
Fluorogenic Detection of Sulfite in Water by Using Copper(II) Azacyclam Complexes

 , ,
, ,  , and
, and 
Abstract
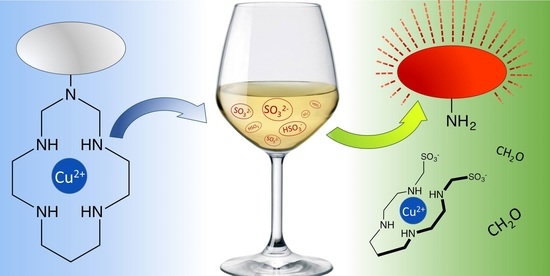
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Crystal Structure of the Azacyclam Complexes
2.2. Decomposition of Azacyclam Complexes in Aqueous Solution
2.3. Decomposition in the Presence of Sulfite
- coordination of HSO3−/SO32− to Cu2+, which could favor the reduction of the coordinated metal center and the formation of poorly stable copper(I) intermediates. The macrocyclic [Cu(N4)]+ species easily undergoes demetalation, as the square-planar coordination environment does not sufficiently stabilize that oxidation state. As a result of this mechanism, the unstable free ligand can be hydrolyzed generating the free sulfonamide LF. Although it could seem plausible, this hypothesis is not completely convincing because sulfite is not effective in the decomposition of macrocyclic copper(II) complexes other than azacyclam ones (e.g., derivatives of [Cu(cyclam)]2+) [83];
- nucleophilic reaction of sulfite and methylenediamine groups of the azacyclam framework, with consequent formation of aminomethanesulfonate derivatives, and release of sulfonamide locking fragments (Scheme 3).
2.4. Quantitative Detection of Sulfite
3. Materials and Methods
3.1. Materials and General Procedures
3.2. Synthesis
3.3. X-ray Crystallographic Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Fazio, T.; Warner, C.R. A Review of Sulphites in Foods: Analytical Methodology and Reported Findings. Food Addit. Contam. 1990, 7, 433–454. [Google Scholar] [CrossRef] [PubMed]
- Krska, R.; Becalski, A.; Braekevelt, E.; Koerner, T.; Cao, X.-L.; Dabeka, R.; Godefroy, S.; Lau, B.; Moisey, J.; Rawn, D.F.K.; et al. Challenges and Trends in the Determination of Selected Chemical Contaminants and Allergens in Food. Anal. Bioanal. Chem. 2012, 402, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Vally, H.; Misso, N.L. Adverse Reactions to the Sulphite Additives. Gastroenterol. Hepatol. Bed Bench 2012, 5, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, M.A.; Trüper, H.G. Sulfite Formation by Wine Yeasts. Arch. Microbiol. 1978, 118, 243–247. [Google Scholar] [CrossRef]
- Stine, C.J.; Boatright, W.L.; Lu, G. Intrinsic Sulfite Content of Isolated Soy Proteins. J. Am. Oil Chem. Soc. 2004, 81, 829. [Google Scholar] [CrossRef]
- Vally, H.; Misso, N.L.A.; Madan, V. Clinical Effects of Sulphite Additives. Clin. Exp. Allergy 2009, 39, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Lester, M.R. Sulfite Sensitivity: Significance in Human Health. J. Am. Coll. Nutr. 1995, 14, 229–232. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Code of Federal Regulations, 21CFR130.9. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=130.9 (accessed on 31 January 2022).
- Lawrence, J.F.; Chadha, R.K.; Ménard, C. Comparison of Three Liquid Chromatographic Methods with FDA Optimized Monier-Williams Method for Determination of Total Sulfite in Foods. J. Assoc. Off. Anal. Chem. 1990, 73, 77–79. [Google Scholar] [CrossRef] [PubMed]
- European Commission (EC) Directive 2000/13/EC on the Approximation of the Laws of the Member States Relating to the Labeling, Presentation, and Advertising of Foodstuffs; EC. 2000. Available online: https://www.fsai.ie/uploadedFiles/Consol_Dir2000_13.pdf (accessed on 31 January 2022).
