
Molecular Simulations and Drug Discovery of Adenosine Receptors


Abstract
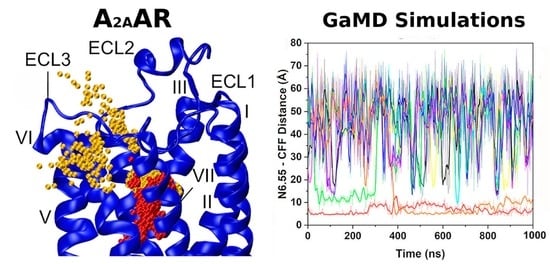
:
1. Introduction
2. Molecular Simulations Revealed Functional Mechanisms of Adenosine Receptors
2.1. Activation of Adenosine Receptors
2.2. Specific G Protein Coupling to Adenosine Receptors
2.3. Biased Agonism of Adenosine Receptors
2.4. Allosteric Modulation of Adenosine Receptors
2.5. Ligand Binding to Adenosine Receptors
2.6. Lipid Interactions with Adenosine Receptors
3. Computer-Aided Drug Discovery of Adenosine Receptors
3.1. Drug Binding Sites of Adenosine Receptors
3.2. Binding Free Energy Calculations of Adenosine Receptors
3.3. Design of Biased Agonists of Adenosine Receptors
3.4. Design of Allosteric Modulators of Adenosine Receptors
3.5. Design of Bitopic Ligands of Adenosine Receptors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADO | Adenosine |
| ARs | Adenosine receptors |
| GPCR | G protein-coupled receptor |
| AC | Adenylyl cyclase |
| PI3K | Phosphoinositide 3 kinase |
| MAPK | Mitogen-activated protein kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PD | Parkinson′s disease |
| NMR | Nuclear Magnetic Resonance |
| cryo-EM | Cryo-electron microscopy |
| MD | Molecular dynamics |
| TM6 | Transmembrane helix 6 |
| H8 | Helix 8 |
| ECL | Extracellular loop |
| NECA | 5′-N-carboxamidoadenosine |
| cMD | Conventional MD |
| MSM | Markov state model |
| GaMD | Gaussian accelerated molecular dynamics |
| MS1 | Metastable state |
| CS1 | Canonical state |
| SuMD | Supervised MD |
| PAM | Positive allosteric modulator |
| NAM | Negative allosteric modulator |
| XAC | Xanthine amine congener |
| HMA | 5-(N,N-hexamethylene) amiloride |
| CFF | Caffeine |
| POPC | 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine |
| POPE | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine |
| DOPG | 1,2-dioleoyl-sn-glycerol-3-phosphoglycerol |
| DOPC | 1,2-dioleoyl-sn-glycerol-3-phosphocholine |
| PIP2 | Phosphatidylinositol bisphosphate 2 |
| SILCS | Site-identification by ligand competitive saturation |
| MM/PBSA | Molecular mechanics Poisson–Boltzmann surface area |
| MM/GBSA | Molecular mechanics generalized Born surface area |
| FEP | Free energy perturbation |
| Average binding energy | |
| Minimum binding energy | |
| AUC | Area under the receiver operating characteristic curves |
| EFs | Enrichment factors |
References
- Jacobson, K.; Gao, Z.-G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmaso, V.; Jacobson, K.A. Purinergic Signaling: Impact of GPCR Structures on Rational Drug Design. ChemMedChem 2020, 15, 1958–1973. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9, eaag1166. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Lasley, R.D. Adenosine receptors and membrane microdomains. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 1284–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, R. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: Different roles, different sources and different receptors. Neurochem. Int. 2001, 38, 107–125. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Abbracchio, M.P.; Burnstock, G.; Daly, J.W.; Harden, T.K.; Jacobson, K.A.; Leff, P.; Williams, M. Nomenclature and classification of purinoceptors. Pharmacol. Rev. 1994, 46, 143–156. [Google Scholar]
- Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef]
- Paganelli, F.; Gaudry, M.; Ruf, J.; Guieu, R. Recent advances in the role of the adenosinergic system in coronary artery disease. Cardiovasc. Res. 2020, 117, 1284–1294. [Google Scholar] [CrossRef]
- Varani, K.; Vincenzi, F.; Merighi, S.; Gessi, S.; Borea, P.A. Biochemical and Pharmacological Role of A1 Adenosine Receptors and Their Modulation as Novel Therapeutic Strategy. In Protein Reviews: Volume 19; Atassi, M.Z., Ed.; Springer: Singapore, 2017; pp. 193–232. [Google Scholar]
- Chandrasekaran, B.; Samarneh, S.; Jaber, A.M.Y.; Kassab, G.; Agrawal, N. Therapeutic Potentials of A2B Adenosine Receptor Ligands: Current Status and Perspectives. Curr. Pharm. Des. 2019, 25, 2741–2771. [Google Scholar] [CrossRef]
- DeOliveira, C.C.; Caria, C.R.E.P.; Gotardo, E.M.F.; Ribeiro, M.; Gambero, A. Role of A1 and A2A adenosine receptor agonists in adipose tissue inflammation induced by obesity in mice. Eur. J. Pharmacol. 2017, 799, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Boison, D.; Yegutkin, G.G. Adenosine Metabolism: Emerging Concepts for Cancer Therapy. Cancer Cell 2019, 36, 582–596. [Google Scholar] [CrossRef]
- Gessi, S.; Merighi, S.; Fazzi, D.; Stefanelli, A.; Varani, K.; Borea, P.A. Adenosine receptor targeting in health and disease. Expert Opin. Investig. Drugs 2011, 20, 1591–1609. [Google Scholar] [CrossRef] [PubMed]
- De Lera Ruiz, M.; Lim, Y.-H.; Zheng, J. Adenosine A2A Receptor as a Drug Discovery Target. J. Med. Chem. 2014, 57, 3623–3650. [Google Scholar] [CrossRef]
- Nazario, L.R.; da Silva, R.S.; Bonan, C.D. Targeting Adenosine Signaling in Parkinson’s Disease: From Pharmacological to Non-pharmacological Approaches. Front. Neurosci. 2017, 11, 658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dungo, R.; Deeks, E.D. Istradefylline: First Global Approval. Drugs 2013, 73, 875–882. [Google Scholar] [CrossRef]
