
Thiazole/Thiadiazole/Benzothiazole Based Thiazolidin-4-One Derivatives as Potential Inhibitors of Main Protease of SARS-CoV-2

 ,
,  ,
,  ,
, 
Abstract
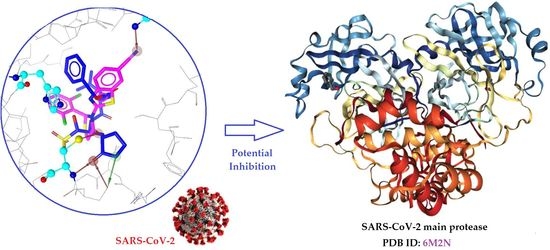
:
1. Introduction
2. Results
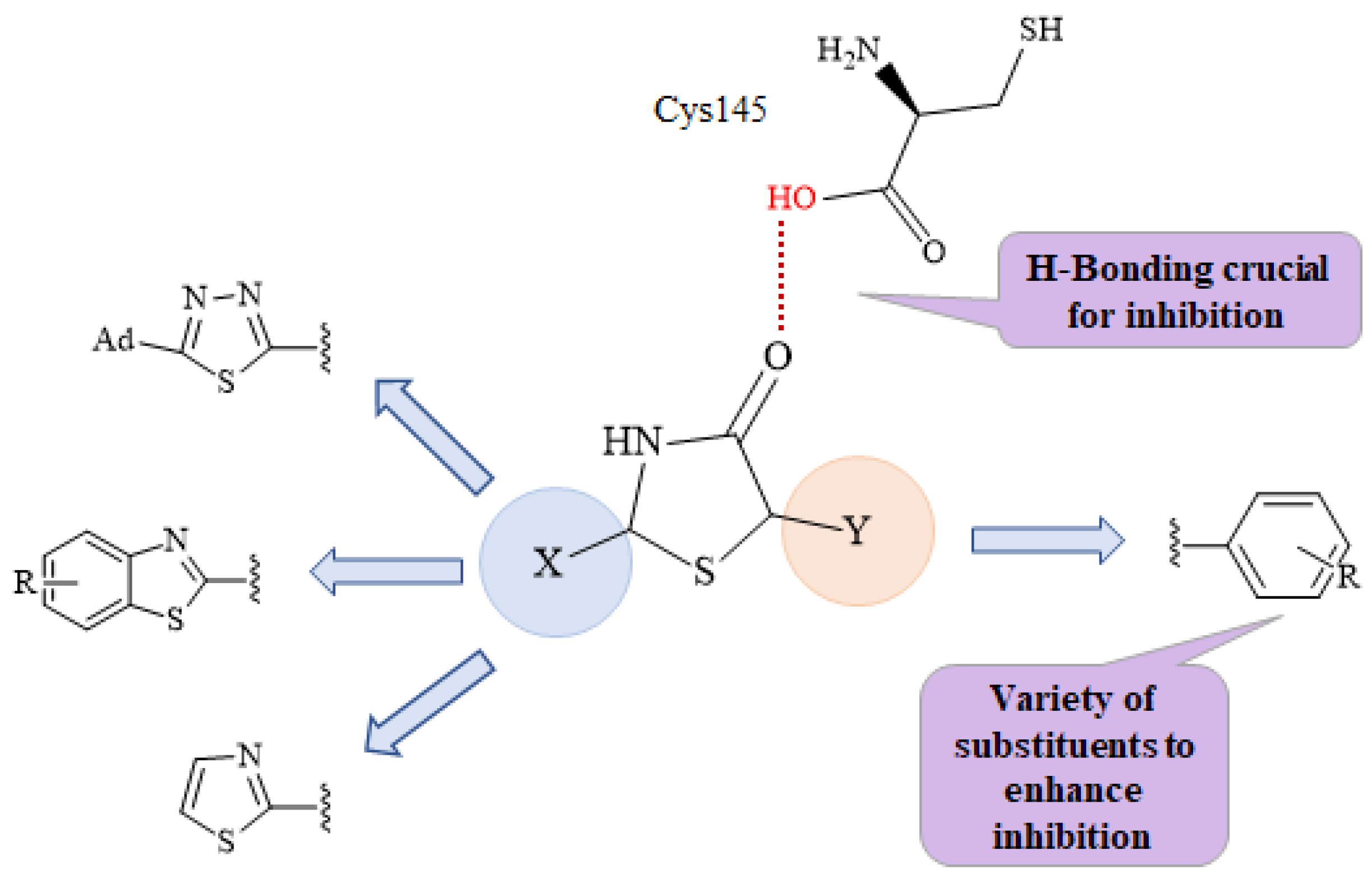
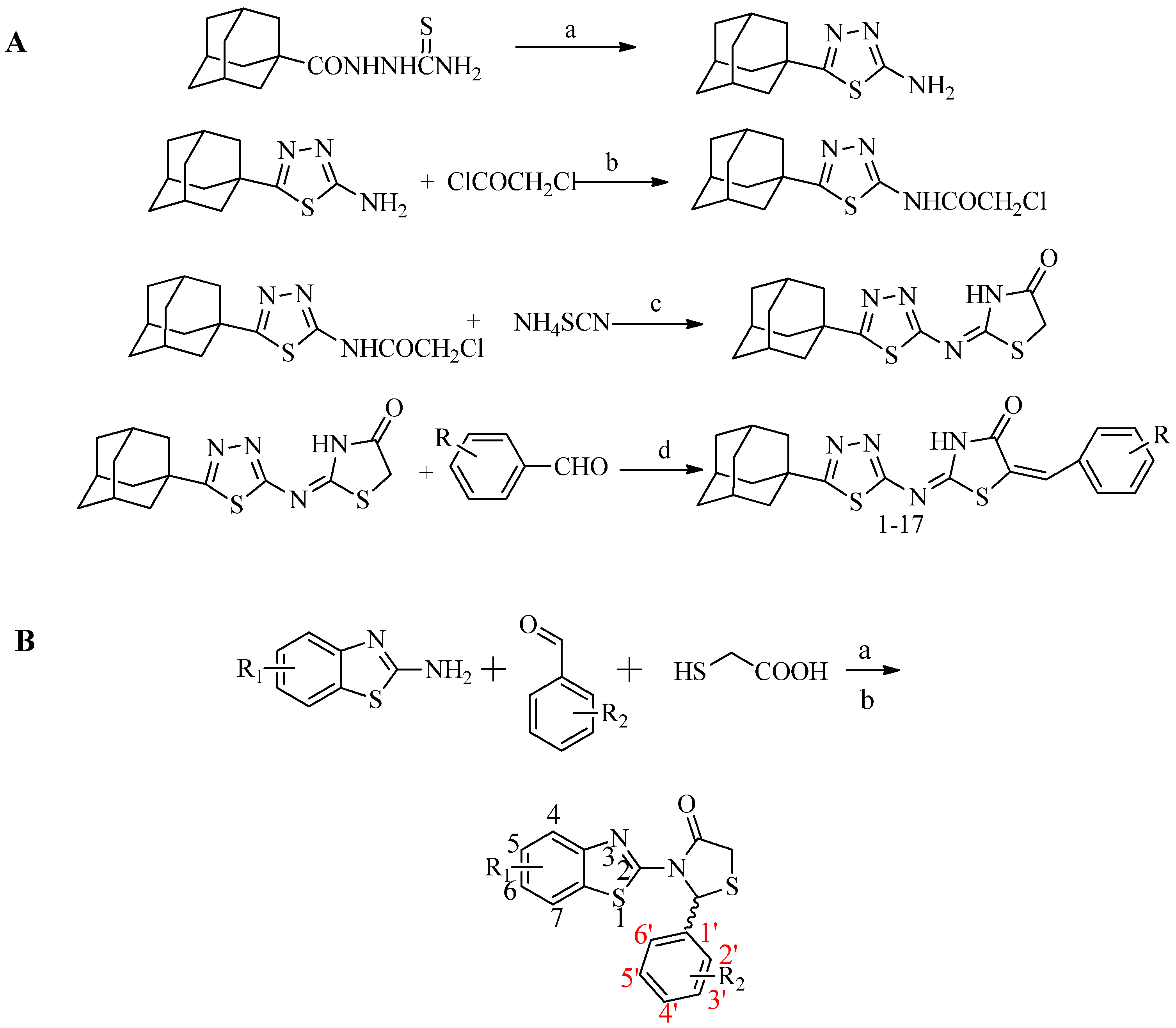
