
Automated Microbial Library Generation Using the Bioinformatics Platform IDBac


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Collection and Processing of Environmental Samples
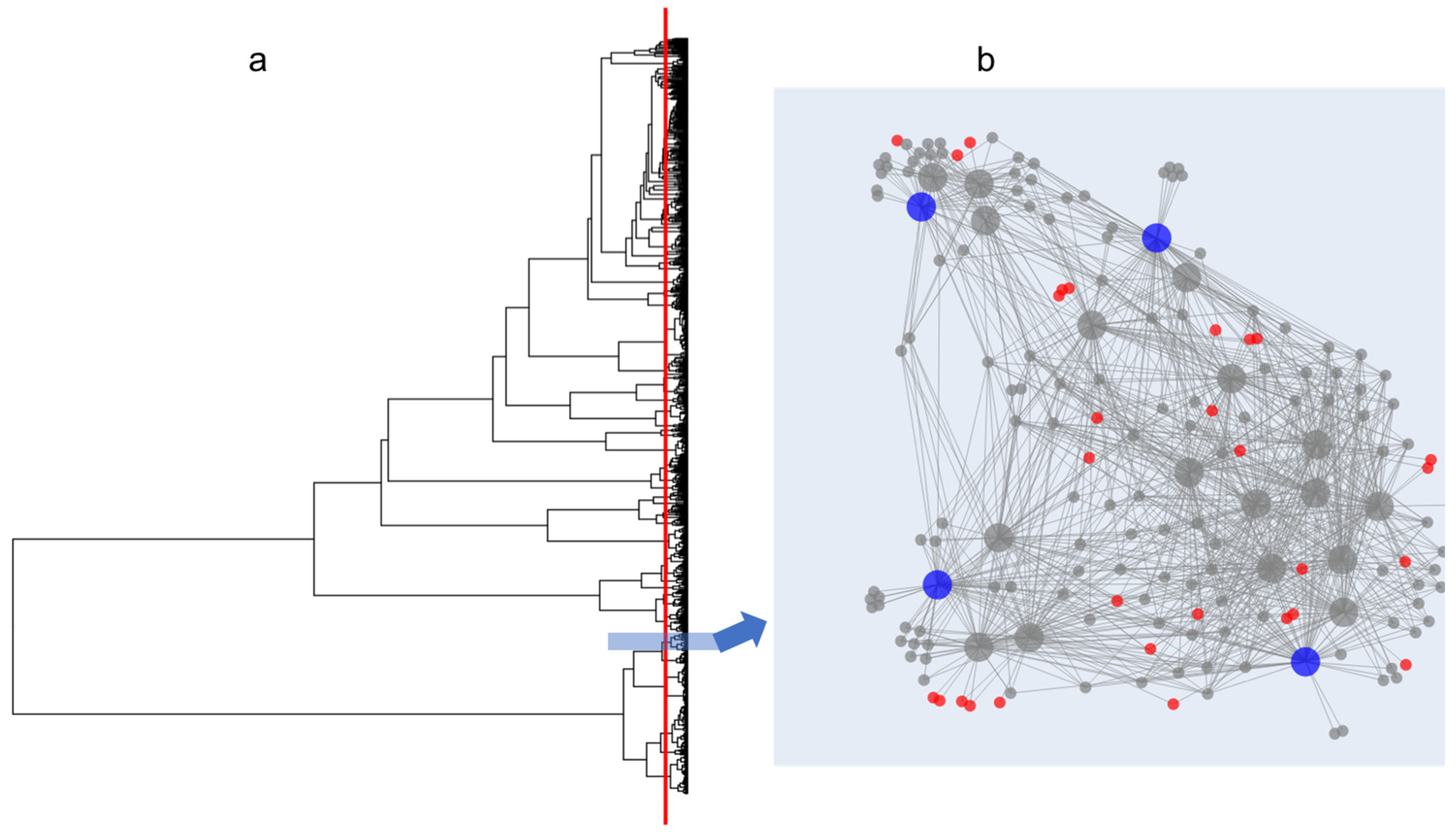
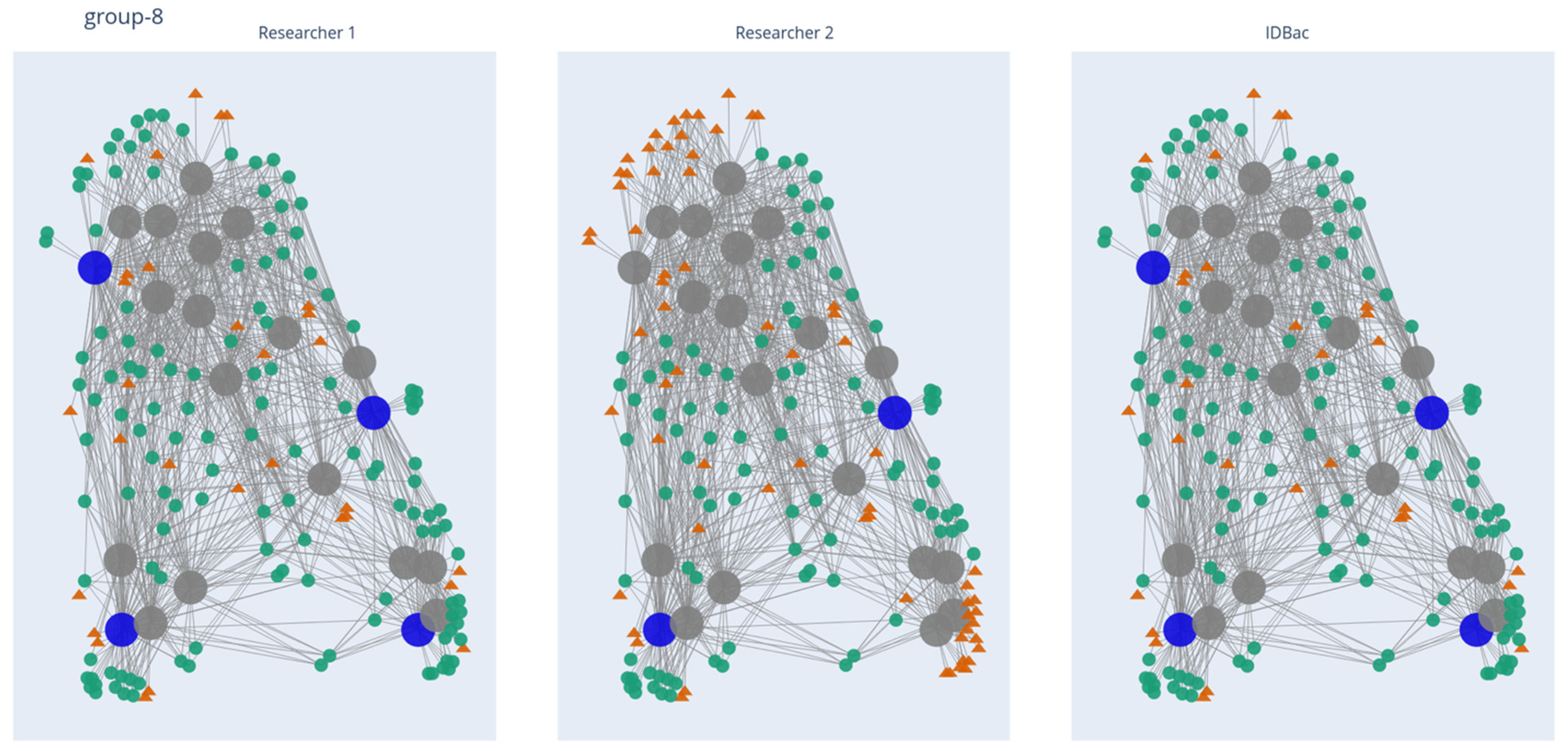
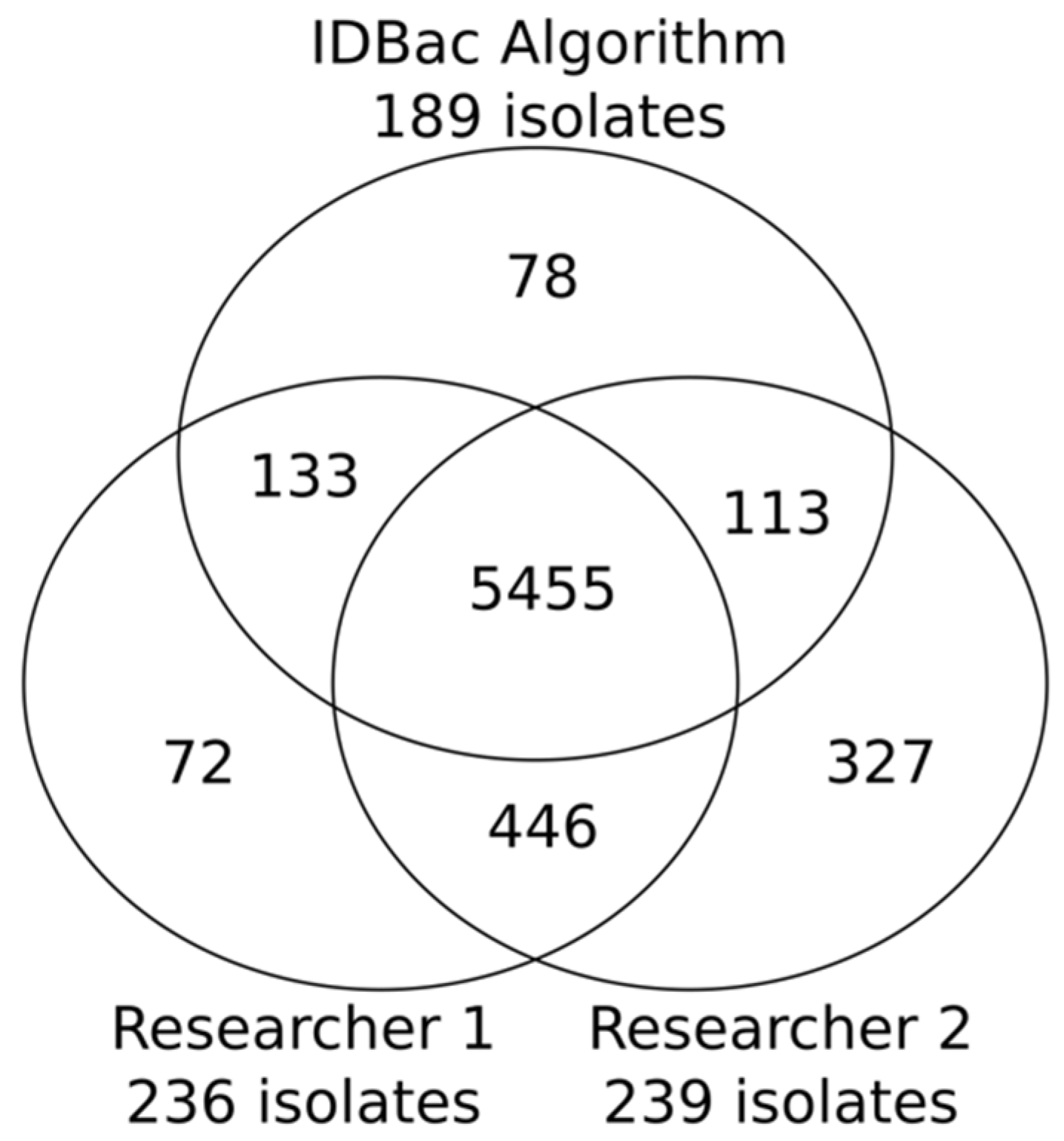
2.2. Application of the Isolate Prioritization Algorithm to a Collection of Unknown Environmental Bacterial Isolates
2.3. Study Limitations and Perspectives
3. Conclusions
4. Experimental Section
Sample Collection and Processing MALDI-TOF MS
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bouslimani, A.; Sanchez, L.M.; Garg, N.; Dorrestein, P.C. Mass spectrometry of natural products: Current, emerging and future technologies. Nat. Prod. Rep. 2014, 31, 718–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Braun, D.R.; Michel, C.R.; Klassen, J.L.; Adnani, N.; Wyche, T.P.; Bugni, T.S. Microbial strain prioritization using metabolomics tools for the discovery of natural products. Anal. Chem. 2012, 84, 4277–4283. [Google Scholar] [PubMed]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular networking as a dereplication strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinski, T.F. Microscale methodology for structure elucidation of natural products. Curr. Opin. Biotechnol. 2010, 21, 819–826. [Google Scholar] [PubMed] [Green Version]
- Bauman, K.D.; Butler, K.S.; Moore, B.S.; Chekan, J.R. Genome mining methods to discover bioactive natural products. Nat. Prod. Rep. 2021, 38, 2100–2129. [Google Scholar]
- Wilson, B.A.P.; Thornburg, C.C.; Henrich, C.J.; Grkovic, T.; O’Keefe, B.R. Creating and screening natural product libraries. Nat. Prod. Rep. 2020, 37, 893–918. [Google Scholar]
