
3,4-Unsubstituted 2-tert-Butyl-pyrrolidine-1-oxyls with Hydrophilic Functional Groups in the Side Chains

 , and
, and
Abstract
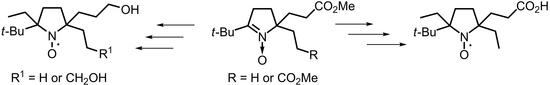
:
1. Introduction
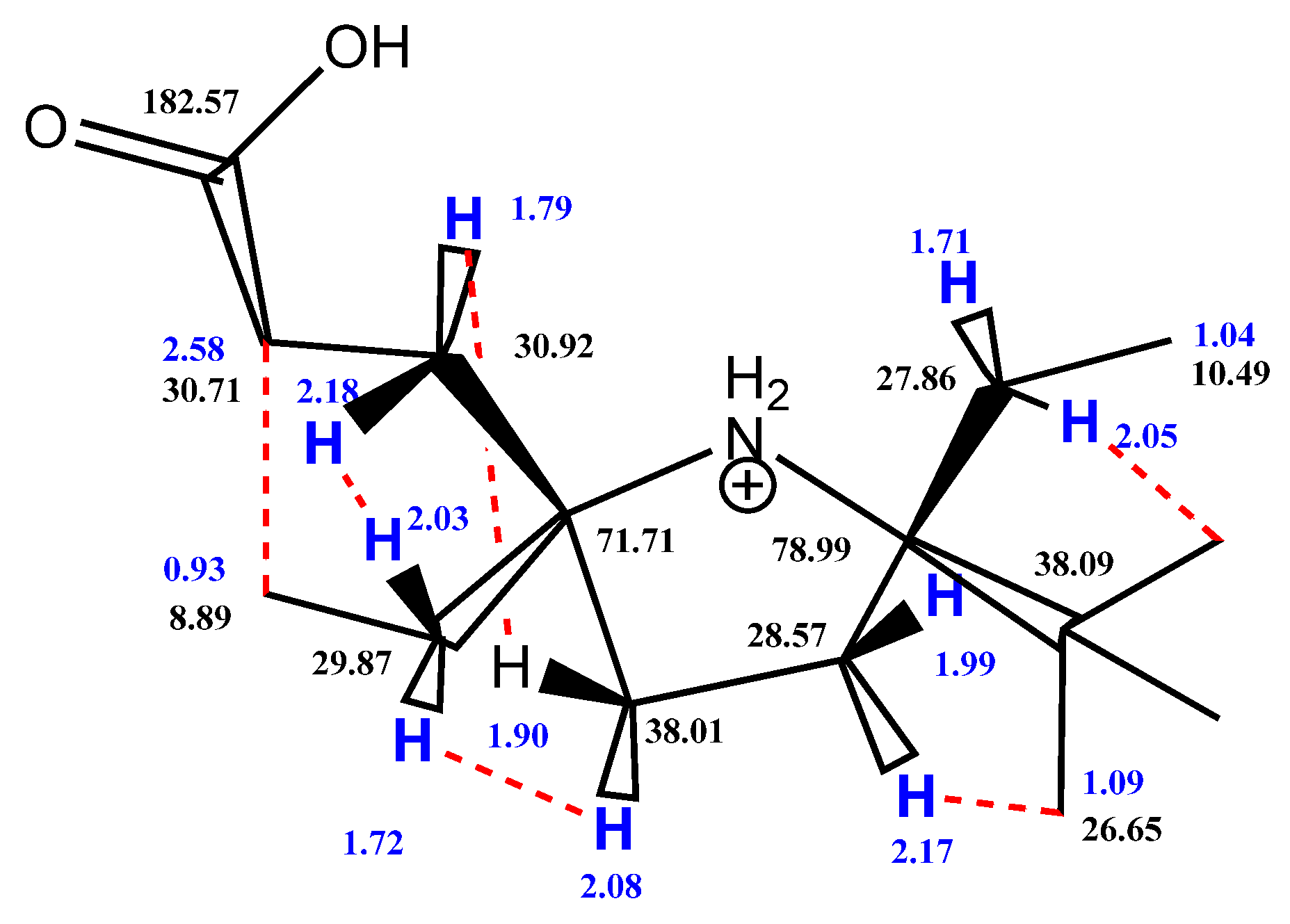
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.1.1. Conditions for Spectral Analysis
3.1.2. Conditions for Kinetics Experiments
3.1.3. Partition Coefficient Measurements
3.2. Synthesis
3.2.1. Synthesis of tert-Butyl Substituted Nitrones (General Method)
3.2.2. 3-(5-tert-Butyl-2-ethyl-1-oxido-3,4-dihydro-2H-pyrrol-2-yl)propanoic Acid (5a)
3.2.3. 3,3′-(5-tert-Butyl-1-oxido-3,4-dihydro-2H-pyrrole-2,2-diyl)dipropanoic Acid (5b)
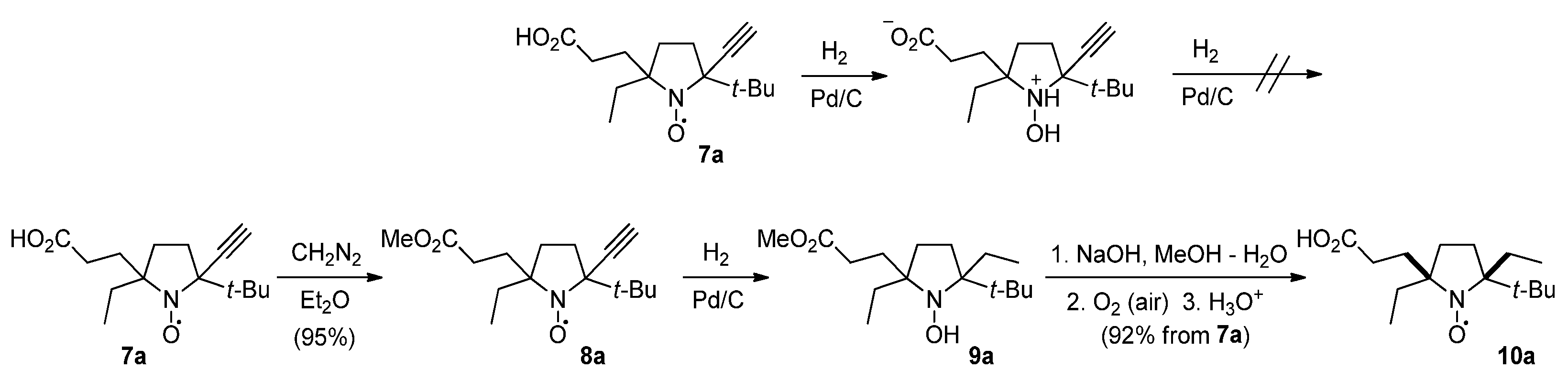
3.2.4. 2-tert-Butyl-5-(2-carboxyethyl)-5-ethyl-2-ethynylpyrrolidine-1-oxyl (7a,a′)
3.2.5. 2-tert-Butyl-5-ethyl-2-ethynyl-5-(3-methoxy-3-oxopropyl)pyrrolidine-1-oxyl (8a)
3.2.6. 2-tert-Butyl-5-(2-carboxyethyl)-2,5-diethylpyrrolidine-1-oxyl (10a)
3.2.7. Synthesis of tert-Butyl Substituted Nitrones, Containing 3-Hydroxypropyl Moiety (General Method)
3.2.8. Reaction of Nitrones 11a and 11b with 2,2-Dimethoxypropane (General Method)
3.2.9. Reaction of Nitrones 12a and 12b with Ethyllithium (General Method)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A


References
- Ouari, O.; Gigmes, D. Nitroxides: Synthesis, Properties and Applications; Royal Society of Chemistry: London, UK, 2021; ISBN 978-1-78801-752-7. [Google Scholar] [CrossRef]
- Kokorin, A.I. (Ed.) Nitroxides—Theory, Experiment and Applications; IntechOpen: London, UK, 2012; ISBN 978-9535107224. [Google Scholar]
- Likhtenshtein, G.I.; Yamauchi, J.; Nakatsuji, S.; Smirnov, A.; Tamura, R. Nitroxides. Applications in Chemistry, Biomedicine and Material Science; Wiley: New York, NY, USA, 2008; ISBN 978-3-527-31889-6. Available online: https://www.intechopen.com/books/2507 (accessed on 17 February 2022).