- Lim, H.-S.; Park, S.-K.; Kim, S.-H.; Song, S.-B.; Jang, S.-J.; Kim, M. Comparison of Four Different Methods for the Determination of Sulfites in Foods Marketed in South Korea. Food Addit. Contam. Part A 2014, 31, 187–196. [Google Scholar] [CrossRef]
- Ensafi, A.A.; Karimi-Maleh, H.; Keyvanfard, M. A New Voltammetric Sensor for the Determination of Sulfite in Water and Wastewater Using Modified-Multiwall Carbon Nanotubes Paste Electrode. Int. J. Environ. Anal. Chem. 2013, 93, 650–660. [Google Scholar] [CrossRef]
- Haskins, J.E.; Kendall, H.; Baird, R.B. A Low Level Spectrophotometric Method for the Determination of Sulfite in Water. Water Res. 1984, 18, 751–753. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, M. Simple Methods for Rapid Determination of Sulfite in Food Products. Food Control. 2006, 17, 975–980. [Google Scholar] [CrossRef]
- Keil, R.; Hampp, R.; Ziegler, H. Cycling Technique for the Determination of Femtomole Amounts of Sulfite. Anal. Chem. 1989, 61, 1755–1758. [Google Scholar] [CrossRef]
- Pundir, C.S.; Rawal, R. Determination of Sulfite with Emphasis on Biosensing Methods: A Review. Anal. Bioanal. Chem. 2013, 405, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- McFeeters, R.F.; Barish, A.O. Sulfite Analysis of Fruits and Vegetables by High-Performance Liquid Chromatography (HPLC) with Ultraviolet Spectrophotometric Detection. J. Agric. Food Chem. 2003, 51, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Robbins, K.S.; Shah, R.; MacMahon, S.; de Jager, L.S. Development of a Liquid Chromatography-Tandem Mass Spectrometry Method for the Determination of Sulfite in Food. J. Agric. Food Chem. 2015, 63, 5126–5132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-J.; Kim, Y.-K. Analysis of Free and Total Sulfites in Food by Ion Chromatography with Electrochemical Detection. J. Food Sci. 2006, 51, 1360–1361. [Google Scholar] [CrossRef]
- Daunoravicius, Z.; Padarauskas, A. Capillary Electrophoretic Determination of Thiosulfate, Sulfide and Sulfite Using in-Capillary Derivatization with Iodine. Electrophoresis 2002, 23, 2439–2444. [Google Scholar] [CrossRef]
- Jankovskiene, G.; Daunoravicius, Z.; Padarauskas, A. Capillary Electrophoretic Determination of Sulfite Using the Zone-Passing Technique of in-Capillary Derivatization. J. Chromatogr. A 2001, 934, 67–73. [Google Scholar] [CrossRef]
- Monier-Williams, G.W. Ministry of Health. The Determination of Sulphur Dioxide in Foods. Analyst 1927, 52, 343–344. [Google Scholar] [CrossRef]
- Vahl, J.; Converse, J. Ripper Procedure for Determining Sulfur Dioxide in Wine: Collaborative Study. J.-Assoc. Off. Anal. Chem. 1980, 63, 194–199. [Google Scholar] [CrossRef] [PubMed]
- AOAC. Optimized Monier-Williams Method. In Official Methods of Analysis; AOAC Int.: Rockville, MD, USA, 2019. [Google Scholar]
- AOAC. Sulfite (Free) in Wines. In Official Methods of Analysis; AOAC Int.: Rockville, MD, USA, 2019. [Google Scholar]
- AOAC. Sulfite (Total) in Foods and Beverages. In Official Methods of Analysis; Method 990.29; AOAC Int.: Rockville, MD, USA, 2019. [Google Scholar]
- AOAC. Sulfites in Foods and Beverages. In Official Methods of Analysis; Method 990.31; AOAC Int.: Rockville, MD, USA, 2019. [Google Scholar]