2.1. Chemistry
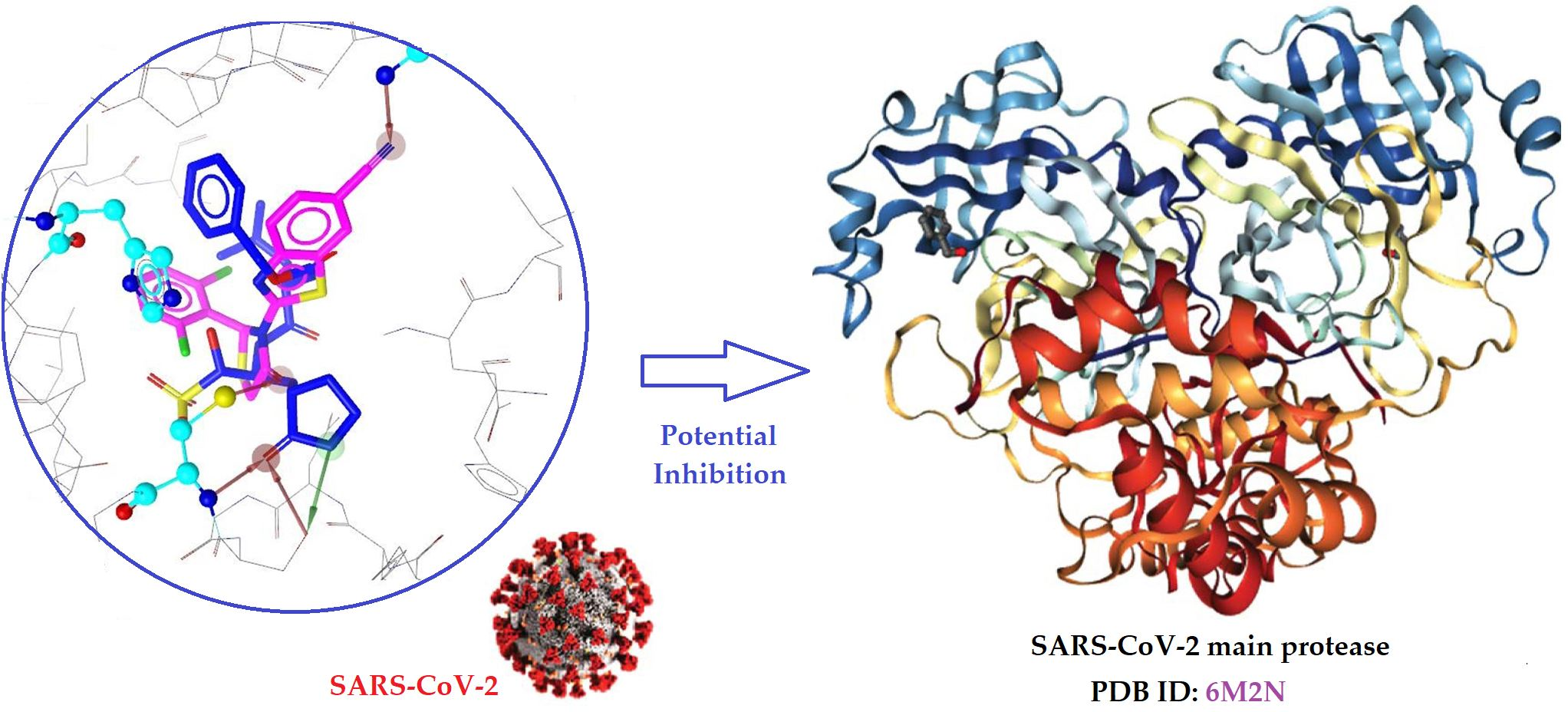
2.2. Molecular Docking Prediction
2.3. Biological Evaluation
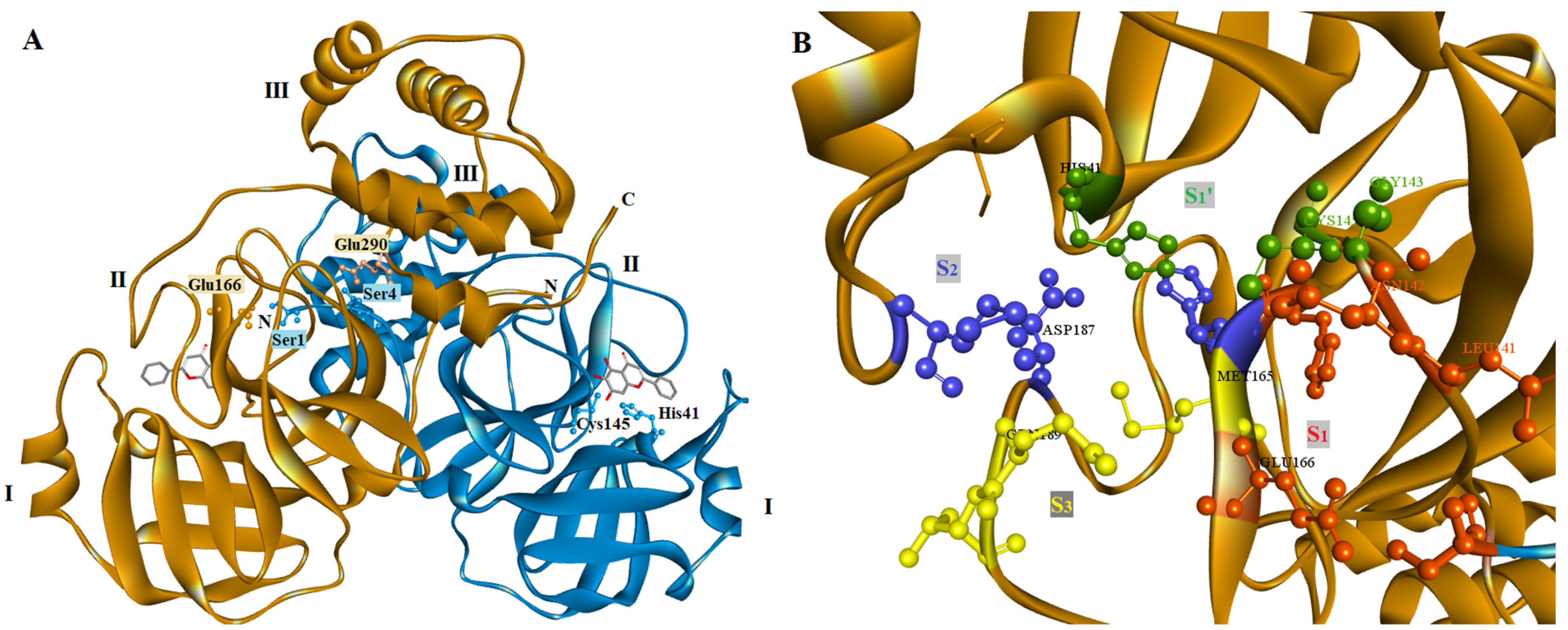
2.4. Docking Studies
2.5. Assessment of Cellular Viability
3. Materials and Methods
3.1. Synthesis
3.2. Molecular Docking
3.3. Inhibition of SARS-CoV-2 3CLpro Enzymatic Activity by Synthesized Compounds
3.4. Evaluation of Cellular Viability by MTT Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- WHO. Novel Coronavirus e China. 2020. Available online: https://www.who.int/csr/don/12-january-2020-novel-coronavirus-china/en/ (accessed on 15 December 2020).
