
Applications of Tandem Mass Spectrometry (MS/MS) in Protein Analysis for Biomedical Research

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. MS/MS Proteomics Experimental Design
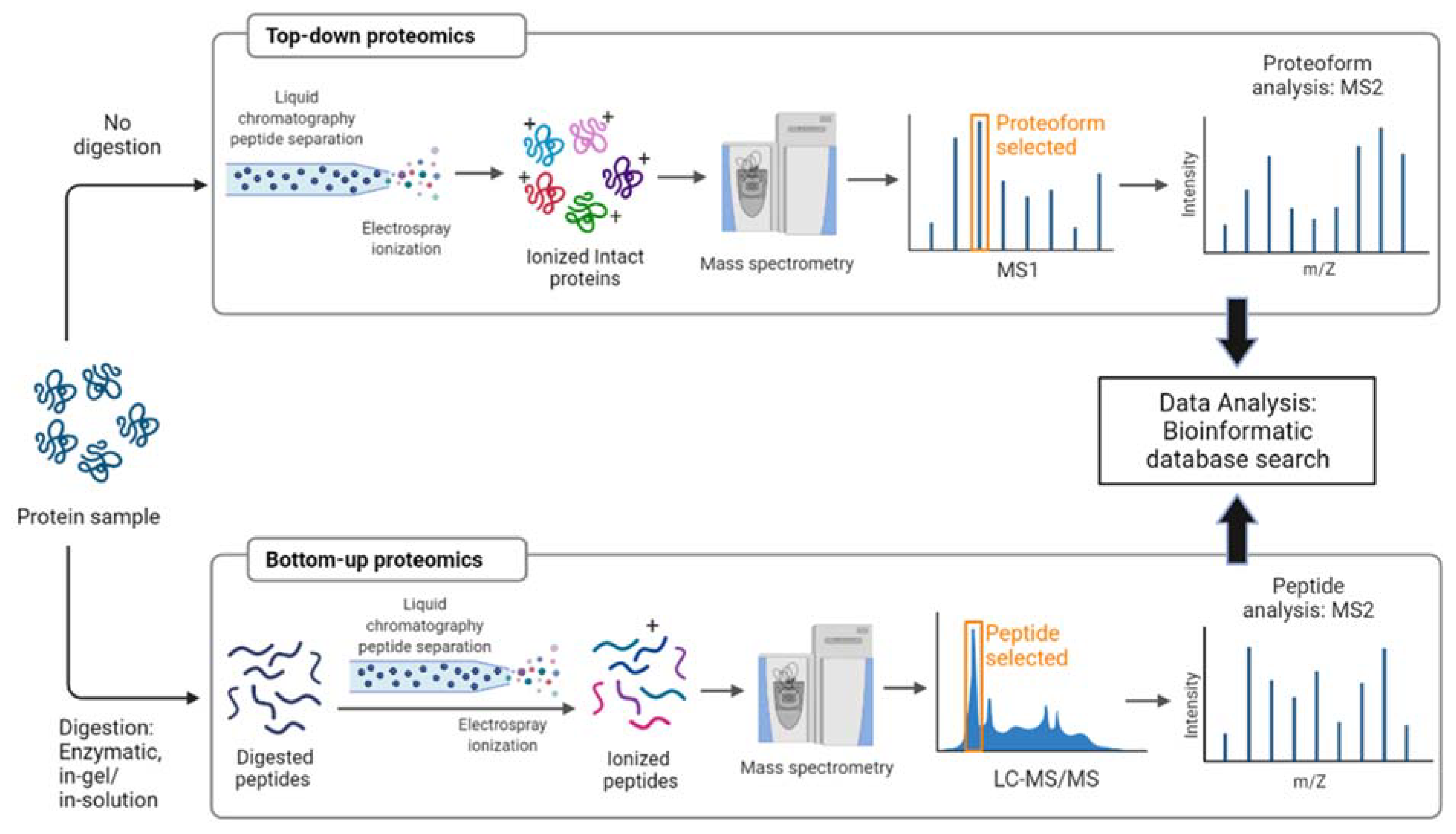
2.1. Sample Preparation
2.2. Sample Analysis (Fractionation, Ionization, and Analysis) and Data Acquisition
2.2.1. HPLC/UPLC-Based Fractionation Prior MS Analysis
2.2.2. Sample Ionization Using MALDI-MS or ESI-MS
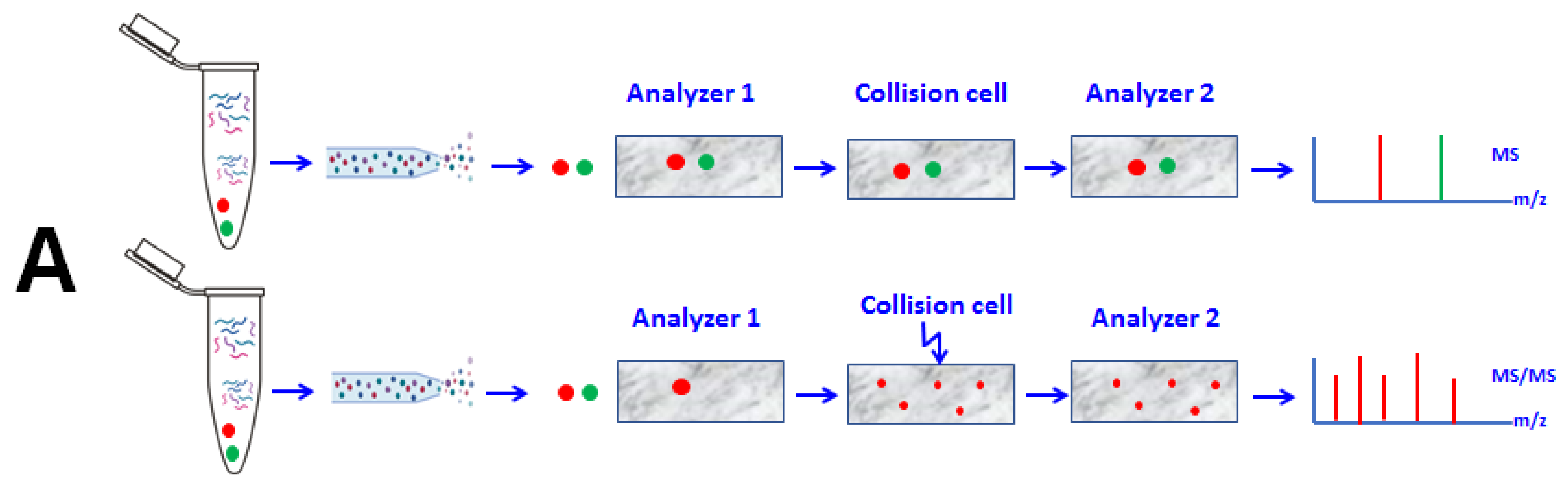
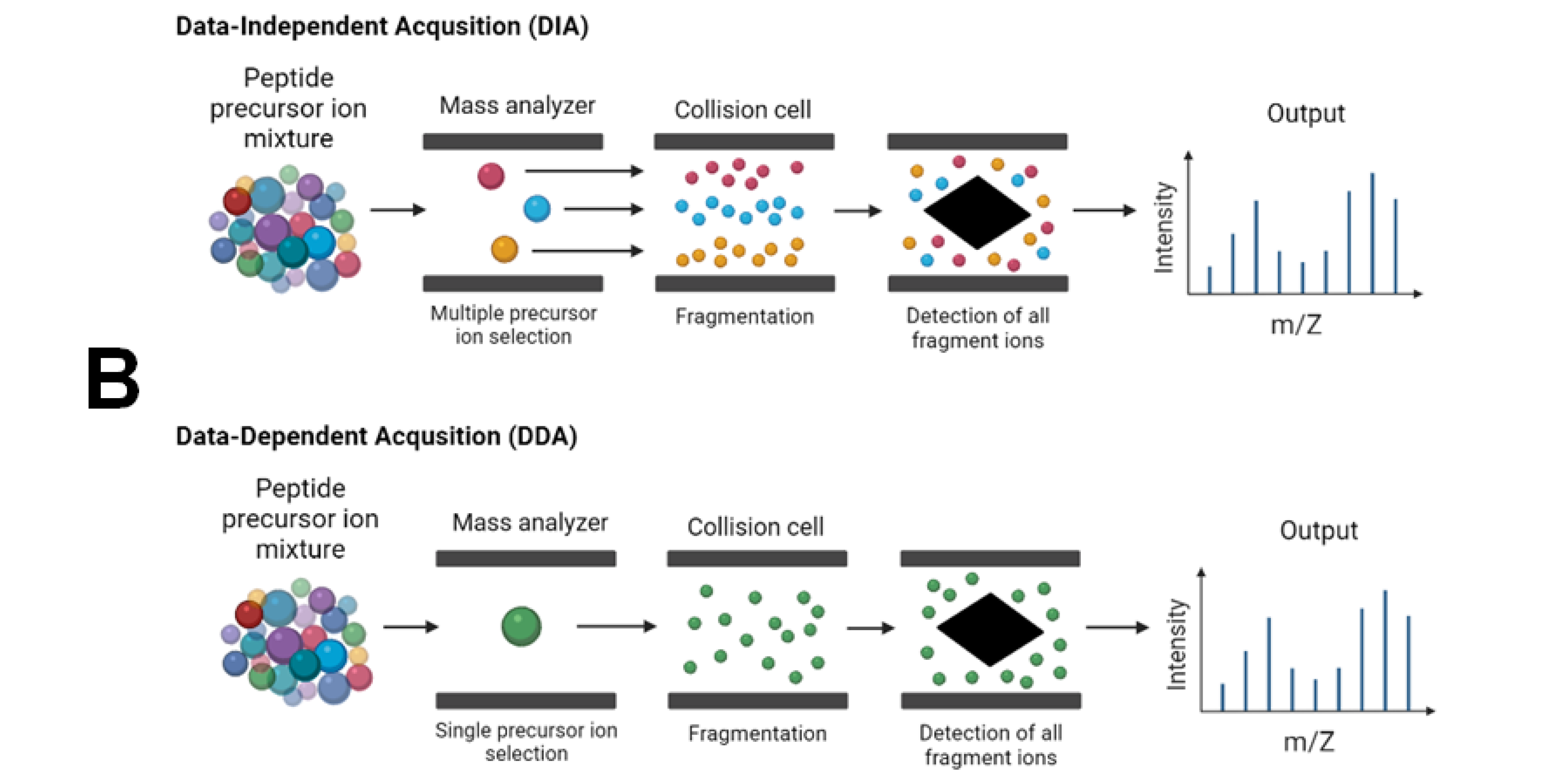
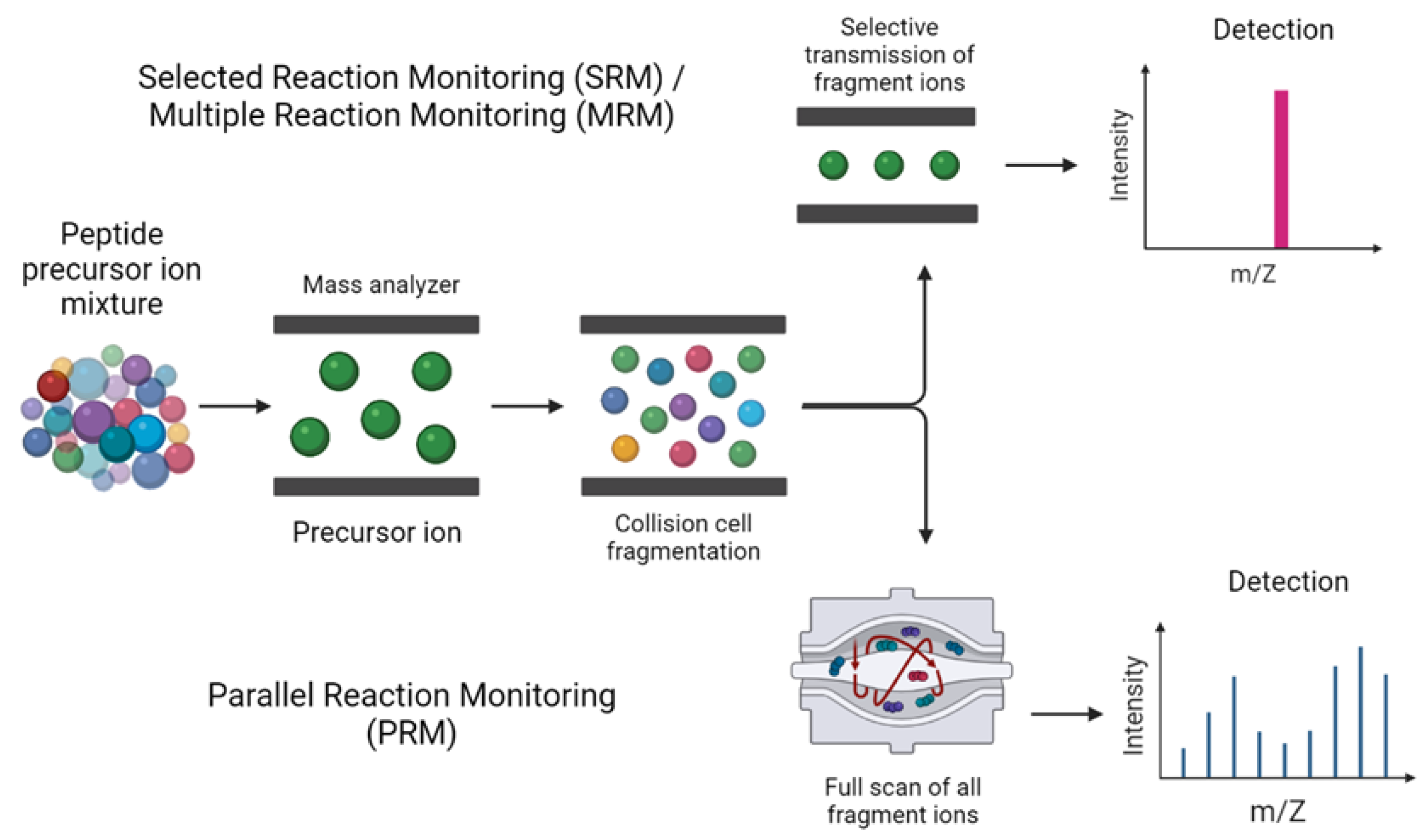
2.2.3. Data Acquisition
2.3. Data Analysis
3. Applications of Tandem Mass Spectrometry in Biomedical Research
3.1. Applications of Tandem MS-Based Proteomics in Systemsbiology
3.2. Applications of Tandem MS in Biofluids Proteomics and Enzyme Activity Assessment
3.3. Applications of Tandem MS in Biomarkers Discovery
3.4. Applications of Tandem MS in Oncoproteomics and Non-Oncologic Diseases
3.5. Applications of Tandem MS in Neuroproteomics
3.6. Tandem Mass Spectrometry Detection and Quantification of PTMs
3.7. MS/MS Applications for Assessment of Quality of Biological Samples and Optimization of Analytical Methods
3.8. Applications of Tandem MS in In Vitro Studies
3.9. Applications of MS/MS in Microbiology and Metaproteomics
3.10. Applications of MS/MS in Foodomics
3.11. Applications of Tandem Mass Spectrometry in Exposomics and Environmental Pollution
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhan, X.; Li, J.; Guo, Y.; Golubnitschaja, O. Mass spectrometry analysis of human tear fluid biomarkers specific for ocular and systemic diseases in the context of 3P medicine. EPMA J. 2021, 12, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S. Empowering Clinical Diagnostics with Mass Spectrometry. ACS Omega 2020, 5, 2041–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Caudillo, L.; Ortega-Lozano, A.J.; Martínez-Batallar, Á.G.; Rosas-Vargas, H.; Minauro-Sanmiguel, F.; Encarnación-Guevara, S. Principal component analysis on LC-MS/MS and 2DE-MALDI-TOF in glioblastoma cell lines reveals that mitochondria act as organelle sensors of the metabolic state in glioblastoma. Oncol. Rep. 2020, 44, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.D. Tandem mass spectroscopy in diagnosis and clinical research. Indian J. Clin. Biochem. 2015, 30, 121–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.S.; Lam, C.W.K.; Chan, M.H.M.; Cheung, R.C.K.; Law, L.K.; Lit, L.C.W.; Ng, K.F.; Suen, M.W.M.; Tai, H.L. Electrospray ionisation mass spectrometry: Principles and clinical applications. Clin. Biochem. Rev. 2003, 24, 3–12. [Google Scholar]
- Jones, L.M. Mass spectrometry-based methods for structural biology on a proteome-wide scale. Biochem. Soc. Trans. 2020, 48, 945–954. [Google Scholar] [CrossRef]
- Fox, A. Mass spectrometry for species or strain identification after culture or without culture: Past, present, and future. J. Clin. Microbiol. 2006, 44, 2677–2680. [Google Scholar] [CrossRef] [Green Version]
- Nakayasu, E.S.; Gritsenko, M.; Piehowski, P.D.; Gao, Y.; Orton, D.J.; Schepmoes, A.A.; Fillmore, T.L.; Frohnert, B.I.; Rewers, M.; Krischer, J.P.; et al. Tutorial: Best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat. Protoc. 2021, 16, 3737–3760. [Google Scholar] [CrossRef]
- Li, Y.; Shan, M.; Zhu, Z.; Mao, X.; Yan, M.; Chen, Y.; Zhu, Q.; Li, H.; Gu, B. Application of MALDI-TOF MS to rapid identification of anaerobic bacteria. BMC Infect. Dis. 2019, 19, 941. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Sun, E.; Wang, Y.; Pan, H.; Zhang, Y.; Li, Y.; Zhang, X.; Li, C.; Du, L.; Wang, C. Rapid Identification and Antimicrobial Susceptibility Testing for Urinary Tract Pathogens by Direct Analysis of Urine Samples Using a MALDI-TOF MS-Based Combined Protocol. Front. Microbiol. 2019, 10, 1182. [Google Scholar] [CrossRef]
- Pourfarzam, M.; Zadhoush, F. Newborn Screening for inherited metabolic disorders; news and views. J. Res. Med. Sci. 2013, 18, 801–808. [Google Scholar]
- Macklin, A.; Khan, S.; Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: Applications to cancer research. Clin. Proteom. 2020, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Kaur, U.; Johnson, D.T.; Chea, E.E.; Deredge, D.J.; Espino, J.A.; Jones, L.M. Evolution of Structural Biology through the Lens of Mass Spectrometry. Anal. Chem. 2019, 91, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Nahar, S.; Miyagi-Shiohira, C.; Kinjo, T.; Toyoda, Z.; Kobayashi, N.; Saitoh, I.; Watanabe, M.; Fujita, J.; Noguchi, H. A Liquid Chromatography with Tandem Mass Spectrometry-Based Proteomic Analysis of the Proteins Secreted by Human Adipose-Derived Mesenchymal Stem Cells. Cell Transplant. 2018, 27, 1469–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabb, D.L.; MacCoss, M.J.; Wu, C.C.; Anderson, S.D.; Yates, J.R. Similarity among Tandem Mass Spectra from Proteomic Experiments: Detection, Significance, and Utility. Anal. Chem. 2003, 75, 2470–2477. [Google Scholar] [CrossRef]
- Finoulst, I.; Pinkse, M.; Van Dongen, W.; Verhaert, P. Sample preparation techniques for the untargeted LC-MS-based discovery of peptides in complex biological matrices. J. Biomed. Biotechnol. 2011, 2011, 245291. [Google Scholar] [CrossRef] [Green Version]
- Duong, A.; Park, J.-M.; Lim, H.-J.; Lee, H. Proteomics in Forensic Analysis: Applications for Human Samples. Appl. Sci. 2021, 11, 3393. [Google Scholar] [CrossRef]
- Wang, W.-Q.; Jensen, O.N.; Møller, I.M.; Hebelstrup, K.H.; Rogowska-Wrzesinska, A. Evaluation of sample preparation methods for mass spectrometry-based proteomic analysis of barley leaves. Plant. Methods 2018, 14, 72. [Google Scholar] [CrossRef]
- Dittrich, J.; Adam, M.; Maas, H.; Hecht, M.; Reinicke, M.; Ruhaak, L.; Cobbaert, C.; Engel, C.; Wirkner, K.; Löffler, M.; et al. Targeted on-line SPE-LC-MS/MS Assay for the quantitation of twelve apolipoproteins from human blood. Proteomics 2017, 18, 1700279. [Google Scholar] [CrossRef]
- Dey, K.K.; Wang, H.; Niu, M.; Bai, B.; Wang, X.; Li, Y.; Cho, J.-H.; Tan, H.; Mishra, A.; High, A.A.; et al. Deep undepleted human serum proteome profiling toward biomarker discovery for Alzheimer’s disease. Clin. Proteom. 2019, 16, 16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Li, M.; Yang, Y.; Guo, Z.; Sun, Y.; Shao, C.; Li, M.; Sun, W.; Gao, Y. A comprehensive analysis and annotation of human normal urinary proteome. Sci. Rep. 2017, 7, 3024. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Garrigues, L.; Van den Toorn, H.; Stahl, B.; Heck, A.J.R. Discovery and Quantification of Nonhuman Proteins in Human Milk. J. Proteome Res. 2019, 18, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Xie, Y.; Ramachandran, P.; Loo, R.; li, Y.; Loo, J.; Wong, D. Large-scale identification of proteins in human salivary proteome by liquid chromatography/mass spectrometry and two-dimensional gel electrophoresis-mass spectrometry. Proteomics 2005, 5, 1714–1728. [Google Scholar] [CrossRef] [PubMed]
- Burat, B.; Reynaerts, A.; Baiwir, D.; Fléron, M.; Eppe, G.; Leal, T.; Mazzucchelli, G. Characterization of the Human Eccrine Sweat Proteome-A Focus on the Biological Variability of Individual Sweat Protein Profiles. Int. J. Mol. Sci. 2021, 22, 10871. [Google Scholar] [CrossRef] [PubMed]
- Ciordia, S.; Alvarez-Sola, G.; Rullan, M.; Urman, J.; Avila, M.; Corrales, F. Digging deeper into bile proteome. J. Proteom. 2020, 230, 103984. [Google Scholar] [CrossRef] [PubMed]
- Papagerakis, P.; Zheng, L.; Kim, D.; Said, R.; Ehlert, A.; Chung, K.; Papagerakis, S. Saliva and Gingival Crevicular Fluid (GCF) Collection for Biomarker Screening: Methods and Protocols; Humana Press: New York, NY, USA, 2019; pp. 549–562. [Google Scholar]
- Lee, J.-H.; Jung, J.H.; Kim, J.; Baek, W.-K.; Rhee, J.; Kim, T.-H.; Kim, S.-H.; Kim, K.P.; Son, C.-N.; Kim, J.-S. Proteomic analysis of human synovial fluid reveals potential diagnostic biomarkers for ankylosing spondylitis. Clin. Proteom. 2020, 17, 20. [Google Scholar] [CrossRef]