- Haskó, G.; Csóka, B.; Németh, Z.H.; Vizi, E.S.; Pacher, P. A2B adenosine receptors in immunity and inflammation. Trends Immunol. 2009, 30, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Kotańska, M.; Szafarz, M.; Mika, K.; Dziubina, A.; Bednarski, M.; Müller, C.E.; Sapa, J.; Kieć-Kononowicz, K. PSB 603—A known selective adenosine A2B receptor antagonist—Has anti-inflammatory activity in mice. Biomed. Pharmacother. 2020, 135, 111164. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic Signaling during Inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Barawkar, D.A.; Ramdas, V.; Patel, M.; Waman, Y.; Panmand, A.; Kumar, S.; Thorat, S.; Naykodi, M.; Goswami, A.; et al. Design and synthesis of novel xanthine derivatives as potent and selective A2B adenosine receptor antagonists for the treatment of chronic inflammatory airway diseases. Eur. J. Med. Chem. 2017, 134, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Tang, G.; Gao, P.; Zhang, B.; Xiao, H.; Si, L.-Y. Activation of adenosine A2B receptor attenuates high glucose-induced apoptosis in H9C2 cells via PI3K/Akt signaling. In Vitro Cell. Dev. Biol. Anim. 2018, 54, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Preti, D.; Borea, P.A.; Varani, K. Medicinal Chemistry of A3 Adenosine Receptor Modulators: Pharmacological Activities and Therapeutic Implications. J. Med. Chem. 2012, 55, 5676–5703. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Klutz, A.M.; Tosh, D.K.; Ivanov, A.A.; Preti, D.; Baraldi, P.G. Medicinal chemistry of the A3 adenosine receptor: Agonists, antagonists, and receptor engineering. In Adenosine Receptors in Health and Disease; Springer: Cham, Switzerland, 2009; pp. 123–159. [Google Scholar]
- Antonioli, L.; Lucarini, E.; Lambertucci, C.; Fornai, M.; Pellegrini, C.; Benvenuti, L.; Mannelli, L.D.C.; Spinaci, A.; Marucci, G.; Blandizzi, C.; et al. The Anti-Inflammatory and Pain-Relieving Effects of AR170, an Adenosine A3 Receptor Agonist, in a Rat Model of Colitis. Cells 2020, 9, 1509. [Google Scholar] [CrossRef]
- Park, C.-W.; Han, C.-T.; Sakaguchi, Y.; Lee, J.; Youn, H.-Y. Safety evaluation of FM101, an A3 adenosine receptor modulator, in rat, for developing as therapeutics of glaucoma and hepatitis. EXCLI J. 2020, 19, 187–200. [Google Scholar]
- Pal, Y.; Bandyopadhyay, N.; Pal, R.S.; Ahmed, S.; Bandopadhyay, S. Perspective and Potential of A2A and A3 Adenosine Receptors as Therapeutic Targets for the Treatment of Rheumatoid Arthritis. Curr. Pharm. Des. 2019, 25, 2859–2874. [Google Scholar] [CrossRef]
- Coppi, E.; Dettori, I.; Cherchi, F.; Bulli, I.; Venturini, M.; Pedata, F.; Pugliese, A.M. New Insight into the Role of Adenosine in Demyelination, Stroke and Neuropathic Pain. Front. Pharmacol. 2021, 11, 625662. [Google Scholar] [CrossRef]
- Wang, J.; Bhattarai, A.; Ahmad, W.I.; Farnan, T.S.; John, K.P.; Miao, Y. Computer-aided GPCR drug discovery. In GPCRs; Jastrzebska, B., Park, P.S.H., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 283–293. [Google Scholar]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of Three-Dimensional Structural Information of Biological Macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [Green Version]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Beauglehole, A.R.; Baker, S.P.; Scammells, P.J. Fluorosulfonyl-Substituted Xanthines as Selective Irreversible Antagonists for the A1-Adenosine Receptor. J. Med. Chem. 2000, 43, 4973–4980. [Google Scholar] [CrossRef] [PubMed]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the Adenosine A1 Receptor Reveals the Basis for Subtype Selectivity. Cell 2017, 168, 867–877.e13. [Google Scholar] [CrossRef] [Green Version]
- Cheng, R.K.; Segala, E.; Robertson, N.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Fiez-Vandal, C.; Marshall, F.H.; Cooke, R.M. Structures of Human A1 and A2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Structure 2017, 25, 1275–1285.e4. [Google Scholar] [CrossRef] [PubMed]
- Peet, N.P.; Lentz, N.L.; Meng, E.C.; Dudley, M.W.; Ogden, A.M.L.; Demeter, D.A.; Weintraub, H., Jr.; Bey, P. A novel synthesis of xanthines: Support for a new binding mode for xanthines with respect to adenosine at adenosine receptors. J. Med. Chem. 1990, 33, 3127–3130. [Google Scholar] [CrossRef]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.-L.; Nguyen, A.T.; Furness, S.G.; Venugopal, H.; Baltos, J.-A.; Plitzko, J.M.; Danev, R. Structure of the adenosine-bound human adenosine A1 receptor–G i complex. Nature 2018, 558, 559–563. [Google Scholar] [CrossRef]
- Draper-Joyce, C.J.; Bhola, R.; Wang, J.; Bhattarai, A.; Nguyen, A.T.N.; Cowie-Kent, I.; O’Sullivan, K.; Chia, L.Y.; Venugopal, H.; Valant, C.; et al. Positive allosteric mechanisms of adenosine A1 receptor-mediated analgesia. Nature 2021, 597, 571–576. [Google Scholar] [CrossRef]
- Minetti, P.; Tinti, M.O.; Carminati, P.; Castorina, M.; Di Cesare, M.A.; Di Serio, S.; Gallo, G.; Ghirardi, O.; Giorgi, F.; Giorgi, L.; et al. 2-n-Butyl-9-methyl-8-[1,2,3]triazol-2-yl-9H-purin-6-ylamine and Analogues as A2A Adenosine Receptor Antagonists. Design, Synthesis, and Pharmacological Characterization. J. Med. Chem. 2005, 48, 6887–6896. [Google Scholar] [CrossRef]
- Martynowycz, M.W.; Shiriaeva, A.; Ge, X.; Hattne, J.; Nannenga, B.L.; Cherezov, V.; Gonen, T. MicroED structure of the human adenosine receptor determined from a single nanocrystal in LCP. Proc. Natl. Acad. Sci. USA 2021, 118, e2106041118. [Google Scholar] [CrossRef] [PubMed]
- Amelia, T.; van Veldhoven, J.P.D.; Falsini, M.; Liu, R.; Heitman, L.H.; van Westen, G.J.P.; Segala, E.; Verdon, G.; Cheng, R.K.Y.; Cooke, R.M.; et al. Crystal Structure and Subsequent Ligand Design of a Nonriboside Partial Agonist Bound to the Adenosine A2A Receptor. J. Med. Chem. 2021, 64, 3827–3842. [Google Scholar] [CrossRef] [PubMed]
- Ihara, K.; Hato, M.; Nakane, T.; Yamashita, K.; Kimura-Someya, T.; Hosaka, T.; Ishizuka-Katsura, Y.; Tanaka, R.; Tanaka, T.; Sugahara, M.; et al. Isoprenoid-chained lipid EROCOC17+4: A new matrix for membrane protein crystallization and a crystal delivery medium in serial femtosecond crystallography. Sci. Rep. 2020, 10, 19305. [Google Scholar] [CrossRef]
- Lee, M.-Y.; Geiger, J.; Ishchenko, A.; Han, G.W.; Barty, A.; White, T.; Gati, C.; Batyuk, A.; Hunter, M.S.; Aquila, A.; et al. Harnessing the power of an X-ray laser for serial crystallography of membrane proteins crystallized in lipidic cubic phase. IUCrJ 2020, 7, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Jespers, W.; Verdon, G.; Azuaje, J.; Majellaro, M.; Keränen, H.; García-Mera, X.; Congreve, M.; Deflorian, F.; De Graaf, C.; Zhukov, A.; et al. X-ray Crystallography and Free Energy Calculations Reveal the Binding Mechanism of A2A Adenosine Receptor Antagonists. Angew. Chem. Int. Ed. 2020, 59, 16536–16543. [Google Scholar] [CrossRef]