- Zhu, N.; Zhang, D.; Wang, W. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; He, W.T.; Wang, L.; Lai, A.; Ji, X.; Zhai, X.; Li, G.; Suchard, M.A.; Tian, J.; Zhou, J. COVID-19: Epidemiology, Evolution, and Cross-Disciplinary Perspectives. Trends Mol. Med. 2020, 26, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Ghanbari, R.; Teimoori, A.; Sadeghi, A.; Mohamadkhani, A.; Rezasoltani, S.; Asadi, E.; Jouyban, A.; Sumner, S.C. Existing antiviral options against SARS-CoV-2 replication in COVID-19 patients. Future Microbiol. 2020, 15, 1747–1758. [Google Scholar] [CrossRef]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417–4423. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Eckstrand, C.; Groutas, W.C.; Bannasch, M.; Meadows, J.M.; Chang, K.O. Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg. 2018, 20, 378–392. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Rathnayake, A.D.; Zheng, J.; Kim, Y.; Perera, K.D.; Mackin, S.; Meyerholz, D.K.; Kashipathy, M.M.; Battaile, K.P.; Lovell, S.; Perlman, S.; et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Transl. Med. 2020, 12, eabc5332. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Yang, W.H.; Yang, C.S.; Hou, M.H.; Tsai, C.L.; Chou, Y.Z.; Hung, M.C.; Chen, Y. Structural basis of SARS-CoV-2 main protease inhibition by a broad-spectrum anti-coronaviral drug. Am. J. Cancer Res. 2020, 10, 2535–2545. [Google Scholar] [PubMed]
- Vuong, W.; Khan, M.B.; Fischer, C. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef] [PubMed]
- Perera, K.D.; Galasiti Kankanamalage, A.C.; Rathnayake, A.D.; Honeyfield, A.; Groutas, W.; Chang, K.O.; Kim, Y. Protease inhibitors broadly effective against feline, ferret and mink coronaviruses. Antivir. Res. 2018, 160, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Song, J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef]
- Tang, B.; He, F.; Liu, D.; Fang, M.; Wu, Z.; Xu, D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Huang, M.; Li, D.M.; Tang, N. Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J. Thromb. Thrombolysis 2021, 51, 1107–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimeno, A.; Ojeda-Montes, M.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [Green Version]
