
Integrated Machine Learning and Chemoinformatics-Based Screening of Mycotic Compounds against Kinesin Spindle ProteinEg5 for Lung Cancer Therapy

 ,
,  , , , and
, , , and
Abstract
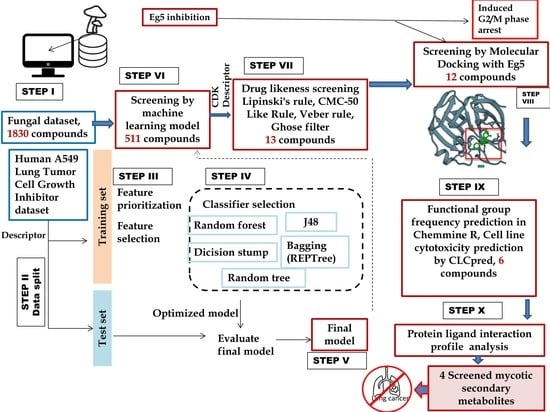
:
1. Introduction
2. Methods
2.1. Machine Learning Model Development and Screening
2.2. Drug-Likeness Screening and Molecular Docking
2.3. Functional Group Analysis and Cell Line Cytotoxicity Prediction
2.4. Molecular Dynamics Simulation
3. Results
3.1. Performance Comparison of Different ML Classifiers and Data Set Screening
3.2. Drug Likeness and Molecular Docking
3.3. Functional Group Cell Line Cytotoxicity and Binding Pattern of Screened Molecules
3.4. Binding Stability Analysis of the Screened Compounds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| AL | Artificial Intelligence |
| AMR | Atom Molar Refractivity |
| CAGR | Compound Annual Growth Rate |
| CDK | Chemistry Development Kit |
| CSF | Correlation-Based Feature Selection |
| ER | Estrogen Receptor |
| GUI | Graphic User Interface |
| HER | Electronic Health Records |
| IHR | Indian Himalayan Region |
| KIF-11 | Kinesin Family Member-11 |
| MCF-7 | Michigan Cancer Foundation-7 |
| MD | Molecular Dynamics |
| MeFSAT | Medicinal Fungi Secondary Metabolite And Therapeutics |
| ML | Machine Learning |
| NIC | National Cancer Institute |
| PDB | Protein Data Bank |
| PLIP | Protein Legend Interaction Profiler |
| QED | Quantitative Estimate of Drug-Likeness |
| QSAR | Quantitative Structure-Activity Relationship |
| RG | Radius of Gyration |
| RMSD | Root Mean Square Deviations |
| ROC | Receiver Operating Characteristic Curve |
| SMILES | Standard Data Format (SDF) To Molecular-Input Line-Entry System |
| TPSA | Total Polar Surface Area |
| USD | United States Dollar |
| WEKA | Waikato Environment for Knowledge Analysis |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Chand, B.; Kuniyal, J.C.; Chand, R. Ambient Air Quality and Its Sources Surrounding to Hydropower Projects in the Satluj Basin, Northwestern Himalaya, India. MAPAN 2019, 34, 495–510. [Google Scholar] [CrossRef]
- Garcia-Saez, I.; Skoufias, D.A. Eg5 Targeting Agents: From new anti-mitotic based inhibitor discovery to cancer therapy and resistance. Biochem. Pharmacol. 2020, 1, 114364. [Google Scholar] [CrossRef] [PubMed]
- Bertran, M.T.; Sdelci, S.; Regué, L.; Avruch, J.; Caelles, C.; Roig, J. Nek9 is a Plk1-activated kinase that controls early centrosome separation through Nek6/7 and Eg5. EMBO J. 2011, 30, 2634–2647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Lu, J.; Ding, K.; Bi, D.; Niu, Z.; Cao, Q.; Zhang, J.; Ding, S. The expression of Eg5 predicts a poor outcome for patients with renal cell carcinoma. Med. Oncol. 2013, 30, 476. [Google Scholar] [CrossRef] [PubMed]
- Nand, M.; Maiti, P.; Pant, R.; Kumari, M.; Chandra, S.; Pande, V. Virtual screening of natural compounds as inhibitors of EGFR 696-1022 T790M associated with non-small cell lung cancer. Bioinformation 2016, 12, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.D.; Kim, E.D.; Liu, L.; Buckley, R.S.; Parameswaran, S.; Kim, S.; Wojcik, E.J. Small molecule allosteric uncoupling of microtubule depolymerase activity from motility in human Kinesin-5 during mitotic spindle assembly. Sci. Rep. 2019, 9, 19900. [Google Scholar] [CrossRef]