- Carlos, K.S.; Treblin, M.; de Jager, L.S. Comparison and Optimization of Three Commercial Methods with an LC-MS/MS Method for the Determination of Sulfites in Food and Beverages. Food Chem. 2019, 286, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Gale, P.A.; Caltagirone, C. Fluorescent and Colorimetric Sensors for Anionic Species. Coord. Chem. Rev. 2018, 354, 2–27. [Google Scholar] [CrossRef]
- Aletti, A.B.; Gillen, D.M.; Gunnlaugsson, T. Luminescent/Colorimetric Probes and (Chemo-) Sensors for Detecting Anions Based on Transition and Lanthanide Ion Receptor/Binding Complexes. Coord. Chem. Rev. 2018, 354, 98–120. [Google Scholar] [CrossRef]
- Kim, D.-S.; Chung, Y.-M.; Jun, M.; Ahn, K.H. Selective Colorimetric Sensing of Anions in Aqueous Media through Reversible Covalent Bonding. J. Org. Chem. 2009, 74, 4849–4854. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Zha, D.; Anslyn, E.V. Recent Advances in Supramolecular Analytical Chemistry Using Optical Sensing. Chem. Rev. 2015, 115, 7840–7892. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, M.; Martínez-Máñez, R.; Ros-Lis, J.V.; Batista, R.M.F.; Costa, S.P.G.; Raposo, M.M.M.; Sancenón, F. A Dual Channel Sulphur-Containing a Macrocycle Functionalised BODIPY Probe for the Detection of Hg(Ii) in a Mixed Aqueous Solution. New J. Chem 2018, 42, 7863–7868. [Google Scholar] [CrossRef] [Green Version]
- Harvey, P.; Nonat, A.; Platas-Iglesias, C.; Natrajan, L.S.; Charbonnière, L.J. Sensing Uranyl(VI) Ions by Coordination and Energy Transfer to a Luminescent Europium(III) Complex. Angew. Chem. Int. Ed. 2018, 57, 9921–9924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhao, Q.; Feng, W.; Li, F. Luminescent Chemodosimeters for Bioimaging. Chem. Rev. 2013, 113, 192–270. [Google Scholar] [CrossRef] [PubMed]
- Sancenón, F.; Pascual, L.; Oroval, M.; Aznar, E.; Martínez-Máñez, R. Gated Silica Mesoporous Materials in Sensing Applications. ChemistryOpen 2015, 4, 418–437. [Google Scholar] [CrossRef]
- Toscani, A.; Marín-Hernández, C.; Robson, J.A.; Chua, E.; Dingwall, P.; White, A.J.P.; Sancenón, F.; de la Torre, C.; Martínez-Máñez, R.; Wilton-Ely, J.D.E.T. Highly Sensitive and Selective Molecular Probes for Chromo-Fluorogenic Sensing of Carbon Monoxide in Air, Aqueous Solution and Cells. Chem. Eur. J. 2019, 25, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Cotruvo, J.A., Jr.; Aron, A.T.; Ramos-Torres, K.M.; Chang, C.J. Synthetic Fluorescent Probes for Studying Copper in Biological Systems. Chem. Soc. Rev. 2015, 44, 4400–4414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.-F.; Zhao, M.; Wang, G. A Rhodamine-Based Fluorescent Probe Selective for Bisulfite Anion in Aqueous Ethanol Media. Sens. Actuators B Chem. 2011, 152, 8–13. [Google Scholar] [CrossRef]
- Sun, Y.-Q.; Wang, P.; Liu, J.; Zhang, J.; Guo, W. A Fluorescent Turn-on Probe for Bisulfite Based on Hydrogen Bond-Inhibited C—N Isomerization Mechanism. Analyst 2012, 137, 3430–3433. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Jia, H.; Feng, J.; Qin, J.; Li, Z. “Reactive” Probe for Hydrogen Sulfite: Good Ratiometric Response and Bioimaging Application. Sens. Actuators B Chem. 2013, 184, 274–280. [Google Scholar] [CrossRef]