- McConkey, B.J.; Sobolev, V.; Edelman, M. The performance of current methods in ligand-protein docking. Curr. Sci. 2002, 83, 845–855. [Google Scholar]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Sci. 2020, 10, 313–319. [Google Scholar]
- Yang, H.T.; Yang, M.J.; Ding, Y. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chemistry 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Fesatidou, M.; Zagaliotis, P.; Camoutsis, C.; Petrou, A.; Ciric, A.; Sokovic, M. 5-Adamantan thiadiazole-based thiazolidinones as antimicrobial agents. Design, synthesis, molecular docking and evaluation. Bioorg. Med. Chem. 2018, 26, 4664–4676. [Google Scholar] [CrossRef]
- Petrou, A.; Eleftheriou, P.; Geronikaki, A.; Akrivou, M.G.; Vizirianakis, I. Novel thiazolidin-4-ones as potential non-nucleoside inhibitors of HIV-1 reverse transcriptase. Molecules 2019, 24, 3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftheriou, P.; Petrou, A.; Geronikaki, A.; Liaras, K.; Dirnali, S.; Anna, M. Prediction of enzyme inhibition and mode of inhibitory action based on calculation of distances between hydrogen bond donor/acceptor groups of the molecule and docking analysis: An application on the discovery of novel effective PTP1B inhibitors. SAR QSAR Environ. Res. 2015, 26, 557–576. [Google Scholar] [CrossRef]
- Eleftheriou, P.; Amanatidou, D.; Petrou, A.; Geronikaki, A. In silico evaluation of the effectivity of approved protease inhibitors against the main protease of the novel SARS-CoV-2 virus. Molecules 2020, 25, 2529. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganou, C.A.; Eleftheriou, P.T.; Theodosis-Nobelos, P.; Geronikaki, A.A.; Lialiaris, T.; Rekka, E.A. Docking analysis targeted to the whole enzyme: An application to the prediction of inhibition of PTP1b by thiomorpholine and thiazolyl derivatives. SAR QSAR Environ. Res. 2018, 29, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Akrivou, M.G.; Demertzidou, V.P.; Theodoroula, N.F.; Chatzopoulou, F.M.; Kyritsis, K.A.; Grigoriadis, N.; Zografos, A.L.; Vizirianakis, I.S. Uncovering the pharmacological response of novel sesquiterpene derivatives that differentially alter gene expression and modulate the cell cycle in cancer cells. Int. J. Oncol. 2018, 53, 2167–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Nο | R1 | R2 | Est. Free Binding Energy (kcal/mol)) S-(−) | Est. Free Binding Energy (kcal/mol) R-(+) | Νο | R1 | R2 | Est. Free Binding Energy (kcal/mol) S-(−) | Est. Free Binding Energy (kcal/mol) R-(+) |