- Nass, K.; Cheng, R.; Vera, L.; Mozzanica, A.; Redford, S.; Ozerov, D.; Basu, S.; James, D.; Knopp, G.; Cirelli, C.; et al. Advances in long-wavelength native phasing at X-ray free-electron lasers. IUCrJ 2020, 7, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Ishchenko, A.; Stauch, B.; Han, G.W.; Batyuk, A.; Shiriaeva, A.; Li, C.; Zatsepin, N.; Weierstall, U.; Liu, W.; Nango, E.; et al. Toward G protein-coupled receptor structure-based drug design using X-ray lasers. IUCrJ 2019, 6, 1106–1119. [Google Scholar] [CrossRef]
- Shimazu, Y.; Tono, K.; Tanaka, T.; Yamanaka, Y.; Nakane, T.; Mori, C.; Kimura, K.T.; Fujiwara, T.; Sugahara, M.; Tanaka, R.; et al. High-viscosity sample-injection device for serial femtosecond crystallography at atmospheric pressure. J. Appl. Crystallogr. 2019, 52, 1280–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borodovsky, A.; Barbon, C.M.; Wang, Y.; Ye, M.; Prickett, L.; Chandra, D.; Shaw, J.; Deng, N.; Sachsenmeier, K.; Clarke, J.D.; et al. Small molecule AZD4635 inhibitor of A2AR signaling rescues immune cell function including CD103+ dendritic cells enhancing anti-tumor immunity. J. Immunother. Cancer 2020, 8, e000417. [Google Scholar] [CrossRef] [PubMed]
- Martin-Garcia, J.M.; Zhu, L.; Mendez, D.; Lee, M.-Y.; Chun, E.; Li, C.; Hu, H.; Subramanian, G.; Kissick, D.; Ogata, C.; et al. High-viscosity injector-based pink-beam serial crystallography of microcrystals at a synchrotron radiation source. IUCrJ 2019, 6, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Volpini, R.; Costanzi, S.; Lambertucci, C.; Portino, F.R.; Taffi, S.; Vittori, S.; Klotz, K.-N.; Cristalli, G. Adenosine receptor agonists: Synthesis and binding affinity of 2- (aryl)alkylthioadenosine derivatives. Arkivoc 2004, 2004, 301–311. [Google Scholar] [CrossRef]
- Nafria, J.G.; Lee, Y.; Bai, X.; Carpenter, B.; Tate, C.G. Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. eLife 2018, 7, e35946. [Google Scholar] [CrossRef]
- White, K.L.; Eddy, M.T.; Gao, Z.-G.; Han, G.W.; Lian, T.; Deary, A.; Patel, N.; Jacobson, K.A.; Katritch, V.; Stevens, R.C. Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 2018, 26, 259–269.e5. [Google Scholar] [CrossRef] [Green Version]
- Rucktooa, P.; Cheng, R.K.Y.; Segala, E.; Geng, T.; Errey, J.C.; Brown, G.A.; Cooke, R.M.; Marshall, F.H.; Doré, A.S. Towards high throughput GPCR crystallography: In Meso soaking of Adenosine A2A Receptor crystals. Sci. Rep. 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Barawkar, D.A.; Thorat, S.; Shejul, Y.D.; Patel, M.; Naykodi, M.; Jain, V.; Salve, Y.; Prasad, V.; Chaudhary, S.; et al. Design, Synthesis of Novel, Potent, Selective, Orally Bioavailable Adenosine A2A Receptor Antagonists and Their Biological Evaluation. J. Med. Chem. 2017, 60, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Andrews, S.P.; Doré, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-Triazine Derivatives as Adenosine A2A Antagonists using Structure Based Drug Design. J. Med. Chem. 2012, 55, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Sams, A.G.; Mikkelsen, G.K.; Larsen, M.; Langgård, M.; Howells, M.E.; Schrøder, T.J.; Brennum, L.T.; Torup, L.; Jørgensen, E.B.; Bundgaard, C.; et al. Discovery of Phosphoric Acid Mono-{2-[(E/Z)-4-(3,3-dimethyl-butyrylamino)-3,5-difluoro-benzoylimino]-thiazol-3-ylmethyl} Ester (Lu AA47070): A Phos-phonooxymethylene Prodrug of a Potent and Selective hA2A Receptor Antagonist. J. Med. Chem. 2011, 54, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Eddy, M.T.; Lee, M.-Y.; Gao, Z.-G.; White, K.L.; Didenko, T.; Horst, R.; Audet, M.; Stanczak, P.; McClary, K.M.; Han, G.W.; et al. Allosteric Coupling of Drug Binding and Intracellular Signaling in the A2A Adenosine Receptor. Cell 2018, 172, 68–80.e12. [Google Scholar] [CrossRef] [Green Version]
- Broecker, J.; Morizumi, T.; Ou, W.-L.; Klingel, V.; Kuo, A.; Kissick, D.J.; Ishchenko, A.; Lee, M.-Y.; Xu, S.; Makarov, O.; et al. High-throughput in situ X-ray screening of and data collection from protein crystals at room temperature and under cryogenic conditions. Nat. Protoc. 2018, 13, 260–292. [Google Scholar] [CrossRef]
- Weinert, T.; Olieric, N.; Cheng, R.; Brünle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef]
- Zhukov, A.; Andrews, S.P.; Errey, J.C.; Robertson, N.; Tehan, B.; Mason, J.S.; Marshall, F.H.; Weir, M.; Congreve, M. Biophysical Mapping of the Adenosine A2A Receptor. J. Med. Chem. 2011, 54, 4312–4323. [Google Scholar] [CrossRef]
- Melnikov, I.; Polovinkin, V.; Kovalev, K.; Gushchin, I.; Shevtsov, M.; Shevchenko, V.; Mishin, A.; Alekseev, A.; Rodriguez-Valera, F.; Borshchevskiy, V.; et al. Fast iodide-SAD phasing for high-throughput membrane protein structure determination. Sci. Adv. 2017, 3, e1602952. [Google Scholar] [CrossRef] [Green Version]
- Martin-Garcia, J.M.; Conrad, C.E.; Nelson, G.; Stander, N.; Zatsepin, N.A.; Zook, J.; Zhu, L.; Geiger, J.; Chun, E.; Kissick, D.; et al. Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 2017, 4, 439–454. [Google Scholar] [CrossRef]
- Sun, B.; Bachhawat, P.; Chu, M.L.-H.; Wood, M.; Ceska, T.; Sands, Z.A.; Mercier, J.; Lebon, F.; Kobilka, T.S.; Kobilka, B.K. Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc. Natl. Acad. Sci. USA 2017, 114, 2066–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batyuk, A.; Galli, L.; Ishchenko, A.; Han, G.W.; Gati, C.; Popov, P.A.; Lee, M.-Y.; Stauch, B.; White, T.A.; Barty, A.; et al. Native phasing of x-ray free-electron laser data for a G protein–coupled receptor. Sci. Adv. 2016, 2, e1600292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeling, S.E.; Albinson, F.; Ayres, B.E.; Butchers, P.R.; Chambers, C.; Cherry, P.C.; Ellis, F.; Ewan, G.B.; Gregson, M.; Knight, J.; et al. The discovery and synthesis of highly potent, A2A receptor agonists. Bioorg. Med. Chem. Lett. 2000, 10, 403–406. [Google Scholar] [CrossRef]