- Yin, C.; Li, X.; Yue, Y.; Chao, J.; Zhang, Y.; Huo, F. A New Fluorescent Material and Its Application in Sulfite and Bisulfite Bioimaging. Sens. Actuators B Chem. 2017, 246, 615–622. [Google Scholar] [CrossRef]
- Sun, Y.-Q.; Liu, J.; Zhang, J.; Yang, T.; Guo, W. Fluorescent Probe for Biological Gas SO2 Derivatives Bisulfite and Sulfite. Chem. Commun. 2013, 49, 2637–2639. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Feng, H.; Li, H.; Wang, Y.; Zhang, Z.; Kang, W.; Jia, H.; Yang, X.; Meng, Q.; Zhang, R. Red-Emission Probe for Ratiometric Fluorescent Detection of Bisulfite and Its Application in Live Animals and Food Samples. ACS Omega 2020, 5, 5452–5459. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; He, T.; Li, K.; Wu, M.-B.; Huang, Z.; Yu, X.-Q. A Real-Time Colorimetric and Ratiometric Fluorescent Probe for Sulfite. Analyst 2013, 138, 3018–3025. [Google Scholar] [CrossRef]
- Santos-Figueroa, L.E.; Giménez, C.; Agostini, A.; Aznar, E.; Marcos, M.D.; Sancenón, F.; Martínez-Máñez, R.; Amorós, P. Selective and Sensitive Chromofluorogenic Detection of the Sulfite Anion in Water Using Hydrophobic Hybrid Organic–Inorganic Silica Nanoparticles. Angew. Chem. 2013, 125, 13957–13961. [Google Scholar] [CrossRef]
- Xu, G.; Wu, H.; Liu, X.; Feng, R.; Liu, Z. A Simple Pyrene-Pyridinium-Based Fluorescent Probe for Colorimetric and Ratiometric Sensing of Sulfite. Dyes Pigment. 2015, 120, 322–327. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.; Ding, S.; Zhang, H.; Feng, G. A Near-Infrared Fluorescent Probe for Rapid, Colorimetric and Ratiometric Detection of Bisulfite in Food, Serum, and Living Cells. Sens. Actuators B Chem. 2015, 211, 377–384. [Google Scholar] [CrossRef]
- Tan, L.; Lin, W.; Zhu, S.; Yuan, L.; Zheng, K. A Coumarin-Quinolinium-Based Fluorescent Probe for Ratiometric Sensing of Sulfite in Living Cells. Org. Biomol. Chem. 2014, 12, 4637–4643. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.G.; Hwang, J.; Eor, S.; Chang, S.-K. Chromogenic and Fluorogenic Signaling of Sulfite by Selective Deprotection of Resorufin Levulinate. Org. Lett. 2010, 12, 5624–5627. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Liu, C.; Zhu, Y.-C.; Zhu, Y.-Z. A Boron-Dipyrromethene-Based Fluorescent Probe for Colorimetric and Ratiometric Detection of Sulfite. J. Agric. Food Chem. 2011, 59, 11935–11939. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Hou, P.; Wang, J.; Song, X. A Highly Sulfite-Selective Ratiometric Fluorescent Probe Based on ESIPT. RSC Adv. 2012, 2, 10869–10873. [Google Scholar] [CrossRef]
- Ma, X.; Liu, C.; Shan, Q.; Wei, G.; Wei, D.; Du, Y. A Fluorescein-Based Probe with High Selectivity and Sensitivity for Sulfite Detection in Aqueous Solution. Sens. Actuators B Chem. 2013, 188, 1196–1200. [Google Scholar] [CrossRef]
- Fabbrizzi, L.; Licchelli, M.; Mosca, L.; Poggi, A. Template Synthesis of Azacyclam Metal Complexes Using Primary Amides as Locking Fragments. Coord. Chem. Rev. 2010, 254, 1628–1636. [Google Scholar] [CrossRef]
- Abbà, F.; De Santis, G.; Fabbrizzi, L.; Licchelli, M.; Manotti Lanfredi, A.M.; Pallavicini, P.; Poggi, A.; Ugozzoli, F. Nickel(II) Complexes of Azacyclams: Oxidation and Reduction Behavior and Catalytic Effects in the Electroreduction of Carbon Dioxide. Inorg. Chem. 1994, 33, 1366–1375. [Google Scholar] [CrossRef]