- Paletta, J.T.; Pink, M.; Foley, B.; Rajca, S.; Rajca, A. Synthesis and reduction kinetics of sterically shielded pyrrolidine nitroxides. Org. Lett. 2012, 14, 5322–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T. Sterically shielded spin labels for in-cell EPR spectroscopy: Analysis of stability in reducing environment. Free Radic. Res. 2015, 49, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Ovcherenko, S.S.; Chinak, O.A.; Chechushkov, A.V.; Dobrynin, S.A.; Kirilyuk, I.A.; Krumkacheva, O.A.; Richter, V.A.; Bagryanskaya, E.G. Uptake of cell-penetrating peptide rl2 by human lung cancer cells: Monitoring by electron paramagnetic resonance and confocal laser scanning microscopy. Molecules 2021, 26, 5442. [Google Scholar] [CrossRef] [PubMed]
- Ketter, S.; Dajka, M.; Rogozhnikova, O.; Dobrynin, S.A.; Tormyshev, V.M.; Bagryanskaya, E.G.; Joseph, B. In situ distance measurements in a membrane transporter using maleimide functionalized orthogonal spin labels and 5-pulse electron-electron double resonance spectroscopy. J. Magn. Reson. Open 2022, 10–11, 100041. [Google Scholar] [CrossRef]
- Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; et al. A Bioresistant Nitroxide Spin Label for In-Cell EPR Spectroscopy: In Vitro and In Oocytes Protein Structural Dynamics Studies. Angew. Chem. Int. Ed. 2018, 57, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Soikkeli, M.; Kettunen, M.I.; Nivajärvi, R.; Olsson, V.; Rönkkö, S.; Laakkonen, J.P.; Lehto, V.P.; Kavakka, J.; Heikkinen, S. Assessment of the Relaxation-Enhancing Properties of a Nitroxide-Based Contrast Agent TEEPO-Glc with in Vivo Magnetic Resonance Imaging. Contrast Media Mol. Imaging 2019, 2019, 5629597. [Google Scholar] [CrossRef] [PubMed]
- Emoto, M.; Mito, F.; Yamasaki, T.; Yamada, K.I.; Sato-Akaba, H.; Hirata, H.; Fujii, H. A novel ascorbic acid-resistant nitroxide in fat emulsion is an efficient brain imaging probe for in vivo EPR imaging of mouse. Free Radic. Res. 2011, 45, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Dobrynin, S.A.; Glazachev, Y.I.; Gatilov, Y.V.; Chernyak, E.I.; Salnikov, G.E.; Kirilyuk, I.A. Synthesis of 3,4-Bis(hydroxymethyl)-2,2,5,5-tetraethylpyrrolidin-1-oxyl via 1,3-Dipolar Cycloaddition of Azomethine Ylide to Activated Alkene. J. Org. Chem. 2018, 83, 5392–5397. [Google Scholar] [CrossRef] [PubMed]
- Lampp, L.; Morgenstern, U.; Merzweiler, K.; Imming, P.; Seidel, R.W. Synthesis and characterization of sterically and electrostatically shielded pyrrolidine nitroxide radicals. J. Mol. Struct. 2019, 1182, 87–94. [Google Scholar] [CrossRef]
- Dobrynin, S.A.; Usatov, M.S.; Zhurko, I.F.; Morozov, D.A.; Polienko, Y.F.; Glazachev, Y.I.; Parkhomenko, D.A.; Tyumentsev, M.A.; Gatilov, Y.V.; Chernyak, E.I.; et al. A Simple Method of Synthesis of 3-Carboxy-2,2,5,5-Tetraethylpyrrolidine-1-oxyl and Preparation of Reduction-Resistant Spin Labels and Probes of Pyrrolidine Series. Molecules 2021, 26, 5761. [Google Scholar] [CrossRef] [PubMed]
