
Covalent Reversible Inhibitors of Cysteine Proteases Containing the Nitrile Warhead: Recent Advancement in the Field of Viral and Parasitic Diseases

 ,
,  ,
,  ,
,  ,
, 
 and
and 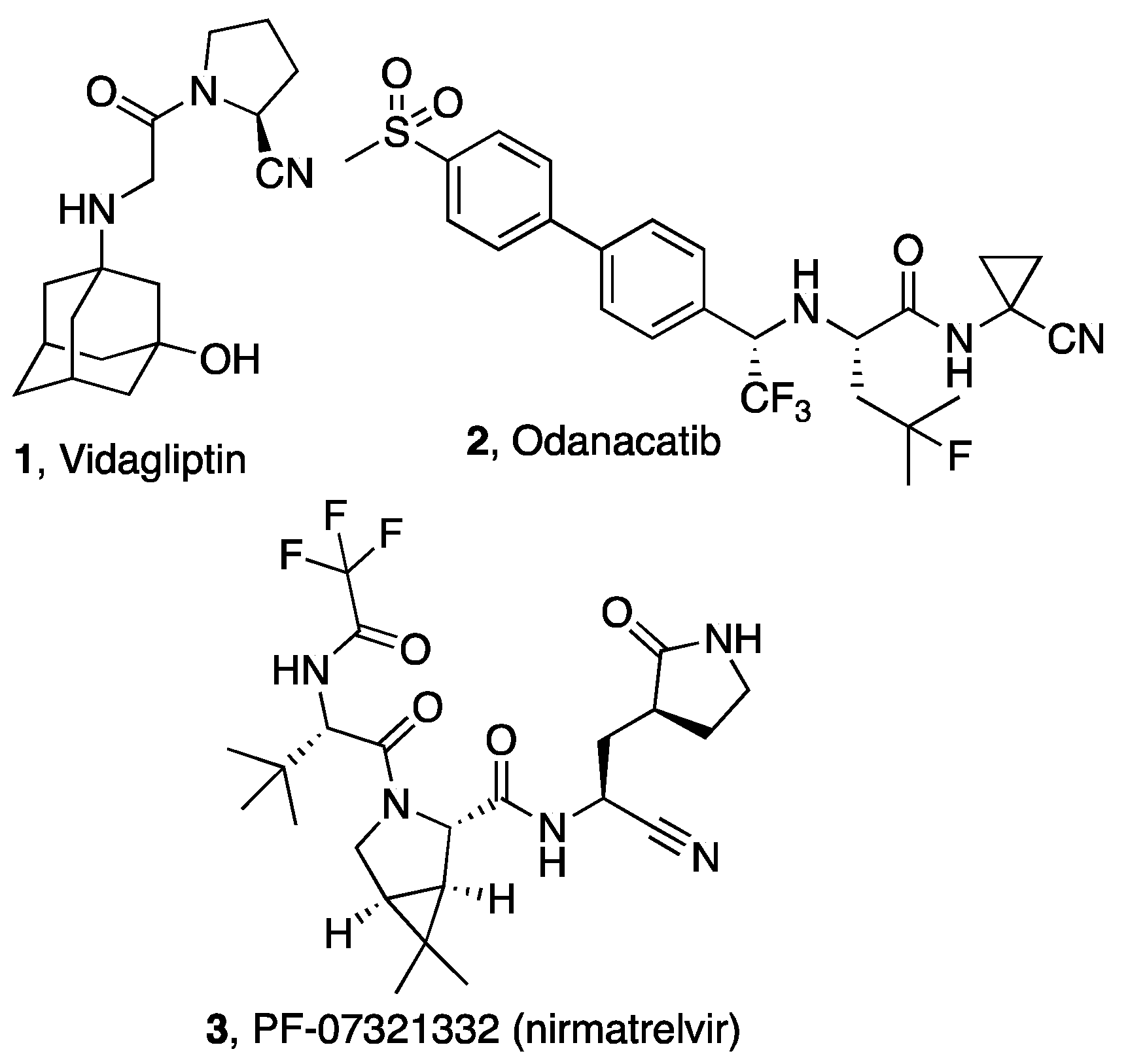{kind=link}
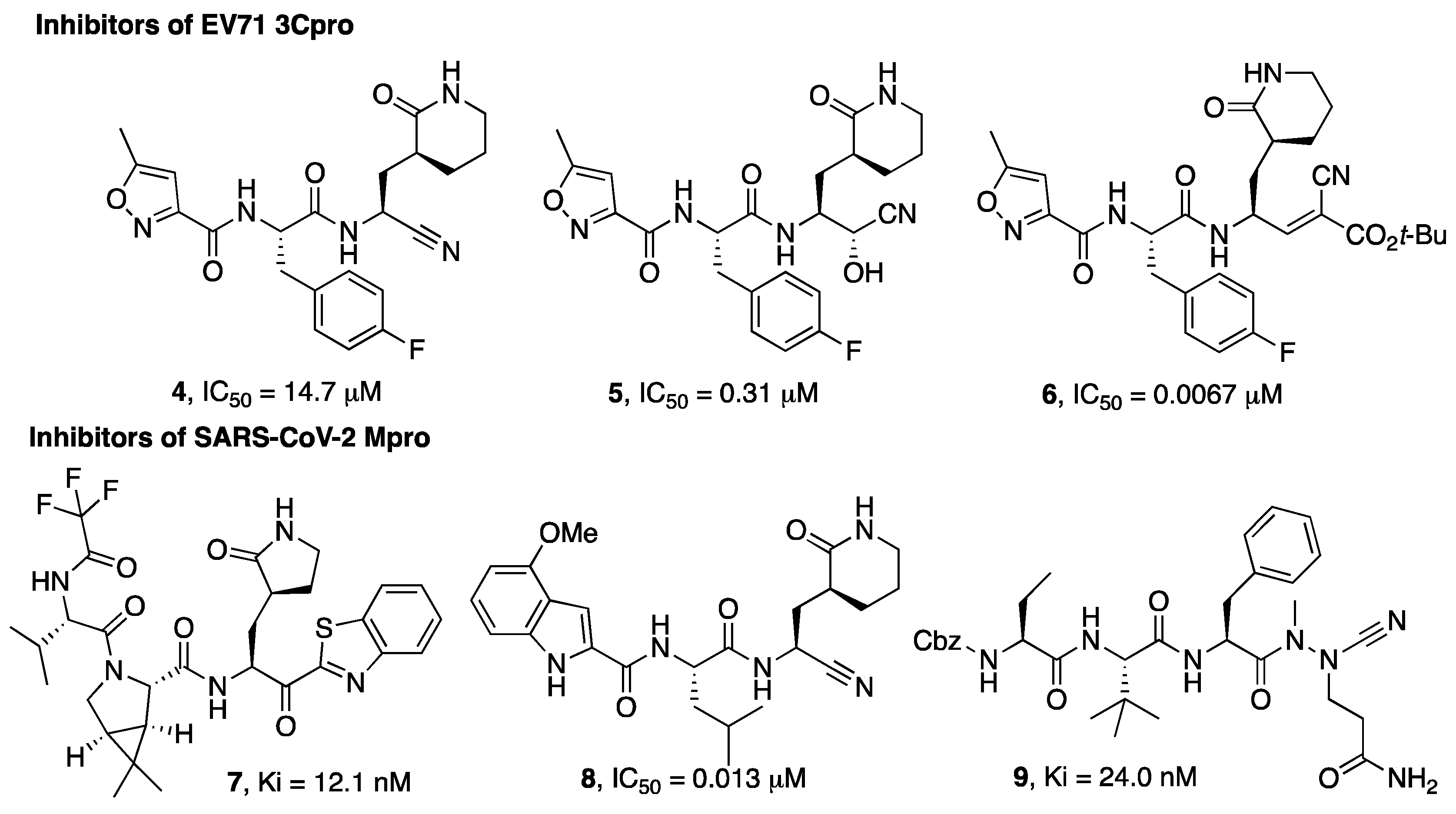{kind=link}
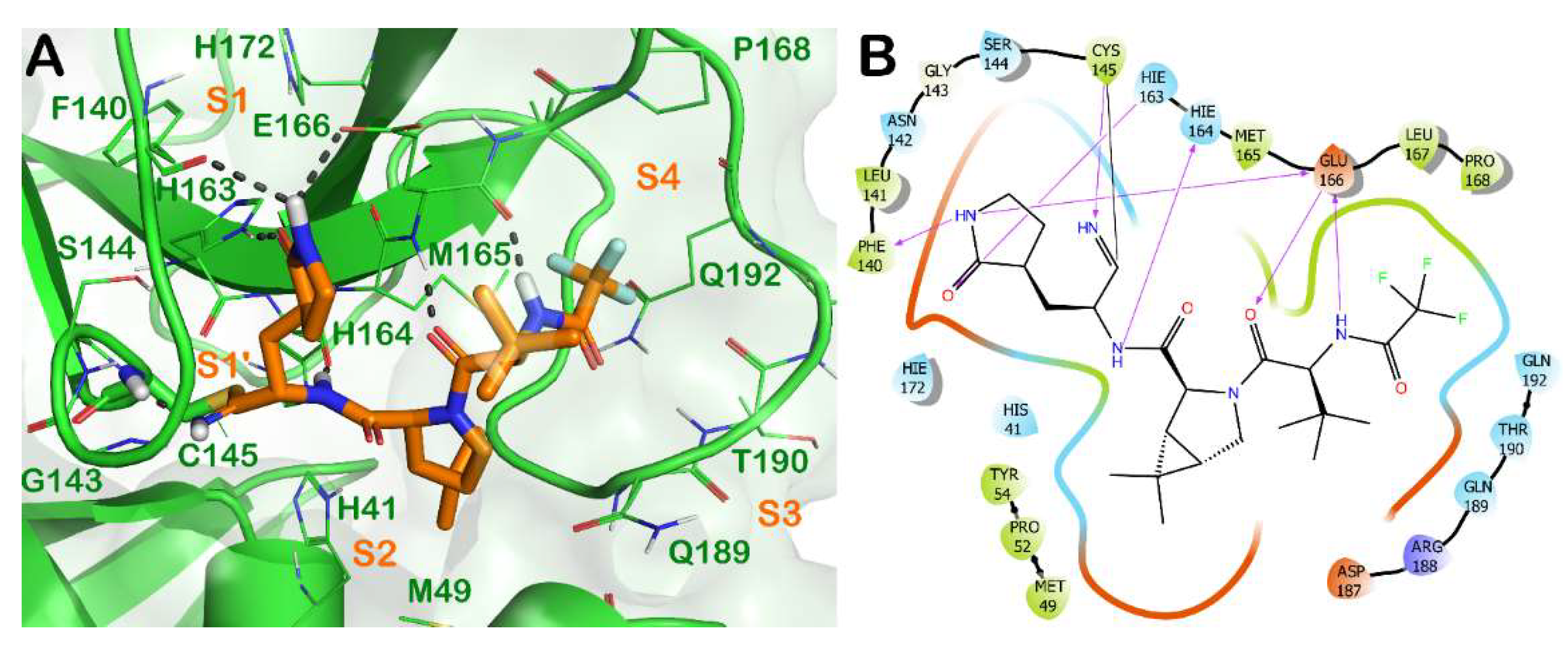{kind=link}