- De Blas, A.; De Santis, G.; Fabbrizzi, L.; Licchelli, M.; Pallavicini, P. Novel Routes to Functionalized Cyclam-like Macrocycles. Pure Appl. Chem. 1993, 65, 455–459. [Google Scholar] [CrossRef]
- Tsymbal, L.V.; Andriichuk, I.L.; Shova, S.; Trzybiński, D.; Woźniak, K.; Arion, V.B.; Lampeka, Y.D. Coordination Polymers of the Macrocyclic Nickel(II) and Copper(II) Complexes with Isomeric Benzenedicarboxylates: The Case of Spatial Complementarity between the Bis-Macrocyclic Complexes and o-Phthalate. Cryst. Growth Des. 2021, 21, 2355–2370. [Google Scholar] [CrossRef]
- Lee, J.H.; Moon, H.R. Structural Diversity of Metal–Organic Frameworks via Employment of Azamacrocycles as a Building Block. J. Incl. Phenom. Macrocycl. Chem. 2018, 92, 237–249. [Google Scholar] [CrossRef]
- Suh, M.P.; Ko, J.W.; Choi, H.J. A Metal−Organic Bilayer Open Framework with a Dynamic Component: Single-Crystal-to-Single-Crystal Transformations. J. Am. Chem. Soc. 2002, 124, 10976–10977. [Google Scholar] [CrossRef] [PubMed]
- Ju, P.; Jiang, L.; Lu, T.-B. An Unprecedented Dynamic Porous Metal–Organic Framework Assembled from Fivefold Interlocked Closed Nanotubes with Selective Gas Adsorption Behaviors. Chem Commun 2013, 49, 1820–1822. [Google Scholar] [CrossRef] [PubMed]
- Stackhouse, C.A.; Ma, S. Azamacrocyclic-Based Metal Organic Frameworks: Design Strategies and Applications. Polyhedron 2018, 145, 154–165. [Google Scholar] [CrossRef]
- Savastano, M.; Monini, V.; Bazzicalupi, C.; Bianchi, A. Bidimensional Polyiodide Netting Stabilized by a Cu(II) Macrocyclic Complex. Inorganics 2022, 10, 12. [Google Scholar] [CrossRef]
- Martínez-Camarena, Á.; Savastano, M.; Blasco, S.; Delgado-Pinar, E.; Giorgi, C.; Bianchi, A.; García-España, E.; Bazzicalupi, C. Assembly of Polyiodide Networks with Cu(II) Complexes of Pyridinol-Based Tetraaza Macrocycles. Inorg. Chem. 2022, 61, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.-C.; Zhong, D.-C.; Lu, T.-B. Non-Noble Metal-Based Molecular Complexes for CO2 Reduction: From the Ligand Design Perspective. EnergyChem 2020, 2, 100034. [Google Scholar] [CrossRef]
- Wang, J.-W.; Zhong, D.-C.; Lu, T.-B. Artificial Photosynthesis: Catalytic Water Oxidation and CO2 Reduction by Dinuclear Non-Noble-Metal Molecular Catalysts. Coord. Chem. Rev. 2018, 377, 225–236. [Google Scholar] [CrossRef]
- Sayed, S.E.; Licchelli, M.; Martínez-Máñez, R.; Sancenón, F. Capped Mesoporous Silica Nanoparticles for the Selective and Sensitive Detection of Cyanide. Chem. Asian J. 2017, 12, 2670–2674. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, S.; Milani, M.; Licchelli, M.; Martínez-Máñez, R.; Sancenón, F. Hexametaphosphate-Capped Silica Mesoporous Nanoparticles Containing CuII Complexes for the Selective and Sensitive Optical Detection of Hydrogen Sulfide in Water. Chem.