- Carpenter, B.; Nehme, R.; Warne, T.; Leslie, A.G.; Tate, C.G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef]
- Segala, E.; Guo, D.; Cheng, R.K.Y.; Bortolato, A.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Heitman, L.H.; Ijzerman, A.P.; Marshall, F.H.; et al. Controlling the Dissociation of Ligands from the Adenosine A2A Receptor through Modulation of Salt Bridge Strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef]
- Matsuda, A.; Shinozaki, M.; Yamaguchi, T.; Homma, H.; Nomoto, R.; Miyasaka, T.; Watanabe, Y.; Abiru, T. Nucleosides and nucleotides. 103. 2-Alkynyladenosines: A novel class of selective adenosine A2 receptor agonists with potent antihypertensive effects. J. Med. Chem. 1992, 35, 241–252. [Google Scholar] [CrossRef]
- Lebon, G.; Edwards, P.C.; Leslie, A.G.W.; Tate, C.G. Molecular Determinants of CGS21680 Binding to the Human Adenosine A2A Receptor. Mol. Pharmacol. 2015, 87, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; Ijzerman, A.P.; et al. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Hino, T.; Arakawa, T.; Iwanari, H.; Yurugi-Kobayashi, T.; Ikeda-Suno, C.; Nakada-Nakura, Y.; Kusano-Arai, O.; Weyand, S.; Shimamura, T.; Nomura, N.; et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 2012, 482, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the Adenosine A2A Receptor in Complex with ZM241385 and the Xanthines XAC and Caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.W.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.-G.; Cherezov, V.; Stevens, R.C. Structure of an Agonist-Bound Human A2A Adenosine Receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaakola, V.-P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.T.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [Green Version]
- Park, P.S.-H.; Lodowski, D.T.; Palczewski, K. Activation of G Protein–Coupled Receptors: Beyond Two-State Models and Tertiary Conformational Changes. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 107–141. [Google Scholar] [CrossRef] [Green Version]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of Molecular Switches in GPCRs—Theoretical and Experimental Studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Eddy, M.T.; Gao, Z.-G.; Mannes, P.; Patel, N.; Jacobson, K.A.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Extrinsic Tryptophans as NMR Probes of Allosteric Coupling in Membrane Proteins: Application to the A2A Adenosine Receptor. J. Am. Chem. Soc. 2018, 140, 8228–8235. [Google Scholar] [CrossRef]
- Sušac, L.; Eddy, M.T.; Didenko, T.; Stevens, R.C.; Wüthrich, K. A2A adenosine receptor functional states characterized by 19F-NMR. Proc. Natl. Acad. Sci. USA 2018, 115, 12733–12738. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, D.; Piñeiro, A.; Gutiérrez-De-Terán, H. Molecular Dynamics Simulations Reveal Insights into Key Structural Elements of Adenosine Receptors. Biochemistry 2011, 50, 4194–4208. [Google Scholar] [CrossRef]
- Caliman, A.D.; Swift, S.E.; Wang, Y.; Miao, Y.; McCammon, J.A. Investigation of the conformational dynamics of the apo A2A adenosine receptor. Protein Sci. 2015, 24, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Filipek, S.; Palczewski, K.; Vogel, H. Activation of G-protein-coupled receptors correlates with the formation of a continuous internal water pathway. Nat. Commun. 2014, 5, 4733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Hu, Z.; Filipek, S.; Vogel, H. W246(6.48) opens a gate for a continuous intrinsic water pathway during activation of the adenosine A2A receptor. Angew. Chem. Int. Ed. 2015, 54, 556–559. [Google Scholar]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.M.; Filizola, M. Showcasing modern molecular dynamics simulations of membrane proteins through G protein-coupled receptors. Curr. Opin. Struct. Biol. 2011, 21, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilardaga, J.-P.; Bünemann, M.; Krasel, C.; Castro, M.; Lohse, M. Measurement of the millisecond activation switch of G protein–coupled receptors in living cells. Nat. Biotechnol. 2003, 21, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Arantes, P.R.; Bhattarai, A.; Hsu, R.V.; Pawnikar, S.; Huang, Y.M.; Palermo, G.; Miao, Y. Gaussian accelerated molecular dynamics (GaMD): Principles and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1521. [Google Scholar] [CrossRef]
- Spiwok, V.; Sucur, Z.; Hosek, P. Enhanced sampling techniques in biomolecular simulations. Biotechnol. Adv. 2015, 33, 1130–1140. [Google Scholar] [CrossRef]
- Lovera, S.; Cuzzolin, A.; Kelm, S.; De Fabritiis, G.; Sands, Z.A. Reconstruction of apo A2A receptor activation pathways reveal ligand-competent intermediates and state-dependent cholesterol hotspots. Sci. Rep. 2019, 9, 14199. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jonsson, A.L.; Beuming, T.; Shelley, J.C.; Voth, G.A. Ligand-Dependent Activation and Deactivation of the Human Adenosine A2A Receptor. J. Am. Chem. Soc. 2013, 135, 8749–8759. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.-G.; Inoue, A.; Jacobson, K.A. On the G protein-coupling selectivity of the native A2B adenosine receptor. Biochem. Pharmacol. 2017, 151, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Cordeaux, Y.; Ijzerman, A.P.; Hill, S.J. Coupling of the human A1adenosine receptor to different heterotrimeric G proteins: Evidence for agonist-specific G protein activation. J. Cereb. Blood Flow Metab. 2004, 143, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeaux, Y.; Briddon, S.; Megson, A.E.; McDonnell, J.; Dickenson, J.; Hill, S. Influence of Receptor Number on Functional Responses Elicited by Agonists Acting at the Human Adenosine A1 Receptor: Evidence for Signaling Pathway-Dependent Changes in Agonist Potency and Relative Intrinsic Activity. Mol. Pharmacol. 2000, 58, 1075–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, G.D.; Valant, C.; Dowell, S.J.; Mijaljica, D.; Devenish, R.J.; Scammells, P.J.; Sexton, P.M.; Christopoulos, A. Determination of Adenosine A1 Receptor Agonist and Antagonist Pharmacology Using Saccharomyces cerevisiae: Implications for Ligand Screening and Functional Selectivity. J. Pharmacol. Exp. Ther. 2009, 331, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Nivedha, A.K.; Tate, C.G.; Vaidehi, N. Dynamic Role of the G Protein in Stabilizing the Active State of the Adenosine A2A Receptor. Structure 2019, 27, 703–712.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Miao, Y. Mechanistic Insights into Specific G Protein Interactions with Adenosine Receptors. J. Phys. Chem. B 2019, 123, 6462–6473. [Google Scholar] [CrossRef] [PubMed]
