthesis of N-protected 1-aminomethylphosphonium salts. Catalysts 2021, 11, 552. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, L. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Mazurkiewicz, R.; Październiok-Holewa, A.; Kononienko, A. A Novel Synthesis of 1-Aminoalkanephosphonic Acid Derivatives from 1-(N-Acylamino)alkyltriphenylphosphonium Salts. Phosphorus Sulfur Silicon 2010, 185, 1986–1992. [Google Scholar] [CrossRef]
- Październiok-Holewa, A.; Adamek, J.; Mazurkiewicz, R.; Zielińska, K. Amidoalkylating Properties of 1-(N-Acylamino)Alkyltriphenylphosphonium Salts. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 205–212. [Google Scholar] [CrossRef]
- Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Mazurkiewicz, R. Comparative Studies on the Amidoalkylating Properties of N-(1-Methoxyalkyl)Amides and 1-(N-Acylamino)Alkyltriphenylphosphonium Salts in the Michaelis–Arbuzov-Like Reaction: A New One-Pot Transformation of N-(1-Methoxyalkyl)Amides into Phosphonic or Phosphinic Analogs of N-Acyl-α-Amino Acids. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 967–980. [Google Scholar] [CrossRef]
- Zielińska, K.; Mazurkiewicz, R.; Szymańska, K.; Jarzębski, A.; Magiera, S.; Erfurt, K. Penicillin G Acylase-Mediated Kinetic Resolution of Racemic 1-(N-Acylamino)alkylphosphonic and 1-(N-Acylamino)alkylphosphinic Acids and Their Esters. J. Mol. Catal. B Enzym. 2016, 132, 31–40. [Google Scholar] [CrossRef]
- Zielińska, K.; Mazurkiewicz, R.; Szymańska, K.; Jarzębski, A. Batch and in-flow kinetic resolution of racemic 1-(N-acylamino)alkylphosphonic and 1-(N-acylamino)alkylphosphinic acids and their esters using immobilized penicillin G acylase. Tetrahedron Asymmetry 2017, 28, 146–152. [Google Scholar] [CrossRef]
- Walęcka-Kurczyk, A.; Walczak, K.; Kuźnik, A.; Stecko, S.; Październiok-Holewa, A. The Synthesis of α-Aminophosphonates via Enantioselective Organocatalytic Reaction of 1-(N-Acylamino)alkylphosphonium Salts with Dimethyl Phosphite. Molecules 2020, 25, 405. [Google Scholar] [CrossRef] [Green Version]
