
Distinct Mechanisms of Cytotoxicity in Novel Nitrogenous Heterocycles: Future Directions for a New Anti-Cancer Agent

 ,
,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
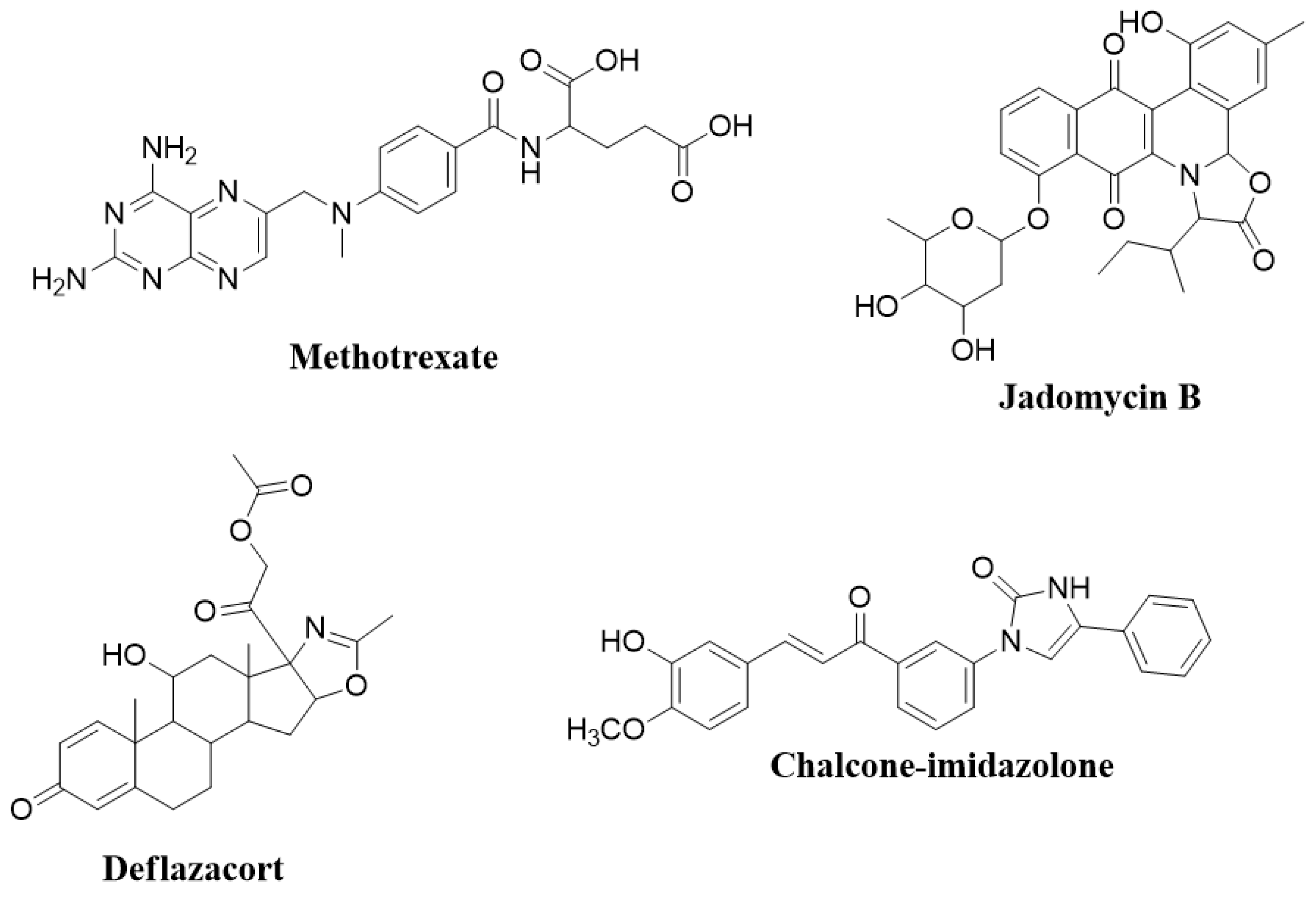
2.1. The Effect of Nitrogen-Based Heterocyclic Derivatives on Cytotoxicity
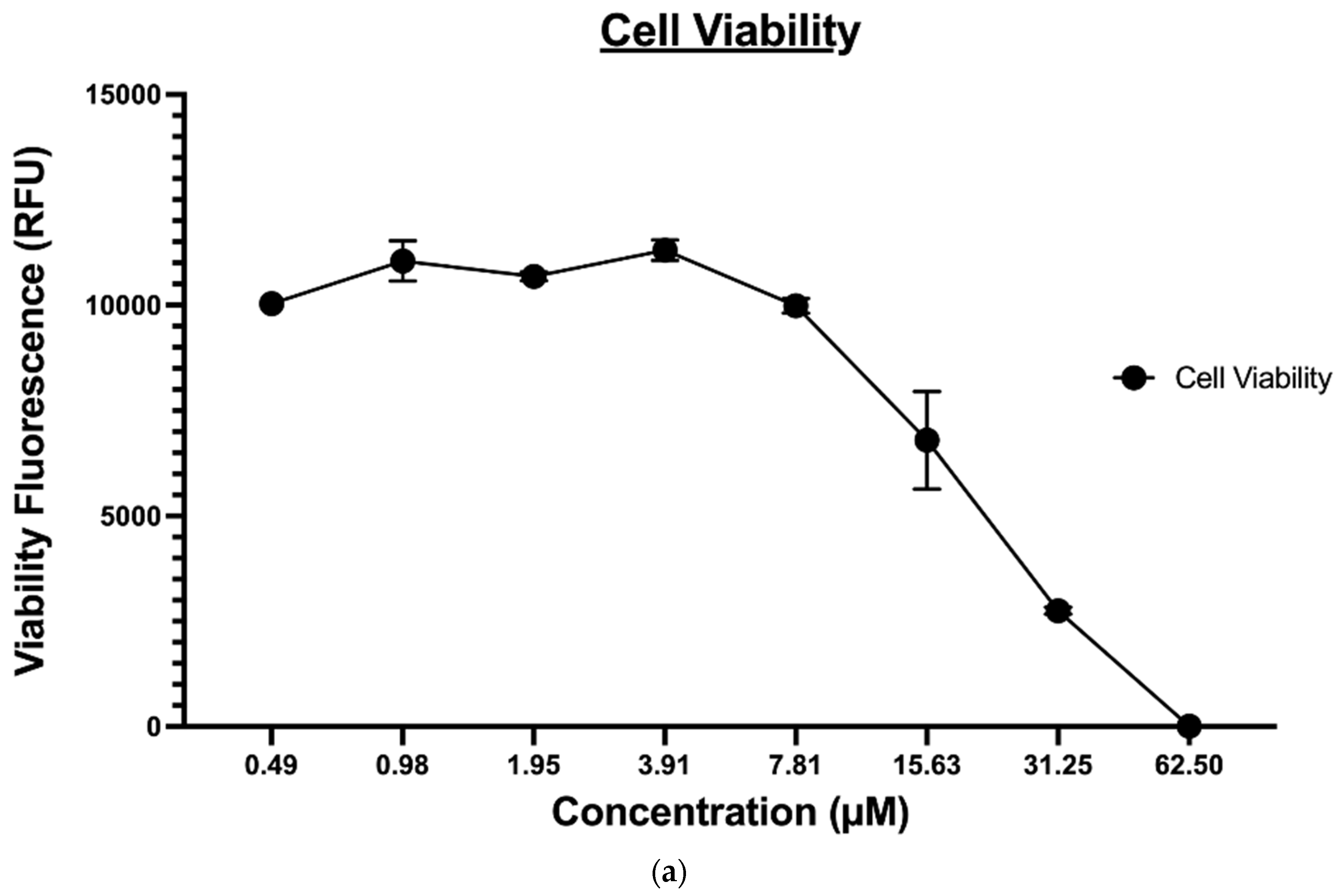
2.1.1. Cell Viability and Proliferation Analysis
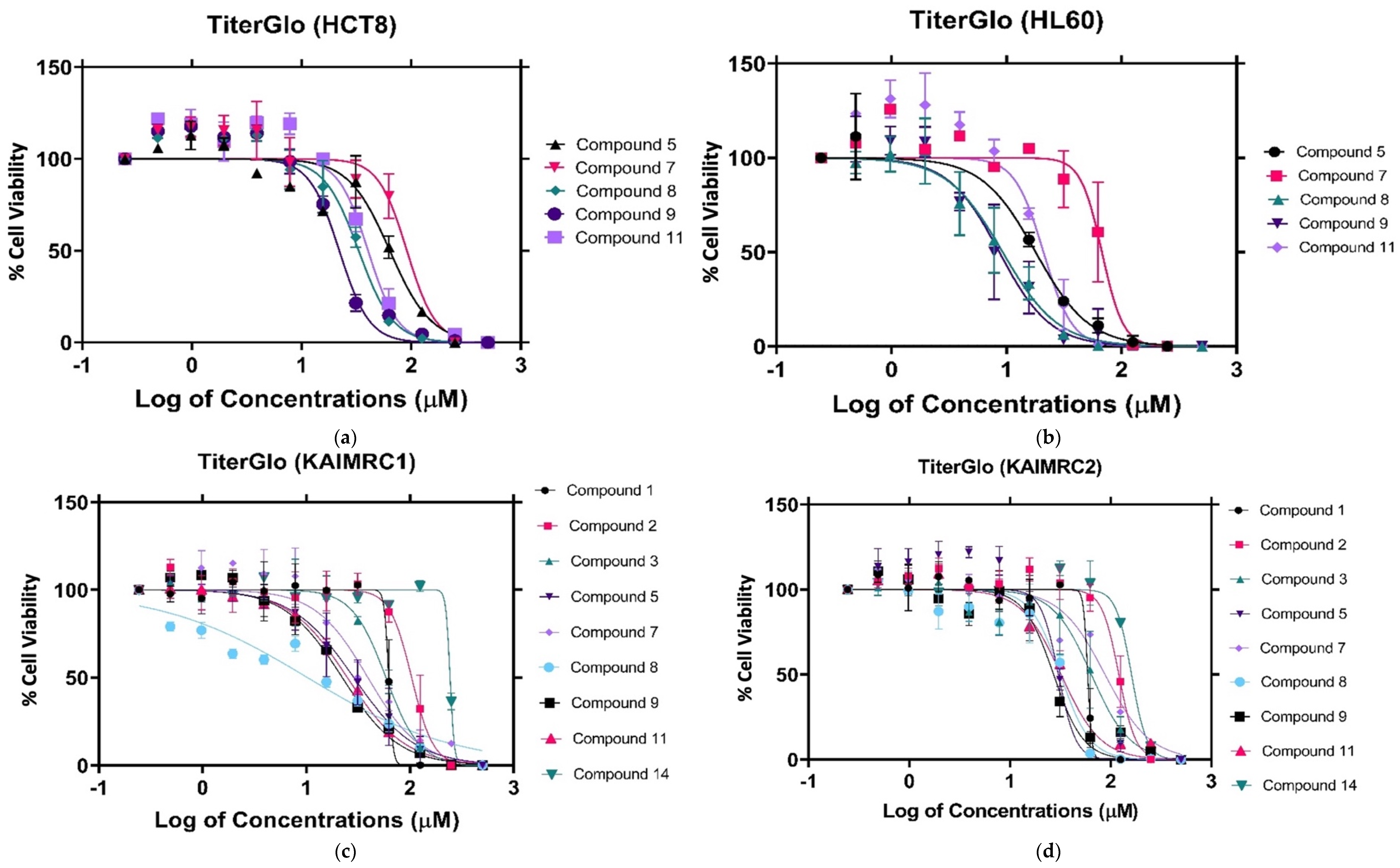
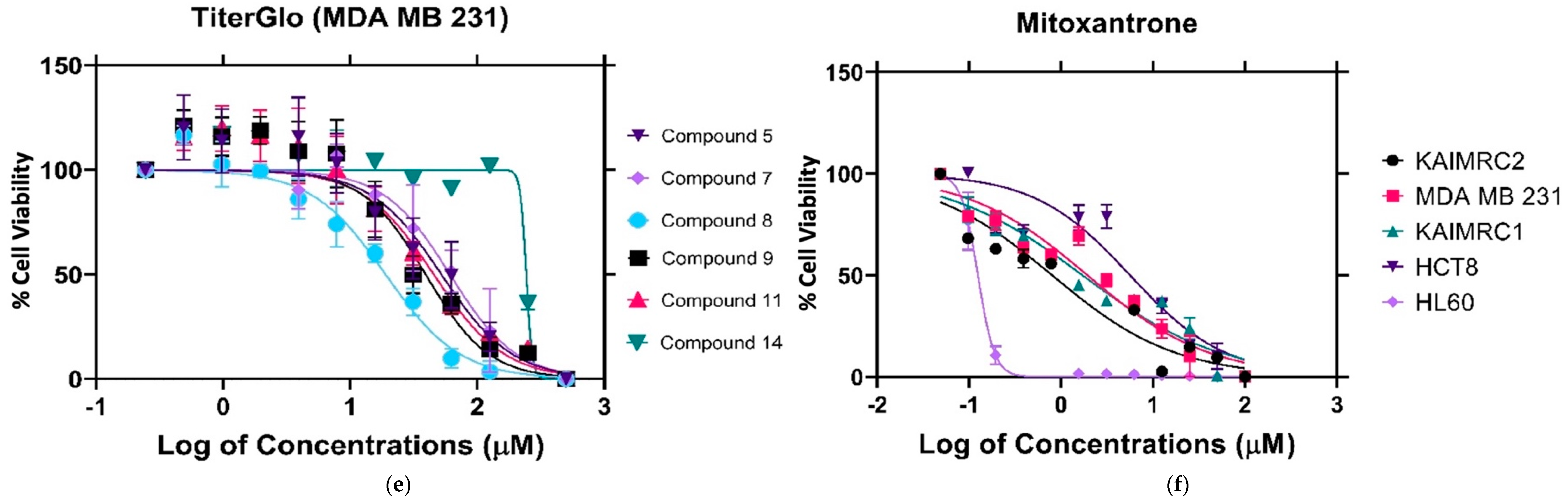
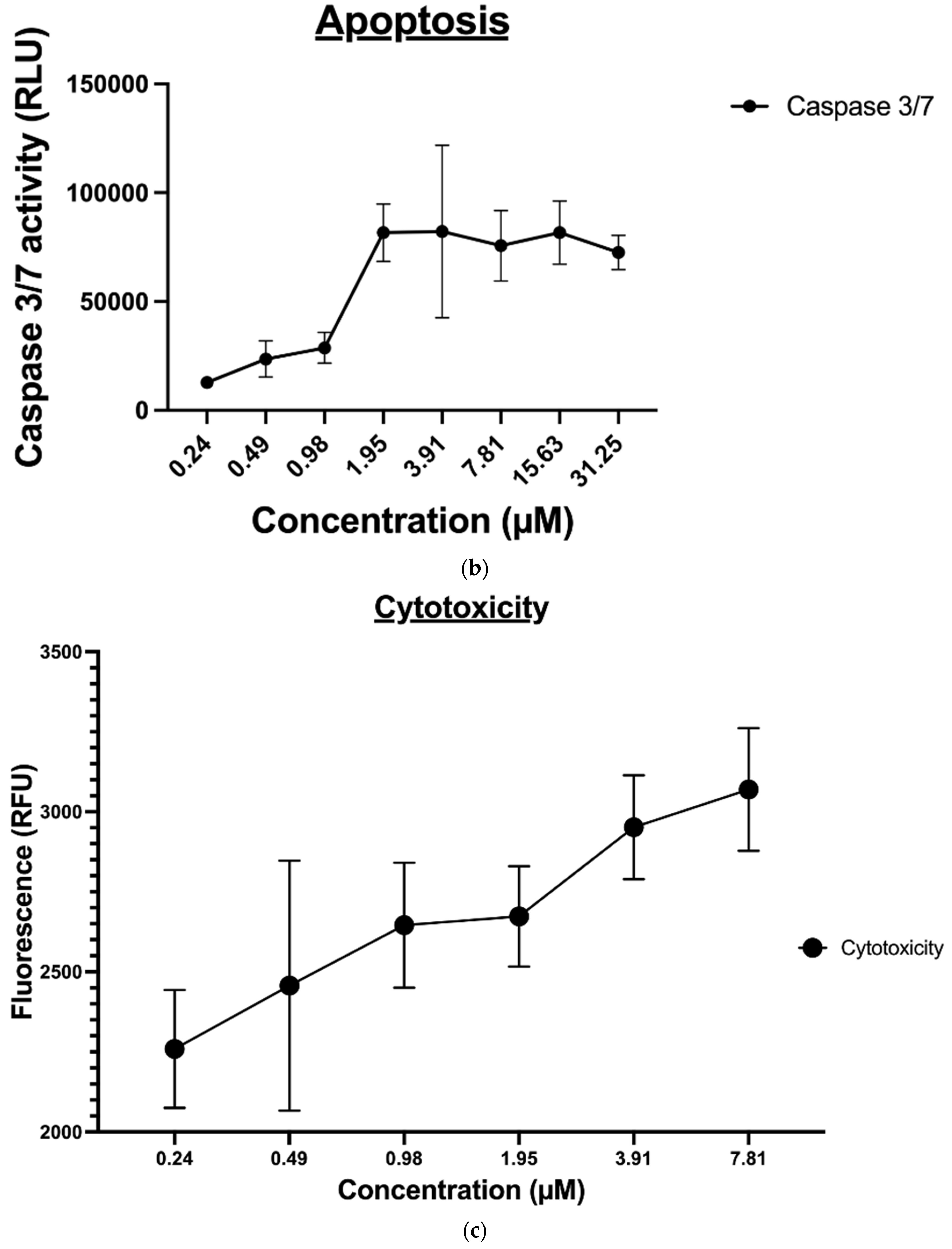
2.1.2. Cytotoxicity Evaluation of Nitrogen-based Derivatives using CellTiter-Glo Assay
2.1.3. High-Content Imaging (HCI)
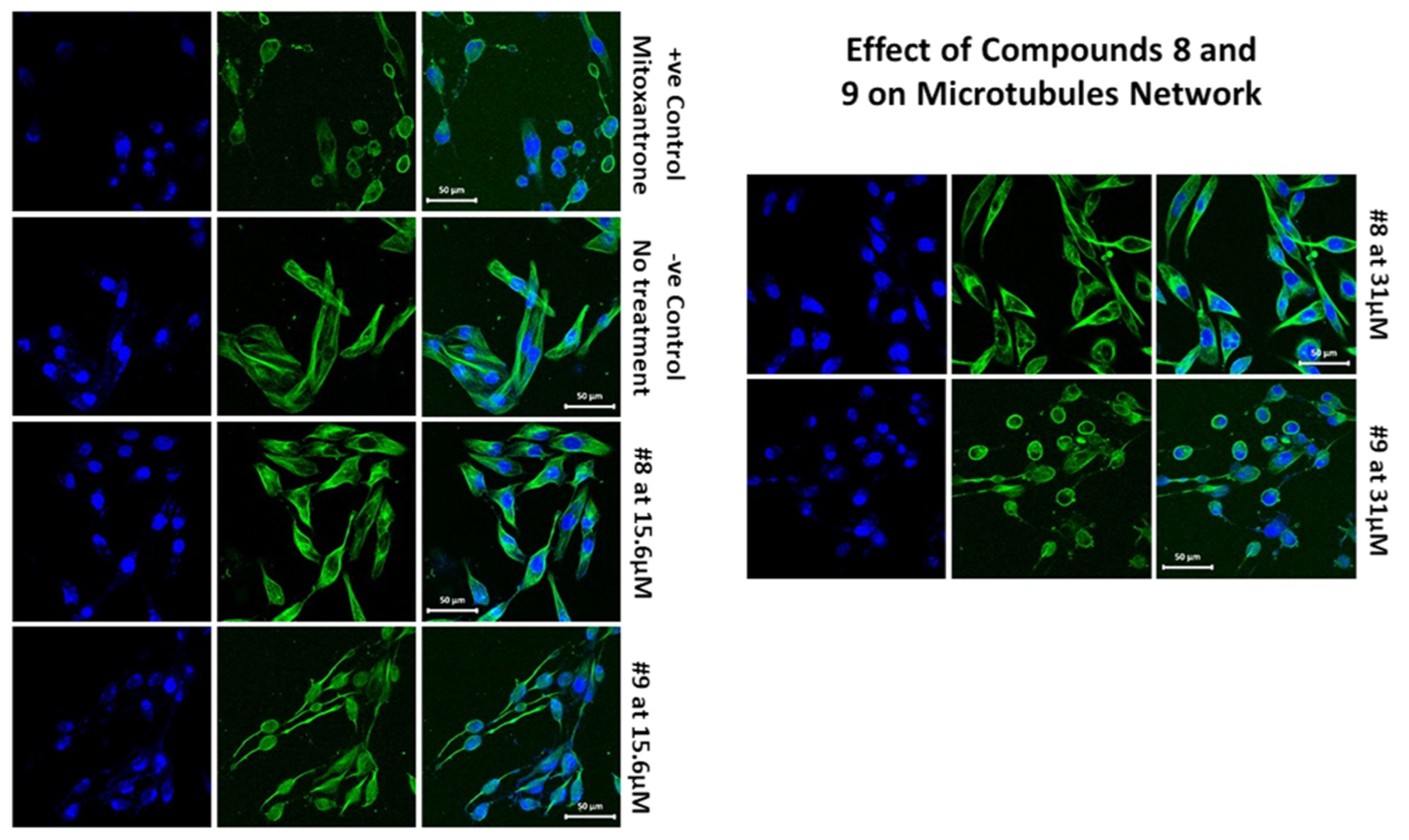
2.1.4. Effects of Compounds 8 and 9 on Microtubular Networks
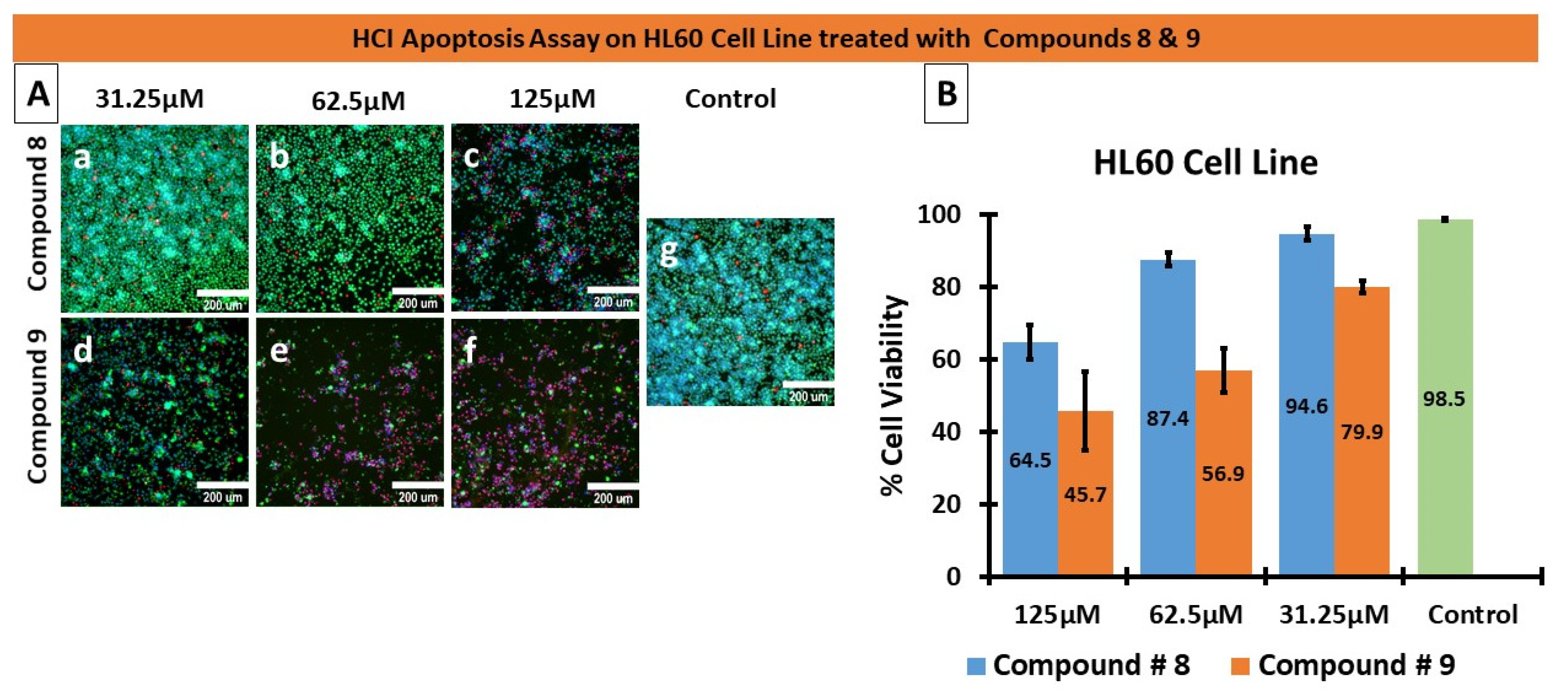
2.1.5. Apoptosis
2.2. Computational Studies
2.2.1. Anti-Cancer Activity and Molecular Target Prediction
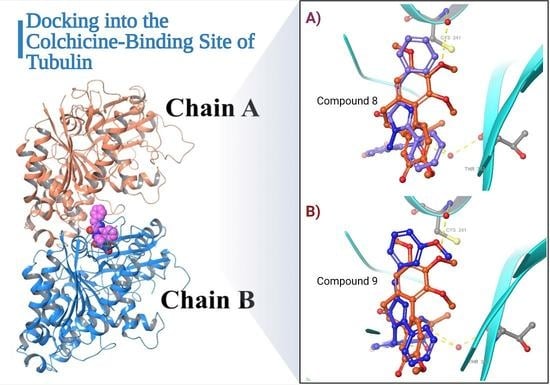
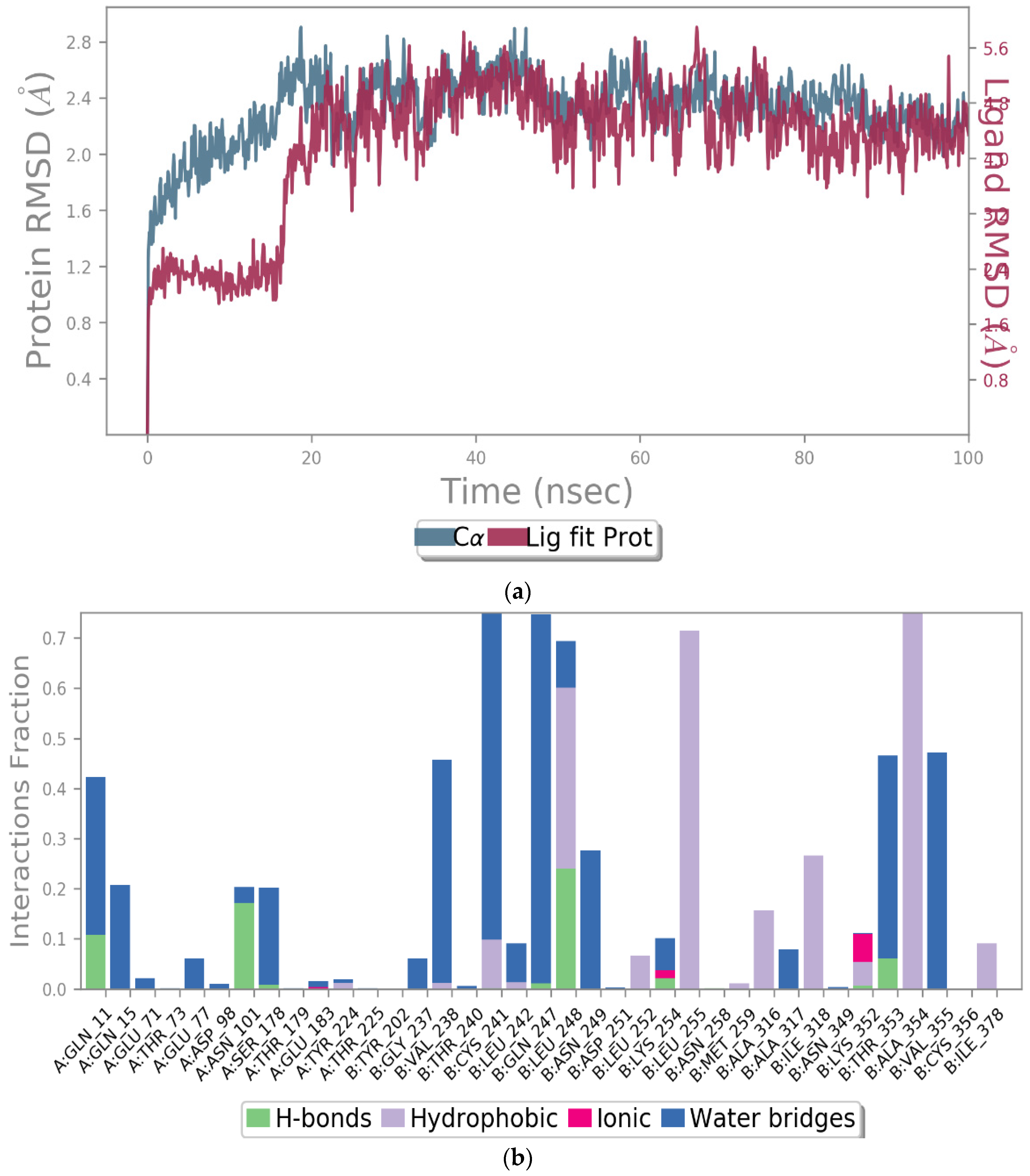
2.2.2. Molecular Docking and Dynamic Simulation with Tubulin Crystal Structure
2.2.3. Predictions of ADME Properties
2.2.4. Safety Profile Analysis—CYP P450 Enzyme Inhibition
2.2.5. Organ and Endpoint Toxicity Analysis
3. Materials and Methods
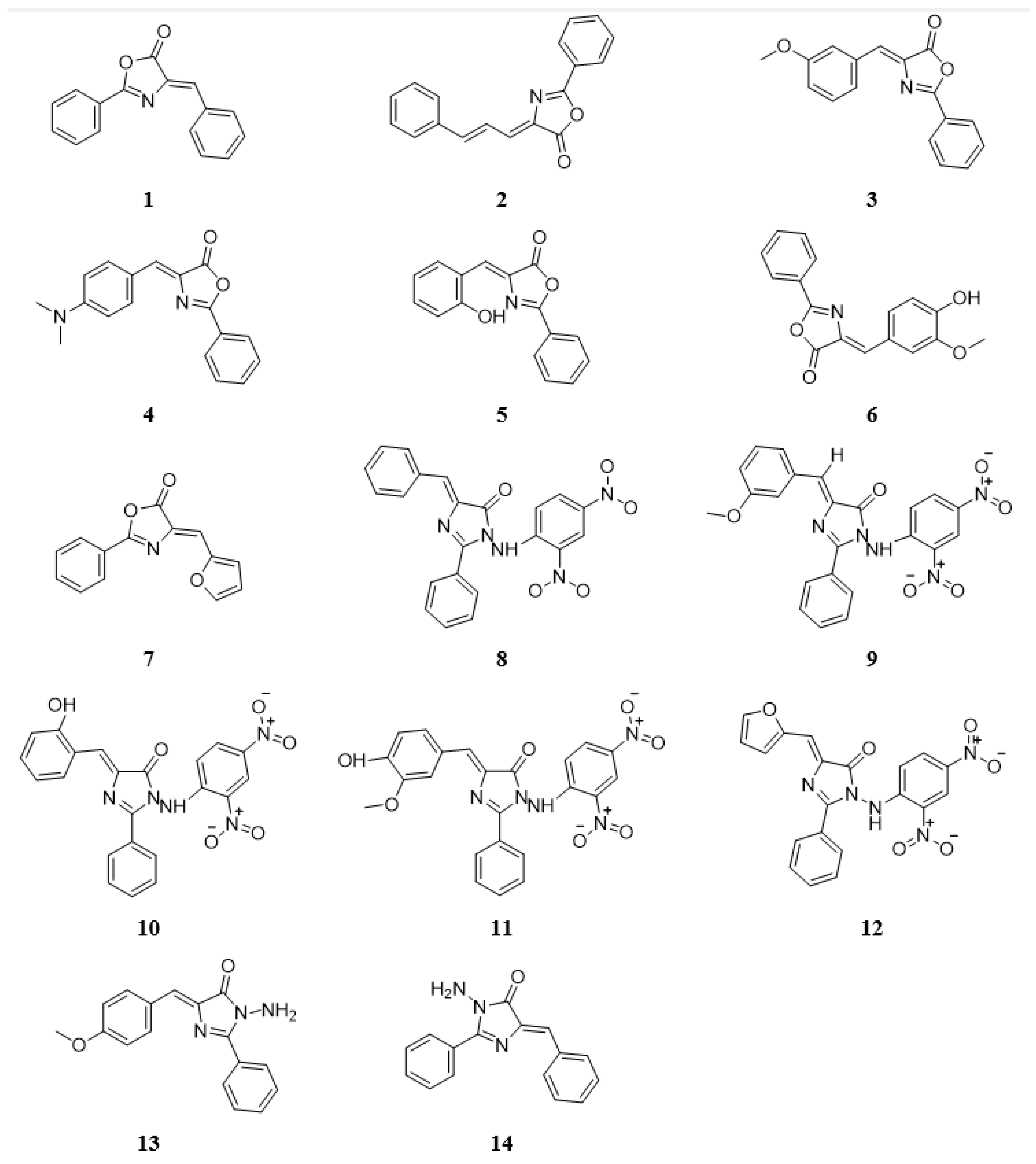
3.1. Nitrogenous Heterocycle Samples
3.2. Anti-Cancer Activity Investigation
3.2.1. MTT Assay
3.2.2. CellTiter-Glo Assay
3.3. High Content Imaging
3.4. Tubulin Staining and Imaging
3.5. Apoptosis
3.6. Computational Methods:
3.6.1. Anti-Cancer Activity Prediction
3.6.2. Molecular Target Predictions
3.6.3. Molecular Docking and MM-GBSA Binding Free-Energy Calculations with Tubulin Crystal Structure
3.6.4. Molecular Dynamic Simulation with Tubulin Crystal Structure
3.6.5. Prediction of ADME/T Properties
3.6.6. Safety Profile Analysis
CYP P450 Enzyme Inhibition
Organ and Endpoint Toxicity Analysis
4. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| ATCC | American Type Culture Collection |
| ATP | Adenosine triphosphate |
| BBB | Blood–brain barrier |
| BCI-2 | B-cell lymphoma 2 |
| CBSI | Colchicine-binding-site inhibitors |
| Log P | Lipophilicity |
| Log S | Solubility |
| MAPS | Microtubule-associated proteins |
| MaxTc | Max Tanimoto coefficients |
| MCF-10A | Human breast epithelial cell line |
| MDA-MB-321 | Human breast adenocarcinoma cell line |
| MMP | Mitochondrial Membrane Potential |
| NA | Not applicable |
| Pa | Probability of being active |
| PASS | Plan for Achieving Self Support |
| Pi | Probability of being inactive |
| PRa | Progesterone receptor alpha |
| RMSD | Root-mean-square deviation |
| SEA | Similarity ensemble approach |
| SP | Standard precision |
| VEGF | Vascular endothelial growth factor |
| XP | Extra precision |
| 3D | Three dimensional |
| ABC | ATP-binding cassette |
| ADME | Absorption, Distribution, Metabolism, and Excretion. |
| AKT-PI3 | protein kinase B/Phosphoinositide 3-kinase |