|---|---|---|---|---|---|---|---|---|---|
| a1 | 7-Cl | 2,6-di-F | −8.57 | −9.27 | i7 | 6-Br | 2,3-di-Cl | −6.19 | −7.04 |
| a2 | 7-Cl | 2-F, 6-Cl | −2.79 | −3.54 | i8 | 6-Br | 2,4-di-Cl | −5.23 | −5.67 |
| a3 | 4,5-di-Cl | 4-F | −9.16 | −9.58 | k1 | 6-CN | 4-NO2 | −4.60 | −4.11 |
| b1 | 6-F | 4-F | −8.57 | −8.93 | k2 | 6-CN | 2,6-di-F | −5.38 | −5.61 |
| b2 | 6-F | 4-NO2 | −6.37 | −6.88 | k3 | 6-CN | 2-F, 6-Cl | −10.44 | −10.78 |
| b3 | 6-F | 4-Cl | −6.91 | −7.12 | k4 | 6-CN | 2,6-di-Cl | −4.37 | −5.60 |
| b4 | 6-F | 4-OCH3 | −5.23 | −6.17 | k5 | 6-CN | 4-F | −6.35 | −6.99 |
| b5 | 6-F | 4-OH | −5.64 | −5.34 | k6 | 6-CN | 2,4-di-Cl | −6.14 | −7.01 |
| b6 | 6-F | 4-Br | −6.92 | −5.80 | l1 | 6-CF3 | 2,6-di-Cl | −5.33 | −5.10 |
| b7 | 6-F | 2,3-di-Cl | −6.93 | −5.86 | l2 | 6-CF3 | 2,6-di-F | −4.03 | −3.96 |
| b8 | 6-F | 2,4-di-Cl | −5.61 | −5.72 | l3 | 6-CF3 | 2-F, 6-Cl | −6.17 | −7.00 |
| c1 | 6-Cl | 4-F | −10.25 | −10.70 | l4 | 6-CF3 | 4-Br | −5.29 | −6.33 |
| c2 | 6-Cl | 4-NO2 | −3.49 | −4.11 | l5 | 6-CF3 | 2,3-di-Cl | −4.24 | −4.58 |
| c3 | 6-Cl | 4-Cl | −3.02 | −4.34 | l6 | 6-CF3 | 2,4-di-Cl | −5.20 | −5.87 |
| c4 | 6-Cl | 4-OCH3 | −4.71 | −5.06 | m1 | 6-Ad | 2,6-di-Cl | −6.33 | −6.32 |
| c5 | 6-Cl | 4-OH | −9.72 | −9.85 | m2 | 6-Ad | 2-F, 6-Cl | −9.57 | −10.16 |
| c6 | 6-Cl | 4-Br | −2.19 | −3.21 | m3 | 6-Ad | 2,6-di-F | −5.44 | −5.72 |
| c7 | 6-Cl | 2,3-di-Cl | −4.25 | −6.14 | m4 | 6-Ad | 2,3-di-Cl | −6.38 | −6.10 |
| c8 | 6-Cl | 2,4-di-Cl | −3.75 | −4.66 | m5 | 6-Ad | 2,4-di-Cl | −5.13 | −6.70 |
| d1 | 4-Cl | 4-F | −8.51 | −8.63 | m6 | 6-Ad | 4-F | −2.16 | −3.88 |
| d2 | 4-Cl | 4-NO2 | −6.13 | −6.45 | m7 | 6-Ad | 4-NO2 | −1.03 | −2.67 |
| d3 | 4-Cl | 4-Cl | −5.29 | −6.74 | m8 | 6-Ad | 4-Cl | −5.30 | −3.46 |
| d4 | 4-Cl | 4-OCH3 | −7.22 | −7.50 | m9 | 6-Ad | 4-OCH3 | −6.90 | −7.10 |
| d5 | 4-Cl | 4-OH | −6.97 | −7.82 | m10 | 6-Ad | 4-OH | −2.88 | −3.67 |
| d6 | 4-Cl | 4-Br | −4.63 | −5.19 | m11 | 6-Ad | 4-Br | −8.91 | −9.03 |
| d7 | 4-Cl | 2,3-di-Cl | −2.94 | −3.68 | n1 | 4-CH3, 6-Ad | 2,6-di-Cl | −7.34 | −7.66 |
| d8 | 4-Cl | 2,4-di-Cl | −5.26 | −6.28 | n2 | 4-CH3, 6-Ad | 2,6-di-F | −10.10 | −10.12 |
| e1 | 4-OCH3 | 4-F | −5.37 | −6.27 | n3 | 4-CH3, 6-Ad | 2-F, 6-Cl | −7.10 | −7.91 |