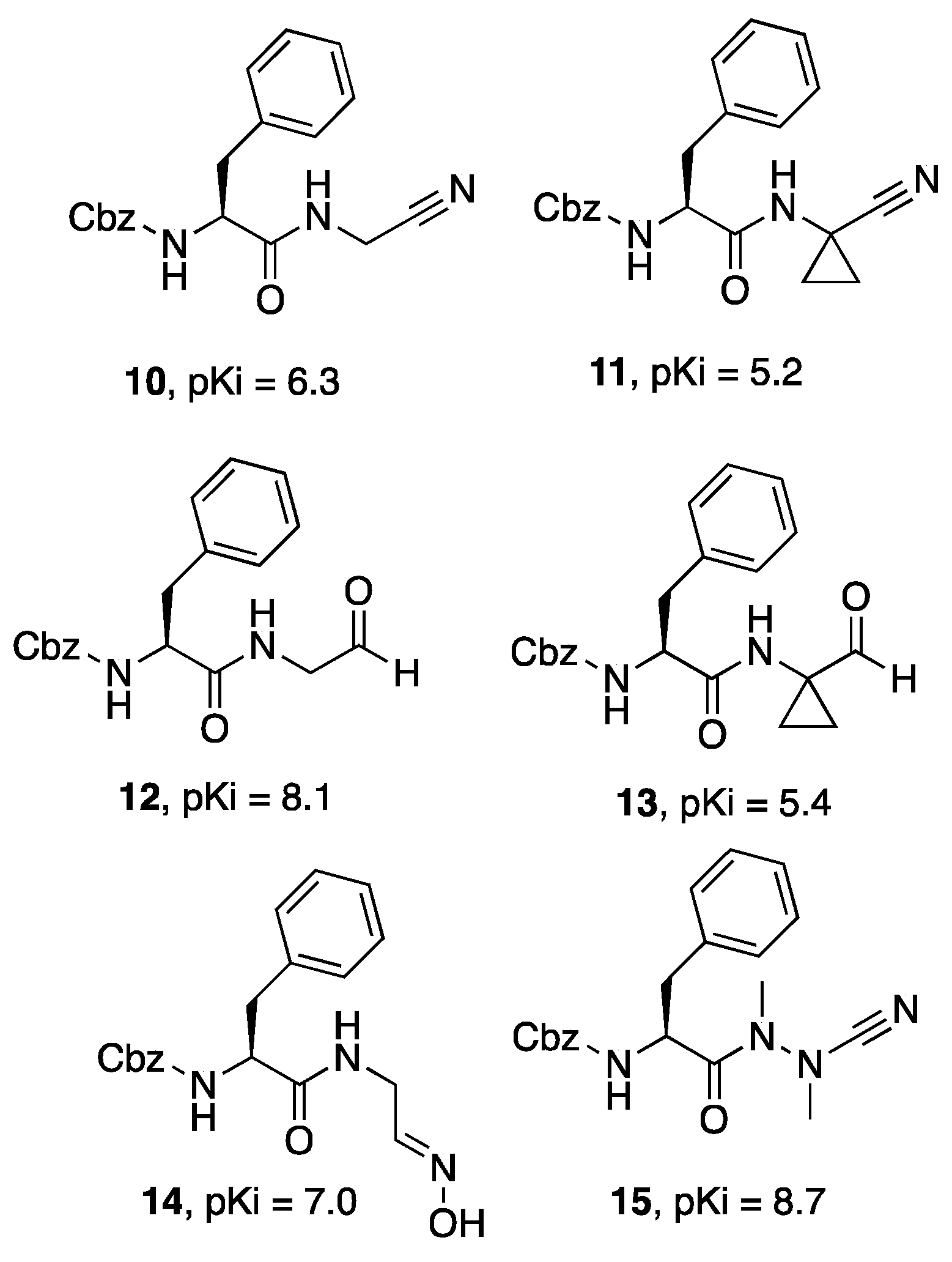{kind=link}
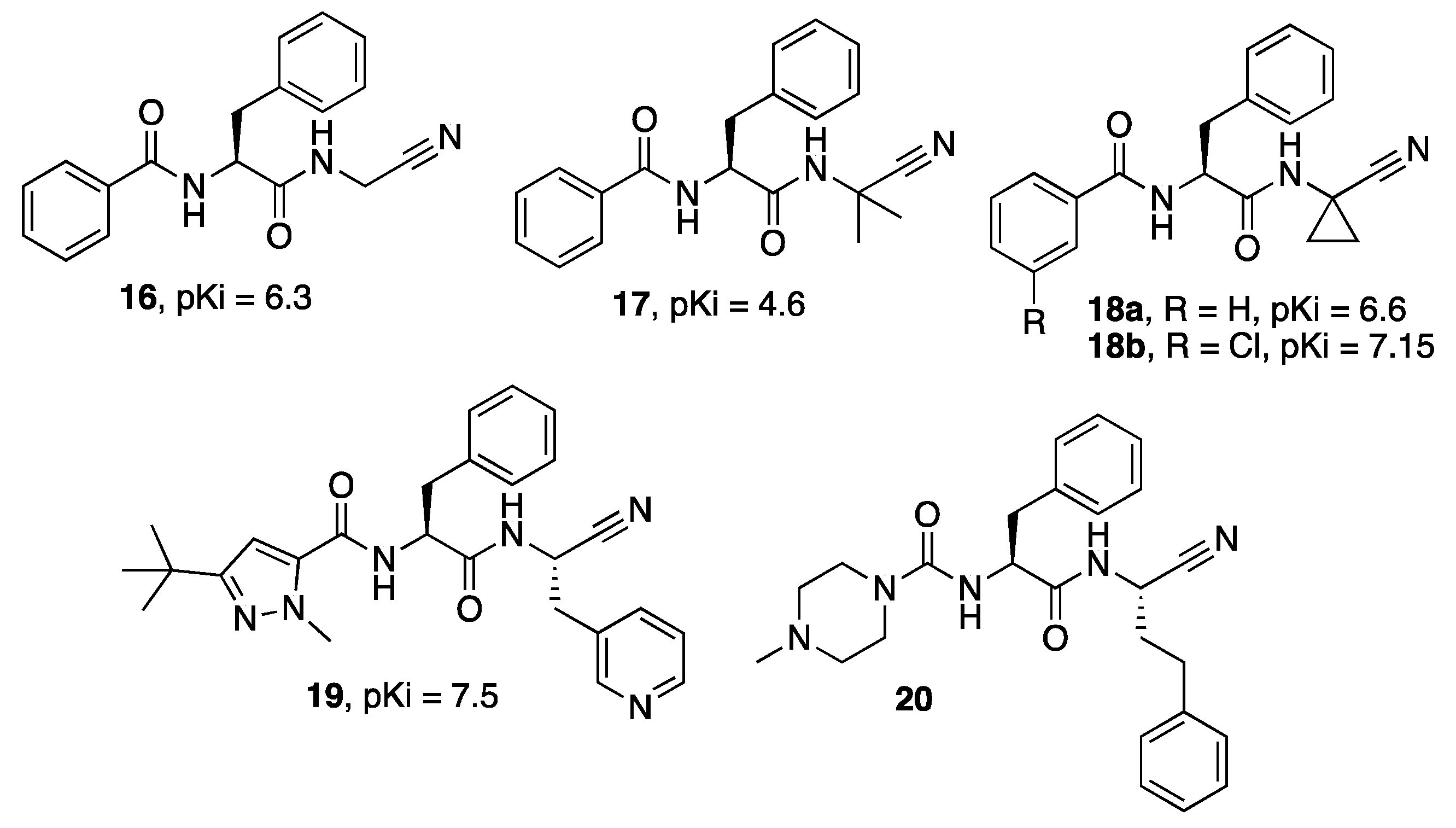{kind=link}
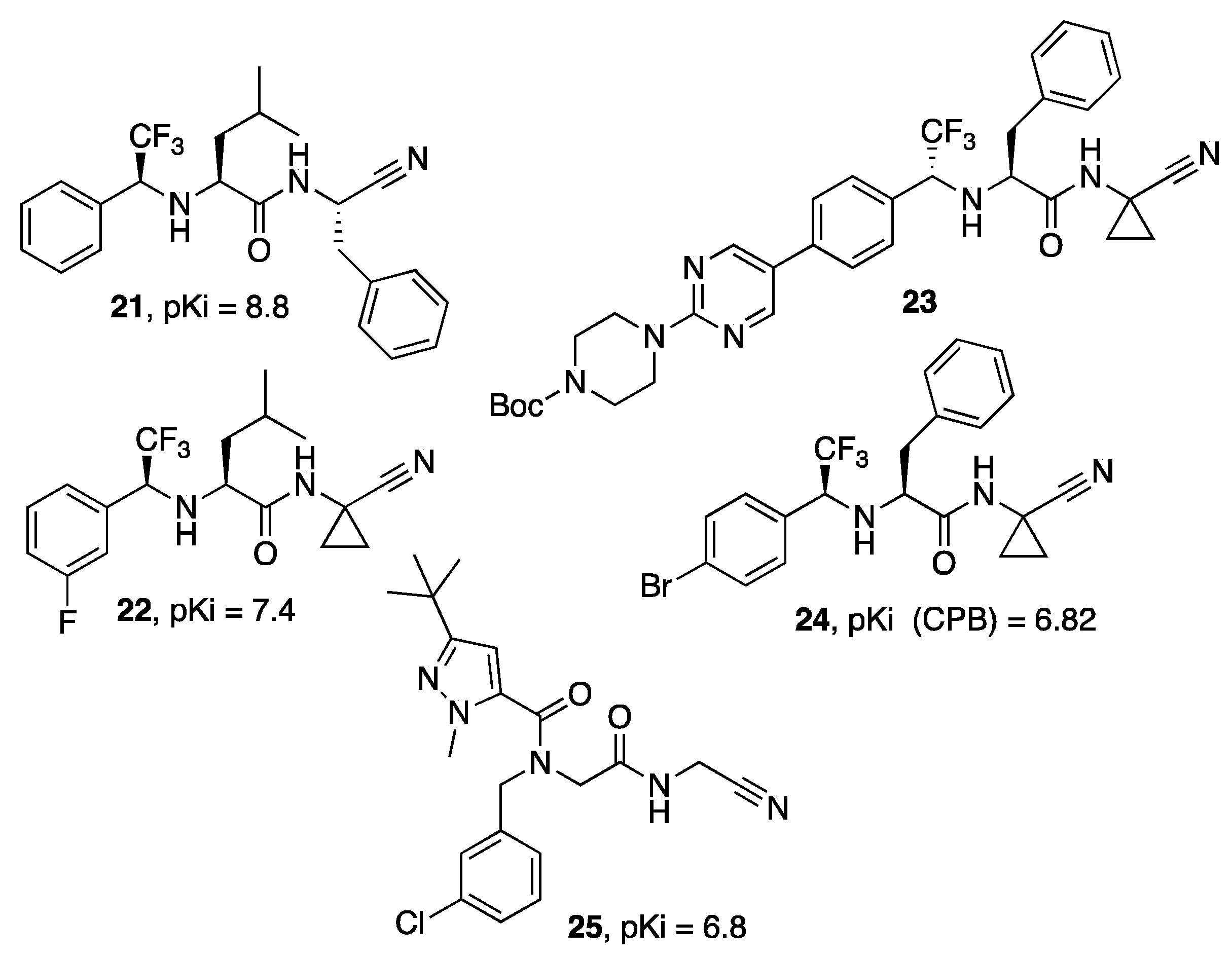{kind=link}