| CYP | Cytochrome P450 |
| DFF-45 | DNA fragmentation factor 45 |
| ER-LBD | Estrogen Receptor Ligand-Binding Domain |
| Era | Estrogen receptor alpha |
| HBSS | Hank’s Balanced Salt Solution |
| HCI | High content imagining |
| HCT8 | Human ileocecal adenocarcinoma cell line |
| HL60 | Human leukemia cell line |
| HTS | High-throughput screening |
| IC50 | Half-maximal inhibitory concentration |
| JAK-STAT | Janus Kinase/Signal Transducer and Activator of Transcription. |
| KAIMRC1 | King Abdullah international medical research center 1 cell line |
| KAIMRC2 | King Abdullah international medical research center 1 cell line |
| LD | Lethal dose |
References
- Ma, G.; Chong, L.; Li, Z.; Cheung, A.H.; Tattersall, M.H. Anticancer activities of sesquiterpene lactones from Cyathocline purpurea in vitro. Cancer Chemother. Pharmacol. 2009, 64, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Gudat, D. Diazaphospholenes: N-Heterocyclic phosphines between molecules and Lewis pairs. Acc. Chem. Res. 2010, 43, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.S.; Ko, S.B.; Walters, N.R.; Kang, Y.; Sauriol, F.; Wang, S. Formation of Azaborines by Photoelimination of B, N-Heterocyclic compounds. Angew. Chem. 2013, 125, 4642–4646. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, G.; Li, Y.; Wang, S.; Lei, A. The synergistic effect of self-assembly and visible-light induced the oxidative C–H acylation of N-heterocyclic aromatic compounds with aldehydes. Chem. Commun. 2018, 54, 5744–5747. [Google Scholar] [CrossRef]
- Matesanz, A.I.; Herrero, J.M.; Quiroga, A.G. Chemical and biological evaluation of thiosemicarbazone-bearing heterocyclic metal complexes. Curr. Top. Med. Chem. 2021, 21, 59–72. [Google Scholar] [CrossRef]
- Bezenšek, J.; Grošelj, U.; Stare, K.; Svete, J.; Stanovnik, B. Transformations of enaminones. A simple one-pot synthesis of imidazolone derivatives. Tetrahedron 2012, 68, 516–522. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Sreekala, C.; Zhang, Z.; Budhraja, A.; Ding, S.; Son, Y.-O.; Wang, X.; Hitron, A.; Hyun-Jung, K.; Wang, L. Cancer prevention with promising natural products: Mechanisms of action and molecular targets. Anti Cancer Agents Med. Chem. 2012, 12, 1159–1184. [Google Scholar] [CrossRef]
- Khwaza, V.; Oyedeji, O.O.; Aderibigbe, B.A. Ursolic acid-based derivatives as potential anti-cancer agents: An update. Int. J. Mol. Sci. 2020, 21, 5920. [Google Scholar] [CrossRef]
- Crews, P.; Clark, D.P.; Tenney, K. Variation in the alkaloids among Indo-Pacific Leucetta sponges. J. Nat. Prod. 2003, 66, 177–182. [Google Scholar] [CrossRef]
- Koźmiński, P.; Halik, P.K.; Chesori, R.; Gniazdowska, E. Overview of dual-acting drug methotrexate in different neurological diseases, autoimmune pathologies and cancers. Int. J. Mol. Sci. 2020, 21, 3483. [Google Scholar] [CrossRef]