- Soria Esponera, J.; Acera, A.; Merayo-Lloves, J.; Durán, J.; González, N.; Rodriguez, S.; Bistolas, N.; Schumacher, S.; Bier, F.; Peter, H.; et al. Tear proteome analysis in ocular surface diseases using label-free LC-MS/MS and multiplexed-microarray biomarker validation. Sci. Rep. 2017, 7, 17487. [Google Scholar] [CrossRef] [Green Version]
- Hsu, T.-Y.; Tsai, K.-W.; Lan, K.-C.; Hung, H.-N.; Lai, Y.-J.; Cheng, H.-H.; Tsai, C.-C.; Li, S.-C. Identifying the potential protein biomarkers of preterm birth in amniotic fluid. Taiwan J. Obstet. Gynecol. 2020, 59, 366–371. [Google Scholar] [CrossRef]
- Kim, Y.E.; Kim, K.; Oh, H.B.; Lee, S.K.; Kang, D. Quantitative proteomic profiling of Cervicovaginal fluid from pregnant women with term and preterm birth. Proteome Sci. 2021, 19, 3. [Google Scholar] [CrossRef]
- Debyser, G.; Mesuere, B.; Clement, L.; Van de Weygaert, J.; Van Hecke, P.; Duytschaever, G.; Aerts, M.; Dawyndt, P.; De Boeck, K.; Vandamme, P.; et al. Faecal proteomics: A tool to investigate dysbiosis and inflammation in patients with cystic fibrosis. J. Cyst. Fibros. 2016, 15, 242–250. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Zhou, T.; Xie, G.; Fu, S.; Gao, L.; Liao, J.; Wu, Y.; Wang, G. Proteomics of exhaled breath condensate in stable COPD and non-COPD controls using tandem mass tags (TMTs) quantitative mass spectrometry: A pilot study. J. Proteom. 2019, 206, 103392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, X.; Li, P.; Zhou, F.; Kong, L.; Qiu, J.; Yuan, Z.; Tan, J. TMT Based Proteomic Analysis of Human Follicular Fluid From Overweight/Obese and Normal-Weight Patients With Polycystic Ovary Syndrome. Front. Endocrinol. 2019, 10, 821. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-C.; Lee, C.-H.; Chiou, S.-H.; Liao, C.-C.; Cheng, C.-W. Proteomic Analysis of Aqueous Humor Proteins in Association with Cataract Risks: Diabetes and Smoking. J. Clin. Med. 2021, 10, 5731. [Google Scholar] [CrossRef] [PubMed]
- Guldbrandsen, A.; Vethe, H.; Farag, Y.; Oveland, E.; Garberg, H.; Berle, M.; Myhr, K.-M.; Opsahl, J.A.; Barsnes, H.; Berven, F.S. In-depth Characterization of the Cerebrospinal Fluid (CSF) Proteome Displayed Through the CSF Proteome Resource (CSF-PR). Mol. Cell. Proteom. 2014, 13, 3152–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, R.; Nakajima, D.; Sato, H.; Endo, Y.; Ohara, O.; Kawashima, Y. A Simple Method for In-Depth Proteome Analysis of Mammalian Cell Culture Conditioned Media Containing Fetal Bovine Serum. Int. J. Mol. Sci. 2021, 22, 2565. [Google Scholar] [CrossRef] [PubMed]
- Kulasingam, V.; Diamandis, E.P. Proteomics Analysis of Conditioned Media from Three Breast Cancer Cell Lines: A Mine for Biomarkers and Therapeutic Targets. Mol. Cell. Proteom. 2007, 6, 1997–2011. [Google Scholar] [CrossRef] [Green Version]
- Jayathirtha, M.; Dupree, E.; Manzoor, Z.; Larose, B.; Sechrist, Z.; Neagu, A.-N.; Brindusa Alina, P.; Darie, C. Mass Spectrometric (MS) Analysis of Proteins and Peptides. Curr. Protein Pept. Sci. 2020, 21, 92–120. [Google Scholar] [CrossRef]
- Coon, J.J.; Syka, J.E.P.; Shabanowitz, J.; Hunt, D.F. Tandem Mass Spectrometry for Peptide and Protein Sequence Analysis. BioTechniques 2005, 38, 519–523. [Google Scholar] [CrossRef]
- Delmonico, L.; Areias, V.R.; Pinto, R.C.; Matos, C.D.S.; Rosa, M.F.F.; De Azevedo, C.M.; Alves, G. Protein identification from dried nipple aspirate fluid on Guthrie cards using mass spectrometry. Mol. Med. Rep. 2015, 12, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, D.; Kawashima, Y.; Shibata, H.; Yasumi, T.; Nishitani-Isa, M.; Izawa, K.; Nishikomori, R.; Heike, T.; Ohara, O. Simple and Sensitive Analysis for Dried Blood Spot Proteins by Sodium Carbonate Precipitation for Clinical Proteomics. J. Proteome Res. 2020, 19, 2821–2827. [Google Scholar] [CrossRef]
- Chu, F.; Mason, K.E.; Anex, D.S.; Jones, A.D.; Hart, B.R. Hair Proteome Variation at Different Body Locations on Genetically Variant Peptide Detection for Protein-Based Human Identification. Sci. Rep. 2019, 9, 7641. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Bartlett, M.G. A review of sample preparation methods for quantitation of small-molecule analytes in brain tissue by liquid chromatography tandem mass spectrometry (LC-MS/MS). Anal. Methods 2014, 6, 6183–6207. [Google Scholar] [CrossRef]
- Hayoun, K.; Gouveia, D.; Grenga, L.; Pible, O.; Armengaud, J.; Alpha-Bazin, B. Evaluation of Sample Preparation Methods for Fast Proteotyping of Microorganisms by Tandem Mass Spectrometry. Front. Microbiol. 2019, 10, 1985. [Google Scholar] [CrossRef] [PubMed]
- Virant-Klun, I.; Leicht, S.; Hughes, C.; Krijgsveld, J. Identification of Maturation-Specific Proteins by Single-Cell Proteomics of Human Oocytes. Mol. Cell. Proteom. 2016, 15, 2616–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, D.; Dehghani, A.; Steen, H. Optimization of Cell Lysis and Protein Digestion Protocols for Protein Analysis by LC-MS/MS; Humana Press: New York, NY, USA, 2015; pp. 259–273. [Google Scholar]
- Saito, S.; Hirao, Y.; Quadery, A.F.; Xu, B.; Elguoshy, A.; Fujinaka, H.; Koma, S.; Yamamoto, K.; Yamamoto, T. The Optimized Workflow for Sample Preparation in LC-MS/MS-Based Urine Proteomics. Methods Protoc. 2019, 2, 46. [Google Scholar] [CrossRef] [Green Version]
- Baecher, S.; Geyer, R.; Lehmann, C.; Vogeser, M. Absorptive chemistry based extraction for LC-MS/MS analysis of small molecule analytes from biological fluids—An application for 25-hydroxyvitamin D. Clin. Chem. Lab. Med. CCLM/FESCC 2013, 52, 1–9. [Google Scholar] [CrossRef]
- Stone, J. A practical guide to sample preparation for liquid chromatography-tandem mass spectrometry in clinical research and toxicology. Spectrosc. Eur. 2018, 30, 15–21. [Google Scholar]
- Mavreli, D.; Evangelinakis, N.; Papantoniou, N.; Kolialexi, A. Quantitative Comparative Proteomics Reveals Candidate Biomarkers for the Early Prediction of Gestational Diabetes Mellitus: A Preliminary Study. In Vivo 2020, 34, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zulkoski, J.P.; Ding, J.; Brown, W.; Addison, T. Liquid chromatography/tandem mass spectrometry sensitivity enhancement via online sample dilution and trapping: Applications in microdosing and dried blood spot (DBS) bioanalysis. Rapid Commun. Mass Spectrom. 2010, 24, 2575–2583. [Google Scholar] [CrossRef]
- Gundry, R.L.; White, M.Y.; Murray, C.I.; Kane, L.A.; Fu, Q.; Stanley, B.A.; Van Eyk, J.E. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr. Protoc. Mol. Biol. 2009, 10, 10–25. [Google Scholar] [CrossRef] [Green Version]
- Nickerson, J.L.; Baghalabadi, V.; Rajendran, S.R.C.K.; Jakubec, P.J.; Said, H.; McMillen, T.S.; Dang, Z.; Doucette, A.A. Recent advances in top-down proteome sample processing ahead of MS analysis. Mass Spectrom. Rev. 2021, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of this Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Wetie, A.; Sokolowska, I.; Woods, A.; Wormwood, K.; Dao, S.; Patel, S.; Clarkson, B.; Darie, C. Automated Mass Spectrometry-Based Functional Assay for the Routine Analysis of the Secretome. J. Lab. Autom. 2012, 18, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.P.; Rawlins, C.M.; DeHart, C.J.; Fornelli, L.; Schachner, L.F.; Lin, Z.; Lippens, J.L.; Aluri, K.C.; Sarin, R.; Chen, B.; et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Uwugiaren, N.; Williams, S.M.; Moore, R.J.; Zhao, R.; Goodlett, D.; Dapic, I.; Paša-Tolić, L.; Zhu, Y. Sensitive Top-Down Proteomics Analysis of a Low Number of Mammalian Cells Using a Nanodroplet Sample Processing Platform. Anal. Chem. 2020, 92, 7087–7095. [Google Scholar] [CrossRef] [PubMed]
- Dau, T.; Bartolomucci, G.; Rappsilber, J. Proteomics Using Protease Alternatives to Trypsin Benefits from Sequential Digestion with Trypsin. Anal. Chem. 2020, 92, 9523–9527. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.; Zampronio, C.; Jones, A.; Hernández-Fernaud, J.R. Updates of the In-Gel Digestion Method for Protein Analysis by Mass Spectrometry. Proteomics 2018, 18, 1800236. [Google Scholar] [CrossRef] [Green Version]
- Medzihradszky, K.F. In-Solution Digestion of Proteins for Mass Spectrometry. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2005; Volume 405, pp. 50–65. [Google Scholar]