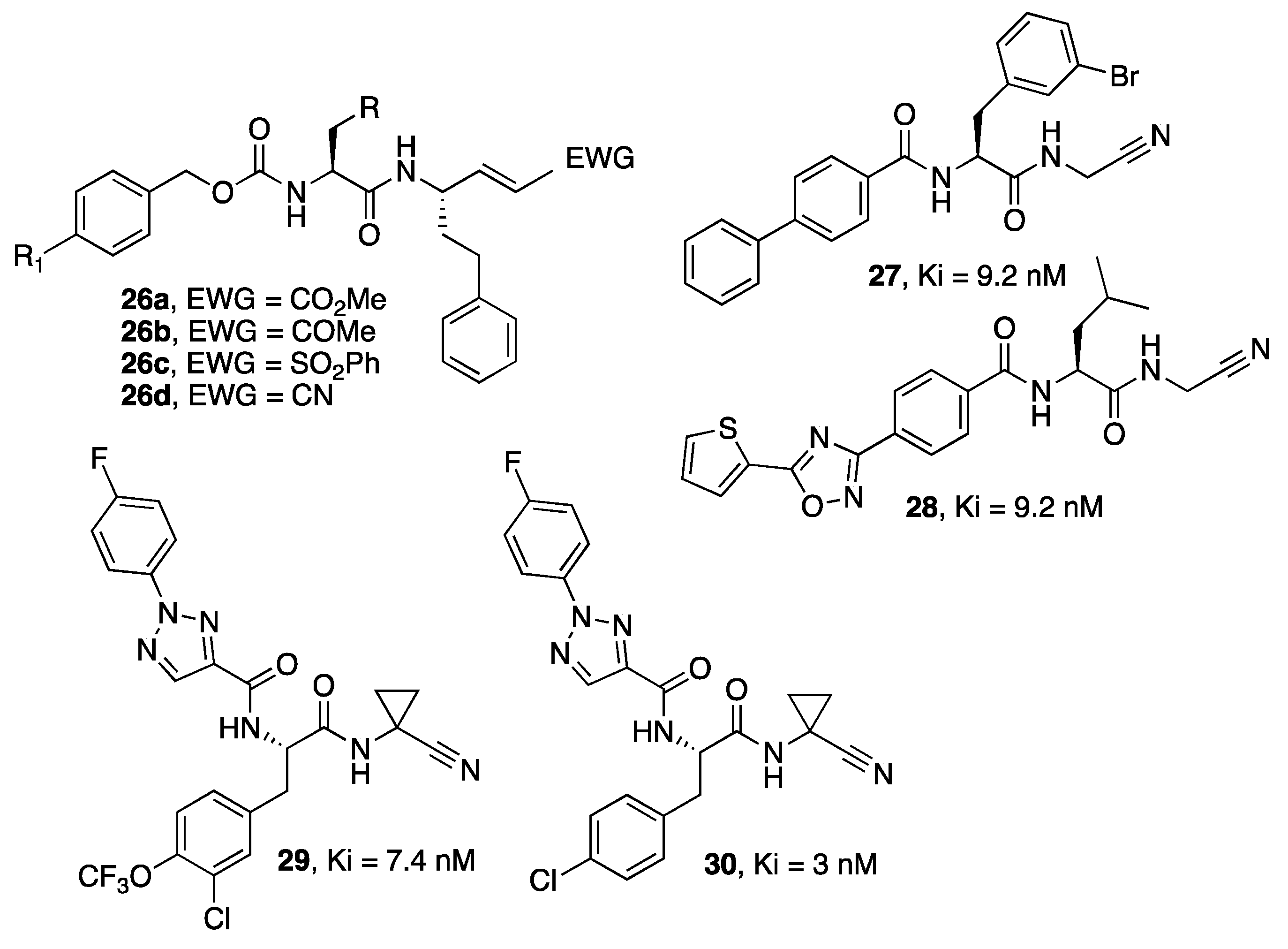{kind=link}
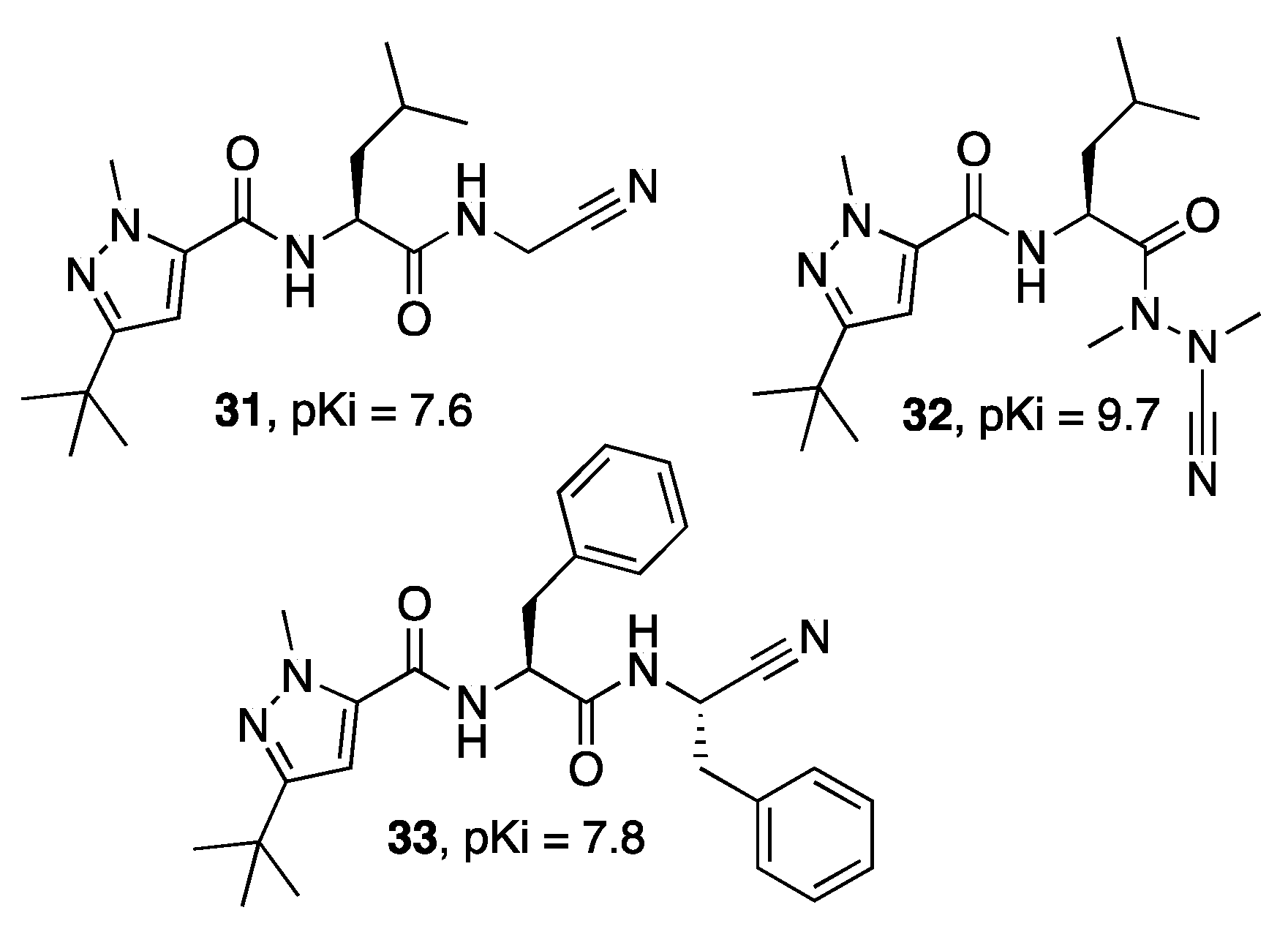{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inhibitors of Viral Cysteine Proteases