- Kuźnik, A.; Mazurkiewicz, R.; Grymel, M.; Zielińska, K.; Adamek, J.; Chmielewska, E.; Bochno, M.; Kubica, S. New method for the synthesis of α-aminoalkylenebisphosphonates and their asymmetric phosphonyl-phosphinyl and phosphonyl-phosphinoyl analogues. Beilstein J. Org. Chem. 2015, 11, 1418–1424. [Google Scholar] [CrossRef] [Green Version]
- Kuźnik, A.; Mazurkiewicz, R.; Zięba, M.; Erfurt, K. 1-(N-Acylamino)-1-triphenylphosphoniumalkylphosphonates: General synthesis and prospects for further synthetic applications. Tetrahedron Lett. 2018, 59, 3307–3310. [Google Scholar] [CrossRef]
- Październiok-Holewa, A.; Adamek, J.; Zielińska, K.; Piernikarczyk, K.; Mazurkiewicz, R. N-(1-acyloaminoalkyl)amidinium salts derived from DBU or related bases as reactive intermediates in α-amidoalkylation reactions. Arkivoc 2012, 4, 314–329. [Google Scholar] [CrossRef] [Green Version]
- Adamek, J.; Mazurkiewicz, R.; Październiok-Holewa, A.; Kuźnik, A.; Grymel, M.; Zielińska, K.; Simka, W. N-[1-(Benzotriazol-1-yl)alkyl]amides from N-acyl-α-amino acids or N-alkylamides. Tetrahedron 2014, 70, 5725–5729. [Google Scholar] [CrossRef]
- Zheng, K.; Shen, D.; Zhang, B.; Hong, R. Stereodivergent Synthesis of Lankacyclinol and Its C2/C18-Congeners Enabled by a Bioinspired Mannich Reaction. J. Org. Chem. 2021, 86, 10991–11005. [Google Scholar] [CrossRef]
- Zheng, K.; Shen, D.; Zhang, B.; Hong, R. Landscape of Lankacidin Biomimetic Synthesis: Structural Revisions and Biogenetic Implications. J. Org. Chem. 2020, 85, 13818–13836. [Google Scholar] [CrossRef]
- Zheng, K.; Hong, R. The Fruit of Gold: Biomimicry in the Syntheses of Lankacidins. Acc. Chem. Res. 2021, 54, 3438–3451. [Google Scholar] [CrossRef]
- Adamek, J.; Węgrzyk, A.; Krawczyk, M.; Erfurt, K. Catalyst-free Friedel-Crafts reaction of 1-(N-acylamino)alkyltriarylphosphonium salts with electron-rich arenes. Tetrahedron 2018, 74, 2575–2583. [Google Scholar] [CrossRef]