- Chamorro-Petronacci, C.; García-García, A.; Lorenzo-Pouso, A.-I.; Gómez-García, F.J.; Padín-Iruegas, M.-E.; Gándara-Vila, P.; Blanco-Carrión, A.; Pérez-Sayáns, M. Management options for low-dose methotrexate-induced oral ulcers: A systematic review. Med. Oral Patol. Oral Cir. Bucal 2019, 24, e181. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Joshi, S.; Singh, D. Imidazole: Having versatile biological activities. J. Chem. 2013, 2013, 3294–3306. [Google Scholar] [CrossRef]
- Takagi, K.; Sugihara, K.; Ohta, J.; Yuki, Y.; Matsuoka, S.-I.; Suzuki, M. Conjugated oligomers containing imidazole in main chain with intramolecular hydrogen bonding. Polym. J. 2008, 40, 614–621. [Google Scholar] [CrossRef] [Green Version]
- Solomatina, A.I.; Chelushkin, P.S.; Krupenya, D.V.; Podkorytov, I.S.; Artamonova, T.O.; Sizov, V.V.; Melnikov, A.S.; Gurzhiy, V.V.; Koshel, E.I.; Shcheslavskiy, V.I. Coordination to imidazole ring switches on phosphorescence of platinum cyclometalated complexes: The route to selective labeling of peptides and proteins via histidine residues. Bioconjug. Chem. 2017, 28, 426–437. [Google Scholar] [CrossRef]
- Adams, C.J.; Haddow, M.F.; Hughes, R.J.; Kurawa, M.A.; Orpen, A.G. Coordination chemistry of platinum and palladium in the solid-state: Synthesis of imidazole and pyrazole complexes. Dalton Trans. 2010, 39, 3714–3724. [Google Scholar] [CrossRef]
- Connors, T.A.; Roberts, J. Platinum Coordination Complexes in Cancer Chemotherapy; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; Volume 48. [Google Scholar]
- Wang, S.; Cai, C.; Shen, Y.; Sun, C.; Shi, Q.; Wu, N.; Zheng, S.; Qian, J.; Zhang, R.; Zhou, H. In vitro activity of contezolid against methicillin-resistant staphylococcus aureus, vancomycin-resistant enterococcus, and strains with linezolid resistance genes from China. Front. Microbiol. 2021, 13, 2408. [Google Scholar] [CrossRef]
- Schecter, G.; Scott, C.; True, L.; Raftery, A.; Flood, J.; Mase, S. Linezolid in the treatment of multidrug-resistant tuberculosis. Clin. Infect. Dis. 2010, 50, 49–55. [Google Scholar] [CrossRef]
- Bala, S.; Saini, M.; Kamboj, S. Methods for synthesis of Oxazolones: A review. Int. J. Chem. Tech. Res. 2011, 3, 1102–1118. [Google Scholar]
- Shalini, K.; Sharma, P.K.; Kumar, N. Imidazole and its biological activities: A review. Der. Chem. Sin. 2010, 1, 36–47. [Google Scholar]
- Liu, J.; Qiu, J.; Wang, M.; Wang, L.; Su, L.; Gao, J.; Gu, Q.; Xu, J.; Huang, S.-L.; Gu, L.-Q. Synthesis and characterization of 1H-phenanthro [9, 10-d] imidazole derivatives as multifunctional agents for treatment of Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1840, 2886–2903. [Google Scholar] [CrossRef]
- Farh, I.K.I. Synthesis of Some Oxazolone and Imidazole Derivatives. Ph.D. Thesis, Sudan University of Science and Technology, Khartoum, Sudan, 2016. [Google Scholar]