3. Inhibitors of Protozoan Parasite Proteases
3.1. Inhibitors of Cruzain (Cz) from Trypanosoma cruzi
3.2. Inhibitors of Rhodesain (RD) from Trypanosoma brucei
3.3. Inhibitors of Cysteine Protease B from Leishmania spp.
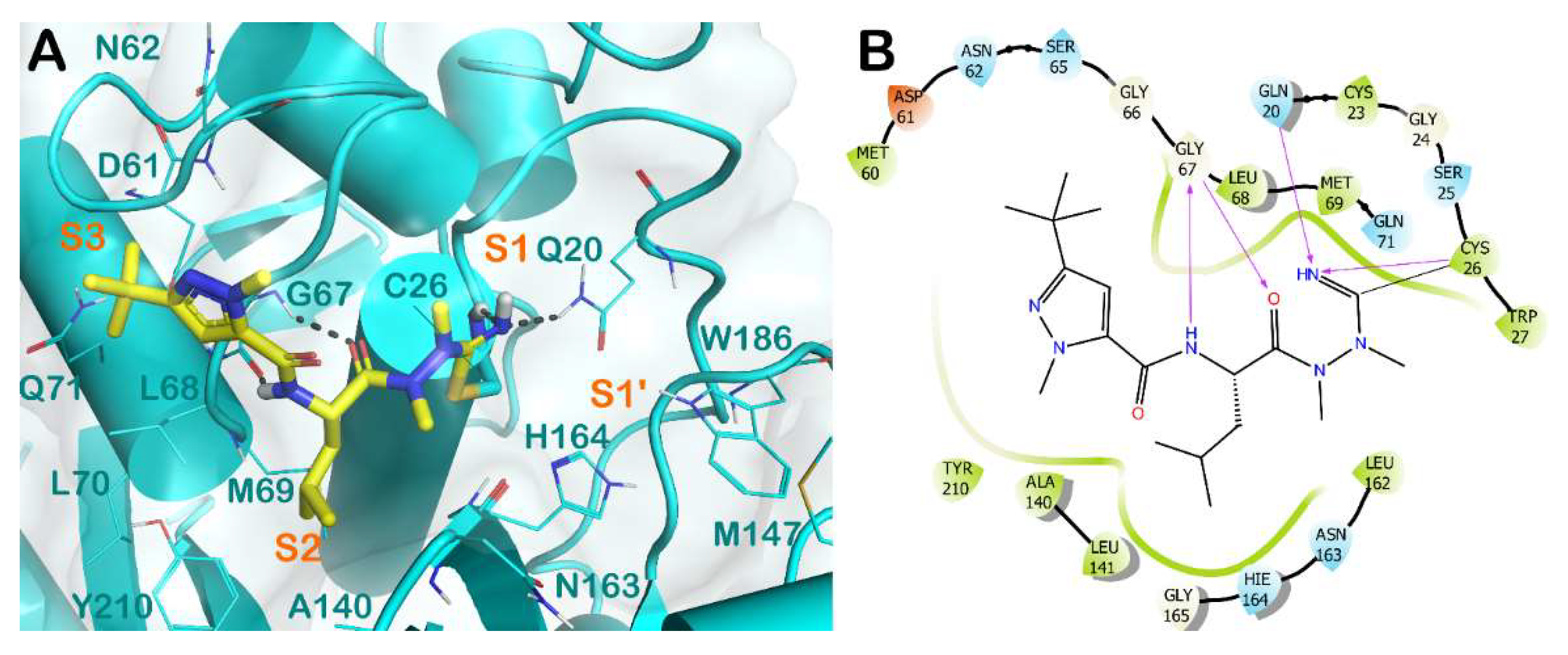
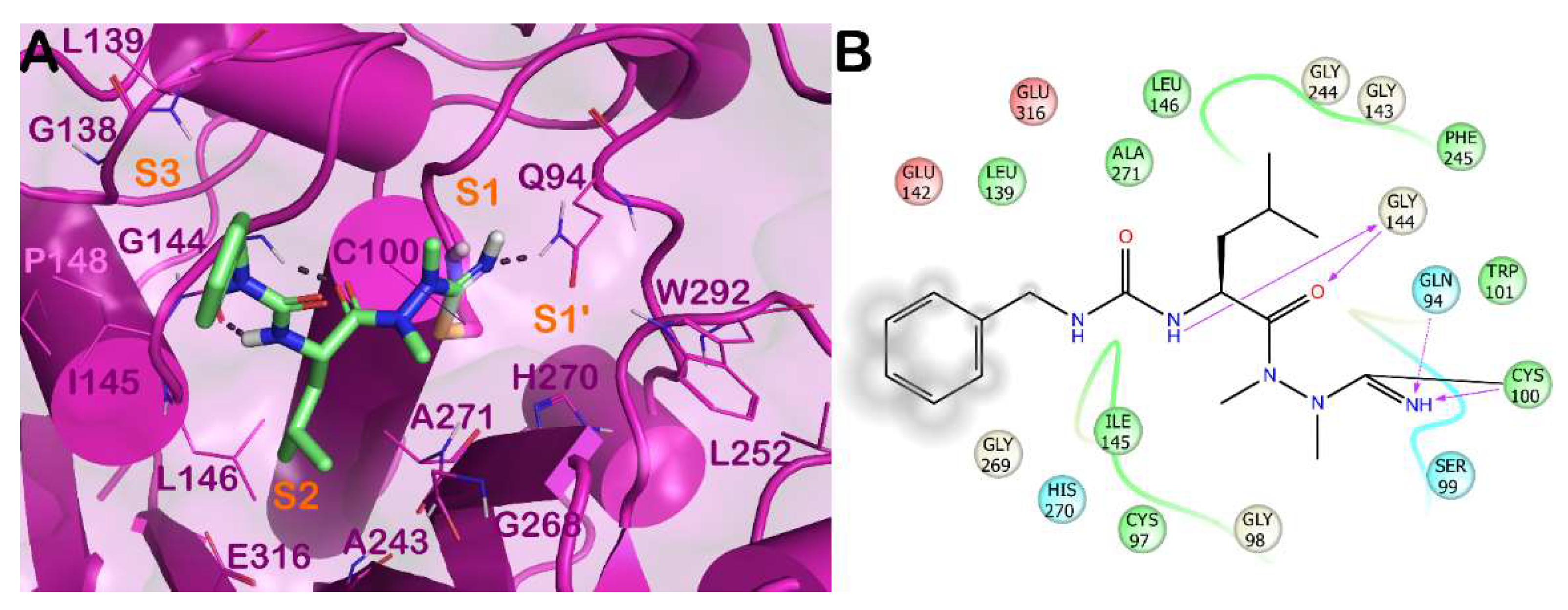
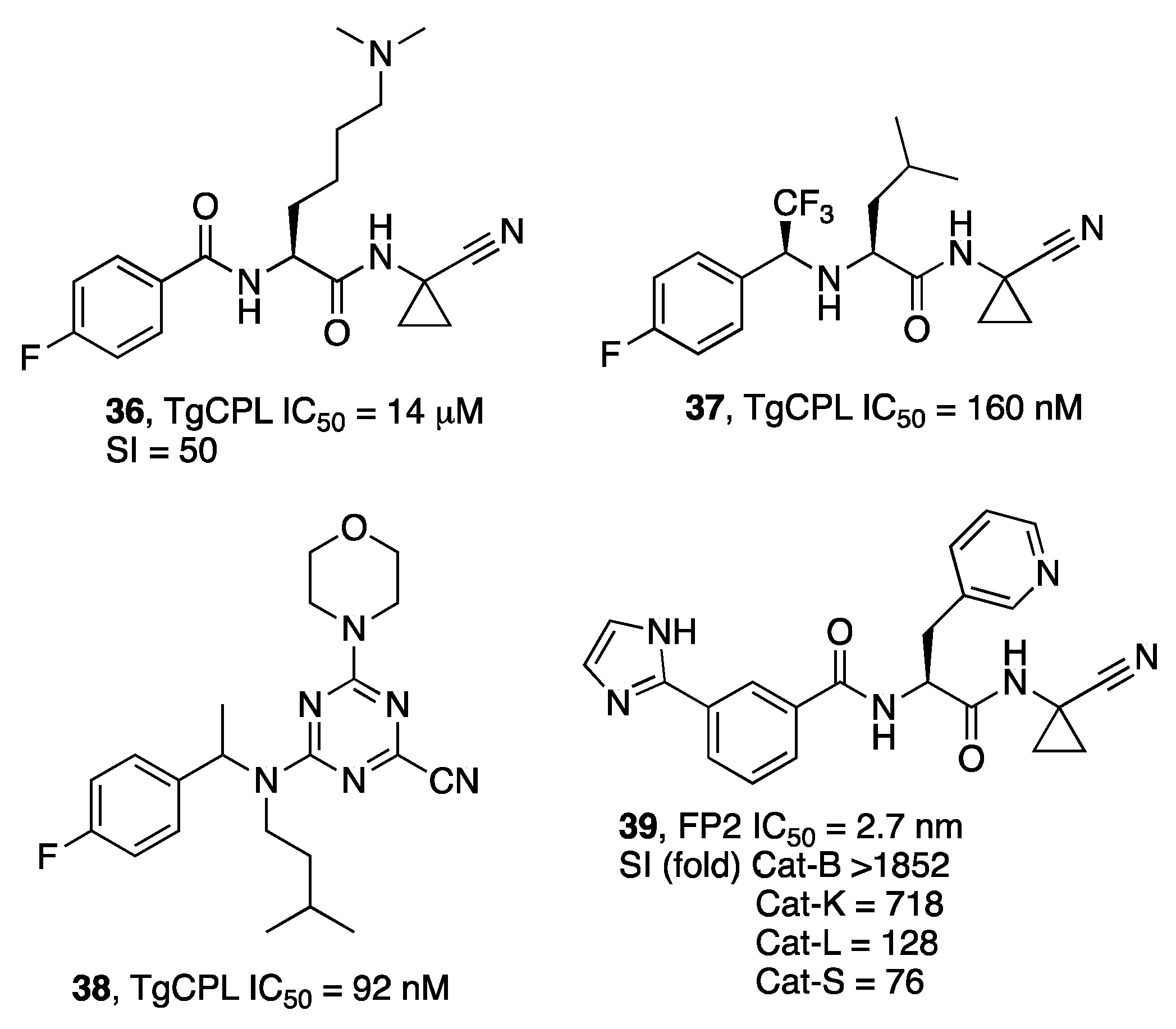
3.4. Apicomplexa Parasites: Toxoplasma gondii and Plasmodium falciparum
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wang, X.; Wang, Y.; Li, X.; Yu, Z.; Song, C.; Du, Y. Nitrile-containing pharmaceuticals: Target, mechanism of action, and their SAR studies. RSC Med. Chem. 2021, 12, 1650–1671. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Du, Y.; Huang, N. A survey of the role of nitrile groups in protein-ligand interactions. Future Med. Chem. 2018, 10, 2713–2728. [Google Scholar] [CrossRef] [PubMed]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114. [Google Scholar] [CrossRef]
- Shindo, N.; Ojida, A. Recent progress in covalent warheads for in vivo targeting of endogenous proteins. Bioorg. Med. Chem. 2021, 47, 116386. [Google Scholar] [CrossRef]
- Wettstein, L.; Kirchhoff, F.; Munch, J. The Transmembrane Protease TMPRSS2 as a Therapeutic Target for COVID-19 Treatment. Int. J. Mol. Sci. 2022, 23, 1351. [Google Scholar] [CrossRef]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Perez-Silanes, S. Examination of multiple Trypanosoma cruzi targets in a new drug discovery approach for Chagas disease. Bioorg. Med. Chem. 2022, 58, 116577. [Google Scholar] [CrossRef]
- Dos Santos, N.I.J.; de Aquino, T.M.; da Silva-Junior, E.F. Cruzain and Rhodesain Inhibitors: Last Decade of Advances in Seeking for New Compounds Against American and African Trypanosomiases. Curr. Top Med. Chem. 2021, 21, 1871–1899. [Google Scholar] [CrossRef]
- Alvarez, V.E.; Iribarren, P.A.; Niemirowicz, G.T.; Cazzulo, J.J. Update on relevant trypanosome peptidases: Validated targets and future challenges. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140577. [Google Scholar] [CrossRef]
- Rosenthal, P.J.; Sijwali, P.S.; Singh, A.; Shenai, B.R. Cysteine proteases of malaria parasites: Targets for chemotherapy. Curr. Pharm. Des. 2002, 8, 1659–1672. [Google Scholar] [CrossRef]
- Tan, M.S.Y.; Blackman, M.J. Malaria parasite egress at a glance. J. Cell Sci. 2021, 134, 134. [Google Scholar] [CrossRef] [PubMed]
- Akaji, K.; Konno, H. Design and Evaluation of Anti-SARS-Coronavirus Agents Based on Molecular Interactions with the Viral Protease. Molecules 2020, 25, 3920. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Gemma, S. Structure-Based Design of Drugs and Other Bioactive Molecules: Tools and Strategies; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Stoian, A.P.; Sachinidis, A.; Stoica, R.A.; Nikolic, D.; Patti, A.M.; Rizvi, A.A. The efficacy and safety of dipeptidyl peptidase-4 inhibitors compared to other oral glucose-lowering medications in the treatment of type 2 diabetes. Metabolism 2020, 109, 154295. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2021, 1–5. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- Serafimova, I.M.; Pufall, M.A.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frodin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. [Google Scholar] [CrossRef]
- Diarimalala, R.O.; Hu, M.; Wei, Y.; Hu, K. Recent advances of enterovirus 71 [Formula: See text] targeting Inhibitors. Virol. J. 2020, 17, 173. [Google Scholar] [CrossRef]