- Potts, M.B.; Kim, H.S.; Fisher, K.W.; Hu, Y.; Carrasco, Y.P.; Bulut, G.B.; Ou, Y.-H.; Herrera-Herrera, M.L.; Cubillos, F.; Mendiratta, S.; et al. Using functional signature ontology (FUSION) to identify mechanisms of action for natural products. Sci. Signal. 2013, 6, ra90. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, A.; Nguyen, L.T.; Dhakal, R.; Murphy, B.T. The need to innovate sample collection and library generation in microbial drug discovery: A focus on academia. Nat. Prod. Rep. 2021, 38, 292–300. [Google Scholar] [CrossRef]
- Liu, X.; Ashforth, E.; Ren, B.; Song, F.; Dai, H.; Liu, M.; Wang, J.; Xie, Q.; Zhang, L. Bioprospecting microbial natural product libraries from the marine environment for drug discovery. J. Antibiot. 2010, 63, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Knight, V.; Sanglier, J.-J.; DiTullio, D.; Braccili, S.; Bonner, P.; Waters, J.; Hughes, D.; Zhang, L. Diversifying microbial natural products for drug discovery. Appl. Microbiol. Biotechnol. 2003, 62, 446–458. [Google Scholar] [CrossRef]
- Okuda, T.; Ando, K.; Bills, G. Fungal germplasm for drug discovery and industrial applications. In Handbook of Industrial Mycology; An, Z., Ed.; CRC Press: Boca Raton, FL, USA, 2004; pp. 123–166. [Google Scholar] [CrossRef]
- Fujimori, F.; Okuda, T. Application of the random amplified polymorphic DNA using the polymerase chain reaction for efficient elimination of duplicate strains in microbial screening. J. Antibiot. 1994, 47, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziemert, N.; Lechner, A.; Wietz, M.; Millán-Aguiñaga, N.; Chavarria, K.L.; Jensen, P.R. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc. Natl. Acad. Sci. USA 2014, 111, E1130-9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevrette, M.G.; Carlos-Shanley, C.; Louie, K.B.; Bowen, B.P.; Northen, T.R.; Currie, C.R. Taxonomic and metabolic incongruence in the ancient genus Streptomyces. Front. Microbiol. 2019, 10, 2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltz, R.H. Marcel Faber Roundtable: Is our antibiotic pipeline unproductive because of starvation, constipation or lack of inspiration? J. Ind. Microbiol. Biotechnol. 2006, 33, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Pye, C.R.; Bertin, M.J.; Lokey, R.S.; Gerwick, W.H.; Linington, R.G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. USA 2017, 114, 5601–5606. [Google Scholar] [CrossRef] [Green Version]
- Clark, C.M.; Costa, M.S.; Sanchez, L.M.; Murphy, B.T. Coupling MALDI-TOF mass spectrometry protein and specialized metabolite analyses to rapidly discriminate bacterial function. Proc. Natl. Acad. Sci. USA 2018, 115, 4981–4986. [Google Scholar] [CrossRef] [Green Version]
- Clark, C.M.; Costa, M.S.; Conley, E.; Li, E.; Sanchez, L.M.; Murphy, B.T. Using the open-source MALDI TOF-MS IDBac pipeline for analysis of microbial protein and specialized metabolite data. J. Vis. Exp. 2019, 147, e59219. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.S.; Clark, C.M.; Ómarsdóttir, S.; Sanchez, L.M.; Murphy, B.T. Minimizing taxonomic and natural product redundancy in microbial libraries using MALDI-TOF MS and the bioinformatics pipeline IDBac. J. Nat. Prod. 2019, 82, 2167–2173. [Google Scholar] [CrossRef]
- Strejcek, M.; Smrhova, T.; Junkova, P.; Uhlik, O. Whole-cell MALDI-TOF MS versus 16S rRNA gene analysis for identification and dereplication of recurrent bacterial isolates. Front. Microbiol. 2018, 9, 1294. [Google Scholar] [CrossRef]