- Październiok-Holewa, A.; Walęcka-Kurczyk, A.; Musioł, S.; Stecko, S. Catalyst-free Mannich-type reaction of 1-(N-acylamino)alkyltriphenylphosphonium salts with silyl enolates. Tetrahedron 2019, 75, 732–742. [Google Scholar] [CrossRef]
- Smolii, O.B.; Brovarets, V.S.; Drach, B.S. Reaction of the Chloride of N-(Triphenylphosphoniomethyl)benzimidoyl chloride with Sodium Rhodanide. Zh. Obshch. Khim. 1987, 57, 2145–2146. [Google Scholar]
- Smolii, O.B.; Brovarets, V.S.; Drach, B.S. Reaction of the Chloride of N-(Triphenylphosphoniomethyl)benzimidoyl chloride with Carboxylic Acid Chlorides. Zh. Obshch. Khim. 1988, 58, 1670–1671. [Google Scholar]
- Dubois, R.J.; Lin, C.-C.; Beisler, J.A. Synthesis and antitumor properties of some isoindolylalkylphosphonium salts. J. Med. Chem. 1978, 21, 303–306. [Google Scholar] [CrossRef]
- Tessier, D.; Filteau, M.; Radu, I. New Antimicrobial Compositions and Uses Thereof. U.S. Patent US 2015/0201622 Al, 23 July 2015. [Google Scholar]
- Tessier, D.; Filteau, M.; Radu, I. Antimicrobial Solution Comprising a Metallic Salt and a Surfactant. International Patent WO 2006105669 A1, 12 October 2006. [Google Scholar]
- Hellmann, H.; Schumacher, O. Quartäre Phosphoniumsalze aus tertiären Phosphinen und quartären Ammoniumsalzen. Justus Liebigs Ann. Chem. 1961, 640, 79–84. [Google Scholar] [CrossRef]
- Enzmann, A.; Eckert, M.; Ponikwar, W.; Polborn, K.; Schneiderbauer, S.; Beller, M.; Beck, W. Aminomethyl and Aminoacetyl Complexes of Palladium(II), Platinum(II), Iron(II) and Rhenium(I) with N-Phthaloyl as Amino Protecting Group and Mechanistic Studies on the Palladium-Catalyzed Amidocarbonylation. Eur. J. Inorg. Chem. 2004, 6, 1330–1340. [Google Scholar] [CrossRef]
- Tan, E.S.; Naylor, J.C.; Groban, E.S.; Bunzow, J.R.; Jacobson, M.P.; Grandy, D.K.; Scanlan, T.S. The Molecular Basis of Species-Specific Ligand Activation of Trace Amine-Associated Receptor 1 (TAAR1). ACS Chem. Biol. 2009, 4, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Adamek, J.; Mazurkiewicz, R.; Węgrzyk, A.; Erfurt, K. 1-Imidoalkylphosphonium salts with modulated Cα-P+ bond strength: Synthesis and application as new active α-imidoalkylating agents. Beilstein J. Org. Chem. 2017, 13, 1446–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavé, G.; Reverte, M.; Vasseur, J.-J.; Smietana, M. Modified internucleoside linkages for nuclease-resistant oligonucleotides. RSC Chem. Biol. 2021, 2, 94–150. [Google Scholar] [CrossRef]
- Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. “Clicktophycin-52”: A Bioactive Cryptophycin-52 Triazole Analogue. Org. Lett. 2010, 12, 1064–1067. [Google Scholar] [CrossRef] [PubMed]
- Adamek, J.; Węgrzyk-Schlieter, A.; Steć, K.; Walczak, K.; Erfurt, K. Michaelis-Arbuzov-Type Reaction of 1-Imidoalkyltriarylphosphonium Salts with Selected Phosphorus Nucleophiles. Molecules 2019, 24, 3405. [Google Scholar] [CrossRef] [Green Version]
- Kober, R.; Steglich, W. Untersuchungen zur Reaktion von Acylaminobrommalonestern und Acylaminobromessigestern mit Trialkylphosphiteneine einfache Synthese von 2-Amino-2-(diethoxyphosphoryl) Essigsäure Ethylester. Liebigs Ann. Chem. 1983, 4, 599–609. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Grymel, M. N-Acyl-α-triphenylphosphonioglycinates: A Novel Cationic Glycine Equivalent and its Reactions with Heteroatom Nucleophiles. Monatsh. Chem. 1999, 130, 597–604. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Grymel, M. A new synthesis of α-amino acid derivatives by reaction of N-acyl-α-triphenylphosphonioglycinates with carbon nucleophiles. Phosporus Sulfur Silicon 2000, 164, 33–43. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Pierwocha, A.W. 4-Phosphoranylidene-5(4H)-oxazolones II. Reactions with alkylating agents. Monatsh. Chem. 1997, 128, 893–900. [Google Scholar] [CrossRef]
- Grymel, M.; Kuźnik, A.; Mazurkiewicz, R. N-Acyl-α-triphenylphosphonio-α-amino acid esters as synthetic equivalents of α-amino acid α-cations. Phosporus Sulfur Silicon 2015, 190, 429–439. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Grymel, M.; Kuźnik, A. Three New in situ Syntheses of N-Acyl-α-triphenylphosphonioglycinates. Monatsh. Chem. 2004, 135, 799–806. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Grymel, M. Reaction of N-Acyl-α-triphenylphosphonio-α-amino Acid Esters with Organic Bases: Mechanism of the Base-Catalyzed Nucleophilic Substitution of the Triphenylphosphonium Group. Monatsh. Chem. 2002, 133, 1197–1204. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Kuźnik, A.; Grymel, M.; Kuźnik, N. N-Acyl-α-triphenylphosphonioglycinates in the Synthesis of α,β-Dehydro-α-amino Acid Derivatives. Monatsh. Chem. 2004, 135, 807–815. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical Modifications Designed to Improve Peptide Stability: Incorporation of Non-Natural Amino Acids, Pseudo-Peptide Bonds, and Cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef]
- Meester, W.J.N.; van Maarseveen, J.H.; Schoemaker, H.E.; Hiemstra, H.; Rutjes, F.P.J.T. Glyoxylates as Versatile Building Blocks for the Synthesis of α-Amino Acid and α-Alkoxy Acid Derivatives via Cationic Intermediates. Eur. J. Org. Chem. 2003, 2003, 2519–2529. [Google Scholar] [CrossRef]
- Heimgartner, H.; Braun, K.; Linden, A. Synthesis and conformational analysis of pentapeptides containing enantiomerically pure 2,2-disubstituted glycines. Helv. Chim. Acta 2008, 91, 526–558. [Google Scholar] [CrossRef] [Green Version]
- Ohfune, Y.; Shinada, T. Enantio- and Diastereoselective Construction of α,α-Disubstituted α-Amino Acids for the Synthesis of Biologically Active Compounds. Eur. J. Org. Chem. 2005, 2005, 5127–5143. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Kuźnik, A. A new convenient synthesis of N-acyl-2-(dimethoxyphosphoryl)glycinates. Tetrahedron Lett. 2006, 47, 3439–3442. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tanaka, K.; Kogen, H. Recent topics of the natural product synthesis by Horner–Wadsworth–Emmons reaction. Tetrahedron Lett. 2018, 59, 568–582. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Kuźnik, A.; Grymel, M.; Październiok-Holewa, A. α-Amino acid derivatives with a Cα-P bond in organic synthesis. Arkivoc 2007, 6, 193–216. [Google Scholar] [CrossRef]