- Berberich, M.; Kowalak, J.; Makusky, A.; Martin, B.; Vullhorst, D.; Buonanno, A.; Markey, S. Sample Preparation in Biological Mass Spectrometry; Springer: Berlin/Heidelberg, Germany, 2011; pp. 109–124. [Google Scholar] [CrossRef]
- Rosenfeld, J.; Capdevielle, J.; Guillemot, J.-C.; Ferrara, P. In-gel Digestion of Proteins for Internal Sequence Analysis After One- or Two-Dimensional Gel Electrophoresis. Anal. Biochem. 1992, 203, 173–179. [Google Scholar] [CrossRef]
- Piersma, S.R.; Warmoes, M.O.; de Wit, M.; de Reus, I.; Knol, J.C.; Jiménez, C.R. Whole gel processing procedure for GeLC-MS/MS based proteomics. Proteome Sci. 2013, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhu, H.-J. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)-Based Proteomics of Drug-Metabolizing Enzymes and Transporters. Molecules 2020, 25, 2718. [Google Scholar] [CrossRef]
- Nomura, F.; Tsuchida, S.; Murata, S.; Satoh, M.; Matsushita, K. Mass spectrometry-based microbiological testing for blood stream infection. Clin. Proteom. 2020, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Vinaiphat, A.; Low, J.K.; Yeoh, K.W.; Chng, W.J.; Sze, S.K. Application of Advanced Mass Spectrometry-Based Proteomics to Study Hypoxia Driven Cancer Progression. Front. Oncol. 2021, 11, 559822. [Google Scholar] [CrossRef] [PubMed]
- Köcher, T.; Pichler, P.; Swart, R.; Mechtler, K. Analysis of protein mixtures from whole-cell extracts by single-run nanoLC–MS/MS using ultralong gradients. Nat. Protoc. 2012, 7, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; The, M.; Giansanti, P.; Mergner, J.; Zheng, R.; Wilhelm, M.; Boychenko, A.; Kuster, B. Identification of 7000–9000 Proteins from Cell Lines and Tissues by Single-Shot Microflow LC–MS/MS. Anal. Chem. 2021, 93, 8687–8692. [Google Scholar] [CrossRef] [PubMed]
- Piehowski, P.D.; Zhu, Y.; Bramer, L.M.; Stratton, K.G.; Zhao, R.; Orton, D.J.; Moore, R.J.; Yuan, J.; Mitchell, H.D.; Gao, Y.; et al. Automated mass spectrometry imaging of over 2000 proteins from tissue sections at 100-μm spatial resolution. Nat. Commun. 2020, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef] [Green Version]
- Kucková, S.; Cejnar, P.; Hynek, R. Characterization of proteins in cultural heritage using MALDi TOF and Lc MS/MS mass spectrometric techniques. Phys. Sci. Rev. 2018, 4. [Google Scholar] [CrossRef]
- Gogichaeva, N.V.; Williams, T.; Alterman, M.A. MALDI TOF/TOF Tandem Mass Spectrometry as a New Tool for Amino Acid Analysis. J. Am. Soc. Mass Spectrom. 2007, 18, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Dufresne, J.; Bowden, P.; Thavarajah, T.; Florentinus-Mefailoski, A.; Chen, Z.Z.; Tucholska, M.; Norzin, T.; Ho, M.T.; Phan, M.; Mohamed, N.; et al. The plasma peptidome. Clin. Proteom. 2018, 15, 39. [Google Scholar] [CrossRef] [Green Version]
- Damodaran, S.; Wood, T.D.; Nagarajan, P.; Rabin, R.A. Evaluating peptide mass fingerprinting-based protein identification. Genom. Proteom. Bioinform. 2007, 5, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Emmott, E.; Jovanovic, M.; Slavov, N. Approaches for Studying Ribosome Specialization. Trends Biochem. Sci. 2019, 44, 478–479. [Google Scholar] [CrossRef]
- Fiorino, G.; Fresch, M.; Brümmer, I.; Losito, I.; Arlorio, M.; Brockmeyer, J.; Monaci, L. Mass Spectrometry-Based Untargeted Proteomics for the Assessment of Food Authenticity: The Case of Farmed Versus Wild-Type Salmon. J. AOAC Int. 2019, 102, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Krasny, L.; Huang, P. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omics 2020, 17, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lopez, C.; Ginebreda, A.; Carrascal, M.; Barcelò, D.; Abian, J.; Tauler, R. Non-target protein analysis of samples from wastewater treatment plants using the regions of interest-multivariate curve resolution (ROIMCR) chemometrics method. J. Environ. Chem. Eng. 2021, 9, 105752. [Google Scholar] [CrossRef]
- Hird, S.; Lau, B.; Schuhmacher, R.; Krska, R. Liquid chromatography-mass spectrometry for the determination of chemical contaminants in food. TrAC Trends Anal. Chem. 2014, 59, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Cilento, E.M.; Jin, L.; Stewart, T.; Shi, M.; Sheng, L.; Zhang, J. Mass spectrometry: A platform for biomarker discovery and validation for Alzheimer’s and Parkinson’s diseases. J. Neurochem. 2019, 151, 397–416. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.; Kaleta, D.; Robinson, E.; Liu, T.; Zhao, R.; Page, J.; Kelly, R.; Moore, R.; Tang, K.; Camp, D.; et al. Enhanced Sensitivity for Selected Reaction Monitoring Mass Spectrometry-based Targeted Proteomics Using a Dual Stage Electrodynamic Ion Funnel Interface. Mol. Cell. Proteom. MCP 2011, 10, S1–S9. [Google Scholar] [CrossRef] [Green Version]
- Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M. Selected Reaction Monitoring Mass Mass Spectrometry (MS) Spectrometry; Springer: Berlin/Heidelberg, Germany, 2020; pp. 53–88. [Google Scholar] [CrossRef]
- Feist, P.; Hummon, A.B. Proteomic challenges: Sample preparation techniques for microgram-quantity protein analysis from biological samples. Int. J. Mol. Sci. 2015, 16, 3537–3563. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, B.K.; Smith, M.; Baghela, A.; Lee, A.H.Y.; Hancock, R.E.W. Systems Biology Approaches to Understanding the Human Immune System. Front. Immunol. 2020, 11, 1683. [Google Scholar] [CrossRef]
- Keller, C.; Wei, P.; Wancewicz, B.; Cross, T.-W.L.; Rey, F.E.; Li, L. Extraction optimization for combined metabolomics, peptidomics, and proteomics analysis of gut microbiota samples. J. Mass Spectrom 2021, 56, e4625. [Google Scholar] [CrossRef] [PubMed]
- Soufi, Y.; Soufi, B. Mass Spectrometry-Based Bacterial Proteomics: Focus on Dermatologic Microbial Pathogens. Front. Microbiol. 2016, 7, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afiuni-Zadeh, S.; Boylan, K.L.M.; Jagtap, P.D.; Griffin, T.J.; Rudney, J.D.; Peterson, M.L.; Skubitz, A.P.N. Evaluating the potential of residual Pap test fluid as a resource for the metaproteomic analysis of the cervical-vaginal microbiome. Sci. Rep. 2018, 8, 10868. [Google Scholar] [CrossRef] [PubMed]
- Morro, B.; Doherty, M.; Balseiro, P.; Handeland, S.; Mackenzie, S.; Sveier, H.; Albalat, A. Plasma proteome profiling of freshwater and seawater life stages of rainbow trout (Oncorhynchus mykiss). PLoS ONE 2020, 15, e0227003. [Google Scholar] [CrossRef] [Green Version]
- Susaki, E.A.; Ukai, H.; Ueda, H.R. Next-generation mammalian genetics toward organism-level systems biology. NPJ Syst. Biol. Appl. 2017, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Witt, N.; Andreotti, S.; Busch, A.; Neubert, K.; Reinert, K.; Tomaso, H.; Meierhofer, D. Rapid and Culture Free Identification of Francisella in Hare Carcasses by High-Resolution Tandem Mass Spectrometry Proteotyping. Front. Microbiol. 2020, 11, 636. [Google Scholar] [CrossRef]
- Fonslow, B.R.; Moresco, J.J.; Tu, P.G.; Aalto, A.P.; Pasquinelli, A.E.; Dillin, A.; Yates, J.R. Mass spectrometry-based shotgun proteomic analysis of C. elegans protein complexes. WormBook Online Rev. C. Elegans Biol. 2014, 1–18. [Google Scholar] [CrossRef]
- Davidson, P.L.; Thompson, J.W.; Foster, M.W.; Moseley, M.A.; Byrne, M.; Wray, G.A. A comparative analysis of egg provisioning using mass spectrometry during rapid life history evolution in sea urchins. Evol. Dev. 2019, 21, 188–204. [Google Scholar] [CrossRef]
- Casas-Vila, N.; Bluhm, A.; Sayols, S.; Dinges, N.; Dejung, M.; Altenhein, T.; Kappei, D.; Altenhein, B.; Roignant, J.-Y.; Butter, F. The developmental proteome of Drosophila melanogaster. Genome Res. 2017, 27, 1273–1285. [Google Scholar] [CrossRef] [Green Version]
- Lößner, C.; Wee, S.; Ler, S.; Li, R.; Carney, T.; Blackstock, W.; Gunaratne, J. Expanding the zebrafish embryo proteome using multiple fractionation approaches and tandem mass spectrometry. Proteomics 2012, 12, 1879–1882. [Google Scholar] [CrossRef]
- Sun, L.; Bertke, M.M.; Champion, M.M.; Zhu, G.; Huber, P.W.; Dovichi, N.J. Quantitative proteomics of Xenopus laevis embryos: Expression kinetics of nearly 4000 proteins during early development. Sci. Rep. 2014, 4, 4365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, C.; Gong, T.; Ni, X.; Li, J.e.; Zhan, D.; Liu, M.; Song, L.; Ding, C.; Xu, J.; et al. A time-resolved multi-omic atlas of the developing mouse stomach. Nat. Commun. 2018, 9, 4910. [Google Scholar] [CrossRef] [PubMed]