- Zhurko, I.F.; Dobrynin, S.; Gorodetskii, A.A.; Glazachev, Y.I.; Rybalova, T.V.; Chernyak, E.I.; Asanbaeva, N.; Bagryanskaya, E.G.; Kirilyuk, I.A. 2-Butyl-2-tert-butyl-5,5-diethylpyrrolidine-1-oxyls: Synthesis and properties. Molecules 2020, 25, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobko, A.A.; Kirilyuk, I.A.; Gritsan, N.P.; Polovyanenko, D.N.; Grigor’ev, I.A.; Khramtsov, V.V.; Bagryanskaya, E.G. EPR and Quantum Chemical Studies of the pH-sensitive Imidazoline and Imidazolidine Nitroxides with Bulky Substituents. Appl. Magn. Reson. 2010, 39, 437–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keana, J.F.W.; Pou, S.; Rosen, G.M. Synthesis and properties of some nitroxide α-carboxylate salts. J. Org. Chem. 1989, 54, 2417–2420. [Google Scholar] [CrossRef]
- Black, D.S.C.; Edwards, G.L.; Evans, R.H.; Keller, P.A.; Laaman, S.M. Synthesis and reactivity of 1-pyrroline-5-carboxylate ester 1-oxides. Tetrahedron 2000, 56, 1889–1897. [Google Scholar] [CrossRef]
- Earla, A.; Walter, E.D.; Braslau, R. Synthesis and spin trapping properties of polystyrene supported trifluoromethylated cyclic nitrones. Free Radic. Res. 2019, 53, 1084–1100. [Google Scholar] [CrossRef] [PubMed]
- Ballini, R.; Bosica, G. Conjugated addition reactions of nitroalkanes with electrophilic alkenes in aqueous media. Eur. J. Org. Chem. 1998, 1998, 355–357. [Google Scholar] [CrossRef]
- Pollini, G.; Barco, A.; De Guili, G. Tetramethylguanidine-catalyzed addition of nitromethane to α,β-unsaturated carboxylic acid esters. Synthesis 1972, 1972, 44–45. [Google Scholar] [CrossRef]
- Golod, E.L.; Bagal, L.I. Destructive nitration of polinitrocarbonylic compounds. I. New method of hexanitroethane synthesis. Russ. J. Org. Chem. 1994, 90, 29–32. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nitroxide | g-Factor | Hp-p, mT (Center) | aN, mT | Partition Coefficient | kred, M−1 s−1 |
|---|---|---|---|---|---|
| 7a,a′ | 2.00571 ± 2 | 0.17 ± 0.005 | 1.61 ± 0.005 | 150 | 0.2 ± 0.05 |
| 8a | 2.00571 ± 2 | 0.17 ± 0.005 | 1.61 ± 0.005 | 600 | 0.6 ± 0.1 |
| 10a | 2.00563 ± 2 | 0.28 ± 0.005 | 1.61 ± 0.005 | 240 | (4 ± 1) × 10−5 |
| 14a | 2.00560 ± 2 | 0.26 ± 0.005 | 1.61 ± 0.005 | 400 | (3 ± 1) × 10−5 |
| 14b | 2.00562 ± 2 | 0.26 ± 0.005 | 1.61 ± 0.005 | 70 | (5 ± 1) × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taratayko, A.I.; Glazachev, Y.I.; Eltsov, I.V.; Chernyak, E.I.; Kirilyuk, I.A. 3,4-Unsubstituted 2-tert-Butyl-pyrrolidine-1-oxyls with Hydrophilic Functional Groups in the Side Chains. Molecules 2022, 27, 1922. https://doi.org/10.3390/molecules27061922
Taratayko AI, Glazachev YI, Eltsov IV, Chernyak EI, Kirilyuk IA. 3,4-Unsubstituted 2-tert-Butyl-pyrrolidine-1-oxyls with Hydrophilic Functional Groups in the Side Chains. Molecules. 2022; 27(6):1922. https://doi.org/10.3390/molecules27061922
Chicago/Turabian StyleTaratayko, Andrey I., Yurii I. Glazachev, Ilia V. Eltsov, Elena I. Chernyak, and Igor A. Kirilyuk. 2022. "3,4-Unsubstituted 2-tert-Butyl-pyrrolidine-1-oxyls with Hydrophilic Functional Groups in the Side Chains" Molecules 27, no. 6: 1922. https://doi.org/10.3390/molecules27061922
APA StyleTaratayko, A. I., Glazachev, Y. I., Eltsov, I. V., Chernyak, E. I., & Kirilyuk, I. A. (2022). 3,4-Unsubstituted 2-tert-Butyl-pyrrolidine-1-oxyls with Hydrophilic Functional Groups in the Side Chains. Molecules, 27(6), 1922. https://doi.org/10.3390/molecules27061922