- Li, Z.; Yu, B.; Qi, F.; Li, F. KIF11 Serves as an Independent Prognostic Factor and Therapeutic Target for Patients with Lung Adenocarcinoma. Front. Oncol. 2021, 11, 660218. [Google Scholar] [CrossRef]
- Saijo, T.; Ishii, G.; Ochiai, A.; Yoh, K.; Goto, K.; Nagai, K.; Kato, H.; Nishiwaki, Y.; Saijo, N. Eg5 expression is closely correlated with the response of advanced non-small cell lung cancer to antimitotic agents combined with platinum chemotherapy. Lung Cancer 2006, 54, 217–225. [Google Scholar] [CrossRef]
- Paier, C.R.K.; Maranhão, S.S.; Carneiro, T.R.; Lima, L.M.; Rocha, D.D.; Santos, R.D.S.; de Farias, K.M.; de Moraes-Filho, M.O.; Pessoa, C. Natural products as new antimitotic compounds for anticancer drug development. Clinics 2018, 73 (Suppl. S1). [Google Scholar] [CrossRef]
- Evidente, A.; Kornienko, A.; Cimmino, A.; Andolfi, A.; Lefranc, F.; Mathieu, V.; Kiss, R. Fungal metabolites with anticancer activity. Nat. Prod. Rep. 2014, 31, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Issa, N.T.; Stathias, V.; Schürer, S.; Dakshanamurthy, S. Machine and Deep Learning Approaches for Cancer Drug Repurposing. Semin. Cancer Biol. 2021, 68, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Global Artificial Intelligence in Drug Discovery Market Size Analysis 2018–2028. Available online: https://Bekryl.com (accessed on 18 December 2021).
- Ponzoni, I.; Sebastián-Pérez, V.; Martínez, M.J.; Roca, C.; Pérez, C.D.L.C.; Cravero, F.; Vazquez, G.; Páez, J.A.; Díaz, M.F.; Campillo, N.E. QSAR Classification Models for Predicting the Activity of Inhibitors of Beta-Secretase (BACE1) Associated with Alzheimer’s Disease. Sci. Rep. 2019, 9, 9102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; de Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, 930–940. [Google Scholar] [CrossRef]
- Vivek-Ananth, R.P.; Sahoo, A.K.; Kumaravel, K.; Mohanraj, K.; Samal, A. MeFSAT: A curated natural product database specific to secondary metabolites of medicinal fungi. RSC Adv. 2021, 11, 2596–2607. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I.H. Data mining in bioinformatics using Weka. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef] [Green Version]
- Puzyn, T.; Leszczynski, J.; Cronin, M.T. (Eds.) Recent Advances in QSAR Studies: Methods and Applications; Springer: Berlin/Heidelberg, Germany, 2010; Volume 8. [Google Scholar] [CrossRef]
- Manallack, D.T.; Prankerd, R.J.; Yuriev, E.; Oprea, T.I.; Chalmers, D.K. The Significance of Acid/Base Properties in Drug Discovery. Chem. Soc. Rev. 2013, 42, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Burden, F.R. A Chemically Intuitive Molecular Index Based on the Eigenvalues of a Modified Adjacency Matrix. Quant. Struct.-Act. Relatsh. 1997, 16, 309–314. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Hall, L.H.; Mohney, B.; Kier, L.B. The Electrotopological State: An Atom Index for QSAR. Quant. Struct. Act. Relatsh. 1991, 10, 43–51. [Google Scholar] [CrossRef]
- Wang, R.; Fu, Y.; Lai, L. A New Atom-Additive Method for Calculating Partition Coefficients. J. Chem. Inf. Comput. Sci. 1997, 37, 615–621. [Google Scholar] [CrossRef]
- He, S.; Ye, T.; Wang, R.; Zhang, C.; Zhang, X.; Sun, G.; Sun, X. An In Silico Model for Predicting Drug-Induced Hepatotoxicity. Int. J. Mol. Sci. 2019, 20, 1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taser, P.Y. Application of Bagging and Boosting Approaches Using Decision Tree-Based Algorithms in Diabetes Risk Prediction. Proceedings 2021, 74, 6. [Google Scholar] [CrossRef]