- Dumolin, C.; Aerts, M.; Verheyde, B.; Schellaert, S.; Vandamme, T.; Van der Jeugt, F.; De Canck, E.; Cnockaert, M.; Wieme, A.D.; Cleenwerck, I.; et al. Introducing SPeDE: High-throughput dereplication and accurate determination of microbial diversity from matrix-assisted laser desorption-ionization time of flight mass spectrometry data. mSystems 2019, 4, e00437-19. [Google Scholar] [CrossRef] [Green Version]
- Chudejova, K.; Bohac, M.; Skalova, A.; Rotova, V.; Papagiannitsis, C.C.; Hanzlickova, J.; Bergerova, T.; Hrabák, J. Validation of a novel automatic deposition of bacteria and yeasts on MALDI target for MALDI-TOF MS-based identification using MALDI Colonyst robot. PLoS ONE 2017, 12, e0190038. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, C.M.; Nguyen, L.; Pham, V.C.; Sanchez, L.M.; Murphy, B.T. Automated Microbial Library Generation Using the Bioinformatics Platform IDBac. Molecules 2022, 27, 2038. https://doi.org/10.3390/molecules27072038
Clark CM, Nguyen L, Pham VC, Sanchez LM, Murphy BT. Automated Microbial Library Generation Using the Bioinformatics Platform IDBac. Molecules. 2022; 27(7):2038. https://doi.org/10.3390/molecules27072038
Chicago/Turabian StyleClark, Chase M., Linh Nguyen, Van Cuong Pham, Laura M. Sanchez, and Brian T. Murphy. 2022. "Automated Microbial Library Generation Using the Bioinformatics Platform IDBac" Molecules 27, no. 7: 2038. https://doi.org/10.3390/molecules27072038
APA StyleClark, C. M., Nguyen, L., Pham, V. C., Sanchez, L. M., & Murphy, B. T. (2022). Automated Microbial Library Generation Using the Bioinformatics Platform IDBac. Molecules, 27(7), 2038. https://doi.org/10.3390/molecules27072038