- Aballo, T.J.; Roberts, D.S.; Melby, J.A.; Buck, K.M.; Brown, K.A.; Ge, Y. Ultrafast and Reproducible Proteomics from Small Amounts of Heart Tissue Enabled by Azo and timsTOF Pro. J. Proteome Res. 2021, 20, 4203–4211. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Kumar, C.; Zougman, A.; Zhang, Y.; Podtelejnikov, A.; Cox, J.; Wiśniewski, J.; Mann, M. Analysis of the Mouse Liver Proteome Using Advanced Mass Spectrometry. J. Proteome Res. 2007, 6, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ying, W.; Song, Y.; Liu, X.; Yang, B.; Wu, S.; Jiang, Y.; Cai, Y.; He, F.; Qian, X. Analysis of human liver proteome using replicate shotgun strategy. Proteomics 2007, 7, 2479–2488. [Google Scholar] [CrossRef]
- Shum, A.; Poljak, A.; Bentley, N.; Turner, N.; Tan, T.; Polly, P. Proteomic profiling of skeletal and cardiac muscle in cancer cachexia: Alterations in sarcomeric and mitochondrial protein expression. Oncotarget 2018, 9, 22001. [Google Scholar] [CrossRef] [Green Version]
- Kruse, A.; Spraggins, J. Uncovering Molecular Heterogeneity in the Kidney with Spatially Targeted Mass Spectrometry. Front. Physiol. 2022, 13, 18. [Google Scholar] [CrossRef]
- Zhao, Y.; Wilmarth, P.A.; Cheng, C.; Limi, S.; Fowler, V.M.; Zheng, D.; David, L.L.; Cvekl, A. Proteome-transcriptome analysis and proteome remodeling in mouse lens epithelium and fibers. Exp. Eye Res. 2019, 179, 32–46. [Google Scholar] [CrossRef]
- Wenke, J.L.; McDonald, W.H.; Schey, K.L. Spatially Directed Proteomics of the Human Lens Outer Cortex Reveals an Intermediate Filament Switch Associated With the Remodeling Zone. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4108–4114. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, T.; Hatano, N.; Aihara, M.; Kawano, H.; Okamoto, M.; Liu, Y.; Izumi, T.; Maekawa, T.; Nakamura, S.; Ishihara, T.; et al. Proteomic analysis of proteins expressing in regions of rat brain by a combination of SDS-PAGE with nano-liquid chromatography-quadrupole-time of flight tandem mass spectrometry. Proteome Sci. 2010, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yu, K.; Tan, H.; Wu, Z.; Cho, J.-H.; Han, X.; Sun, H.; Beach, T.G.; Peng, J. 27-Plex Tandem Mass Tag Mass Spectrometry for Profiling Brain Proteome in Alzheimer’s Disease. Anal. Chem. 2020, 92, 7162–7170. [Google Scholar] [CrossRef]
- Di Paolo, A.; Farias, J.; Garat, J.; Macklin, A.; Ignatchenko, V.; Kislinger, T.; Sotelo-Silveira, J. Rat Sciatic Nerve Axoplasm Proteome Is Enriched with Ribosomal Proteins during Regeneration Processes. J. Proteome Res. 2021, 20, 2506–2520. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, J.; Wang, Y.; Xin, C.; Ma, J.; Xu, S.; Wang, X.; Gao, J.; Zhang, X.; Yang, S. Non-invasive proteome-wide quantification of skin barrier-related proteins using label-free LC-MS/MS analysis. Mol. Med. Rep. 2020, 21, 2227–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, C.; Wu, X.; Bian, J.; Ma, X.; Zhang, G.; Guo, Z.; Wang, Y.; Ci, Y.; Wang, Q.; Xiang, H.; et al. Differential proteomic analysis of fetal and geriatric lumbar nucleus pulposus: Immunoinflammation and age-related intervertebral disc degeneration. BMC Musculoskelet. Disord. 2020, 21, 339. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Zhang, B.; Choi, C.; Yang, S.; Zhou, J.; Harlan, R.; Tian, Y.; Zhang, Z.; Chan, D.W.; Zhang, H. Tissue proteomics using chemical immobilization and mass spectrometry. Anal. Biochem. 2015, 469, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.; Jeong, J.E.; Lee, H.K.; Yun, K.N.; An, H.J.; Lee, B.; Paik, Y.-K.; Jeong, T.S.; Yee, G.T.; Kim, J.Y.; et al. Identification of Missing Proteins in Human Olfactory Epithelial Tissue by Liquid Chromatography–Tandem Mass Spectrometry. J. Proteome Res. 2018, 17, 4320–4324. [Google Scholar] [CrossRef]
- Hesselager, M.; Codrea, M.; Bendixen, E. Evaluation of preparation methods for MS-based analysis of intestinal epithelial cell proteomes. Proteomics 2015, 15, 2350–2357. [Google Scholar] [CrossRef]
- Pedicini, L.; Wiktor, S.D.; Simmons, K.J.; Money, A.; McKeown, L. Affinity-based proteomics reveals novel binding partners for Rab46 in endothelial cells. Sci. Rep. 2021, 11, 4054. [Google Scholar] [CrossRef]
- Ye, T.-M.; Pang, R.; Leung, C.; Chiu, J.-F.; Yeung, W. Two-dimensional liquid chromatography with tandem mass spectrometry–based proteomic characterization of endometrial luminal epithelial surface proteins responsible for embryo implantation. Fertil. Steril. 2015, 103, 853–861. [Google Scholar] [CrossRef]
- Cui, Y.-H.; Liu, Q.; Xu, Z.-Y.; Li, J.-H.; Hu, Z.-X.; Li, M.-J.; Zheng, W.-L.; Li, Z.-J.; Pan, H.-W. Quantitative proteomic analysis of human corneal epithelial cells infected with HSV-1. Exp. Eye Res. 2019, 185, 107664. [Google Scholar] [CrossRef]
- Hinz, S.; Manousopoulou, A.; Miyano, M.; Sayaman, R.W.; Aguilera, K.Y.; Todhunter, M.E.; Lopez, J.C.; Sohn, L.L.; Wang, L.D.; LaBarge, M.A. Deep proteome profiling of human mammary epithelia at lineage and age resolution. iScience 2021, 24, 103026. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Broszczak, D.; Broadbent, J.; Singh, D.; Steck, R.; Parker, T.; Peake, J. Comparative label-free mass spectrometric analysis of temporal changes in the skeletal muscle proteome after impact trauma in rats. Am. J. Physiol. Endocrinol. Metab. 2020, 318, 1022–1037. [Google Scholar] [CrossRef] [PubMed]
- Al-Menhali, A.; Anderson, C.; Gourine, A.; Abramov, A.; D’Souza, A.; Jaganjac, M. Proteomic Analysis of Cardiac Adaptation to Exercise by High Resolution Mass Spectrometry. Front. Mol. Biosci. 2021, 8, 801. [Google Scholar] [CrossRef]
- Xu, M.; Saxena, N.; Vrana, M.; Zhang, H.; Kumar, V.; Billington, S.; Khojasteh, C.; Heyward, S.; Unadkat, J.; Prasad, B. Targeted LC-MS/MS Proteomics-Based Strategy To Characterize in Vitro Models Used in Drug Metabolism and Transport Studies. Anal. Chem. 2018, 90, 11873–11882. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sørensen, K.D.; Høyer, S.; Ørntoft, T.F.; Andersen, C.L.; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4, 587–599.e584. [Google Scholar] [CrossRef] [Green Version]
- Cantrell, L.S.; Rose, K.; Schey, K. Development of Single-Cell Proteomics for Lens Fibers using Tandem Mass Spectrometry. Investig. Ophthalmol. Vis. Sci. 2020, 61, 787. [Google Scholar]
- Weerakoon, H.; Potriquet, J.; Shah, A.K.; Reed, S.; Jayakody, B.; Kapil, C.; Midha, M.K.; Moritz, R.L.; Lepletier, A.; Mulvenna, J.; et al. A primary human T-cell spectral library to facilitate large scale quantitative T-cell proteomics. Sci. Data 2020, 7, 412. [Google Scholar] [CrossRef] [PubMed]
- Lombard-Banek, C.; Reddy, S.; Moody, S.; Nemes, P. Label-free Quantification of Proteins in Single Embryonic Cells with Neural Fate in the Cleavage-Stage Frog (Xenopus laevis) Embryo using CE-ESI-HRMS. Mol. Cell. Proteom. 2016, 15, 2756–2768. [Google Scholar] [CrossRef] [Green Version]
- van de Waterbeemd, M.; Tamara, S.; Fort, K.L.; Damoc, E.; Franc, V.; Bieri, P.; Itten, M.; Makarov, A.; Ban, N.; Heck, A.J.R. Dissecting ribosomal particles throughout the kingdoms of life using advanced hybrid mass spectrometry methods. Nat. Commun. 2018, 9, 2493. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Panner Selvam, M.K.; Baskaran, S. Proteomic Analyses of Human Sperm Cells: Understanding the Role of Proteins and Molecular Pathways Affecting Male Reproductive Health. Int. J. Mol. Sci. 2020, 21, 1621. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Bottinelli, D.; Lisacek, F.; Luban, J.; Strambio-De-Castillia, C.; Varesio, E.; Hopfgartner, G. Optimization of human dendritic cell sample preparation for mass spectrometry-based proteomic studies. Anal. Biochem. 2015, 484, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Veenstra, T.D. Analysis of biofluids for biomarker research. PROTEOMICS–Clin. Appl. 2008, 2, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Matysiak, J.; Hajduk, J.; Mayer, F.; Hebeler, R.; Kokot, Z. Hyphenated LC-MALDI-ToF/ToF and LC-ESI-QToF approach in proteomic characterization of honeybee venom. J. Pharm. Biomed. Anal. 2016, 121, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yi, F.; Kumar, A.B.; Kumar Chennamaneni, N.; Hong, X.; Scott, C.R.; Gelb, M.H.; Turecek, F. Multiplex Tandem Mass Spectrometry Enzymatic Activity Assay for Newborn Screening of the Mucopolysaccharidoses and Type 2 Neuronal Ceroid Lipofuscinosis. Clin. Chem. 2017, 63, 1118–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, D.; Fritz, A.; Partecke, L.I.; Heidecke, C.-D.; Oswald, S. LC–MS/MS method for the simultaneous quantification of intestinal CYP and UGT activity. J. Pharm. Biomed. Anal. 2018, 155, 194–201. [Google Scholar] [CrossRef]