- Tolosa, L.; Donato, M.T.; Gómez-Lechón, M.J. General cytotoxicity assessment by means of the MTT assay. In Protocols in In Vitro Hepatocyte Research; Springer: Berlin/Heidelberg, Germany, 2015; pp. 333–348. [Google Scholar]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. Assay Guidance Manual. 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK144065/ (accessed on 19 March 2018).
- Chacon, E.; Acosta, D.; Lemasters, J.J. Primary cultures of cardiac myocytes as in vitro models for pharmacological and toxicological assessments. In Vitro Methods Pharm. Res. 1997, 209–223. [Google Scholar] [CrossRef]
- Riss, T. Is Your MTT Assay Really the Best Choice. (Promega™, Madison, Wisconsin, United States). 2017. Available online: http://www.promega.in/resources/pubhub/is-your-mtt-assay-really-the-best-choice/ (accessed on 19 March 2018).
- Eckert, L.B.; Repasky, G.A.; Ülkü, A.S.; McFall, A.; Zhou, H.; Sartor, C.I.; Der, C.J. Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res. 2004, 64, 4585–4592. [Google Scholar] [CrossRef] [Green Version]
- Ali, R.; Samman, N.; Al Zahrani, H.; Nehdi, A.; Rahman, S.; Khan, A.L.; Al Balwi, M.; Alriyees, L.A.; Alzaid, M.; Al Askar, A. Isolation and characterization of a new naturally immortalized human breast carcinoma cell line, KAIMRC1. BMC Cancer 2017, 17, 803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, R.; Al Zahrani, H.; Barhoumi, T.; Alhallaj, A.; Mashhour, A.; Alshammari, M.A.; Alshawakir, Y.A.; Baz, O.; Alanazi, A.H.; Khan, A.L. Isolation and establishment of a highly proliferative, cancer stem cell-like, and naturally immortalized triple-negative breast cancer cell line, KAIMRC2. Cells 2021, 10, 1303. [Google Scholar] [CrossRef]
- Ali, I.; Lone, M.N.; Aboul-Enein, H.Y. Imidazoles as potential anticancer agents. Med. Chem. Comm. 2017, 8, 1742–1773. [Google Scholar] [CrossRef]
- Hamel, E. Natural products which interact with tubulin in the vinca domain: Maytansine, rhizoxin, phomopsin A, dolastatins 10 and 15 and halichondrin B. Pharmacol. Ther. 1992, 55, 31–51. [Google Scholar] [CrossRef]
- Jordan, A.; Hadfield, J.A.; Lawrence, N.J.; McGown, A.T. Tubulin as a target for anticancer drugs: Agents which interact with the mitotic spindle. Med. Res. Rev. 1998, 18, 259–296. [Google Scholar] [CrossRef]
- Komuraiah, B.; Ren, Y.; Xue, M.; Cheng, B.; Liu, J.; Liu, Y.; Chen, J. Design, synthesis and biological evaluation of benz-fused five-membered heterocyclic compounds as tubulin polymerization inhibitors with anticancer activities. Chem. Biol. Drug Des. 2021, 97, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Srinivasa, V.; Rangappa, S.; Mervin, L.; Mohan, S.; Paricharak, S.; Baday, S.; Li, F.; Shanmugam, M.K.; Chinnathambi, A. Trisubstituted-imidazoles induce apoptosis in human breast cancer cells by targeting the oncogenic PI3K/Akt/mTOR signaling pathway. PLoS ONE 2016, 11, e0153155. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, K.; Usui, S.; Ishida, R.; Hirano, K. Imidazole-induced cell death, associated with intracellular acidification, caspase-3 activation, DFF-45 cleavage, but not oligonucleosomal DNA fragmentation. Apoptosis 2002, 7, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Patel, P.; Zhang, L.; Evans, H.; Westwell, A.D.; Fischer, P.M.; Chan, S.; Martin, S. 2-[(1-methylpropyl) dithio]-1H-imidazole inhibits tubulin polymerization through cysteine oxidation. Mol. Cancer Ther. 2008, 7, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, F.; Eugenia Garcia-Rubino, M.; Lozano-Lopez, C.; Kawano, D.; Eifler-Lima, V.; von Poser, G.; Campos, J. Imidazoles and benzimidazoles as tubulin-modulators for anti-cancer therapy. Curr. Med. Chem. 2015, 22, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Quan, D.; Chen, J.; Ding, J.; Zhao, J.; Lv, L.; Chen, J. Design, synthesis, and biological evaluation of 1-substituted-2-aryl imidazoles targeting tubulin polymerization as potential anticancer agents. Eur. J. Med. Chem. 2019, 184, 111732. [Google Scholar] [CrossRef]
- Alghamdi, S.S.; Suliman, R.S.; Almutairi, K.; Kahtani, K.; Aljatli, D. Imidazole as a promising medicinal scaffold: Current status and future direction. Drug Des. Dev. Ther. 2021, 15, 3289. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of tau as a microtubule-associated protein: Structural and functional aspects. Front. Aging Neurosci. 2019, 204. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today: Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- O’Connor, R. The pharmacology of cancer resistance. Anticancer. Res. 2007, 27, 1267–1272. [Google Scholar]
- Lynch, T.; Neff, A.P. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, P.; Bari, S.; Surana, S.; Shirkhedkar, A.; Wakode, S.; Shelar, S.; Racharla, S.; Ugale, V.; Ghodke, M. Logical synthetic strategies and structure-activity relationship of indolin-2-one hybrids as small molecule anticancer agents: An overview. J. Mol. Struct. 2022, 1247, 131280. [Google Scholar] [CrossRef]
- Yusof, E.N.B.M. Synthesis, Structural Characterization and Cytotoxicity Study of Tin (Iv) Compounds Containing ONS Schiff Bases. Ph.D. Thesis, The University of Newcastle, Newcastle, Australia, 2019. [Google Scholar]
- Erwin, W.M.; Islam, D.; Inman, R.D.; Fehlings, M.G.; Tsui, F.W. Notochordal cells protect nucleus pulposus cells from degradation and apoptosis: Implications for the mechanisms of intervertebral disc degeneration. Arthritis Res. Ther. 2011, 13, R215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargazi, A.; Shiri, F.; Keikha, S.; Majd, M.H. Hyaluronan magnetic nanoparticle for mitoxantrone delivery toward CD44-positive cancer cells. Colloids Surf. B Biointerfaces 2018, 171, 150–158. [Google Scholar] [CrossRef]