- Pfefferkorn, J.A.; Nugent, R.A.; Gross, R.J.; Greene, M.L.; Mitchell, M.A.; Reding, M.T.; Funk, L.A.; Anderson, R.; Wells, P.A.; Shelly, J.A.; et al. Inhibitors of HCV NS5B polymerase. Part 2: Evaluation of the northern region of (2Z)-2-benzoylamino-3-(4-phenoxy-phenyl)-acrylic acid. Bioorg. Med. Chem. Lett. 2005, 15, 2812–2818. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, N.; Joullié, M.M. Total synthesis of isoroquefortine E and phenylahistin. Tetrahedron Lett. 2009, 50, 6755–6757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Xiong, C.; Zhang, J.; Hruby, V.J. Practical, asymmetric synthesis of aromatic-substituted bulky and hydrophobic tryptophan and phenylalanine derivatives. Tetrahedron 2002, 58, 3101–3110. [Google Scholar] [CrossRef]
- Cativiela, C.; Diaz de Villegas, M.D.; Gálvez, J.A.; Su, G. Horner-Wadsworth-Emmons reaction for the synthesis of unusual alpha,beta-didehydroamino acids with a chiral axis. Arkivoc 2004, 4, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Aguado, G.P.; Moglioni, A.G.; Ortuño, R.M. Enantiodivergent synthesis of cyclobutyl-(Z)-α,β-dehydro-α-amino acid derivatives from (−)-cis-pinononic acid. Tetrahedron Asymmetry 2003, 14, 217–223. [Google Scholar] [CrossRef]
- Etayo, P.; Vidal-Ferran, A. Rhodium-catalysed asymmetric hydrogenation as a valuable synthetic tool for the preparation of chiral drugs. Chem. Soc. Rev. 2013, 42, 728–754. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, M.; Akireddy, S.R.; Reddy, R.E. Nonproteinogenic amino acids: An efficient asymmetric synthesis of (S)-(−)-acromelobic acid and (S)-(−)-acromelobinic acid. Tetrahedron 2002, 58, 6951–6963. [Google Scholar] [CrossRef]
- Blaskovich, M.A. Handbook on Syntheses of Amino Acids, General Routes to Amino Acids; American Chemical Society & Oxford University Press: New York, NY, USA, 2010. [Google Scholar]
- Yasuno, Y.; Mizutani, I.; Sueuchi, Y.; Wakabayashi, Y.; Yasuo, N.; Shimamoto, K.; Shinada, T. Catalytic Asymmetric Hydrogenation of Dehydroamino Acid Esters with Biscarbamate Protection and Its Application to the Synthesis of xCT Inhibitors. Chem. Eur. J. 2019, 25, 5145–5148. [Google Scholar] [CrossRef]