- Rey, M.; Yang, M.; Lee, L.; Zhang, Y.; Sheff, J.G.; Sensen, C.W.; Mrazek, H.; Halada, P.; Man, P.; McCarville, J.L.; et al. Addressing proteolytic efficiency in enzymatic degradation therapy for celiac disease. Sci. Rep. 2016, 6, 30980. [Google Scholar] [CrossRef]
- Chen, H.; Wilkerson, C.G.; Kuchar, J.A.; Phinney, B.S.; Howe, G.A. Jasmonate-inducible plant enzymes degrade essential amino acids in the herbivore midgut. Proc. Natl. Acad. Sci. USA 2005, 102, 19237–19242. [Google Scholar] [CrossRef] [Green Version]
- Khorramnejad, A.; Gomis-Cebolla, J.; Talaei-Hassanlouei, R.; Bel, Y.; Escriche, B. Genomics and Proteomics Analyses Revealed Novel Candidate Pesticidal Proteins in a Lepidopteran-Toxic Bacillus thuringiensis Strain. Toxins 2020, 12, 673. [Google Scholar] [CrossRef]
- Seger, C.; Salzmann, L. After another decade: LC–MS/MS became routine in clinical diagnostics. Clin. Biochem. 2020, 82, 2–11. [Google Scholar] [CrossRef]
- Fleszar, M.G.; Fortuna, P.; Zawadzki, M.; Kosyk, B.; Krzystek-Korpacka, M. Simultaneous LC-MS/MS-Based Quantification of Free 3-Nitro-l-tyrosine, 3-Chloro-l-tyrosine, and 3-Bromo-l-tyrosine in Plasma of Colorectal Cancer Patients during Early Postoperative Period. Molecules 2020, 25, 5158. [Google Scholar] [CrossRef]
- Zhou, M.; Kong, Y.; Wang, X.; Li, W.; Chen, S.; Wang, L.; Wang, C.; Zhang, Q. LC-MS/MS-Based Quantitative Proteomics Analysis of Different Stages of Non-Small-Cell Lung Cancer. BioMed Res. Int. 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Lu, M.; He, J.; Whitelegge, J.; Faull, K.; Saxton, R.; Chang, H. Mass spectrometry (LC-MS/MS) analysis of proximal fluid and cell membrane proteins for breast cancer biomarkers. Cancer Res. 2008, 68, 4436. [Google Scholar]
- Whelan, S.A.; He, J.; Lu, M.; Souda, P.; Saxton, R.E.; Faull, K.F.; Whitelegge, J.P.; Chang, H.R. Mass spectrometry (LC-MS/MS) identified proteomic biosignatures of breast cancer in proximal fluid. J. Proteome Res. 2012, 11, 5034–5045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, N.; Wang, K.; Fang, S.; Zhao, X.; Huang, T.; Chen, H.; Yan, F.; Tang, Y.; Zhou, H.; Zhu, J. Discovery of a Potential Plasma Protein Biomarker Panel for Acute-on-Chronic Liver Failure Induced by Hepatitis B Virus. Front. Physiol. 2017, 8, 1009. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wei, D.; Deng, M. Comparative Analysis of Serum Proteins Between Hepatitis B Virus Genotypes B and C Infection by DIA-Based Quantitative Proteomics. Infect. Drug Resist. 2021, 14, 4701–4715. [Google Scholar] [CrossRef] [PubMed]
- George, A.L.; Shaheed, S.U.; Sutton, C.W. High-Throughput Proteomic Profiling of Nipple Aspirate Fluid from Breast Cancer Patients Compared with Non-Cancer Controls: A Step Closer to Clinical Feasibility. J. Clin. Med. 2021, 10, 2243. [Google Scholar] [CrossRef]
- Ishikawa, S.; Ishizawa, K.; Tanaka, A.; Kimura, H.; Kitabatake, K.; Sugano, A.; Edamatsu, K.; Ueda, S.; Iino, M. Identification of Salivary Proteomic Biomarkers for Oral Cancer Screening. In Vivo 2021, 35, 541–547. [Google Scholar] [CrossRef]
- Ploypetch, S.; Roytrakul, S.; Phaonakrop, N.; Kittisenachai, S.; Leetanasaksakul, K.; Pisamai, S.; Kalpravidh, C.; Rungsipipat, A.; Suriyaphol, G. In-gel digestion coupled with mass spectrometry (GeLC-MS/MS)-based salivary proteomic profiling of canine oral tumors. BMC Vet. Res. 2020, 16, 335. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, Y.; Kim, Y.; Kim, S.; Kim, J.J.; Kim, K.M.; Yoshizawa, J.; Fan, L.-Y.; Cao, C.-X.; Wong, D.T.W. Differential Proteomic Analysis of Human Saliva using Tandem Mass Tags Quantification for Gastric Cancer Detection. Sci. Rep. 2016, 6, 22165. [Google Scholar] [CrossRef]
- Salazar, M.G.; Jehmlich, N.; Murr, A.; Dhople, V.M.; Holtfreter, B.; Hammer, E.; Völker, U.; Kocher, T. Identification of periodontitis associated changes in the proteome of whole human saliva by mass spectrometric analysis. J. Clin. Periodontol. 2013, 40, 825–832. [Google Scholar] [CrossRef]
- Beretov, J.; Wasinger, V.C.; Millar, E.K.A.; Schwartz, P.; Graham, P.H.; Li, Y. Proteomic Analysis of Urine to Identify Breast Cancer Biomarker Candidates Using a Label-Free LC-MS/MS Approach. PLoS ONE 2015, 10, e0141876. [Google Scholar] [CrossRef] [PubMed]
- Laputková, G.; Talian, I.; Schwartzová, V.; Schwartzová, Z. MALDI-TOF MS profiling in the discovery and identification of salivary proteomic patterns of temporomandibular joint disorders. Open Chem. 2020, 18, 1173–1180. [Google Scholar] [CrossRef]
- Abdulwahab, R.A.; Alaiya, A.; Shinwari, Z.; Allaith, A.A.A.; Giha, H.A. LC-MS/MS proteomic analysis revealed novel associations of 37 proteins with T2DM and notable upregulation of immunoglobulins. Int. J. Mol. Med. 2019, 43, 2118–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, K.P.; Han, T.-L.; Tong, C.; Baker, P.N. Mass Spectrometry-Based Proteomics for Pre-Eclampsia and Preterm Birth. Int. J. Mol. Sci. 2015, 16, 10952–10985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Lee, J.E.; Kim, Y.M.; Park, Y.; Choi, J.-W.; Park, K.H. Identifying potential biomarkers related to pre-term delivery by proteomic analysis of amniotic fluid. Sci. Rep. 2020, 10, 19648. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, H.; Che, G.; Li, Y.; Gao, J.; Yang, Q.; Zhou, B.; Gao, L.; Tao, W.; Liang, Y.; et al. Placental protein 14 as a potential biomarker for diagnosis of preterm premature rupture of membranes. Mol. Med. Rep. 2018, 18, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Pernemalm, M.; Sandberg, A.; Zhu, Y.; Boekel, J.; Tamburro, D.; Schwenk, J.M.; Björk, A.; Wahren-Herlenius, M.; Åmark, H.; Östenson, C.-G.; et al. In-depth human plasma proteome analysis captures tissue proteins and transfer of protein variants across the placenta. eLife 2019, 8, e41608. [Google Scholar] [CrossRef]
- Sun, R.-J.; Yin, D.-M.; Yuan, D.; Liu, S.-Y.; Zhu, J.-J.; Shan, N.-N. Quantitative LC–MS/MS uncovers the regulatory role of autophagy in immune thrombocytopenia. Cancer Cell Int. 2021, 21, 548. [Google Scholar] [CrossRef]
- Letunica, N.; Cai, T.; Cheong, J.L.Y.; Doyle, L.W.; Monagle, P.; Ignjatovic, V. The use of proteomics for blood biomarker research in premature infants: A scoping review. Clin. Proteom. 2021, 18, 13. [Google Scholar] [CrossRef]
- Madan, A.; El-Ferzli, G.; Carlson, S.M.; Whitin, J.C.; Schilling, J.; Najmi, A.; Yu, T.T.-S.; Lau, K.; Dimmitt, R.A.; Cohen, H.J. A Potential Biomarker in the Cord Blood of Preterm Infants Who Develop Retinopathy of Prematurity. Pediatric Res. 2007, 61, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, G.; Carota, G.; Li Volti, G.; Giuffrè, M. Biomarkers of Oxidative Stress for Neonatal Lung Disease. Front. Pediatr. 2021, 9, 618867. [Google Scholar] [CrossRef] [PubMed]
- Agakidou, E.; Agakidis, C.; Gika, H.; Sarafidis, K. Emerging Biomarkers for Prediction and Early Diagnosis of Necrotizing Enterocolitis in the Era of Metabolomics and Proteomics. Front. Pediatr. 2020, 8, 602255. [Google Scholar] [CrossRef] [PubMed]
- Chatziioannou, C.A.; Wolters, J.; Sarafidis, K.; Thomaidou, A.; Agakidis, C.; Govorukhina, N.; Kuivenhoven, J.; Bischoff, R.; Theodoridis, G. Targeted LC-MS/MS for the evaluation of proteomics biomarkers in the blood of neonates with necrotizing enterocolitis and late-onset sepsis. Anal. Bioanal. Chem. 2018, 410, 7163–7175. [Google Scholar] [CrossRef] [PubMed]