- Pavlica, S.; Gaunitz, F.; Gebhardt, R. Comparative in vitro toxicity of seven zinc-salts towards neuronal PC12 cells. Toxicol. In Vitro 2009, 23, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Badmus, J.; Ekpo, O.; Hussein, A.; Meyer, M.; Hiss, D. Antiproliferative and apoptosis induction potential of the methanolic leaf extract of Holarrhena floribunda (G. Don). Evid. Based Complementary Altern. Med. 2015, 2015, 74–88. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, H.; Akaki, J.; Nakamura, S.; Okazaki, Y.; Kojima, H.; Tamesada, M.; Yoshikawa, M. Apoptosis-inducing effects of sterols from the dried powder of cultured mycelium of Cordyceps sinensis. Chem. Pharm. Bull. 2009, 57, 411–414. [Google Scholar] [CrossRef] [Green Version]
- Filimonov, D.; Lagunin, A.; Gloriozova, T.; Rudik, A.; Druzhilovskii, D.; Pogodin, P.; Poroikov, V.J.C. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K.J.N. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Alghamdi, S.S.; Suliman, R.S.; Alsaeed, A.S.; Almutairi, K.K.; Aljammaz, N.A.; Altolayyan, A.; Ali, R.; Alhallaj, A.J.D.D. Novel anti-tubulin compounds from Trigonella foenum-graecum seeds; Insights into in-vitro and molecular docking studies. Drug Des. Dev. Ther. 2021, 15, 4195. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V.J.S. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Names | Breast Cancer | Non-Malignant Breast Epithelial Cells | Colorectal Cancer |
|---|---|---|---|
| (MDA-MB-231) | (MCF-10A) | (HCT8) | |
| 1 | 22.88 | N/A | 51.15 |
| 2 | 43.55 | 65.5 | 72.22 |
| 3 | 39.95 | NA | N/A |
| 4 | 69.44 | NA | N/A |
| 5 | 39.56 | 34.20 | 39.06 |
| 6 | 50.65 | 50.38 | 70.83 |
| 7 | 22.96 | 62.99 | 73.55 |
| 8 | 4.759 | 30.66 | 29.53 |
| 9 | 17.02 | 9.516 | 13.20 |
| 10 | 56.02 | N/A | N/A |
| 11 | 57.15 | N/A | N/A |
| 12 | 50.75 | N/A | N/A |
| 13 | 78.46 | N/A | N/A |
| 14 | 138.7 | N/A | N/A |
| Mitoxantrone | 3.171 | 2.898 | 0.7113 |
| Compound. | Leukemia | Breast Cancer | Colorectal Cancer | ||
|---|---|---|---|---|---|
| HL60 | MDA-MB-231 | KAIMRC1 | KAIMRC2 | HCT8 | |
| 1 | N/A | N/A | 62.21 | 59.21 | N/A |
| 2 | N/A | N/A | 103.6 | 120.6 | 121.3 |
| 3 | N/A | N/A | 58.56 | 64.32 | N/A |
| 4 | N/A | N/A | N/A | N/A | N/A |
| 5 | 17.86 | 52.13 | 28.64 | 30.30 | 63.70 |
| 6 | NA | N/A | N/A | N/A | N/A |
| 7 | 67.43 | 60.91 | 39.18 | 87.93 | 91.22 |
| 8 | 9.237 | 18.97 | 10.20 | 31.64 | 33.11 |
| 9 | 8.632 | 39.19 | 22.18 | 27.11 | 22.05 |
| 10 | N/A | N/A | N/A | N/A | N/A |
| 11 | 20.92 | 45.42 | 24.81 | 32.16 | 40.35 |
| 12 | N/A | N/A | N/A | N/A | N/A |
| 13 | N/A | N/A | N/A | N/A | N/A |
| 14 | N/A | 242.7 | 243.4 | 161.2 | N/A |
| Mitoxantrone | 0.1252 | 1.936 | 1.713 | 0.8008 | 5.618 |
| Compound | PASS Online | SEA Search Server | ||
|---|---|---|---|---|
| Anticancer | Microtubule-Associated Protein Tau | |||
| Pa | Pi | p Value | MaxTC | |
| 1 | Inactive | Inactive | Inactive | |
| 2 | Inactive | Inactive | 9.42 × 10−7 | 0.37 |
| 3 | 0.258 | 0.180 | Inactive | |
| 4 | 0.242 | 0.192 | 1.864 × 10−38 | 0.43 |