- Baltos, J.-A.; Gregory, K.J.; White, P.J.; Sexton, P.; Christopoulos, A.; May, L.T. Quantification of adenosine A1 receptor biased agonism: Implications for drug discovery. Biochem. Pharmacol. 2015, 99, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, E.A.; Baltos, J.; Nguyen, A.T.N.; Christopoulos, A.; White, P.J.; May, L.T. New paradigms in adenosine receptor pharmacology: Allostery, oligomerization and biased agonism. J. Cereb. Blood Flow Metab. 2018, 175, 4036–4046. [Google Scholar] [CrossRef] [Green Version]
- McNeill, S.M.; Baltos, J.-A.; White, P.J.; May, L.T. Biased agonism at adenosine receptors. Cell. Signal. 2021, 82, 109954. [Google Scholar] [CrossRef]
- Jarpe, M.B.; Knall, C.; Mitchell, F.M.; Buhl, A.M.; Duzic, E.; Johnson, G.L. [d-Arg1,d-Phe5,d-Trp7,9,Leu11]Substance P Acts as a Biased Agonist toward Neuropeptide and Chemokine Receptors. J. Biol. Chem. 1998, 273, 3097–3104. [Google Scholar] [CrossRef] [Green Version]
- Nagi, K.; Onaran, H.O. Biased agonism at G protein-coupled receptors. Cell. Signal. 2021, 83, 109981. [Google Scholar] [CrossRef]
- Langemeijer, E.V.; Verzijl, D.; Dekker, S.J.; IJzerman, A.P. Functional selectivity of adenosine A1 receptor ligands? Purinergic Signal. 2013, 9, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, M.J.; Hill, E.; Huckstepp, R.; Barkan, K.; Deganutti, G.; Leuenberger, M.; Preti, B.; Winfield, I.; Wei, H.; Imlach, W.; et al. A biased adenosine A1R agonist elicits analgesia without cardiorespiratory depression. bioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.04.04.023945v3.full (accessed on 31 January 2022). [CrossRef] [Green Version]
- Kato, H.E.; Zhang, Y.; Hu, H.; Suomivuori, C.-M.; Kadji, F.M.N.; Aoki, J.; Kumar, K.K.; Fonseca, R.; Hilger, D.; Huang, W. Conformational transitions of a neurotensin receptor 1–Gi1 complex. Nature 2019, 572, 80–85. [Google Scholar] [CrossRef]
- Liu, X.; Xu, X.; Hilger, D.; Aschauer, P.; Tiemann, J.; Du, Y.; Liu, H.; Hirata, K.; Sun, X.; Guixà-González, R.; et al. Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell 2019, 177, 1243–1251.e12. [Google Scholar] [CrossRef] [PubMed]
- Valant, C.; May, L.T.; Aurelio, L.; Chuo, C.H.; White, P.J.; Baltos, J.-A.; Sexton, P.M.; Scammells, P.J.; Christopoulos, A. Separation of on-target efficacy from adverse effects through rational design of a bitopic adenosine receptor agonist. Proc. Natl. Acad. Sci. USA 2014, 111, 4614–4619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deganutti, G.; Barkan, K.; Ladds, G.; Reynolds, C.A. Multisite Model of Allosterism for the Adenosine A1 Receptor. J. Chem. Inf. Model. 2021, 61, 2001–2015. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Toti, K.S.; Campbell, R.; Suresh, R.R.; Yang, H.; Jacobson, K.A. Allosteric Antagonism of the A2A Adenosine Receptor by a Series of Bitopic Ligands. Cells 2020, 9, 1200. [Google Scholar] [CrossRef]
- Miao, Y.; Bhattarai, A.; Nguyen, A.T.N.; Christopoulos, A.; May, L.T. Structural Basis for Binding of Allosteric Drug Leads in the Adenosine A1 Receptor. Sci. Rep. 2018, 8, 16836. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T., III; Cisneros, G.; Cruzeiro, V.; Darden, T. Amber 2021; University of California Press: Berkeley, CA, USA, 2021. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.N.; Vecchio, E.A.; Thomas, T.; Nguyen, T.D.; Aurelio, L.; Scammells, P.J.; White, P.J.; Sexton, P.M.; Gregory, K.J.; May, L.T.; et al. Role of the Second Extracellular Loop of the Adenosine A1 Receptor on Allosteric Modulator Binding, Signaling, and Cooperativity. Mol. Pharmacol. 2016, 90, 715–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deganutti, G.; Cuzzolin, A.; Ciancetta, A.; Moro, S. Understanding allosteric interactions in G protein-coupled receptors using Supervised Molecular Dynamics: A prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000. Bioorg. Med. Chem. 2015, 23, 4065–4071. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, M.; Bolcato, G.; Deganutti, G.; Sturlese, M.; Moro, S. Revisiting the Allosteric Regulation of Sodium Cation on the Binding of Adenosine at the Human A2A Adenosine Receptor: Insights from Supervised Molecular Dynamics (SuMD) Simulations. Molecules 2019, 24, 2752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Pan, A.C.; Dror, R.O.; Mocking, T.; Liu, R.; Heitman, L.H.; Shaw, D.E.; Ijzerman, A.P. Molecular Basis of Ligand Dissociation from the Adenosine A2A Receptor. Mol. Pharmacol. 2016, 89, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Deganutti, G.; Welihinda, A.; Moro, S. Comparison of the Human A2A Adenosine Receptor Recognition by Adenosine and Inosine: New Insight from Supervised Molecular Dynamics Simulations. ChemMedChem 2017, 12, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- Sabbadin, D.; Ciancetta, A.; Deganutti, G.; Cuzzolin, A.; Moro, S. Exploring the recognition pathway at the human A2A adenosine receptor of the endogenous agonist adenosine using supervised molecular dynamics simulations. MedChemComm 2015, 6, 1081–1085. [Google Scholar] [CrossRef]
- Bolcato, G.; Bissaro, M.; Deganutti, G.; Sturlese, M.; Moro, S. New Insights into Key Determinants for Adenosine 1 Receptor Antagonists Selectivity Using Supervised Molecular Dynamics Simulations. Biomolecules 2020, 10, 732. [Google Scholar] [CrossRef]