- Cabral, E.; Soares, H.; Vitorino, R.; Ferreira, R.; Henriques-Coelho, T. Prediction of cardiovascular risk in preterm neonates through urinary proteomics: An exploratory study. Porto Biomed. J. 2017, 2, 287–292. [Google Scholar] [CrossRef]
- Myers, M.V.; Maxwell, S.E.; Wang, X. Quantification of XRCC and DNA-PK proteins in cancer cell lines and human tumors by LC–MS/MS. Bioanalysis 2014, 6, 2969–2983. [Google Scholar] [CrossRef] [Green Version]
- Al-Wajeeh, A.S.; Salhimi, S.M.; Al-Mansoub, M.A.; Khalid, I.A.; Harvey, T.M.; Latiff, A.; Ismail, M.N. Comparative proteomic analysis of different stages of breast cancer tissues using ultra high performance liquid chromatography tandem mass spectrometer. PLoS ONE 2020, 15, e0227404. [Google Scholar] [CrossRef] [Green Version]
- Goto, R.; Nakamura, Y.; Takami, T.; Sanke, T.; Tozuka, Z. Quantitative LC-MS/MS Analysis of Proteins Involved in Metastasis of Breast Cancer. PLoS ONE 2015, 10, e0130760. [Google Scholar] [CrossRef] [Green Version]
- Rong, D.; Lin, X.; Luo, Y.; Mok, T.S.; Wang, Q.; Wang, H.; Zhang, T. Identification of the differentially expressed proteins in nasopharyngeal carcinoma by proteomics. Transl. Cancer Res. 2019, 9, 21–29. [Google Scholar] [CrossRef]
- Keshamouni, V.; Jagtap, P.; Michailidis, G.; Strahler, J.; Kuick, R.; Reka, A.; Papoulias, P.; Krishnapuram, R.; Srirangam, A.; Standiford, T.; et al. Temporal Quantitative Proteomics by iTRAQ 2D-LC-MS/MS and Corresponding mRNA Expression Analysis Identify Post-Transcriptional Modulation of Actin-Cytoskeleton Regulators During TGF-β-Induced Epithelial-Mesenchymal Transition. J. Proteome Res. 2009, 8, 35–47. [Google Scholar] [CrossRef]
- Zhan, X.; Huang, Y.; Long, Y. Two-dimensional Gel Electrophoresis Coupled with Mass Spectrometry Methods for an Analysis of Human Pituitary Adenoma Tissue Proteome. J. Vis. Exp. JoVE 2018, 134, 56739. [Google Scholar] [CrossRef]
- Qiu, F.; Chen, F.; Liu, D.; Xu, J.; He, J.; Xiao, J.; Cao, L.; Huang, X. LC-MS/MS-based screening of new protein biomarkers for cervical precancerous lesions and cervical cancer. Nan Fang Yi Ke Da Xue Xue Bao = J. South. Med. Univ. 2019, 39, 13–22. [Google Scholar] [CrossRef]
- Chokchaichamnankit, D.; Watcharatanyatip, K.; Subhasitanont, P.; Weeraphan, C.; Keeratichamroen, S.; Sritana, N.; Kantathavorn, N.; Diskul-Na-Ayudthaya, P.; Saharat, K.; Chantaraamporn, J.; et al. Urinary biomarkers for the diagnosis of cervical cancer by quantitative label-free mass spectrometry analysis. Oncol. Lett. 2019, 17, 5453–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslebagh, R.; Channaveerappa, D.; Arcaro, K.F.; Darie, C.C. Proteomics analysis of human breast milk to assess breast cancer risk. Electrophoresis 2018, 39, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Aslebagh, R.; Channaveerappa, D.; Arcaro, K.F.; Darie, C.C. Comparative two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) of human milk to identify dysregulated proteins in breast cancer. Electrophoresis 2018, 39, 1723–1734. [Google Scholar] [CrossRef]
- Aslebagh, R.; Channaveerappa, D.; Pentecost, B.T.; Arcaro, K.F.; Darie, C.C. Combinatorial Electrophoresis and Mass Spectrometry-Based Proteomics in Breast Milk for Breast Cancer Biomarker Discovery. Adv. Exp. Med. Biol. 2019, 1140, 451–467. [Google Scholar] [CrossRef]
- Zhang, J.; Rector, J.; Lin, J.Q.; Young, J.H.; Sans, M.; Katta, N.; Giese, N.; Yu, W.; Nagi, C.; Suliburk, J.; et al. Nondestructive tissue analysis for ex vivo and in vivo cancer diagnosis using a handheld mass spectrometry system. Sci. Transl. Med. 2017, 9, eaan3968. [Google Scholar] [CrossRef] [Green Version]
- King, M.E.; Zhang, J.; Lin, J.Q.; Garza, K.Y.; DeHoog, R.J.; Feider, C.L.; Bensussan, A.V.; Sans, M.; Krieger, A.; Badal, S.P.; et al. Rapid diagnosis and tumor margin assessment during pancreatic cancer surgery with the MasSpec Pen technology. Proc. Natl. Acad. Sci. USA 2021, 118, e2104411118. [Google Scholar] [CrossRef]
- Sobolev, V.; Mezentsev, A.; Ziganshin, R.; Soboleva, A.; Denieva, M.; Korsunskaya, I.; Svitich, O. LC-MS/MS analysis of lesional and normally looking psoriatic skin reveals significant changes in protein metabolism and RNA processing. PLoS ONE 2021, 16, e0240956. [Google Scholar] [CrossRef]
- Chen, A.; Chen, Z.; Xia, Y.; Lu, D.; Jia, J.; Hu, K.; Sun, A.; Zou, Y.; Qian, J.; Ge, J. Proteomics Analysis of Myocardial Tissues in a Mouse Model of Coronary Microembolization. Front. Physiol. 2018, 9, 1318. [Google Scholar] [CrossRef]
- Li, K.W.; Gonzalez-Lozano, M.A.; Koopmans, F.; Smit, A.B. Recent Developments in Data Independent Acquisition (DIA) Mass Spectrometry: Application of Quantitative Analysis of the Brain Proteome. Front. Mol. Neurosci. 2020, 13, 564446. [Google Scholar] [CrossRef]
- Kim, T.-H.; Choi, J.; Kim, H.G.; Kim, H. Quantification of Neurotransmitters in Mouse Brain Tissue by Using Liquid Chromatography Coupled Electrospray Tandem Mass Spectrometry. J. Anal. Methods Chem. 2014, 2014, 506870. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, F.; Ferland, C.; Vachon, P. Identification, characterization and quantification of specific neuropeptides in rat spinal cord by liquid chromatography electrospray quadrupole ion trap mass spectrometry. Biomed. Chromatogr. BMC 2009, 23, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Meibohm, B.; Yates, R. LC/MS/MS in drug development: Targeting the brain. BioTechniques 2005, 38, S19S–S23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seino, Y.; Nakamura, T.; Harada, T.; Nakahata, N.; Kawarabayashi, T.; Ueda, T.; Takatama, M.; Shoji, M. Quantitative Measurement of Cerebrospinal Fluid Amyloid-β Species by Mass Spectrometry. J. Alzheimers Dis. 2021, 79, 573–584. [Google Scholar] [CrossRef]
- Kirmess, K.M.; Meyer, M.R.; Holubasch, M.S.; Knapik, S.S.; Hu, Y.; Jackson, E.N.; Harpstrite, S.E.; Verghese, P.B.; West, T.; Fogelman, I.; et al. The PrecivityAD™ test: Accurate and reliable LC-MS/MS assays for quantifying plasma amyloid beta 40 and 42 and apolipoprotein E proteotype for the assessment of brain amyloidosis. Clin. Chim. Acta 2021, 519, 267–275. [Google Scholar] [CrossRef]
- Drepper, F.; Biernat, J.; Kaniyappan, S.; Meyer, H.; Mandelkow, E.; Warscheid, B.; Mandelkow, E. A combinatorial native MS and LC-MS/MS approach reveals high intrinsic phosphorylation of human Tau but minimal levels of other key modifications. J. Biol. Chem. 2020, 295, 18213–18225. [Google Scholar] [CrossRef]
- Rotunno, M.S.; Lane, M.; Zhang, W.; Wolf, P.; Oliva, P.; Viel, C.; Wills, A.-M.; Alcalay, R.N.; Scherzer, C.R.; Shihabuddin, L.S.; et al. Cerebrospinal fluid proteomics implicates the granin family in Parkinson’s disease. Sci. Rep. 2020, 10, 2479. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.A.; An, J.; Hood, B.L.; Conrads, T.P.; Bowser, R.P. Label-Free LC-MS/MS Proteomic Analysis of Cerebrospinal Fluid Identifies Protein/Pathway Alterations and Candidate Biomarkers for Amyotrophic Lateral Sclerosis. J. Proteome Res. 2015, 14, 4486–4501. [Google Scholar] [CrossRef]
- Yan, X.; Mai, L.; Lin, C.; He, W.; Yin, G.; Yu, J.; Huang, L.; Pan, S. CSF-Based Analysis for Identification of Potential Serum Biomarkers of Neural Tube Defects. Neurosci. Bull. 2017, 33, 436–444. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Song, W.; Tian, X.; Liu, Y.; Wang, Y.; Ma, J.; Wang, C.; Yan, G. A proteomic analysis of urine biomarkers in autism spectrum disorder. J. Proteom. 2021, 242, 104259. [Google Scholar] [CrossRef]
- Ngounou Wetie, A.G.; Wormwood, K.L.; Russell, S.; Ryan, J.P.; Darie, C.C.; Woods, A.G. A Pilot Proteomic Analysis of Salivary Biomarkers in Autism Spectrum Disorder. Autism Res. 2015, 8, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Su, H.; Hung, C.-C.; Lane, H.-Y.; Shiea, J. Characterization of Potential Protein Biomarkers for Major Depressive Disorder Using Matrix-Assisted Laser Desorption Ionization/Time-of-Flight Mass Spectrometry. Molecules 2021, 26, 4457. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, X.; Liu, W.; Jiang, M.; Wang, Z.; Tao, J. Proteomics Analysis of the Spinal Dorsal Horn in Diabetic Painful Neuropathy Rats With Electroacupuncture Treatment. Front. Endocrinol. 2021, 12, 608183. [Google Scholar] [CrossRef] [PubMed]