| 5 | 0.224 | 0.208 | Inactive | |
| 6 | 0.364 | 0.119 | 2.801 × 10−33 | 0.44 |
| 7 | Inactive | Inactive | Inactive | |
| 8 | 0.413 | 0.099 | 1.544 × 10−6 | 0.32 |
| 9 | 0.434 | 0.092 | 1.137 × 10−6 | 0.34 |
| 10 | 0.411 | 0.100 | Inactive | |
| 11 | 0.498 | 0.072 | 3.343 × 10−26 | 0.36 |
| 12 | Inactive | Inactive | Inactive | |
| 13 | 0.495 | 0.073 | 7.011 × 10−10 | 0.34 |
| 14 | 0.493 | 0.074 | Inactive | |
| Compound Name | Docking Scores (Kcal/mol) | MMGBSA dG Bind (Kcal/mol) |
|---|---|---|
| Colchicine | −10.10 | −88.61 |
| Mitoxantrone | −10.42 | −42.31 |
| Compound 8 | −7.56 | −22.47 |
| Compound 9 | −7.40 | −42.35 |
| Compound | GI Absorption | BBB Penetration | P-gp Substrate | Log Po/w | Log S | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| SwissADME | Qikprop (% Absorption) | SwissADME | Qikprop | SwissADME | Qikprop | SwissADME | Qikprop | SwissADME | Qikprop | |
| 1 | High | 100 | Yes | 0 | No | N/A | 2.54 | 2.87 | −5.44 | −3.529 |
| 2 | N/A | 100 | N/A | 0 | N/A | N/A | 3.10 | 3.586 | N/A | −4.385 |
| 3 | High | 100 | Yes | 0 | No | N/A | 2.55 | 2.869 | −5.56 | −3.383 |
| 4 | High | 100 | Yes | 0 | No | N/A | 2.61 | 3.249 | −5.54 | −4.305 |
| 5 | High | 92.21 | Yes | −1 | No | N/A | 2.25 | 2.81 | −4.86 | −3.657 |
| 6 | High | 91.67 | Yes | −1 | No | N/A | 2.26 | 2.459 | −4.98 | −3.721 |
| 7 | High | 100 | Yes | 0 | No | N/A | 2.13 | 2.105 | −4.66 | −2.525 |
| 8 | Low | 80.14 | No | −2 | No | N/A | 3.10 | 3.049 | −6.26 | −5.222 |
| 9 | Low | 67.92 | No | −2 | No | N/A | 3.11 | 3.174 | −6.35 | −5.164 |
| 10 | Low | 52.27 | No | −2 | No | N/A | 2.80 | 2.607 | −5.67 | −5.677 |
| 11 | Low | 55.65 | No | −2 | No | N/A | 2.81 | 2.520 | −5.76 | −4.877 |
| 12 | Low | 62.87 | No | −2 | No | N/A | 2.69 | 2.382 | −5.48 | −4.231 |
| 13 | High | 95.79 | Yes | −1 | No | N/A | 1.33 | 2.452 | −4.79 | −3.936 |
| 14 | High | 96.06 | Yes | 0 | No | N/A | 1.32 | 2.501 | −4.68 | −3.766 |
| Compound | CYP 1A2 | CYP 2C19 | CYP 2C9 | CYP 2D6 | CYP 3A4 |
|---|---|---|---|---|---|
| 1 | Yes | Yes | Yes | No | No |
| 2 | N/A | N/A | N/A | N/A | N/A |
| 3 | Yes | Yes | Yes | No | No |
| 4 | Yes | Yes | Yes | No | No |
| 5 | Yes | No | No | No | No |
| 6 | Yes | Yes | Yes | No | Yes |
| 7 | Yes | Yes | No | No | No |
| 8 | No | Yes | Yes | No | No |
| 9 | No | Yes | Yes | No | Yes |
| 10 | No | Yes | Yes | No | No |
| 11 | No | Yes | Yes | No | Yes |
| 12 | No | Yes | Yes | No | Yes |
| 13 | Yes | No | No | No | No |
| 14 | Yes | No | No | No | No |
| Compound | Oral Toxicity | Prediction of Active Organ Toxicity and Toxicity Endpoints | Probability | |
|---|---|---|---|---|
| Predicted LD50 (mg/kg) | Predicted Toxicity Class | |||
| 1 | 1190 | 4 | Hepatotoxicity | 0.69 |
| Immunotoxicity | 0.96 | |||
| Aromatase | 1.0 | |||
| Estrogen Receptor Alpha (ER) | 0.99 | |||
| Estrogen Receptor Ligand-Binding Domain (ER-LBD) | 1.0 | |||
| 2 | 3700 | 5 | Hepatotoxicity | 0.59 |