| e2 | 4-OCH3 | 4-NO2 | −8.62 | −8.90 | n4 | 4-CH3, 6-Ad | 2,3-di-Cl | −6.52 | −7.04 |
| e3 | 4-OCH3 | 4-Cl | −6.49 | 7.13 | n5 | 4-CH3, 6-Ad | 2,4-di-Cl | −8.33 | −8.42 |
| e4 | 4-OCH3 | 4-OCH3 | −5.41 | −6.82 | n6 | 4-CH3, 6-Ad | 4-F | −6.27 | −6.44 |
| e5 | 4-OCH3 | 4-OH | −5.34 | −5.33 | n7 | 4-CH3, 6-Ad | 4-NO2 | −5.12 | −5.49 |
| e6 | 4-OCH3 | 4-Br | −6.27 | −6.64 | n8 | 4-CH3, 6-Ad | 4-Cl | −4.67 | −6.74 |
| f1 | 6-OCH3 | 4-F | −5.22 | −6.39 | o1 | 5,6-di-CH3 | 4-F | −5.14 | −6.30 |
| f2 | 6-OCH3 | 4-NO2 | −5.10 | −6.82 | o2 | 5,6-di-CH3 | 4-NO2 | −2.83 | −2.61 |
| f3 | 6-OCH3 | 4-Cl | −3.78 | −4.09 | o3 | 5,6-di-CH3 | 4-Cl | −3.66 | −4.72 |
| f4 | 6-OCH3 | 4-OCH3 | −3.38 | −3.56 | o4 | 5,6-di-CH3 | 4-OCH3 | −3.28 | −4.56 |
| h1 | 6-OCF3 | 2,6-di-Cl | −5.13 | −6.72 | q1 | 4-CH3 | 4-F | −5.67 | −5.91 |
| h2 | 6-OCF3 | 2,6-di-F | −6.23 | −4.97 | q2 | 4-CH3 | 4-NO2 | −7.05 | −7.40 |
| h3 | 6-OCF3 | 2-F, 6-Cl | −1.37 | −4.06 | q3 | 4-CH3 | 4-Cl | −6.55 | −6.81 |
| h4 | 6-OCF3 | 2,3-di-Cl | −5.62 | −4.09 | q4 | 4-CH3 | 4-OCH3 | −5.00 | −5.37 |
| h5 | 6-OCF3 | 2,4-di-Cl | −2.88 | −1.86 | q5 | 4-CH3 | 4-OH | −4.10 | −6.89 |
| h6 | 6-OCF3 | 4-F | −9.05 | −9.17 | q6 | 4-CH3 | 4-Br | −2.56 | −2.88 |
| h7 | 6-OCF3 | 4-NO2 | −5.42 | −5.69 | q7 | 4-CH3 | 2,3-di-Cl | −3.65 | −4.78 |
| h8 | 6-OCF3 | 4-Cl | −8.63 | −8.80 | q8 | 4-CH3 | 2,4-di-Cl | −2.31 | −3.79 |
| h9 | 6-OCF3 | 4-OCH3 | −4.37 | −4.66 | r1 | 6-CH3 | 4-F | −5.30 | −6.71 |
| 6M2N Initial inhibitor * | −10.45 | GC376 | −10.35 | ||||||

| No | R1 | R2 | % Inhibition at 50 μM | IC50 (μΜ) |
|---|---|---|---|---|
| a3 | 4,5-di-Cl | 4-F | 0 | >50 |
| a1 | 7-Cl | 2,6-di-F | 0 | >50 |
| b1 | 6-F | 4-F | 0 | >50 |
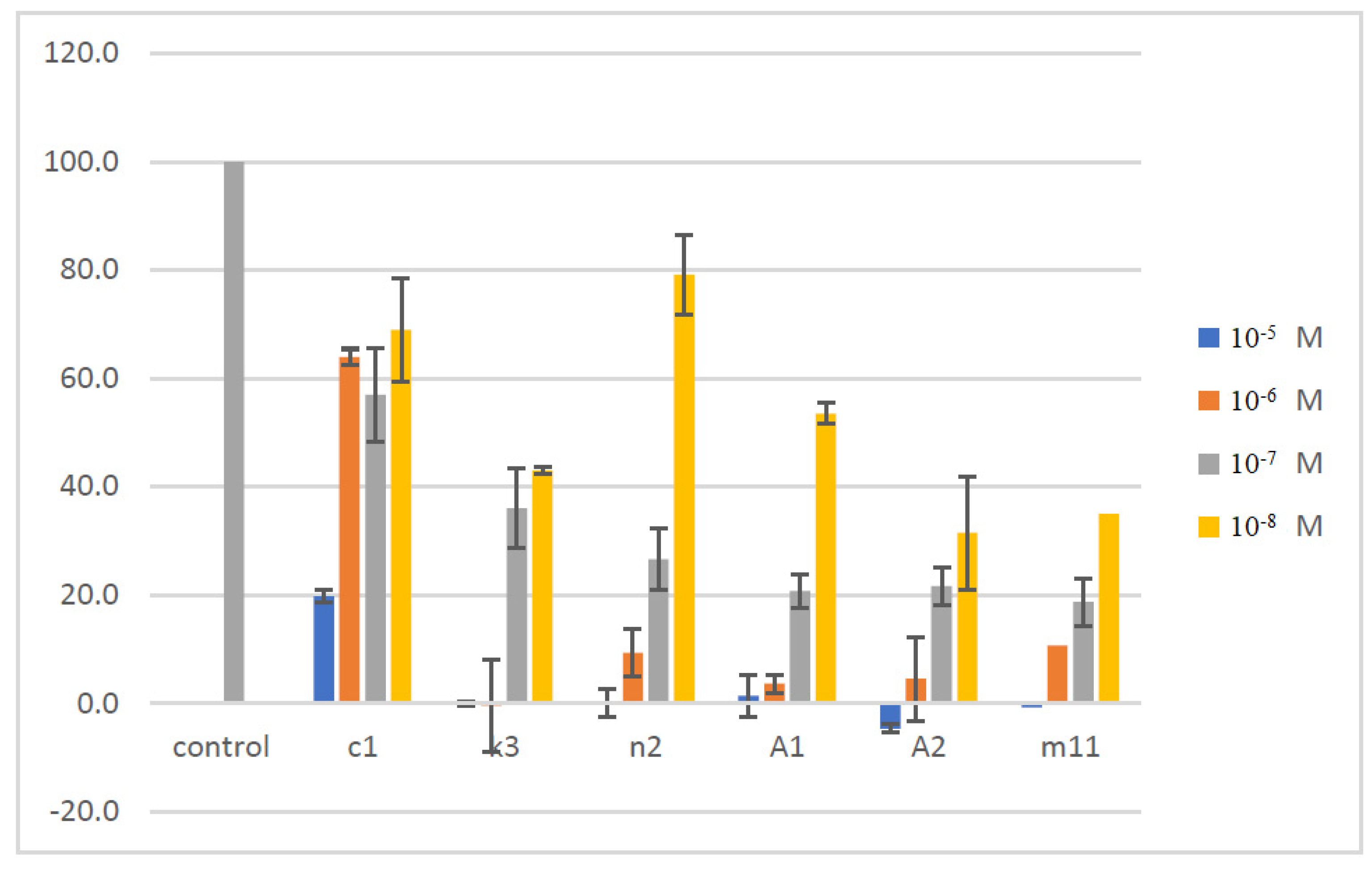
| c1 | 6-Cl | 4-F | 88.03 | 4.736 |
| c5 | 6-Cl | 4-OH | 7.87 | >50 |
| d1 | 4-Cl | 4-F | 6.72 | >50 |
| e2 | 4-OCH3 | 4-NO2 | 31.30 | >50 |
| h6 | 6-OCF3 | 4-F | 25.18 | >50 |
| h8 | 6-OCF3 | 4-Cl | 3.04 | >50 |
| k3 | 6-CN | 2-F, 6-Cl | 100 | 0.010 |
| n2 | 4-Me, 6-Ad | 2-F, 6-Cl | 100 | 9.984 |
| m2 | 6-Ad | 2-F, 6-Cl | 2.79 | >50 |
| m11 | 6-Ad | 4-Br | 45.00 | >50 |
| A1 | 6-Ad | 4-NO2 | 94.11 | 34.4 |
| A2 | 6-Ad | 2,6-di-F | 91.34 | 13.21 |
| GC376 | 100 at 100 μM | 0.439 |
| No | Est. Free Binding Energy (kcal/mol) | I-H | Residues Involved in Hydrogen Bonds | Hydrophobic Interactions |