- Lučić, B.; Batista, J.; Bojović, V.; Lovrić, M.; Kržić, A.S.; Bešlo, D.; Nadramija, D.; Vikić-Topić, D. Estimation of Random Accuracy and its Use in Validation of Predictive Quality of Classification Models within Predictive Challenges. Croat. Chem. Acta 2019, 92, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Geete, A.; Damre, M.; Kokkula, A. Drug Likeness Tool (DruLiTo) Chemistry Development Kit (CDK), Department of Pharmacoinformatics NIPER, Mohali. Available online: http://www.niper.gov.in (accessed on 22 July 2021).
- Steinbeck, C.; Han, Y.Q.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E.L. The Chemistry Development Kit (CDK): An open-source Java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Nand, M.; Maiti, P.; Joshi, T.; Chandra, S.; Pande, V.; Kuniyal, J.C.; Ramakrishnan, M.A. Virtual screening of anti-HIV1 compounds against SARS-CoV-2: Machine learning modeling, chemoinformatics and molecular dynamics simulation based analysis. Sci. Rep. 2021, 10, 20397. [Google Scholar] [CrossRef]
- Talapatra, S.K.; Anthony, N.G.H.; Mackay, S.P.; Kozielski, F. Mitotic Kinesin Eg5 Overcomes Inhibition to the Phase I/II Clinical Candidate SB743921 by an Allosteric Resistance Mechanism. J. Med. Chem. 2013, 56, 6317–6329. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. Publ. Protein Soc. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, 530–534. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. A language and environment for statistical computing: Reference index. Vienna: R Foundation for Statistical Computing. 2010. Available online: http://www.polsci.wvu.edu (accessed on 26 July 2021).
- Cao, Y.; Charisi, A.; Cheng, L.-C.; Jiang, T.; Girke, T. ChemmineR: Acompound mining framework for R. Bioinformatics 2008, 24, 1733–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, P.; Nand, M.; Joshi, T.; Ramakrishnan, M.A.; Chandra, S. Identification of luteolin -7-glucoside and epicatechin gallate from Vernonia cinerea, as novel EGFR L858R kinase inhibitors against lung cancer: Docking and simulation-based study. J. Biomol. Struct. Dyn. 2020, 5048–5057. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.A.; Dubovskaja, V.I.; Rudik, A.; Pogodin, P.V.; Druzhilovskiy, D.; Gloriozova, T.A.; Filimonov, D.; Sastry, N.G.; Poroikov, V.V. CLC-Pred: A freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS ONE 2018, 25, e0191838. [Google Scholar] [CrossRef] [Green Version]
- Filimonov, D.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. Available online: http://www.way2drug.com/PASSonline (accessed on 8 June 2021). [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Shahbaaz, M.; Nkaule, A.; Christoffels, A. Designing novel possible kinase inhibitor derivatives as therapeutics against Mycobacterium tuberculosis: An in silico study. Sci. Rep. 2019, 9, 4405. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Jukic, S.; Saracevic, M.; Subasi, A.; Kevric, J. Comparison of Ensemble Machine Learning Methods for Automated Classification of Focal and Non-Focal Epileptic EEG Signals. Mathematics 2020, 8, 1481. [Google Scholar] [CrossRef]
- Patrick, G.L. An Introduction to Medicinal Chemistry; Oxford University Press: New York, NY, USA, 2017; pp. 1–832. Available online: https://global.oup.com (accessed on 26 August 2021).