- Pellesi, L.; Guerzoni, S.; Baraldi, C.; Cainazzo, M.; Pini, L.; Bellei, E. Identification of candidate proteomic markers in the serum of medication overuse headache patients: An exploratory study. Cephalalgia 2020, 40, 033310242092184. [Google Scholar] [CrossRef]
- Ji, G.; Diep, C.; Hu, S.; Li, H.; Merrill, R.; Messadi, D.; Hu, S. Potential protein biomarkers for burning mouth syndrome discovered by quantitative proteomics. Mol. Pain 2017, 13, 174480691668679. [Google Scholar] [CrossRef] [Green Version]
- Perchey, R.T.; Tonini, L.; Tosolini, M.; Fournié, J.-J.; Lopez, F.; Besson, A.; Pont, F. PTMselect: Optimization of protein modifications discovery by mass spectrometry. Sci. Rep. 2019, 9, 4181. [Google Scholar] [CrossRef] [Green Version]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Larsen, M.R.; Trelle, M.B.; Thingholm, T.E.; Jensen, O.N. Analysis of posttranslational modifications of proteins by tandem mass spectrometry. BioTechniques 2006, 40, 790–798. [Google Scholar] [CrossRef]
- Duan, G.; Walther, D. The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput. Biol. 2015, 11, e1004049. [Google Scholar] [CrossRef]
- Dunphy, K.; Dowling, P.; Bazou, D.; O’Gorman, P. Current Methods of Post-Translational Modification Analysis and Their Applications in Blood Cancers. Cancers 2021, 13, 1930. [Google Scholar] [CrossRef]
- Whelan, S.A.; Lu, M.; He, J.; Yan, W.; Saxton, R.E.; Faull, K.F.; Whitelegge, J.P.; Chang, H.R. Mass spectrometry (LC-MS/MS) site-mapping of N-glycosylated membrane proteins for breast cancer biomarkers. J. Proteome Res. 2009, 8, 4151–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Xu, Y.; Wang, G.; Fang, C.; Bao, H.; Zhang, Y.; Lu, H. FluoroTRAQ: Quantitative Analysis of Protein S-Nitrosylation through Fluorous Solid-Phase Extraction Combining with iTRAQ by Mass Spectrometry. Anal. Chem. 2020, 92, 15317–15322. [Google Scholar] [CrossRef] [PubMed]
- Udeshi, N.; Mertins, P.; Svinkina, T.; Carr, S. Large-Scale Identification of Ubiquitination Sites by Mass Spectrometry. Nat. Protoc. 2013, 8, 1950–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, S.; Zhan, X.; Lu, M.; Li, N.; Long, Y.; Li, X.; Desiderio, D.M.; Zhan, X. Quantitative Analysis of Ubiquitinated Proteins in Human Pituitary and Pituitary Adenoma Tissues. Front. Endocrinol. 2019, 10, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hristova, V.; Sun, S.; Zhang, H.; Chan, D.W. Proteomic analysis of degradation ubiquitin signaling by ubiquitin occupancy changes responding to 26S proteasome inhibition. Clin. Proteom. 2020, 17, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, G.M.; Vogel, C. Mass spectrometry analysis of K63-ubiquitinated targets in response to oxidative stress. Data Brief. 2015, 4, 130–134. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Day, N.J.; Feng, S.; Gaffrey, M.J.; Lin, T.-D.; Paurus, V.L.; Monroe, M.E.; Moore, R.J.; Yang, B.; Xian, M.; et al. Mass spectrometry-based direct detection of multiple types of protein thiol modifications in pancreatic beta cells under endoplasmic reticulum stress. Redox Biol. 2021, 46, 102111. [Google Scholar] [CrossRef]
- Tikhonov, D.; Kulikova, L.; Kopylov, A.T.; Rudnev, V.; Stepanov, A.; Malsagova, K.; Izotov, A.; Kulikov, D.; Zulkarnaev, A.; Enikeev, D.; et al. Proteomic and molecular dynamic investigations of PTM-induced structural fluctuations in breast and ovarian cancer. Sci. Rep. 2021, 11, 19318. [Google Scholar] [CrossRef]
- Sanda, M.; Ahn, J.; Kozlik, P.; Goldman, R. Analysis of site and structure specific core fucosylation in liver cirrhosis using exoglycosidase-assisted data-independent LC-MS/MS. Sci. Rep. 2021, 11, 23273. [Google Scholar] [CrossRef]
- Kendrick, N.; Powers, G.; Johansen, J.; Hoelter, M.; Koll, A.; Carlson, S.; Channaveerappa, D.; Darie, C.C. Preparation of a phosphotyrosine-protein standard for use in semiquantitative western blotting with enhanced chemiluminescence. PLoS ONE 2020, 15, e0234645. [Google Scholar] [CrossRef]
- Sturtz, L.A.; Wang, G.; Shah, P.; Searfoss, R.; Raj-Kumar, P.-K.; Hooke, J.A.; Fantacone-Campbell, J.L.; Deyarmin, B.; Cutler, M.L.; Sarangarajan, R.; et al. Comparative analysis of differentially abundant proteins quantified by LC–MS/MS between flash frozen and laser microdissected OCT-embedded breast tumor samples. Clin. Proteom. 2020, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Rossouw, S.; Bendou, H.; Blignaut, R.; Bell, L.; Rigby, J.; Christoffels, A.; Ladányi, A. Evaluation of Protein Purification Techniques and Effects of Storage Duration on LC-MS/MS Analysis of Archived FFPE Human CRC Tissues Evaluation of Protein Purification Techniques and Effects of Storage Duration on LC-MS/MS Analysis of Archived FFPE Human CRC Tissues. Pathol. Oncol. Res. 2021, 27, 622855. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.E.; Hedrick, V.; Paschoal Sobreira, T.J.; Dykhuizen, E.C.; Aryal, U.K. Analysis of Human Nuclear Protein Complexes by Quantitative Mass Spectrometry Profiling. Proteomics 2018, 18, e1700427. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Franquesa, A.; Stocks, B.; Chubanava, S.; Hattel, H.B.; Moreno-Justicia, R.; Peijs, L.; Treebak, J.T.; Zierath, J.R.; Deshmukh, A.S. Mass-spectrometry-based proteomics reveals mitochondrial supercomplexome plasticity. Cell Rep. 2021, 35, 109180. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Karnovsky, A.; Sans, M.D.; Andrews, P.C.; Williams, J.A. Molecular characterization of the endoplasmic reticulum: Insights from proteomic studies. Proteomics 2010, 10, 4040–4052. [Google Scholar] [CrossRef]
- Thelen, M.; Winter, D.; Braulke, T.; Gieselmann, V. SILAC-Based Comparative Proteomic Analysis of Lysosomes from Mammalian Cells Using LC-MS/MS; Humana Press: New York, NY, USA, 2017; Volume 1594, pp. 1–18. [Google Scholar]
- Mosen, P.; Sanner, A.; Singh, J.; Winter, D. Targeted Quantification of the Lysosomal Proteome in Complex Samples. Proteomes 2021, 9, 4. [Google Scholar] [CrossRef]
- Gronemeyer, T.; Wiese, S.; Ofman, R.; Bunse, C.; Pawlas, M.; Hayen, H.; Eisenacher, M.; Stephan, C.; Meyer, H.E.; Waterham, H.R.; et al. The proteome of human liver peroxisomes: Identification of five new peroxisomal constituents by a label-free quantitative proteomics survey. PLoS ONE 2013, 8, e57395. [Google Scholar] [CrossRef]
- Choi, S.; Kelber, J.; Jiang, X.; Strnadel, J.; Fujimura, K.; Pasillas, M.; Coppinger, J.; Klemke, R. Procedures for the biochemical enrichment and proteomic analysis of the cytoskeletome. Anal. Biochem. 2014, 446, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Vrana, M.; Zhang, H.; Prasad, B. An LC-MS/MS proteomics based methodology to simultaneously evaluate the recovery, enrichment and purity of microsomal and cytosolic fractions isolated from tissues for in vitro drug metabolism studies. Drug Metab. Pharmacokinet. 2018, 33, S17. [Google Scholar] [CrossRef]
- Smolarz, M.; Pietrowska, M.; Matysiak, N.; Mielańczyk, Ł.; Widlak, P. Proteome Profiling of Exosomes Purified from a Small Amount of Human Serum: The Problem of Co-Purified Serum Components. Proteomes 2019, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Lai, X. Reproducible method to enrich membrane proteins with high purity and high yield for an LC-MS/MS approach in quantitative membrane proteomics. Electrophoresis 2013, 34, 809–817. [Google Scholar] [CrossRef] [Green Version]
- León, I.R.; Schwämmle, V.; Jensen, O.N.; Sprenger, R.R. Quantitative Assessment of In-solution Digestion Efficiency Identifies Optimal Protocols for Unbiased Protein Analysis. Mol. Cell. Proteom. 2013, 12, 2992–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapek, J.D., Jr.; McGrath, J.L.; Ricke, W.A.; Friedman, A.E. LC/LC-MS/MS of an innovative prostate human epithelial cancer (PHEC) in vitro model system. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 893–894, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, J.; Timms, J.F. Proteomic profiling of ovarian cancer models using TMT-LC-MS/MS. Methods Mol. Biol. 2013, 1049, 271–284. [Google Scholar]