- Deganutti, G.; Barkan, K.; Preti, B.; Leuenberger, M.; Wall, M.; Frenguelli, B.G.; Lochner, M.; Ladds, G.; Reynolds, C.A. Deciphering the Agonist Binding Mechanism to the Adenosine A1 Receptor. ACS Pharmacol. Transl. Sci. 2021, 4, 314–326. [Google Scholar] [CrossRef]
- Do, H.N.; Akhter, S.; Miao, Y. Pathways and Mechanism of Caffeine Binding to Human Adenosine A2A Receptor. Front. Mol. Biosci. 2021, 8, 673170. [Google Scholar] [CrossRef]
- Khelashvili, G.; Grossfield, A.; Feller, S.E.; Pitman, M.C.; Weinstein, H. Structural and dynamic effects of cholesterol at preferred sites of interaction with rhodopsin identified from microsecond length molecular dynamics simulations. Proteins 2009, 76, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Khelashvili, G.; Albornoz, P.B.C.; Johner, N.; Mondal, S.; Caffrey, M.; Weinstein, H. Why GPCRs behave differently in cubic and lamellar lipidic mesophases. J. Am. Chem. Soc. 2012, 134, 15858–15868. [Google Scholar] [CrossRef]
- Mondal, S.; Johnston, J.M.; Wang, H.; Khelashvili, G.; Filizola, M.; Weinstein, H. Membrane Driven Spatial Organization of GPCRs. Sci. Rep. 2013, 3, srep02909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chachisvilis, M.; Zhang, Y.-L.; Frangos, J.A. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15463–15468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattarai, A.; Wang, J.; Miao, Y. G-Protein-Coupled Receptor-Membrane Interactions Depend on the Receptor Activation State. J. Comput. Chem. 2020, 41, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Perly, B.; Smith, I.C.P.; Jarrell, H.C. Acyl chain dynamics of phosphatidylethanolamines containing oleic acid and dihydrosterculic acid: Deuteron NMR relaxation studies. Biochemistry 1985, 24, 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Rossetti, G.; Bauer, A.; Carioni, P. Binding of the Antagonist Caffeine to the Human Adenosine Receptor hA2AR in Nearly Physiological Conditions. PLoS ONE 2015, 10, e0126833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruzzese, A.; Dalton, J.A.R.; Giraldo, J. Insights into adenosine A2A receptor activation through cooperative modulation of agonist and allosteric lipid interactions. PLoS Comput. Biol. 2020, 16, e1007818. [Google Scholar] [CrossRef]
- Leonard, A.N.; Lyman, E. Activation of G-protein-coupled receptors is thermodynamically linked to lipid solvation. Biophys. J. 2021, 120, 1777–1787. [Google Scholar] [CrossRef]
- Ma, N.; Lee, S.; Vaidehi, N. Activation Microswitches in Adenosine Receptor A2A Function as Rheostats in the Cell Membrane. Biochemistry 2020, 59, 4059–4071. [Google Scholar] [CrossRef]
- Song, W.; Yen, H.-Y.; Robinson, C.V.; Sansom, M.S. State-dependent Lipid Interactions with the A2A Receptor Revealed by MD Simulations Using In Vivo-Mimetic Membranes. Structure 2018, 27, 392–403.e3. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.K.; Almurad, O.; Pejana, R.J.; Morrison, Z.A.; Pandey, A.; Picard, L.-P.; Nitz, M.; Sljoka, A.; Prosser, R.S. Allosteric modulation of the adenosine A2A receptor by cholesterol. eLife 2022, 11, e73901. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhu, W. Molecular docking for drug discovery and development: A widely used approach but far from perfect. Future Med. Chem. 2016, 8, 1707–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, D.; Gao, Z.-G.; Moss, S.M.; Jacobson, K.A.; Carlsson, J. Molecular Docking Screening Using Agonist-Bound GPCR Structures: Probing the A2A Adenosine Receptor. J. Chem. Inf. Model. 2015, 55, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Caliman, A.D.; Miao, Y.; McCammon, J.A. Mapping the allosteric sites of the A2A adenosine receptor. Chem. Biol. Drug Des. 2017, 91, 5–16. [Google Scholar] [CrossRef]
- Yu, W.; Lakkaraju, S.K.; Raman, E.P.; Fang, L.; MacKerell, A.D., Jr. Pharmacophore modeling using site-identification by ligand competitive saturation (SILCS) with multiple probe molecules. J. Chem. Inf. Model. 2015, 55, 407–420. [Google Scholar] [CrossRef]
- Raman, E.P.; Yu, W.; Lakkaraju, S.K.; MacKerell, A. Inclusion of Multiple Fragment Types in the Site Identification by Ligand Competitive Saturation (SILCS) Approach. J. Chem. Inf. Model. 2013, 53, 3384–3398. [Google Scholar] [CrossRef]
- Wang, X.; Jespers, W.; Prieto-Díaz, R.; Majellaro, M.; Ijzerman, A.P.; van Westen, G.J.P.; Sotelo, E.; Heitman, L.H.; Gutiérrez-De-Terán, H. Identification of V6.51L as a selectivity hotspot in stereoselective A2B adenosine receptor antagonist recognition. Sci. Rep. 2021, 11, 14171. [Google Scholar] [CrossRef]
- Ferenczy, G.G.; Keserű, G.M. Thermodynamics guided lead discovery and optimization. Drug Discov. Today 2010, 15, 919–932. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Lenselink, E.B.; Beuming, T.; Van Veen, C.; Massink, A.; Sherman, W.; Van Vlijmen, H.W.T.; Ijzerman, A.P. In search of novel ligands using a structure-based approach: A case study on the adenosine A2A receptor. J. Comput. Mol. Des. 2016, 30, 863–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keränen, H.; de Terán, H.G.; Åqvist, J. Structural and Energetic Effects of A2A Adenosine Receptor Mutations on Agonist and Antagonist Binding. PLoS ONE 2014, 9, e108492. [Google Scholar] [CrossRef]
- Matricon, P.; Vo, D.D.; Gao, Z.-G.; Kihlberg, J.; Jacobson, K.A.; Carlsson, J. Fragment-based design of selective GPCR ligands guided by free energy simulations. Chem. Commun. 2021, 57, 12305–12308. [Google Scholar] [CrossRef] [PubMed]
- Matricon, P.; Suresh, R.R.; Gao, Z.-G.; Panel, N.; Jacobson, K.A.; Carlsson, J. Ligand design by targeting a binding site water. Chem. Sci. 2020, 12, 960–968. [Google Scholar] [CrossRef]
- Deflorian, F.; Perez-Benito, L.; Lenselink, E.B.; Congreve, M.; Van Vlijmen, H.W.T.; Mason, J.S.; De Graaf, C.; Tresadern, G. Accurate Prediction of GPCR Ligand Binding Affinity with Free Energy Perturbation. J. Chem. Inf. Model. 2020, 60, 5563–5579. [Google Scholar] [CrossRef]