- Connelly, P.R.; Thomson, J.A. Heat capacity changes and hydrophobic interactions in the binding of FK506 and rapamycin to the FK506 binding protein. Proc. Natl. Acad. Sci. USA 1992, 89, 4781–4785. [Google Scholar] [CrossRef] [Green Version]
- Kostal, J. Chapter Four - Computational Chemistry in Predictive Toxicology. Adv. Mol. Toxicol. 2016, 10, 139–186. [Google Scholar] [CrossRef]
- Ogunwa, T.H.; Laudadio, E.; Galeazzi, R.; Miyanishi, T. Insights into the Molecular Mechanisms of Eg5 Inhibition by (+)-Morelloflavone. Pharmaceuticals 2019, 12, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banyal, A.; Thakur, V.; Thakur, R.; Kumar, P. Endophytic Microbial Diversity: A New Hope for the Production of Novel Anti-tumor and Anti-HIV Agents as Future Therapeutics. Curr. Microbiol. 2021, 78, 1699–1717. [Google Scholar] [CrossRef] [PubMed]
- Gauchan, D.P.; Vélëz, H.; Acharya, A.; Östman, J.R.; Lundén, K.; Elfstrand, M.; García-Gil, M.R. Annulohypoxylon sp. Strain MUS1, an endophytic fungus isolated from Taxus wallichiana Zucc., produces taxol and other bioactive metabolites. 3 Biotech 2021, 11, 152. [Google Scholar] [CrossRef]
- Peng, X.-R.; Lu, S.-Y.; Shao, L.-D.; Zhou, L.; Qiu, M.-H. Structural Elucidation and Biomimetic Synthesis of (±)-Cochlactone A with Anti-Inflammatory Activity. J. Org. Chem. 2018, 83, 5516–5522. [Google Scholar] [CrossRef]
- Bijalwan, A.; Bahuguna, K.; Vasishth, A.; Singh, A.; Chaudhary, S.; Dongariyal, A.; Thakur, T.K.; Kaushik, S.; Ansari, M.J.; Alfarraj, S.; et al. Growth performance of Ganoderma lucidum using billet method in Garhwal Himalaya, India. Saudi Biol. Sci. 2021, 28, 2709–2717. [Google Scholar] [CrossRef]
- Mo, S.; Wang, S.; Zhou, G.; Yang, Y.; Li, Y.; Chen, A.X.; Shi, J. Phelligridins C-F: Cytotoxic pyrano [4,3-c][2]benzopyran-1,6-dione and furo[3,2-c]pyran-4-one derivatives from the fungus Phellinus igniarius. J. Nat. Prod. 2004, 67, 823–828. [Google Scholar] [CrossRef]
- Azeem, U.; Dhingra, G.S.; Shri, R. Pharmacological potential of wood inhabiting fungi of genus Phellinus quél.: An overview. J. Pharmacogn. Phytochem. 2018, 7, 1161–1171. [Google Scholar]
- Wang, B.T.; Qi, Q.Y.; Ma, K.; Pei, Y.F.; Han, J.J.; Xu, W.; Li, E.W.; Liu, H.W. Depside α-glucosidase inhibitors from a culture of the mushroom. Stereum hirsutum. Planta Med. 2014, 80, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Vabeikhokhei, J.M.; Mangaiha, Z.; Zothanzama, J.; Lalrinawmi, H. Diversity Study of Wood Rotting Fungi from Two different Forests in Mizoram, India. Int. J. Curr. Microbiol. Appl. Sci. 2019, 8, 2775–2785. [Google Scholar] [CrossRef]