- Adamek, J.; Mrowiec-Białon, J.; Październiok-Holewa, A.; Mazurkiewicz, R. Thermogravimetrical investigations of the dealkoxycarbonylation of N-acyl-α-triphenylphosphonioglycinates. Thermochim. Acta 2011, 512, 22–27. [Google Scholar] [CrossRef]
- Gorewoda, T.; Mazurkiewicz, R.; Simka, W.; Mlostoń, G.; Schroeder, G.; Kubicki, M.; Kuźnik, N. 3-Triphenylphosphonio-2,5-piperazinediones as new chiral glycine cation equivalents. Tetrahedron Asymmetry 2011, 22, 823–833. [Google Scholar] [CrossRef]











































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
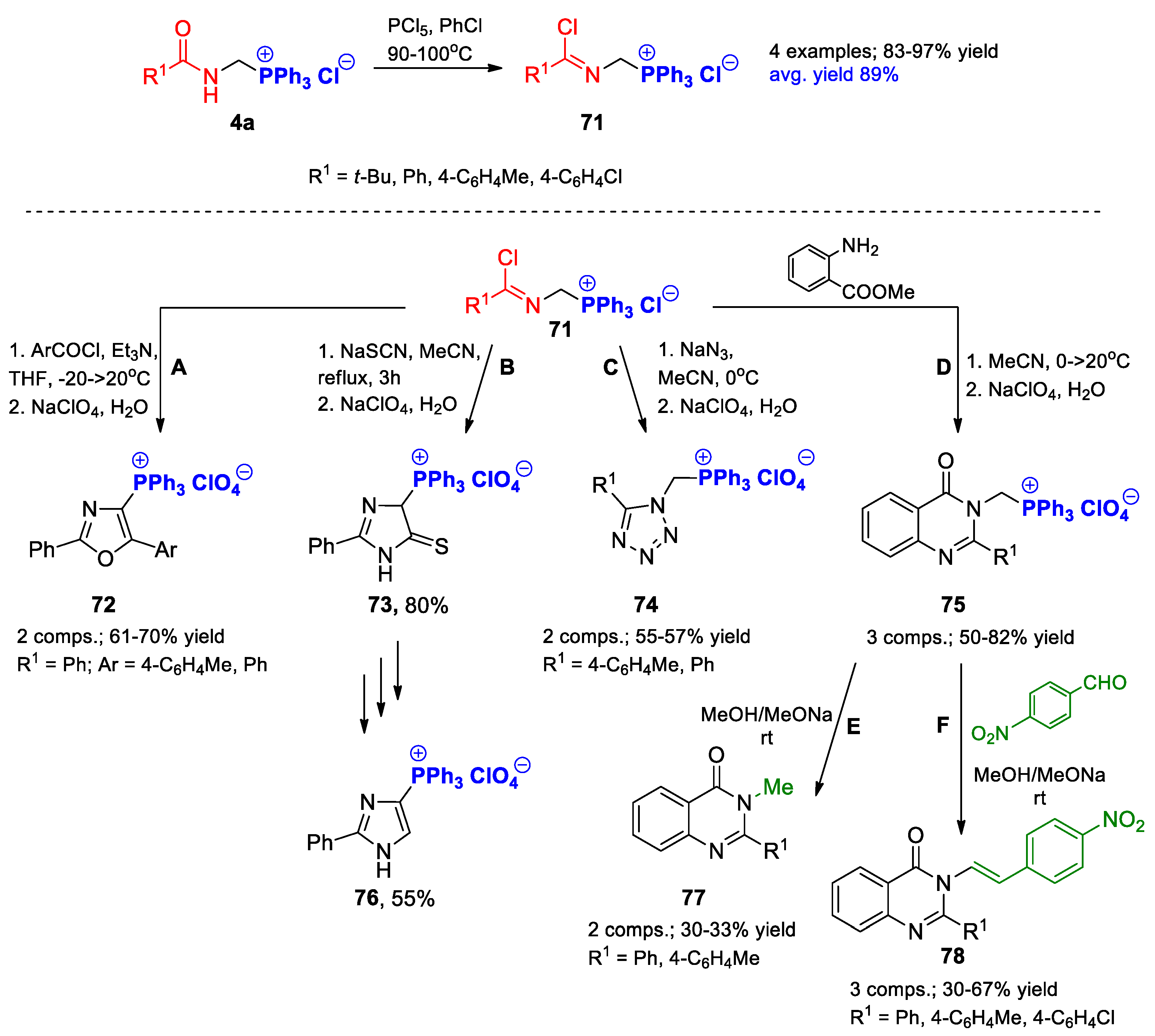{kind=link}
{kind=link}
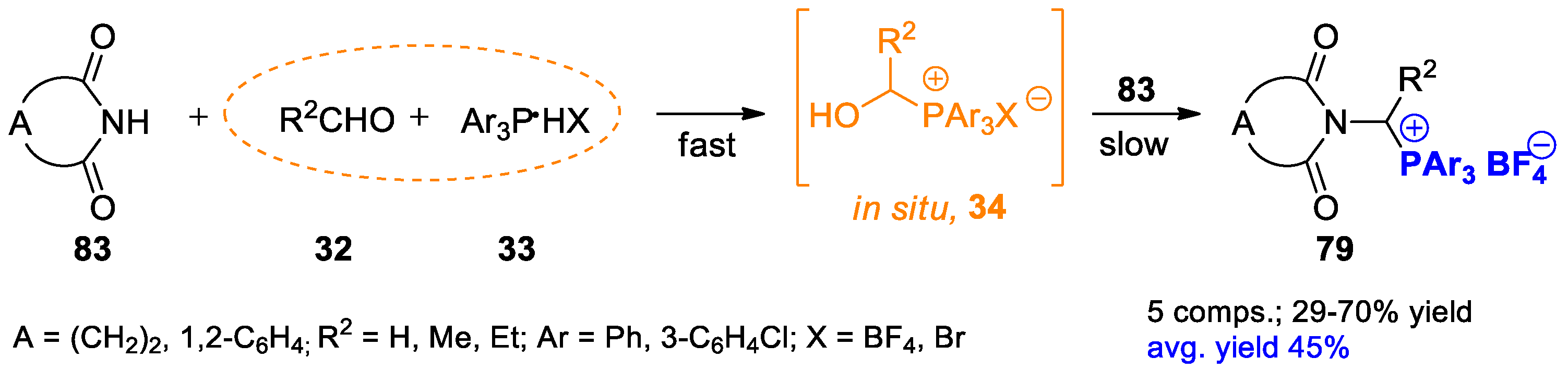{kind=link}
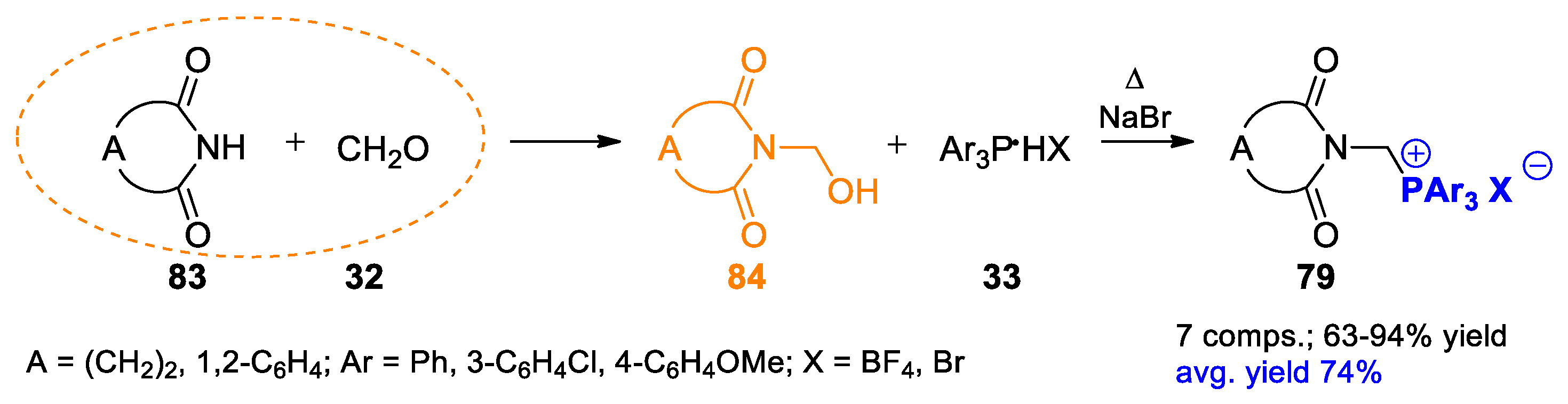{kind=link}
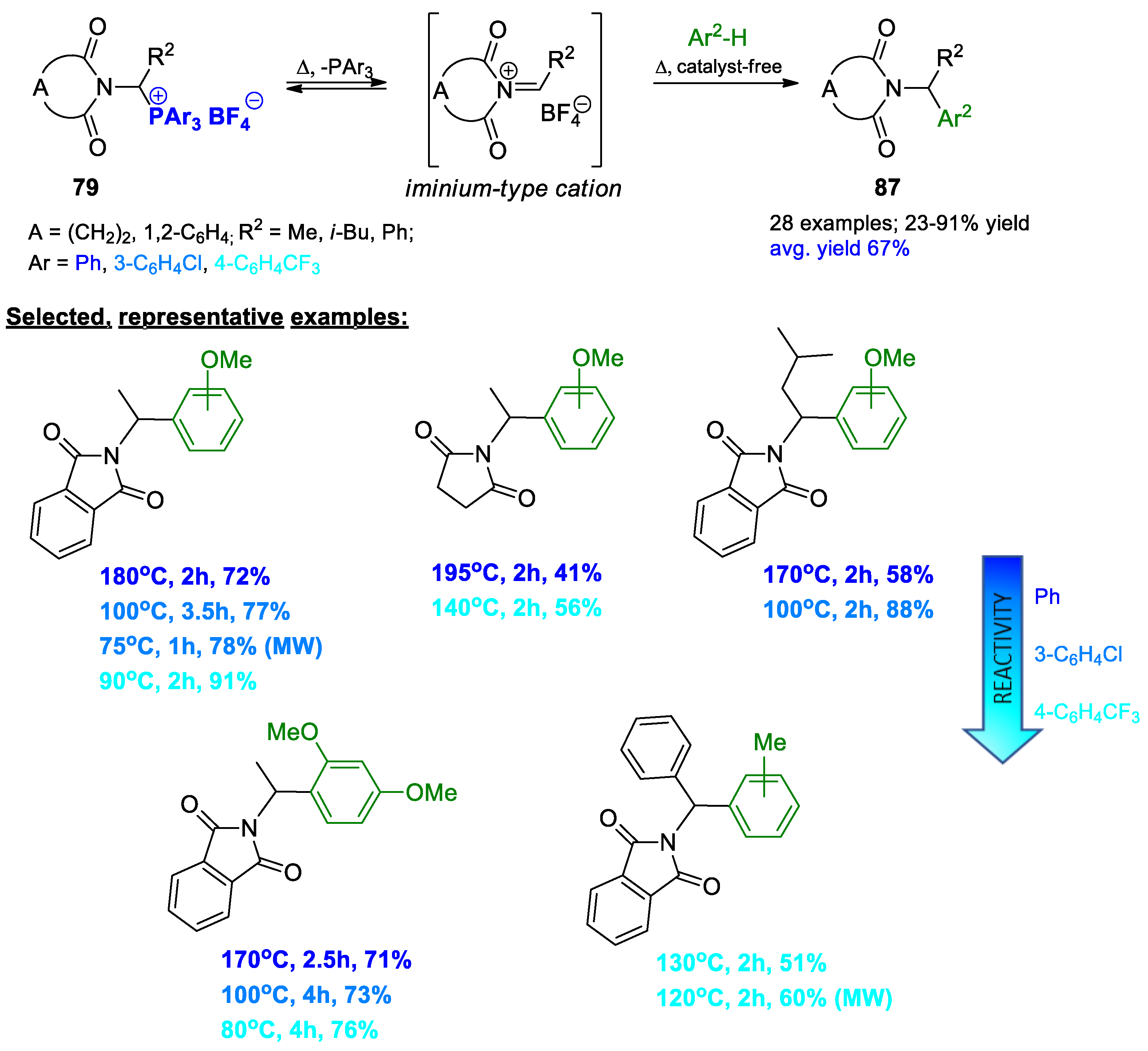{kind=link}
{kind=link}
{kind=link}
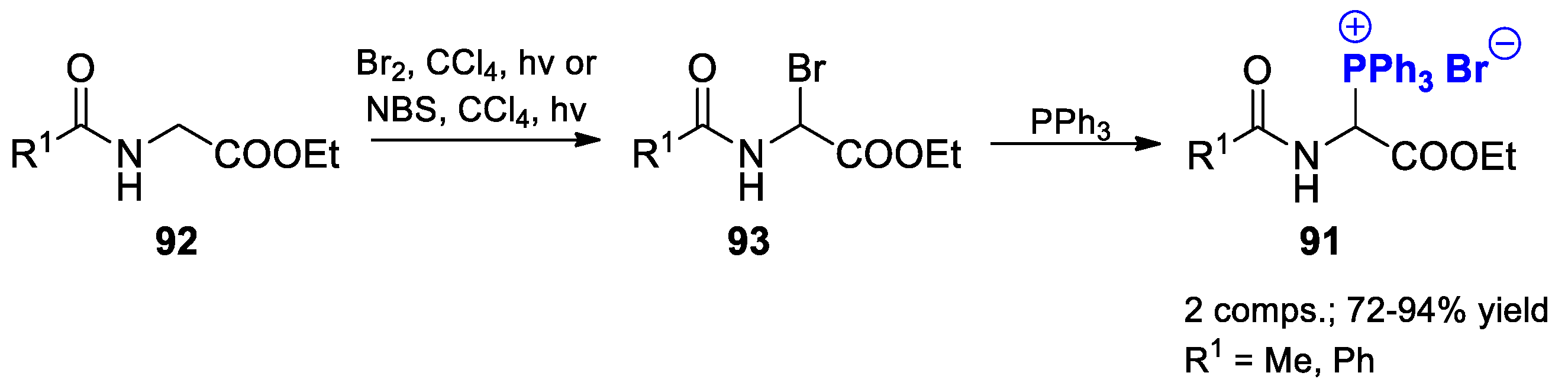{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
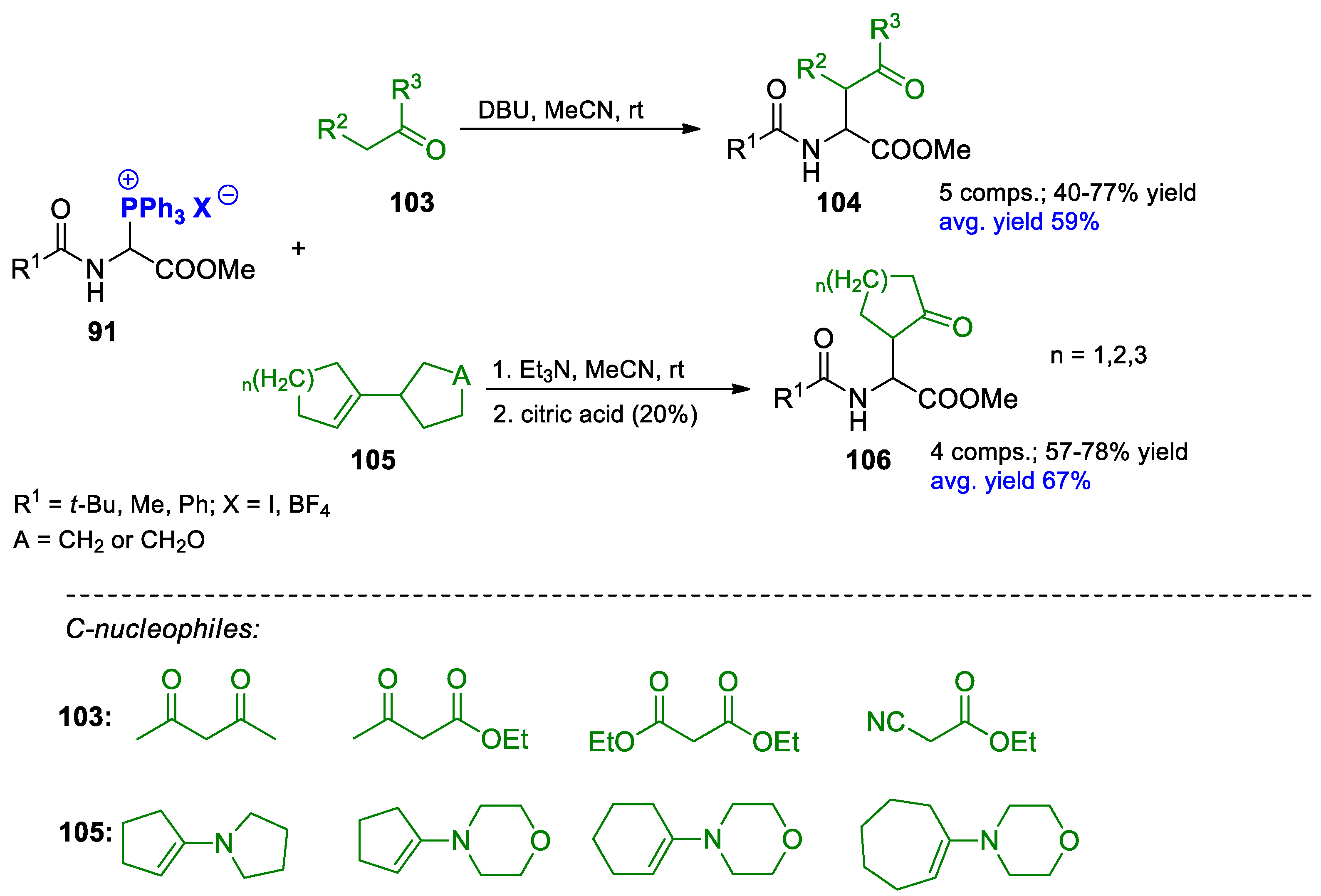{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure of Precursor | Summary of Characteristics | Examples of Use in α-Amidoalkylation (Selected Research or Review Literature) a |
|---|---|---|
 | limited structural diversity, limited reactivity, parent compounds for the other α-amidoalkylating agents, activation with acidic catalysts, synthesis from amides (or imides) and aldehydes (mostly in situ)—only N-hydoxymethylamides (or -imides) can be easily isolated | [3,4,6,7,8,9,10,11,12] |
 | limited reactivity, high structural diversity, activation with acidic catalysts, main synthesis methods based on electrochemical alkoxylation | [5,6,7,8,9,12,13,14] |
 | high reactivity, rather low yields in α-amidoalkylation reactions (lots of by-products), difficulties in the preparation, purification and storage | [6,7,8,9,12] |
 | high reactivity (good leaving group), high structural diversity, activation with acidic catalysts, easy to use and storage, diverse methods of synthesis, broad scope of application | [8,9,12,16,17,18,19] |