-Eur. J. 2015, 21, 7002–7006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrizzi, L.; Licchelli, M.; Manotti Lanfredi, A.M.; Vassalli, O.; Ugozzoli, F. Template Synthesis of a Tetraaza Macrocycle Which Involves Benzaldehyde Rather Than Formaldehyde as a Building Block. Isolation and Structure Determination of the Open-Chain Schiff Base Intermediate Complex. Inorg. Chem. 1996, 35, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Fabbrizzi, L.; Lanfredi, A.M.M.; Pallavicini, P.; Perotti, A.; Taglietti, A.; Ugozzoli, F. Crystal and Molecular Structure and Solution Behaviour of Low-Spin (3-Methyl-1,3,5,8,12-Pentaazacyclotetradecane)Κ4N1,N5,N8,N12)Nickel(II) Diperchlorate. J. Chem Soc. Dalton Trans. 1991, 3263–3269. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Lampeka, Y.D. A New One-Pot Synthesis of Bis(Macrocyclic) Complexes: Preparation and Characterization of Nickel Complexes with Bis(Pentaazamacrocyclic) Ligands. J. Chem. Soc. Chem. Commun. 1991, 1077–1079. [Google Scholar] [CrossRef]
- Hay, R.W.; Danby, A.; Lightfoot, P.; Lampeka, Y.D. Padlock Macrocyclic Complexes. The Synthesis of a Range of Nickel(II) Complexes of N-Alkyl Azacyclams and the Crystal Structure of (3-Ethyl-1,3,5,8,12-Penta-Azacyclotetradecane) Nickel(II) Perchlorate. Polyhedron 1997, 16, 2777–2783. [Google Scholar] [CrossRef]
- Vengatajalabathy Gobi, K.; Ohsaka, T. Electrochemical and Spectral Properties of Novel Dinickel(II) and Dicopper(II) Complexes with N,N-Linked Bis(Pentaazacyclotetradecane). Electrochim. Acta 1998, 44, 269–278. [Google Scholar] [CrossRef]
- Bernhardt, P.V.; Hayes, E.J. Aminotriazines as Locking Fragments in Macrocyclic Synthesis. Inorg. Chem. 1998, 37, 4214–4219. [Google Scholar] [CrossRef] [PubMed]
- De Blas, A.; De Santis, G.; Fabbrizzi, L.; Licchelli, M.; Lanfredi, A.M.M.; Morosini, P.; Pallavicini, P.; Ugozzoli, F. Amides and Sulfonamides: Efficient Molecular Padlocks for the Template Synthesis of Azacyclam (1,3,5,8,12-Pentaazacyclotetradecane) Macrocycles. J. Chem. Soc. Dalton Trans. 1993, 1411–1416. [Google Scholar] [CrossRef]
- De Santis, G.; Fabbrizzi, L.; Licchelli, M.; Mangano, C.; Pallavicini, P. The Copper(I) Complex of a Metallocyclam-Functionalized Phenanthroline: A Poorly Stable Species That Is Very Resistant to Oxidation. Inorg. Chem. 1993, 32, 3385–3387. [Google Scholar] [CrossRef]
- De Blas, A.; De Santis, G.; Fabbrizzi, L.; Licchelli, M.; Lanfredi, A.M.M.; Pallavicini, P.; Poggi, A.; Ugozzoli, F. Pyridines with an Appended Metallocyclam Subunit. Versatile Building Blocks to Supramolecular Multielectron Redox Systems. Inorg. Chem. 1993, 32, 106–113. [Google Scholar] [CrossRef]
- De Santis, G.; Fabbrizzi, L.; Licchelli, M.; Mangano, C.; Pallavicini, P.; Poggi, A. Ferrocene-Metallocyclam Conjugates: New Redox Systems Whose Two-Electron Activity Can Be Modulated through the Medium. Inorg. Chem. 1993, 32, 854–860. [Google Scholar] [CrossRef]
- Fabbrizzi, L.; Licchelli, M.; De Santis, G.; Sardone, N.; Velders, A.H. Fluorescence Redox Switching Systems Operating through Metal Centres: The NiIII/NiII Couple. Chem. Eur. J. 1996, 2, 1243–1250. [Google Scholar] [CrossRef]
- Boiocchi, M.; Fabbrizzi, L.; Garolfi, M.; Licchelli, M.; Mosca, L.; Zanini, C. Templated Synthesis of Copper(II) Azacyclam Complexes Using Urea as a Locking Fragment and Their Metal-Enhanced Binding Tendencies towards Anions. Chem. Eur. J. 2009, 15, 11288–11297. [Google Scholar] [CrossRef] [PubMed]