- Kang, H.-S.; Jun, E.-M.; Park, S.-H.; Heo, S.-J.; Lee, T.-S.; Yoo, I.-D.; Kim, J.-P. Cyathusals A, B, and C, Antioxidants from the Fermented Mushroom Cyathus stercoreus. J. Nat. Prod. 2007, 70, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.M. Genus Cyathus Haller ex Pers. (Agaricomycetes) from Eastern Himalaya. KAVAKA 2016, 47, 20–26. Available online: http://www.fungiindia.co.in (accessed on 23 October 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classifier Name | Correctly Classified Instances % (Value) | Kappa Statistic | Mean Absolute Error | Root Mean Square Error | MCC | ROC Area |
|---|---|---|---|---|---|---|
| Random forest | 97.0588 | 0.9401 | 0.08 | 0.1731 | 0.942 | 0.989 |
| J48 | 96.7914 | 0.9346 | 0.05 | 0.175 | 0.937 | 0.964 |
| Decision stump | 96.7914 | 0.9346 | 0.06 | 0.175 | 0.937 | 0.947 |
| Random tree | 92.7807 | 0.8544 | 0.07 | 0.2687 | 0.855 | 0.928 |
| Bagging (REP tree) | 96.5241 | 0.9292 | 0.06 | 0.1844 | 0.931 | 0.96 |
| Title * | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pharmacological Indices | |||||||||||||
| MW | 358 | 358 | 372 | 372 | 330 | 346 | 378 | 378 | 380 | 364 | 386 | 370 | 358 |
| logp | 4 | 4 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 4 | 4 | 2 |
| Alogp | 1 | 1 | 1 | 1 | 0 | −1 | 1 | 1 | 0 | 1 | 3 | 3 | 0 |
| HBA | 5 | 5 | 6 | 6 | 7 | 8 | 6 | 6 | 8 | 7 | 7 | 6 | 7 |
| HBD | 2 | 2 | 3 | 3 | 2 | 3 | 4 | 4 | 4 | 3 | 4 | 3 | 3 |
| TPSA | 84 | 84 | 96 | 96 | 102 | 123 | 107 | 107 | 134 | 113 | 124 | 104 | 113 |
| AMR | 98 | 98 | 105 | 105 | 90 | 91 | 113 | 113 | 106 | 104 | 109 | 108 | 87 |
| nRB | 3 | 3 | 4 | 4 | 3 | 3 | 3 | 3 | 2 | 2 | 6 | 6 | 5 |
| nAtom | 52 | 52 | 51 | 51 | 38 | 39 | 46 | 46 | 40 | 39 | 50 | 49 | 55 |
| nAcidicGroup | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| RC | 4 | 4 | 3 | 3 | 3 | 3 | 4 | 4 | 4 | 4 | 2 | 2 | 2 |
| nRigidB | 26 | 26 | 25 | 25 | 23 | 24 | 28 | 28 | 29 | 28 | 23 | 22 | 21 |
| nAromRing | 1 | 1 | 1 | 1 | 1 | 1 | 3 | 3 | 2 | 2 | 2 | 2 | 0 |
| nHB | 7 | 7 | 9 | 9 | 9 | 11 | 10 | 10 | 12 | 10 | 11 | 9 | 10 |
| SAlerts | 4 | 4 | 5 | 5 | 5 | 5 | 0 | 0 | 4 | 4 | 3 | 4 | 2 |
| Ligand Name | Hydrophobic Interactions | Hydrogen Bond | Other | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Residue | AA | Distance | Residue | AA | Distance | Residue | AA | Distance | |
| (−)-Cochlactone-A | 79B | ILE | 3.85 | 286A | GLY | 1.85 | Salt Bridges | ||
| - | - | - | 286A | GLY | 2.16 | 138B | ARG | 4.45 | |
| 131B | PRO | 3.53 | 297A | ARG | 3.13 | 141B | HIS | 5.14 | |
| - | - | - | - | - | - | - | - | - | |
| 285A | ALA | 3.42 | - | - | - | - | - | - | |
| - | - | - | - | - | - | - | - | - | |