 | high reactivity (good leaving group), high structural diversity, activation with acidic catalysts, easy to use and storage, diverse methods of synthesis, broad scope of application, currently the most popular and convenient | [8,9,12,20,21,22,23] |

| Entry | Substrate 80 | Solvent | Conditions | Yield of 79, % | Refs. |
|---|---|---|---|---|---|
| X | |||||
| 1 | (Me3N)+ I- | methanol | reflux, 4 h | 58 | [70] |
| 2 | Cl | benzene | reflux, 2 h | - | [70] |
| 3 | Br | acetone | reflux, 3 min | 80 | [24] |
| 4 | Br | benzene | reflux, 22 h | 68 | [67,71] a |
| 5 | Br | toluene | reflux, 24 h | - | [72] |

| Phosphonium Salt 79 | Carbonyl Components | Conditions | Intermediate Compound | Targeted Compound |
|---|---|---|---|---|
 |  | KHMDS, THF, 0 °C→RT, 86% |  | 85 |
 | KHMDS, THF, 0 °C→RT, 77% |  | 86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamek, J.; Grymel, M.; Kuźnik, A.; Październiok-Holewa, A. 1-Aminoalkylphosphonium Derivatives: Smart Synthetic Equivalents of N-Acyliminium-Type Cations, and Maybe Something More: A Review. Molecules 2022, 27, 1562. https://doi.org/10.3390/molecules27051562
Adamek J, Grymel M, Kuźnik A, Październiok-Holewa A. 1-Aminoalkylphosphonium Derivatives: Smart Synthetic Equivalents of N-Acyliminium-Type Cations, and Maybe Something More: A Review. Molecules. 2022; 27(5):1562. https://doi.org/10.3390/molecules27051562
Chicago/Turabian StyleAdamek, Jakub, Mirosława Grymel, Anna Kuźnik, and Agnieszka Październiok-Holewa. 2022. "1-Aminoalkylphosphonium Derivatives: Smart Synthetic Equivalents of N-Acyliminium-Type Cations, and Maybe Something More: A Review" Molecules 27, no. 5: 1562. https://doi.org/10.3390/molecules27051562
APA StyleAdamek, J., Grymel, M., Kuźnik, A., & Październiok-Holewa, A. (2022). 1-Aminoalkylphosphonium Derivatives: Smart Synthetic Equivalents of N-Acyliminium-Type Cations, and Maybe Something More: A Review. Molecules, 27(5), 1562. https://doi.org/10.3390/molecules27051562