- Wen, W.; Qi, Z.; Wang, J. The Function and Mechanism of Enterovirus 71 (EV71) 3C Protease. Curr. Microbiol. 2020, 77, 1968–1975. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, L.; Zhai, Y.; Ma, J.; Nie, Q.; Li, T.; Yin, Z.; Sun, Y.; Shang, L. Inhibition of enterovirus 71 replication by an α-hydroxy-nitrile derivative NK-1.9 k. Antivir. Res. 2017, 141, 91–100. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhao, X.; Cui, Z.; Wang, M.; Wang, Y.; Li, L.; Sun, Q.; Yang, X.; Zeng, D.; Liu, Y.; et al. Cyanohydrin as an Anchoring Group for Potent and Selective Inhibitors of Enterovirus 71 3C Protease. J. Med. Chem. 2015, 58, 9414–9420. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, L.; He, S.; Shang, C.; Sun, Y.; Liu, N.; Meek, T.D.; Wang, Y.; Shang, L. Application of Dually Activated Michael Acceptor to the Rational Design of Reversible Covalent Inhibitor for Enterovirus 71 3C Protease. J. Med. Chem. 2019, 62, 6146–6162. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xu, B.; Ma, Y.; Shang, L.; Ye, S.; Wang, Y. Reversible covalent inhibitors suppress enterovirus 71 infection by targeting the 3C protease. Antivir. Res. 2021, 192, 105102. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Yu, B.; Wang, Y.; Wang, F. SARS-CoV-2: Origin, Evolution, and Targeting Inhibition. Front. Cell. Infect. Microbiol. 2021, 11, 676451. [Google Scholar] [CrossRef]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 M(pro): A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef]
- Banerjee, R.; Perera, L.; Tillekeratne, L.M.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discov. Today 2021, 26, 804–816. [Google Scholar] [CrossRef]
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629. [Google Scholar] [CrossRef]
- Bai, B.; Arutyunova, E.; Khan, M.B.; Lu, J.; Joyce, M.A.; Saffran, H.A.; Shields, J.A.; Kandadai, A.S.; Belovodskiy, A.; Hena, M. Peptidomimetic nitrile warheads as SARS-CoV-2 3CL protease inhibitors. RSC Med. Chem. 2021, 12, 1722–1730. [Google Scholar] [CrossRef]
- Breidenbach, J.; Lemke, C.; Pillaiyar, T.; Schäkel, L.; Al Hamwi, G.; Diett, M.; Gedschold, R.; Geiger, N.; Lopez, V.; Mirza, S.; et al. Targeting the Main Protease of SARS-CoV-2: From the Establishment of High Throughput Screening to the Design of Tailored Inhibitors. Angew. Chem. Int. Ed. Engl. 2021, 60, 10423–10429. [Google Scholar] [CrossRef]
- Cazzulo, J.J.; Stoka, V.; Turk, V. Cruzipain, the major cysteine proteinase from the protozoan parasite Trypanosoma cruzi. Biol. Chem. 1997, 378, 1–10. [Google Scholar]
- Perez-Molina, J.A.; Crespillo-Andujar, C.; Bosch-Nicolau, P.; Molina, I. Trypanocidal treatment of Chagas disease. Enferm. Infecc. Microbiol. Clin. (Engl. Ed.) 2021, 39, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Abras, A.; Ballart, C.; Fernandez-Arevalo, A.; Pinazo, M.J.; Gascon, J.; Munoz, C.; Gallego, M. Worldwide Control and Management of Chagas Disease in a New Era of Globalization: A Close Look at Congenital Trypanosoma cruzi Infection. Clin. Microbiol. Rev. 2022, 35, e0015221. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.G.; Ribeiro, J.F.R.; De Vita, D.; Cianni, L.; Franco, C.H.; Freitas-Junior, L.H.; Moraes, C.B.; Rocha, J.R.; Burtoloso, A.C.B.; Kenny, P.W.; et al. A comparative study of warheads for design of cysteine protease inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5031–5035. [Google Scholar] [CrossRef] [PubMed]
- Cianni, L.; Lemke, C.; Gilberg, E.; Feldmann, C.; Rosini, F.; Rocho, F.D.R.; Ribeiro, J.F.R.; Tezuka, D.Y.; Lopes, C.D.; de Albuquerque, S.; et al. Mapping the S1 and S1’ subsites of cysteine proteases with new dipeptidyl nitrile inhibitors as trypanocidal agents. PLoS Negl. Trop. Dis. 2020, 14, e0007755. [Google Scholar] [CrossRef]
- Cianni, L.; Sartori, G.; Rosini, F.; De Vita, D.; Pires, G.; Lopes, B.R.; Leitão, A.; Burtoloso, A.C.B.; Montanari, C.A. Leveraging the cruzain S3 subsite to increase affinity for reversible covalent inhibitors. Bioorg. Chem. 2018, 79, 285–292. [Google Scholar] [CrossRef]