| Phelligridin-C | 79B | ILE | 3.92 | 83B | ARG | 3.27 | π–Cation Interactions | ||
| 125B | TYR | 3.72 | 142B | GLN | 2.55 | 83B | ARG | 4.97 | |
| - | - | - | 286A | GLY | 1.92 | ||||
| 131B | PRO | 3.83 | 290A | GLN | 2.5 | Salt Bridges | |||
| 285A | ALA | 3.29 | 297A | ARG | 2.2 | 138B | ARG | 4.1 | |
| Sterenin-E | 79B | ILE | 3.43 | 83B | ARG | 3.09 | π–Cation Interactions | ||
| 82B | TYR | 3.52 | 141B | HIS | 2.69 | 83B | ARG | 5.19 | |
| - | - | - | 142B | GLN | 2.72 | Salt Bridges | |||
| - | - | - | 142B | GLN | 2.66 | 138B | ARG | 4.74 | |
| 293A | LEU | 3.38 | 287A | ASN | 2.55 | 141B | HIS | 5.08 | |
| - | - | - | 290A | GLN | 2.66 | - | - | - | |
| - | - | - | 297A | ARG | 3.36 | - | - | - | |
| Cyathusal-A | 79B | ILE | 3.73 | 83B | ARG | 3.15 | π–Cation Interactions | ||
| - | - | - | 138B | ARG | 2.37 | 83B | ARG | 5.11 | |
| 285A | ALA | 3.92 | 142B | GLN | 2.51 | - | - | - | |
| - | - | - | 290A | GLN | 2.5 | - | - | - | |
| - | - | - | 297A | ARG | 2.45 | - | - | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiti, P.; Sharma, P.; Nand, M.; Bhatt, I.D.; Ramakrishnan, M.A.; Mathpal, S.; Joshi, T.; Pant, R.; Mahmud, S.; Simal-Gandara, J.; et al. Integrated Machine Learning and Chemoinformatics-Based Screening of Mycotic Compounds against Kinesin Spindle ProteinEg5 for Lung Cancer Therapy. Molecules 2022, 27, 1639. https://doi.org/10.3390/molecules27051639
Maiti P, Sharma P, Nand M, Bhatt ID, Ramakrishnan MA, Mathpal S, Joshi T, Pant R, Mahmud S, Simal-Gandara J, et al. Integrated Machine Learning and Chemoinformatics-Based Screening of Mycotic Compounds against Kinesin Spindle ProteinEg5 for Lung Cancer Therapy. Molecules. 2022; 27(5):1639. https://doi.org/10.3390/molecules27051639
Chicago/Turabian StyleMaiti, Priyanka, Priyanka Sharma, Mahesha Nand, Indra D. Bhatt, Muthannan Andavar Ramakrishnan, Shalini Mathpal, Tushar Joshi, Ragini Pant, Shafi Mahmud, Jesus Simal-Gandara, and et al. 2022. "Integrated Machine Learning and Chemoinformatics-Based Screening of Mycotic Compounds against Kinesin Spindle ProteinEg5 for Lung Cancer Therapy" Molecules 27, no. 5: 1639. https://doi.org/10.3390/molecules27051639
APA StyleMaiti, P., Sharma, P., Nand, M., Bhatt, I. D., Ramakrishnan, M. A., Mathpal, S., Joshi, T., Pant, R., Mahmud, S., Simal-Gandara, J., Alshehri, S., Ghoneim, M. M., Alruwaily, M., Awadh, A. A. A., Alshahrani, M. M., & Chandra, S. (2022). Integrated Machine Learning and Chemoinformatics-Based Screening of Mycotic Compounds against Kinesin Spindle ProteinEg5 for Lung Cancer Therapy. Molecules, 27(5), 1639. https://doi.org/10.3390/molecules27051639