| Carcinogenicity | 0.56 | |||
| 3 | 1400 | 4 | Hepatotoxicity | 0.55 |
| Carcinogenicity | 0.57 | |||
| 4 | 5000 | 5 | Inactive | - |
| 5 | 3700 | 5 | Hepatotoxicity | 0.61 |
| Carcinogenicity | 0.58 | |||
| 6 | 978 | 4 | Hepatotoxicity | 0.56 |
| Carcinogenicity | 0.57 | |||
| 7 | 3500 | 5 | Hepatotoxicity | 0.59 |
| Carcinogenicity | 0.54 | |||
| 8 | 600 | 4 | Hepatotoxicity | 0.63 |
| Carcinogenicity | 0.68 | |||
| Mutagenicity | 0.82 | |||
| 9 | 600 | 4 | Hepatotoxicity | 0.62 |
| Carcinogenicity | 0.55 | |||
| Immunotoxicity | 0.62 | |||
| Mutagenicity | 0.78 | |||
| Mitochondrial Membrane Potential (MMP) | 0.57 | |||
| 10 | 600 | 4 | Hepatotoxicity | 0.63 |
| Carcinogenicity | 0.55 | |||
| Mutagenicity | 0.80 | |||
| Mitochondrial Membrane Potential (MMP) | 0.55 | |||
| 11 | 600 | 4 | Hepatotoxicity | 0.64 |
| Carcinogenicity | 0.54 | |||
| Immunotoxicity | 0.70 | |||
| Mutagenicity | 0.77 | |||
| Mitochondrial Membrane Potential (MMP) | 0.55 | |||
| 12 | 600 | 4 | Hepatotoxicity | 0.64 |
| Carcinogenicity | 0.73 | |||
| Mutagenicity | 0.83 | |||
| Mitochondrial Membrane Potential (MMP) | 0.57 | |||
| 13 | 800 | 4 | Hepatotoxicity | 0.66 |
| Carcinogenicity | 0.58 | |||
| Mutagenicity | 0.51 | |||
| 14 | 800 | 4 | Hepatotoxicity | 0.67 |
| Carcinogenicity | 0.71 | |||
| Color key | ||||
| Class 4: | Harmful if swallowed (300 < LD50 ≤ 2000) | |||
| Class 5: | It may be harmful if swallowed (2000 < LD50 ≤ 5000) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suliman, R.S.; Alghamdi, S.S.; Ali, R.; Rahman, I.; Alqahtani, T.; Frah, I.K.; Aljatli, D.A.; Huwaizi, S.; Algheribe, S.; Alehaideb, Z.; et al. Distinct Mechanisms of Cytotoxicity in Novel Nitrogenous Heterocycles: Future Directions for a New Anti-Cancer Agent. Molecules 2022, 27, 2409. https://doi.org/10.3390/molecules27082409
Suliman RS, Alghamdi SS, Ali R, Rahman I, Alqahtani T, Frah IK, Aljatli DA, Huwaizi S, Algheribe S, Alehaideb Z, et al. Distinct Mechanisms of Cytotoxicity in Novel Nitrogenous Heterocycles: Future Directions for a New Anti-Cancer Agent. Molecules. 2022; 27(8):2409. https://doi.org/10.3390/molecules27082409
Chicago/Turabian StyleSuliman, Rasha Saad, Sahar Saleh Alghamdi, Rizwan Ali, Ishrat Rahman, Tariq Alqahtani, Ibrahim K. Frah, Dimah A. Aljatli, Sarah Huwaizi, Shatha Algheribe, Zeyad Alehaideb, and et al. 2022. "Distinct Mechanisms of Cytotoxicity in Novel Nitrogenous Heterocycles: Future Directions for a New Anti-Cancer Agent" Molecules 27, no. 8: 2409. https://doi.org/10.3390/molecules27082409
APA StyleSuliman, R. S., Alghamdi, S. S., Ali, R., Rahman, I., Alqahtani, T., Frah, I. K., Aljatli, D. A., Huwaizi, S., Algheribe, S., Alehaideb, Z., & Islam, I. (2022). Distinct Mechanisms of Cytotoxicity in Novel Nitrogenous Heterocycles: Future Directions for a New Anti-Cancer Agent. Molecules, 27(8), 2409. https://doi.org/10.3390/molecules27082409