- Cappel, D.; Hall, M.L.; Lenselink, E.B.; Beuming, T.; Qi, J.; Bradner, J.; Sherman, W. Relative Binding Free Energy Calculations Applied to Protein Homology Models. J. Chem. Inf. Model. 2016, 56, 2388–2400. [Google Scholar] [CrossRef] [Green Version]
- Baltos, J.-A.; Paoletta, S.; Nguyen, A.T.N.; Gregory, K.J.; Tosh, D.K.; Christopoulos, A.; Jacobson, K.A.; May, L.T. Structure-Activity Analysis of Biased Agonism at the Human Adenosine A3 Receptor. Mol. Pharmacol. 2016, 90, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Aurelio, L.; Baltos, J.-A.; Ford, L.; Nguyen, A.T.N.; Jörg, M.; Devine, S.M.; Valant, C.; White, P.J.; Christopoulos, A.; May, L.T.; et al. A Structure–Activity Relationship Study of Bitopic N6-Substituted Adenosine Derivatives as Biased Adenosine A1 Receptor Agonists. J. Med. Chem. 2018, 61, 2087–2103. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2005, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonvin, A.M. Flexible protein–protein docking. Curr. Opin. Struct. Biol. 2006, 16, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, C.; Bonvin, A.M.; Winkler, G.S.; van Schaik, F.M.; Timmers, H.T.M.; Boelens, R. Structural Model of the UbcH5B/CNOT4 Complex Revealed by Combining NMR, Mutagenesis, and Docking Approaches. Structure 2004, 12, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Novoa, E.M.; De Pouplana, L.R.; Barril, X.; Orozco, M.; López, M.O. Ensemble Docking from Homology Models. J. Chem. Theory Comput. 2010, 6, 2547–2557. [Google Scholar] [CrossRef]
- Miao, Y.; Goldfeld, D.A.; Von Moo, E.; Sexton, P.M.; Christopoulos, A.; McCammon, J.A.; Valant, C. Accelerated structure-based design of chemically diverse allosteric modulators of a muscarinic G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E5675–E5684. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, A.; Wang, J.; Miao, Y. Retrospective ensemble docking of allosteric modulators in an adenosine G-protein-coupled receptor. Biochim. Biophys. Acta (BBA) Gen. Subj. 2020, 1864, 129615. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Narlawar, R.; Lane, J.R.; Doddareddy, M.; Lin, J.; Brussee, J.; Ijzerman, A.P. Hybrid Ortho/Allosteric Ligands for the Adenosine A1 Receptor. J. Med. Chem. 2010, 53, 3028–3037. [Google Scholar] [CrossRef]
- Schuetz, D.A.; de Witte, W.; Wong, Y.C.; Knasmueller, B.; Richter, L.; Kokh, D.B.; Sadiq, S.K.; Bosma, R.; Nederpelt, I.; Heitman, L.H.; et al. Kinetics for Drug Discovery: An industry-driven effort to target drug residence time. Drug Discov. Today 2017, 22, 896–911. [Google Scholar] [CrossRef] [Green Version]
- Tonge, P.J. Drug–Target Kinetics in Drug Discovery. ACS Chem. Neurosci. 2017, 9, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Pawnikar, S.; Bhattarai, A.; Wang, J.; Miao, Y. Binding Analysis Using Accelerated Molecular Dynamics Simulations and Future Perspectives. Adv. Appl. Bioinform. Chem. 2022, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Adams, P.J.; Azaria, A.; Bank, J.A.; Batson, B.; Bell, A.; Bergdorf, M.; Bhatt, J.; Butts, J.A.; Correia, T. Anton 3: Twenty microseconds of molecular dynamics simulation before lunch. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, St. Louis, MO, USA, 14–19 November 2021; pp. 1–11. [Google Scholar]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.-T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Miao, Y. Peptide Gaussian accelerated molecular dynamics (Pep-GaMD): Enhanced sampling and free energy and kinetics calculations of peptide binding. J. Chem. Phys. 2020, 153, 154109. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Bhattarai, A.; Wang, J. Ligand Gaussian Accelerated Molecular Dynamics (LiGaMD): Characterization of Ligand Binding Thermodynamics and Kinetics. J. Chem. Theory Comput. 2020, 16, 5526–5547. [Google Scholar] [CrossRef]
- Zhao, L.; Ciallella, H.L.; Aleksunes, L.M.; Zhu, H. Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug Discov. Today 2020, 25, 1624–1638. [Google Scholar] [CrossRef]
- Wang, M.; Hou, S.; Wei, Y.; Li, D.; Lin, J. Discovery of novel dual adenosine A1/A2A receptor antagonists using deep learning, pharmacophore modeling and molecular docking. PLoS Comput. Biol. 2021, 17, e1008821. [Google Scholar] [CrossRef]
- Liu, X.; Ye, K.; Van Vlijmen, H.W.T.; Ijzerman, A.P.; Van Westen, G.J.P. An exploration strategy improves the diversity of de novo ligands using deep reinforcement learning: A case for the adenosine A2A receptor. J. Cheminf. 2019, 11, 35. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | A1AR | A2AAR | A2BAR | A3AR | ||
| G protein coupling | Gi, Go | Gs | Gs, Gq/11 | Go, Gq/11 | ||
| Downstream signaling | ↓AC | ↑AC | ↑AC | ↓AC | ||
| ↑Phospholipase C | ↑MAP Kinase | ↑Phospholipase C | ↑Phospholipase C | |||
| ↑K+ channel, ↓Ca2+ channel | ↑PKA | ↑PKA | ↑Ca2+ channel | |||
| ↑PI3K | ↑PKC | ↑MAP Kinase | ↑PI3K | |||
| ↑MAP Kinase | ↑PKC | |||||
| ↑MAP kinase | ||||||
| Adenosine binding affinity | 5.10 nM | 30.9 nM | 1000 nM | 100 nM | ||
| Approved drugs | Drug | Therapeutic use | Drug | Therapeutic use | ||
| Adenosine (agonist) | Paroxysmal supraventricular tachycardia | Adenosine (agonist) | Myocardial perfusion imaging | |||
| Regadenoson (antagonist) | Asthma | Istradefylline (antagonist) | Parkinson’s disease | |||
| Theophylline (antagonist) | Asthma | |||||
| Doxofylline (antagonist) | Asthma | |||||
| Bamifylline (antagonist) | Asthma | |||||
| PDB Code | Resolution (Å) | Binding Affinity | Receptor′s States | Ligand | Reference |
|---|---|---|---|---|---|
| A1AR | |||||
| 5UEN | 3.2 | IC50: 24.9 nM [34] | Inactive | Antagonist (DU172) | Glukhova et al. [35] |
| 5N2S | 3.3 | Ki: 0.7 nM [36] | Inactive | Antagonist (PSB36) | Cheng et al. [36] |
| 6D9H | 3.6 | Ki: 5.10 nM [37] | Active | Agonist (adenosine) | Draper-Joyce et al. [38] |
| 7LD3 | 3.2 | Ki: 5.10 nM [37] | Active | Agonist (adenosine) | Draper-Joyce et al. [39] |
| 7LD4 | 3.3 | - | Active | Agonist and PAM (adenosine and MIPS521) | Draper-Joyce et al. [39] |
| A2AAR | |||||
| 7MR5 | 2.8 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Martynowycz et al. [41] |