- Dos Santos, A.M.; Cianni, L.; De Vita, D.; Rosini, F.; Leitão, A.; Laughton, C.A.; Lameira, J.; Montanari, C.A. Experimental study and computational modelling of cruzain cysteine protease inhibition by dipeptidyl nitriles. Phys. Chem. Chem. Phys. 2018, 20, 24317–24328. [Google Scholar] [CrossRef]
- Cianni, L.; Rocho, F.D.R.; Rosini, F.; Bonatto, V.; Ribeiro, J.F.R.; Lameira, J.; Leitão, A.; Shamim, A.; Montanari, C.A. Optimization strategy of single-digit nanomolar cross-class inhibitors of mammalian and protozoa cysteine proteases. Bioorg. Chem. 2020, 101, 104039. [Google Scholar] [CrossRef]
- Silva, J.R.A.; Cianni, L.; Araujo, D.; Batista, P.H.J.; de Vita, D.; Rosini, F.; Leitão, A.; Lameira, J.; Montanari, C.A. Assessment of the Cruzain Cysteine Protease Reversible and Irreversible Covalent Inhibition Mechanism. J. Chem. Inf. Model 2020, 60, 1666–1677. [Google Scholar] [CrossRef]
- Gomes, J.C.; Cianni, L.; Ribeiro, J.; Dos Reis Rocho, F.; da Costa Martins Silva, S.; Batista, P.H.J.; Moraes, C.B.; Franco, C.H.; Freitas-Junior, L.H.G.; Kenny, P.W.; et al. Synthesis and structure-activity relationship of nitrile-based cruzain inhibitors incorporating a trifluoroethylamine-based P2 amide replacement. Bioorg. Med. Chem. 2019, 27, 115083. [Google Scholar] [CrossRef]
- Quilles, J.C., Jr.; Shamim, A.; Tezuka, D.Y.; Batista, P.H.J.; Lopes, C.D.; de Albuquerque, S.; Montanari, C.A.; Leitão, A. Dipeptidyl nitrile derivatives suppress the Trypanosoma cruzi in vitro infection. Exp. Parasitol. 2020, 219, 108032. [Google Scholar] [CrossRef]
- Fonseca Lameiro, R.D.; Shamim, A.; Rosini, F.; Cendron, R.; Jatai Batista, P.H.; Montanari, C.A. Synthesis, biochemical evaluation and molecular modeling studies of nonpeptidic nitrile-based fluorinated compounds. Future Med. Chem. 2021, 13, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Matos, T.K.B.; Batista, P.H.J.; Dos Reis Rocho, F.; de Vita, D.; Pearce, N.; Kellam, B.; Montanari, C.A.; Leitão, A. Synthesis and matched molecular pair analysis of covalent reversible inhibitors of the cysteine protease CPB. Bioorg. Med. Chem. Lett. 2020, 30, 127439. [Google Scholar] [CrossRef] [PubMed]
- Alves, L.; Santos, D.A.; Cendron, R.; Rocho, F.R.; Matos, T.K.; Leitão, A.; Montanari, C.A. Nitrile-based peptoids as cysteine protease inhibitors. Bioorg. Med. Chem. 2021, 41, 116211. [Google Scholar] [CrossRef]
- Kourbeli, V.; Chontzopoulou, E.; Moschovou, K.; Pavlos, D.; Mavromoustakos, T.; Papanastasiou, I.P. An Overview on Target-Based Drug Design against Kinetoplastid Protozoan Infections: Human African Trypanosomiasis, Chagas Disease and Leishmaniases. Molecules 2021, 26, 4629. [Google Scholar] [CrossRef]
- Previti, S.; Ettari, R.; Cosconati, S.; Amendola, G.; Chouchene, K.; Wagner, A.; Hellmich, U.A.; Ulrich, K.; Krauth-Siegel, R.L.; Wich, P.R.; et al. Development of Novel Peptide-Based Michael Acceptors Targeting Rhodesain and Falcipain-2 for the Treatment of Neglected Tropical Diseases (NTDs). J. Med. Chem. 2017, 60, 6911–6923. [Google Scholar] [CrossRef] [PubMed]
- Schirmeister, T.; Schmitz, J.; Jung, S.; Schmenger, T.; Krauth-Siegel, R.L.; Gütschow, M. Evaluation of dipeptide nitriles as inhibitors of rhodesain, a major cysteine protease of Trypanosoma brucei. Bioorg. Med. Chem. Lett. 2017, 27, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Giroud, M.; Kuhn, B.; Saint-Auret, S.; Kuratli, C.; Martin, R.E.; Schuler, F.; Diederich, F.; Kaiser, M.; Brun, R.; Schirmeister, T.; et al. 2 H-1,2,3-Triazole-Based Dipeptidyl Nitriles: Potent, Selective, and Trypanocidal Rhodesain Inhibitors by Structure-Based Design. J. Med. Chem. 2018, 61, 3370–3388. [Google Scholar] [CrossRef]
- Pradhan, S.; Schwartz, R.A.; Patil, A.; Grabbe, S.; Goldust, M. Treatment options for leishmaniasis. Clin. Exp. Dermatol. 2022, 47, 516–521. [Google Scholar] [CrossRef]