- Meng, F.; Zou, L.; Zhang, T.; Jiang, L.; Ding, Y.; Yu, P.; Peng, J. Using LC-MS/MS-based targeted proteomics to monitor the pattern of ABC transporters expression in the development of drug resistance. Cancer Manag. Res. 2018, 10, 2859–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, R.; Han, X.; Xiao, D.; He, L.; Gong, Y.; Sun, L.; Fan, D.; You, Y.; Wang, T.; Yan, X.; et al. Identification of Functional Interactome of Gastric Cancer Cells with Helicobacter pylori Outer Membrane Protein HpaA by HPLC-MS/MS. BioMed Res. Int. 2020, 2020, 1052926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; He, Y.; Zheng, N.; Yan, X.; Wang, J. Pipeline for Targeted Meta-Proteomic Analyses to Assess the Diversity of Cattle Rumen Microbial Urease. Front. Microbiol. 2020, 11, 2241. [Google Scholar] [CrossRef] [PubMed]
- De Re, V.; Repetto, O.; Zanussi, S.; Casarotto, M.; Caggiari, L.; Canzonieri, V.; Cannizzaro, R. Protein signature characterizing Helicobacter pylori strains of patients with autoimmune atrophic gastritis, duodenal ulcer and gastric cancer. Infect. Agents Cancer 2017, 12, 22. [Google Scholar] [CrossRef]
- Karlsson, R.; Thorell, K.; Hosseini, S.; Kenny, D.; Sihlbom, C.; Sjöling, Å.; Karlsson, A.; Nookaew, I. Comparative Analysis of Two Helicobacter pylori Strains using Genomics and Mass Spectrometry-Based Proteomics. Front. Microbiol. 2016, 7, 1757. [Google Scholar] [CrossRef]
- Oom, A.L.; Stoneham, C.A.; Lewinski, M.K.; Richards, A.; Wozniak, J.M.; Gonzalez, D.J.; Krogan, N.J.; Guatelli, J. Proteomic Organellar Mapping Reveals Modulation of Peroxisomes by HIV-1. bioRxiv 2021. [Google Scholar] [CrossRef]
- Li, X.; Qi, W.; Gao, Y.; Qi, X.; Wang, Y.; Gao, H.; Gao, Y.; Wang, X. Quantitative iTRAQ LC-MS/MS Proteomics Reveals the Proteome Profiles of DF-1 Cells after Infection with Subgroup J Avian Leukosis Virus. BioMed Res. Int. 2015, 2015, 395307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, O.; Zvi, A.; Rosen, O.; Achdout, H.; Ben-Shmuel, A.; Shifman, O.; Yitzhaki, S.; Laskar, O.; Feldberg, L. Specific and Rapid SARS-CoV-2 Identification Based on LC-MS/MS Analysis. ACS Omega 2021, 6, 3525–3534. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, J.L.; DeGrasse, J.A.; McFarland, M.A.; Tall, E.A.; Shefcheck, K.J.; Ward, J.L.; Bannerman, D.D. The proteomic advantage: Label-free quantification of proteins expressed in bovine milk during experimentally induced coliform mastitis. Vet. Immunol. Immunopathol. 2010, 138, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Addis, M.F.; Maffioli, E.M.; Ceciliani, F.; Tedeschi, G.; Zamarian, V.; Tangorra, F.; Albertini, M.; Piccinini, R.; Bronzo, V. Influence of subclinical mastitis and intramammary infection by coagulase-negative staphylococci on the cow milk peptidome. J. Proteom. 2020, 226, 103885. [Google Scholar] [CrossRef] [PubMed]
- Abril, A.G.; Carrera, M.; Böhme, K.; Barros-Velázquez, J.; Cañas, B.; Rama, J.L.R.; Villa, T.G.; Calo-Mata, P. Characterization of Bacteriophage Peptides of Pathogenic Streptococcus by LC-ESI-MS/MS: Bacteriophage Phylogenomics and Their Relationship to Their Host. Front. Microbiol. 2020, 11, 1241. [Google Scholar] [CrossRef]
- Lasch, P.; Uhlig, S.; Uhlig, C.; Wilhelm, C.; Bergmann, N.; Wittke, S. Development and In-House Validation of an LC–MS and LC–MS/MS Assay for the Determination of Food Fraud for Different Fish Species. J. AOAC Int. 2019, 102, 1330–1338. [Google Scholar] [CrossRef]
- Carrera, M.; Pazos, M.; Gasset, M. Proteomics-Based Methodologies for the Detection and Quantification of Seafood Allergens. Foods 2020, 9, 1134. [Google Scholar] [CrossRef]
- Jira, W.; Schwägele, F. HPLC-MS/MS-Detection of Caseins and Whey Proteins in Meat Products. Procedia Food Sci. 2015, 5, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Spörl, J.; Speer, K.; Jira, W. A UHPLC-MS/MS Method for the Detection of Meat Substitution by Nine Legume Species in Emulsion-Type Sausages. Foods 2021, 10, 947. [Google Scholar] [CrossRef]
- Montowska, M.; Spychaj, A. Quantification of species-specific meat proteins in cooked and smoked sausages using infusion mass spectrometry. J. Food Sci. Technol. 2018, 55, 4984–4993. [Google Scholar] [CrossRef] [Green Version]
- Fredriksson, S.-Å.; Hulst, A.G.; Artursson, E.; de Jong, A.L.; Nilsson, C.; van Baar, B.L.M. Forensic Identification of Neat Ricin and of Ricin from Crude Castor Bean Extracts by Mass Spectrometry. Anal. Chem. 2005, 77, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Alves, T.O.; D’Almeida, C.T.S.; Scherf, K.A.; Ferreira, M.S.L. Modern Approaches in the Identification and Quantification of Immunogenic Peptides in Cereals by LC-MS/MS. Front. Plant. Sci. 2019, 10, 1470. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ochoa, Á.; Leyva-Jiménez, F.J.; De la Luz Cádiz-Gurrea, M.; Pimentel-Moral, S.; Segura-Carretero, A. The Role of High-Resolution Analytical Techniques in the Development of Functional Foods. Int. J. Mol. Sci. 2021, 22, 3220. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Lee, J.; Han, M.-J. Comprehensive analysis of the cardiac proteome in a rat model of myocardial ischemia-reperfusion using a TMT-based quantitative proteomic strategy. Proteome Sci. 2020, 18, 2. [Google Scholar] [CrossRef]
- Liu, Z.; Lin, Y.; Zhang, S.; Liang, Q.; Luo, G. Comparative proteomic analysis using 2DE-LC-MS/MS reveals the mechanism of Fuzhuan brick tea extract against hepatic fat accumulation in rats with non-alcoholic fatty liver disease. Electrophoresis 2015, 36, 2002–2016. [Google Scholar] [CrossRef]
- Campos, G.A.F.; Kruizenga, J.G.K.T.; Sagu, S.T.; Schwarz, S.; Homann, T.; Taubert, A.; Rawel, H.M. Effect of the Post-Harvest Processing on Protein Modification in Green Coffee Beans by Phenolic Compounds. Foods 2022, 11, 159. [Google Scholar] [CrossRef]
- Yamamoto, K.; Hayashi, M.; Murakami, Y.; Araki, Y.; Otsuka, Y.; Kashiwagi, T.; Shimamura, T.; Ukeda, H. Development of LC-MS/MS analysis of cyclic dipeptides and its application to tea extract. Biosci. Biotechnol. Biochem. 2015, 80, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Cheng, X.; Liao, B.; Xu, J.; Han, X.; Zhang, J.; Lin, Z.; Hu, L. Spatial protein expression of Panax ginseng by in-depth proteomic analysis for ginsenoside biosynthesis and transportation. J. Ginseng Res. 2021, 45, 58–65. [Google Scholar] [CrossRef]
- Müller, A.; Goedecke, C.; Eisentraut, P.; Piechotta, C.; Braun, U. Microplastic analysis using chemical extraction followed by LC-UV analysis: A straightforward approach to determine PET content in environmental samples. Environ. Sci. Eur. 2020, 32, 85. [Google Scholar] [CrossRef]
- Bartkova, S.; Kahru, A.; Heinlaan, M.; Scheler, O. Techniques Used for Analyzing Microplastics, Antimicrobial Resistance and Microbial Community Composition: A Mini-Review. Front. Microbiol. 2021, 12, 603967. [Google Scholar] [CrossRef]
- Parkinson, E.; Skipp, P.; Aleksic, M.; Garrow, A.; Dadd, T.; Hughes, M.; Clough, G.; O’Connor, C.D. Proteomic analysis of the human skin proteome after in vivo treatment with sodium dodecyl sulphate. PLoS ONE 2014, 9, e97772. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neagu, A.-N.; Jayathirtha, M.; Baxter, E.; Donnelly, M.; Petre, B.A.; Darie, C.C. Applications of Tandem Mass Spectrometry (MS/MS) in Protein Analysis for Biomedical Research. Molecules 2022, 27, 2411. https://doi.org/10.3390/molecules27082411
Neagu A-N, Jayathirtha M, Baxter E, Donnelly M, Petre BA, Darie CC. Applications of Tandem Mass Spectrometry (MS/MS) in Protein Analysis for Biomedical Research. Molecules. 2022; 27(8):2411. https://doi.org/10.3390/molecules27082411
Chicago/Turabian StyleNeagu, Anca-Narcisa, Madhuri Jayathirtha, Emma Baxter, Mary Donnelly, Brindusa Alina Petre, and Costel C. Darie. 2022. "Applications of Tandem Mass Spectrometry (MS/MS) in Protein Analysis for Biomedical Research" Molecules 27, no. 8: 2411. https://doi.org/10.3390/molecules27082411
APA StyleNeagu, A. -N., Jayathirtha, M., Baxter, E., Donnelly, M., Petre, B. A., & Darie, C. C. (2022). Applications of Tandem Mass Spectrometry (MS/MS) in Protein Analysis for Biomedical Research. Molecules, 27(8), 2411. https://doi.org/10.3390/molecules27082411