| 7ARO | 3.1 | - | Inactive | Partial agonist (LUF5833) | Amelia et al. [42] |
| 6LPL | 2.0 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Ihara et al. [43] |
| 6LPK | 1.8 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Ihara et al. [43] |
| 6LPJ | 1.8 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Ihara et al. [43] |
| 6WQA | 2.0 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Lee et al. [44] |
| 6ZDV | 2.1 | pKD: 5.90 [45] | Inactive | Antagonist (PubChem CID 984073) | Jespers et al. [45] |
| 6ZDR | 1.9 | pKD: 8.60 [45] | Inactive | Antagonist (PubChem CID 740769) | Jespers et al. [45] |
| 6S0L | 2.7 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Nass et al. [46] |
| 6S0Q | 2.7 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Nass et al. [46] |
| 6PS7 | 1.9 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Ishchenkoa et al. [47] |
| 6JZH | 2.3 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Shimazu et al. [48] |
| 6GT3 | 2.0 | Ki: 1.7 nM [49] | Inactive | Antagonist (AZD4635) | Borodovsky et al. [49] |
| 6MH8 | 4.2 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Martin-Garcia et al. [50] |
| 6GDG | 4.1 | Ki: 20 nM [51] | Active | Agonist (NECA) | García-Nafría et al. [52] |
| 5WF5 | 2.6 | Ki: 17.3 nM [53] | Active | Agonist (UK-432097) | White et al. [53] |
| 5WF6 | 2.9 | Ki: 17.3 nM [53] | Active | Agonist (UK-432097) | White et al. [53] |
| 5OLH | 2.6 | pkD: 9.0 [54] | Inactive | Antagonist (Vipadenant) | Rucktooa et al. [54] |
| 5OM1 | 2.1 | pkD: 9.6 [54] | Inactive | Antagonist (PubChem CID 135566609) | Rucktooa et al. [54] |
| 5OLG | 1.9 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Rucktooa et al. [54] |
| 5OLO | 3.1 | Ki: 0.3 nM [55] | Inactive | Antagonist (Tozadenant) | Rucktooa et al. [54] |
| 5OM4 | 2.0 | Ki: 1.41 nM [56] | Inactive | Antagonist (PubChem CID 135566609) | Rucktooa et al. [54] |
| 5OLV | 2.0 | Ki: 5.9 nM [57] | Inactive | Antagonist (CHEMBL 1671936) | Rucktooa et al. [54] |
| 5OLZ | 1.9 | Ki: 1.51 nM [56] | Inactive | Antagonist (PubChem CID 135566609) | Rucktooa et al. [54] |
| 6AQF | 2.5 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Eddy et al. [58] |
| 5VRA | 2.4 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Broecker et al. [59] |
| 5NM2 | 2.0 | Ki: 0.1 nM [40] | Inactive | Antagonist (ZM241385) | Weinert et al. [60] |
| 5NM4 | 1.7 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Weinert et al. [60] |
| 5NLX | 2.1 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Weinert et al. [60] |
| 5N2R | 2.8 | - | Inactive | Antagonist (PSB36) | Cheng et al. [36] |
| 5MZP | 2.1 | Ki: 5011.87 nM [61] | Inactive | Caffeine | Cheng et al. [36] |
| 5MZJ | 2.0 | Ki: 0.60 nM [62] | Inactive | Theophylline | Cheng et al. [36] |
| 5JTB | 2.8 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Melnikov et al. [62] |
| 5UVI | 3.2 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Martin-Garcia et al. [63] |
| 5UIG | 3.5 | - | Inactive | Antagonist (PubChem CID 124081196) | Sun et al. [64] |
| 5K2A | 2.5 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Batyuk et al. [65] |
| 5K2D | 1.9 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Batyuk et al. [65] |
| 5K2C | 1.9 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Batyuk et.al. [65] |
| 5K2B | 2.5 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Batyuk et al. [65] |
| 5G53 | 3.4 | Ki: 1.00 nM [66] | Active | Agonist (NECA) | Carpenter et al. [67] |
| 5IU4 | 1.7 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Segala et al. [68] |
| 5IU8 | 2.0 | Ki: 18 nM [68] | Inactive | Antagonist (Q27456347) | Segala et al. [68] |
| 5IUB | 2.1 | Ki: 0.35 nM [68] | Inactive | Antagonist (Q27456347) | Segala et al. [68] |
| 5IUA | 2.2 | Ki: 1.5 nM [68] | Inactive | Antagonist (6DX) | Segala et al. [68] |
| 5IU7 | 1.9 | Ki: 1.1 nM [68] | Inactive | Antagonist (6DX) | Segala et al. [68] |
| 4UG2 | 2.6 | Ki: 8.80 nM [69] | Active | Agonist (CGS-21680) | Lebon et al. [70] |
| 4UHR | 2.6 | Ki: 8.80 nM [69] | Active | Agonist (CGS-21680) | Lebon et al. [70] |
| 4EIY | 1.8 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Liu et al. [71] |
| 3UZC | 3.3 | Kd: 0.25 nM [56] | Inactive | Antagonist (PubChem CID 135566609) | Congreve et al. [56] |
| 3UZA | 3.3 | Ki: 7.76 [56] | Inactive | Antagonist (PubChem CID 56844240) | Congreve et al. [56] |
| 3VGA | 3.1 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Hino et al. [72] |
| 3VG9 | 2.7 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Hino et al. [72] |
| 3PWH | 3.3 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Dore et al. [73] |
| 3RFM | 3.6 | Ki: 5011.87 nM [61] | Inactive | Antagonist (Caffeine) | Dore et al. [73] |
| 3REY | 3.3 | Kd: 10 nM [73] | Inactive | Antagonist (XAC) | Dore et al. [73] |
| 2YDO | 3.0 | Ki: 30.9 nM [74] | Active | Agonist (adenosine) | Lebon et al. [56] |
| 2YDV | 2.6 | Ki: 13.8 nM [74] | Active | Agonist (NECA) | Lebon et al. [56] |
| 3QAK | 2.7 | Ki: 4.75 nM [75] | Active | Agonist (UK-432097) | Xu et al. [75] |
| 3EML | 2.6 | Ki: 0.10 nM [40] | Inactive | Antagonist (ZM241385) | Jaakola et al. [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Bhattarai, A.; Do, H.N.; Akhter, S.; Miao, Y. Molecular Simulations and Drug Discovery of Adenosine Receptors. Molecules 2022, 27, 2054. https://doi.org/10.3390/molecules27072054
Wang J, Bhattarai A, Do HN, Akhter S, Miao Y. Molecular Simulations and Drug Discovery of Adenosine Receptors. Molecules. 2022; 27(7):2054. https://doi.org/10.3390/molecules27072054
Chicago/Turabian StyleWang, Jinan, Apurba Bhattarai, Hung N. Do, Sana Akhter, and Yinglong Miao. 2022. "Molecular Simulations and Drug Discovery of Adenosine Receptors" Molecules 27, no. 7: 2054. https://doi.org/10.3390/molecules27072054
APA StyleWang, J., Bhattarai, A., Do, H. N., Akhter, S., & Miao, Y. (2022). Molecular Simulations and Drug Discovery of Adenosine Receptors. Molecules, 27(7), 2054. https://doi.org/10.3390/molecules27072054