- Amendola, V.; Fabbrizzi, L.; Foti, F.; Licchelli, M.; Mangano, C.; Pallavicini, P.; Poggi, A.; Sacchi, D.; Taglietti, A. Light-Emitting Molecular Devices Based on Transition Metals. Coord. Chem. Rev. 2006, 250, 273–299. [Google Scholar] [CrossRef]
- Boiocchi, M.; Fabbrizzi, L.; Licchelli, M.; Sacchi, D.; Vázquez, M.; Zampa, C. A Two-Channel Molecular Dosimeter for the Optical Detection of Copper (II). Chem. Commun. 2003, 1812–1813. [Google Scholar] [CrossRef] [PubMed]
- Sasakura, K.; Hanaoka, K.; Shibuya, N.; Mikami, Y.; Kimura, Y.; Komatsu, T.; Ueno, T.; Terai, T.; Kimura, H.; Nagano, T. Development of a Highly Selective Fluorescence Probe for Hydrogen Sulfide. J. Am. Chem. Soc. 2011, 133, 18003–18005. [Google Scholar] [CrossRef] [PubMed]
- Santos-Figueroa, L.E.; de la Torre, C.; El Sayed, S.; Sancenón, F.; Martínez-Máñez, R.; Costero, A.M.; Gil, S.; Parra, M. Highly Selective Fluorescence Detection of Hydrogen Sulfide by Using an Anthracene-Functionalized Cyclam–CuII Complex. Eur. J. Inorg. Chem. 2014, 2014, 41–45. [Google Scholar] [CrossRef]
- Mirra, S.; Strianese, M.; Pellecchia, C. A Cyclam-Based Fluorescent Ligand as a Molecular Beacon for Cu2+ and H2S Detection. Eur. J. Inorg. Chem. 2017, 2017, 3900–3907. [Google Scholar] [CrossRef]
- Hathaway, B.J. A new look at the stereochemistry and electronic properties of complexes of the copper(II) ion. In Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 1984; pp. 55–118. [Google Scholar]
- Licchelli, M.; Milani, M.; Pizzo, S.; Poggi, A.; Sacchi, D.; Boiocchi, M. Synthesis of Novel Diazacyclam Copper(II) Complexes by Template Reaction Involving Sulphonamides as Locking Fragments. Inorg. Chim. Acta 2012, 384, 210–218. [Google Scholar] [CrossRef]
- Thoem, V.J.; Boeyens, J.C.A.; McDougall, G.J.; Hancock, R.D. Origin of the High Ligand Field Strength and Macrocyclic Enthalpy in Complexes of Nitrogen-Donor Macrocycles. J. Am. Chem. Soc. 1984, 106, 3198–3207. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Cabbiness, D.K.; Margerum, D.W. Effect of Macrocyclic Structures on the Rate of Formation and Dissociation of Copper(II) Complexes. J. Am. Chem. Soc. 1970, 92, 2151–2153. [Google Scholar] [CrossRef]
- Busch, D.H. Distinctive Coordination Chemistry and Biological Significance of Complexes with Macrocyclic Ligands. Acc. Chem. Res. 1978, 11, 392–400. [Google Scholar] [CrossRef]
- Mansour, A.M. Crystal Structure, DFT, Spectroscopic and Biological Activity Evaluation of Analgin Complexes with Co(Ii), Ni(II) and Cu(II). Dalton Trans. 2014, 43, 15950–15957. [Google Scholar] [CrossRef]
- Borman, P.; Elder, D. Q2(R1) Validation of Analytical Procedures. ICH Qual. Guidel. 2017, 127–166. [Google Scholar]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- van der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Perez, F.; Granger, B.E. IPython: A System for Interactive Scientific Computing. Comput. Sci. Eng. 2007, 9, 21–29. [Google Scholar] [CrossRef]
- Newville, M.; Otten, R.; Nelson, A.; Ingargiola, A.; Stensitzki, T.; Allan, D.; Fox, A.; Carter, F.; Michał; Pustakhod, D.; et al. Lmfit/Lmfit-Py 0.9.13. Available online: https://doi.org/10.5281/zenodo.2620617 (accessed on 31 January 2022).