- Caridha, D.; Vesely, B.; van Bocxlaer, K.; Arana, B.; Mowbray, C.E.; Rafati, S.; Uliana, S.; Reguera, R.; Kreishman-Deitrick, M.; Sciotti, R.; et al. Route map for the discovery and pre-clinical development of new drugs and treatments for cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2019, 11, 106–117. [Google Scholar] [CrossRef]
- Ribeiro, J.F.R.; Cianni, L.; Li, C.; Warwick, T.G.; de Vita, D.; Rosini, F.; Dos Reis Rocho, F.; Martins, F.C.P.; Kenny, P.W.; Lameira, J.; et al. Crystal structure of Leishmania mexicana cysteine protease B in complex with a high-affinity azadipeptide nitrile inhibitor. Bioorg. Med. Chem. 2020, 28, 115743. [Google Scholar] [CrossRef]
- Pittman, K.J.; Aliota, M.T.; Knoll, L.J. Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genom. 2014, 15, 806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, E.T.; Parussini, F.; Huynh, M.H.; Giebel, J.D.; Kelley, A.M.; Zhang, L.; Bogyo, M.; Merritt, E.A.; Carruthers, V.B. Toxoplasma gondii cathepsin L is the primary target of the invasion-inhibitory compound morpholinurea-leucyl-homophenyl-vinyl sulfone phenyl. J. Biol. Chem. 2009, 284, 26839–26850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwicker, J.D.; Diaz, N.A.; Guerra, A.J.; Kirchhoff, P.D.; Wen, B.; Sun, D.; Carruthers, V.B.; Larsen, S.D. Optimization of dipeptidic inhibitors of cathepsin L for improved Toxoplasma gondii selectivity and CNS permeability. Bioorg. Med. Chem. Lett. 2018, 28, 1972–1980. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, J.D.; Smith, D.; Guerra, A.J.; Hitchens, J.R.; Haug, N.; Vander Roest, S.; Lee, P.; Wen, B.; Sun, D.; Wang, L.; et al. Discovery and Optimization of Triazine Nitrile Inhibitors of Toxoplasma gondii Cathepsin L for the Potential Treatment of Chronic Toxoplasmosis in the CNS. ACS Chem. Neurosci. 2020, 11, 2450–2463. [Google Scholar] [CrossRef]
- Ettari, R.; Previti, S.; Di Chio, C.; Zappala, M. Falcipain-2 and Falcipain-3 Inhibitors as Promising Antimalarial Agents. Curr. Med. Chem. 2021, 28, 3010–3031. [Google Scholar] [CrossRef]
- Nizi, E.; Sferrazza, A.; Fabbrini, D.; Nardi, V.; Andreini, M.; Graziani, R.; Gennari, N.; Bresciani, A.; Paonessa, G.; Harper, S. Peptidomimetic nitrile inhibitors of malarial protease falcipain-2 with high selectivity against human cathepsins. Bioorg. Med. Chem. Lett. 2018, 28, 1540–1544. [Google Scholar] [CrossRef]
- Hernández González, J.E.; Hernández Alvarez, L.; Leite, V.B.P.; Pascutti, P.G. Water Bridges Play a Key Role in Affinity and Selectivity for Malarial Protease Falcipain-2. J. Chem. Inf. Model. 2020, 60, 5499–5512. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brogi, S.; Ibba, R.; Rossi, S.; Butini, S.; Calderone, V.; Gemma, S.; Campiani, G. Covalent Reversible Inhibitors of Cysteine Proteases Containing the Nitrile Warhead: Recent Advancement in the Field of Viral and Parasitic Diseases. Molecules 2022, 27, 2561. https://doi.org/10.3390/molecules27082561
Brogi S, Ibba R, Rossi S, Butini S, Calderone V, Gemma S, Campiani G. Covalent Reversible Inhibitors of Cysteine Proteases Containing the Nitrile Warhead: Recent Advancement in the Field of Viral and Parasitic Diseases. Molecules. 2022; 27(8):2561. https://doi.org/10.3390/molecules27082561
Chicago/Turabian StyleBrogi, Simone, Roberta Ibba, Sara Rossi, Stefania Butini, Vincenzo Calderone, Sandra Gemma, and Giuseppe Campiani. 2022. "Covalent Reversible Inhibitors of Cysteine Proteases Containing the Nitrile Warhead: Recent Advancement in the Field of Viral and Parasitic Diseases" Molecules 27, no. 8: 2561. https://doi.org/10.3390/molecules27082561
APA StyleBrogi, S., Ibba, R., Rossi, S., Butini, S., Calderone, V., Gemma, S., & Campiani, G. (2022). Covalent Reversible Inhibitors of Cysteine Proteases Containing the Nitrile Warhead: Recent Advancement in the Field of Viral and Parasitic Diseases. Molecules, 27(8), 2561. https://doi.org/10.3390/molecules27082561