|---|---|---|---|---|
| a1 | −9.27 | 1 | Arg188 | Asn142, Gly143, His164 |
| a3 | −9.58 | 1 | Asn142 | Met49, Glu166, Leu167 |
| b1 | −8.93 | 1 | Gln192 | His164, Asp187, Gln189 |
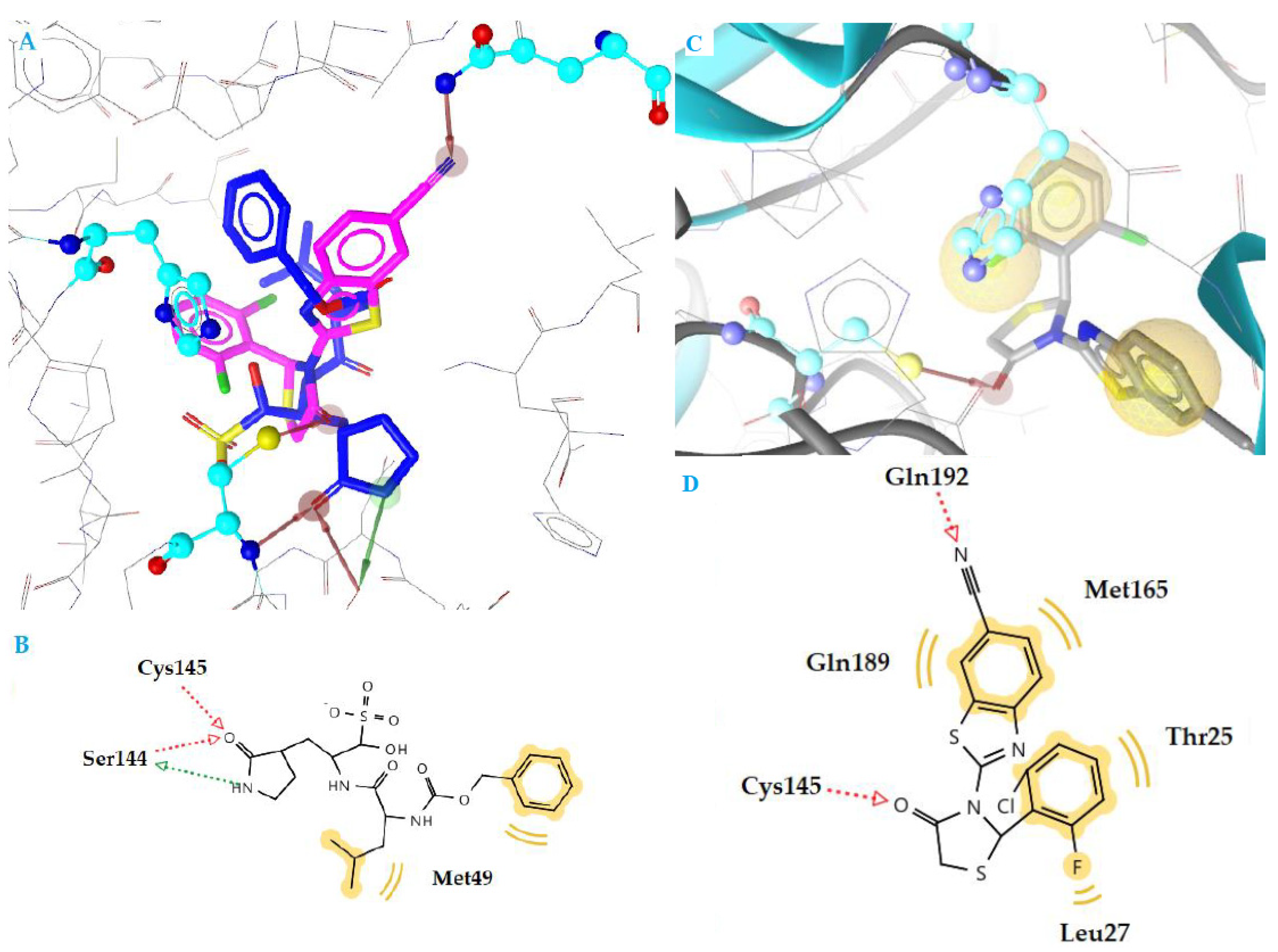
| c1 | −10.70 | 1 | Glu166 | Met49, Met165, Leu167, Arg188, Gln189 |
| c5 | −9.85 | 1 | Glu166 | Leu27, His164, Gln189 |
| d1 | −8.63 | 1 | Gln192 | Thr25, Leu27, Gln189 |
| e2 | −8.90 | 1 | Gln192 | Leu27, Asp187 |
| h6 | −9.17 | 1 | Glu166 | Leu27, Tyr54, Arg188 |
| h8 | −8.80 | 1 | Gln192 | Thr25, Met165 |
| k3 | −10.78 | 2 | Cys145, Gln192 | Thr25, Leu27, Met165, Gln189 |
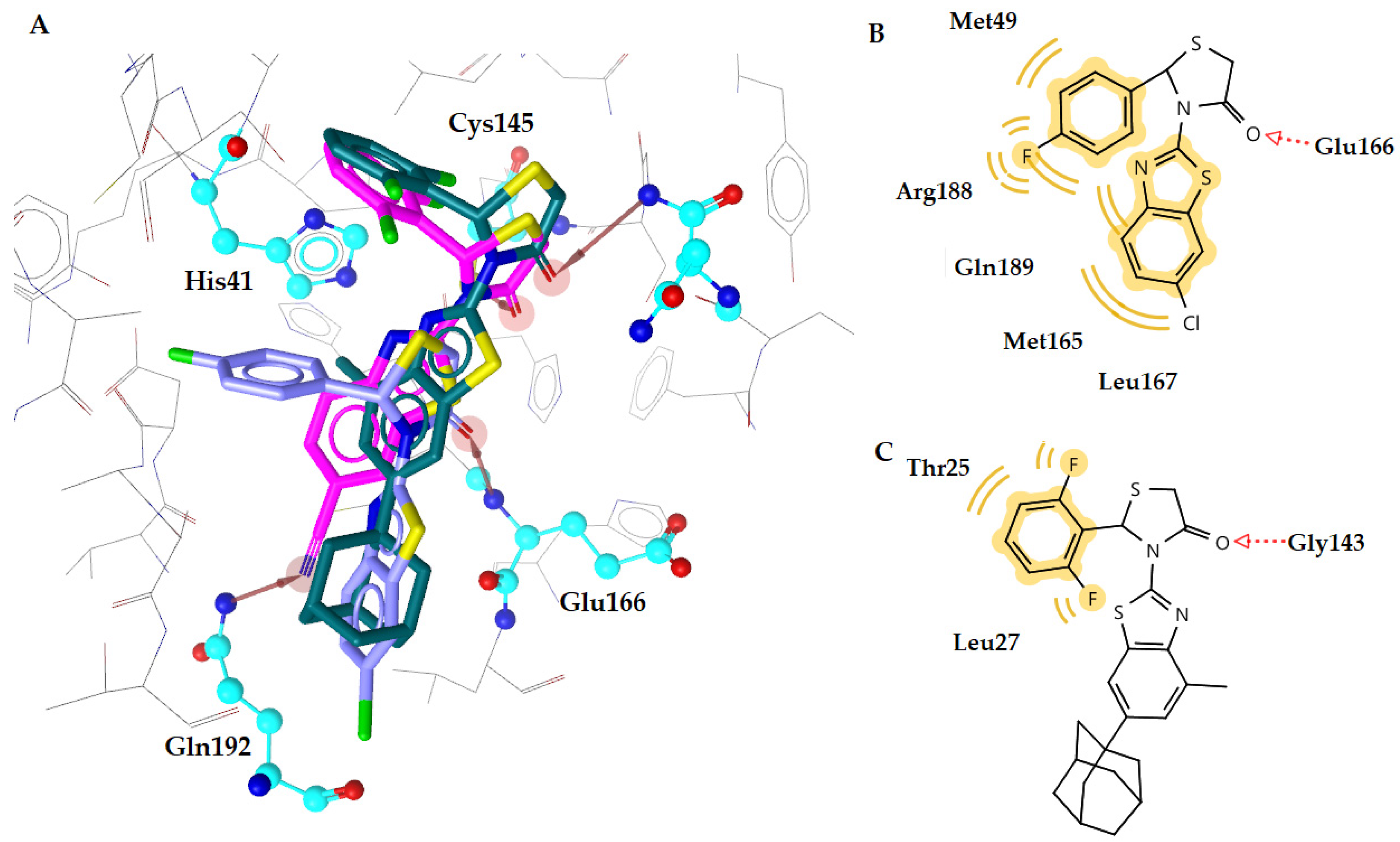
| n2 | −10.12 | 1 | Gly143 | Thr25, Leu27 |
| m2 | −10.16 | 1 | Glu166 | Leu27, Met165 |
| m11 | −9.03 | 1 | Glu166 | Leu27, Gln189 |
| 6M2N Initial inhibitor * | −10.45 | 2 | Gly143, Glu166 | Leu27, Tyr54, Asn42, His164, Gln189, Arg188, Asp187 |
| GC376 | −11.35 | 3 | Ser144, Cys145 | Met49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrou, A.; Zagaliotis, P.; Theodoroula, N.F.; Mystridis, G.A.; Vizirianakis, I.S.; Walsh, T.J.; Geronikaki, A. Thiazole/Thiadiazole/Benzothiazole Based Thiazolidin-4-One Derivatives as Potential Inhibitors of Main Protease of SARS-CoV-2. Molecules 2022, 27, 2180. https://doi.org/10.3390/molecules27072180
Petrou A, Zagaliotis P, Theodoroula NF, Mystridis GA, Vizirianakis IS, Walsh TJ, Geronikaki A. Thiazole/Thiadiazole/Benzothiazole Based Thiazolidin-4-One Derivatives as Potential Inhibitors of Main Protease of SARS-CoV-2. Molecules. 2022; 27(7):2180. https://doi.org/10.3390/molecules27072180
Chicago/Turabian StylePetrou, Anthi, Panagiotis Zagaliotis, Nikoleta F. Theodoroula, George A. Mystridis, Ioannis S. Vizirianakis, Thomas J. Walsh, and Athina Geronikaki. 2022. "Thiazole/Thiadiazole/Benzothiazole Based Thiazolidin-4-One Derivatives as Potential Inhibitors of Main Protease of SARS-CoV-2" Molecules 27, no. 7: 2180. https://doi.org/10.3390/molecules27072180
APA StylePetrou, A., Zagaliotis, P., Theodoroula, N. F., Mystridis, G. A., Vizirianakis, I. S., Walsh, T. J., & Geronikaki, A. (2022). Thiazole/Thiadiazole/Benzothiazole Based Thiazolidin-4-One Derivatives as Potential Inhibitors of Main Protease of SARS-CoV-2. Molecules, 27(7), 2180. https://doi.org/10.3390/molecules27072180