- Farrugia, L.J. WinGX and It ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- North, A.C.T.; Phillips, D.C.; Mathews, F.S. A Semi-Empirical Method of Absorption Correction. Acta Crystallogr. Sect. A 1968, 24, 351–359. [Google Scholar] [CrossRef]
- Brucker. SAINT Software Reference Manual, Version 6; Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.; Polidori, G.; Spagna, R. SIR97: A New Tool for Crystal Structure Determination and Refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λ (nm) | ε (M−1cm−1) |
|---|---|---|
| [Cu(1)]2+ | 231 265(sh) 518 | 19,500 9000 79 |
| [Cu(2)]2+ | 232 265 327 519 | 75,000 12,000 1500 83 |
| [Cu(3)]2+ | 250 334 517 | 23,000 4000 76 |
| [Cu(2.3.2-tet)]2+ | 526 | 73 |
| 3[[Cu(1)](NO3)2] | 3[[Cu(2)](NO3)2]·2H2O] | [Cu(3)](ClO4)2 | |
|---|---|---|---|
| Formula | C48H87Cu3N21O24S3 | C57H91Cu3N21O26S3 | C21H34Cl2CuN6O10S |
| M | 1629.21 | 1773.31 | 697.05 |
| Crystal system | orthorhombic | orthorhombic | monoclinic |
| Space group | Pbca (no. 61) | Pbca (no. 61) | P 21/c (no. 14) |
| a [Å] | 22.803(3) | 24.4069(11) | 13.4017(9) |
| b [Å] | 21.832(6) | 21.8999(10) | 26.1598(18) |
| c [Å] | 27.537(5) | 28.2847(13) | 8.2937(6) |
| β [°] | 90 | 90 | 101.2578(11) |
| V [ų] | 13709(5) | 15118.4(12) | 2851.7(3) |
| Z | 8 | 8 | 4 |
| ρcalcd [g cm−3] | 1.579 | 1.558 | 1.624 |
| µ MoKα [mm−1] | 1.105 | 1.011 | 1.090 |
| Scan type | ω scans | ω scans | ω scans |
| θ range [°] | 2.0–25.1 | 1.4–25.1 | 1.7–25.0 |
| Measured reflections | 12185 | 132295 | 26809 |
| Unique reflections | 12185 | 13361 | 5016 |
| Rint | - | 0.032 | 0.025 |
| Strong data [IO > 2σ(IO)] | 10116 | 10033 | 4364 |
| Refined parameters | 928 | 1027 | 384 |
| R1, wR2 (strong data) | 0.0598, 0.1177 | 0.0613, 0.1579 | 0.0324, 0.0859 |
| R1, wR2 (all data) | 0.0724, 0.1262 | 0.0839, 0.1765 | 0.0380, 0.0904 |
| GOF | 1.134 | 1.042 | 1.061 |
| Max/min residuals [eÅ−3] | 0.48/−0.37 | 1.76/−0.66 | 0.48/−0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciarrocchi, C.; Sacchi, D.; Boiocchi, M.; Weththimuni, M.L.; Orbelli Biroli, A.; Licchelli, M. Fluorogenic Detection of Sulfite in Water by Using Copper(II) Azacyclam Complexes. Molecules 2022, 27, 1852. https://doi.org/10.3390/molecules27061852
Ciarrocchi C, Sacchi D, Boiocchi M, Weththimuni ML, Orbelli Biroli A, Licchelli M. Fluorogenic Detection of Sulfite in Water by Using Copper(II) Azacyclam Complexes. Molecules. 2022; 27(6):1852. https://doi.org/10.3390/molecules27061852
Chicago/Turabian StyleCiarrocchi, Carlo, Donatella Sacchi, Massimo Boiocchi, Maduka Lankani Weththimuni, Alessio Orbelli Biroli, and Maurizio Licchelli. 2022. "Fluorogenic Detection of Sulfite in Water by Using Copper(II) Azacyclam Complexes" Molecules 27, no. 6: 1852. https://doi.org/10.3390/molecules27061852
APA StyleCiarrocchi, C., Sacchi, D., Boiocchi, M., Weththimuni, M. L., Orbelli Biroli, A., & Licchelli, M. (2022). Fluorogenic Detection of Sulfite in Water by Using Copper(II) Azacyclam Complexes. Molecules, 27(6), 1852. https://doi.org/10.3390/molecules27061852