
Decoding an Amino Acid Sequence to Extract Information on Protein Folding


Abstract
:1. Background
- (1)
- How is the information on the 3D native structure of a protein encoded in its D-amino acid sequence (i.e., its folding code)?
- (2)
- How can a protein fold rapidly despite the innumerous possible conformations of the polypeptide chain?
- (3)
- The 3D structure prediction of a protein from its sequence.
2. Outline of the Methods
- (1)
- Inter-residue average distances were computed from the data of proteins with known 3D structures in advance. In the computations of the average distances, the distance between two residues, i and j, along the sequence was classified in the following way. In each category, called the “range”, the average value of inter-Cα residue distances were calculated for a pair of amino acid types. The definition of “range” is as follows. That is, with k = |i − j|, when 1 ≤ k ≤ 8, the range M is defined as 1, and in a similar way, 9 ≤ k ≤ 20, 21 ≤ k ≤ 30, 31 ≤ k ≤ 40, 41 ≤ k ≤ 50, and so on to define the range M = 2, 3, 4, …, respectively.
- (2)
- Thus, an average distance of a pairs of residue types A and B is expressed as d(A, B, M).
- (3)
- A kind of contact map taking inter-residue average distance statistics into account is constructed from only the amino acid sequence. A plot is made when the average distance of a residue pair at the range is less than a cutoff distance determined in advance for each range.
- (4)
- A cutoff distance is tuned so that the density of plots on a map constructed based on inter-residue average distances is close to that of the contact map constructed from the 3D structure of the protein under consideration.
- (5)
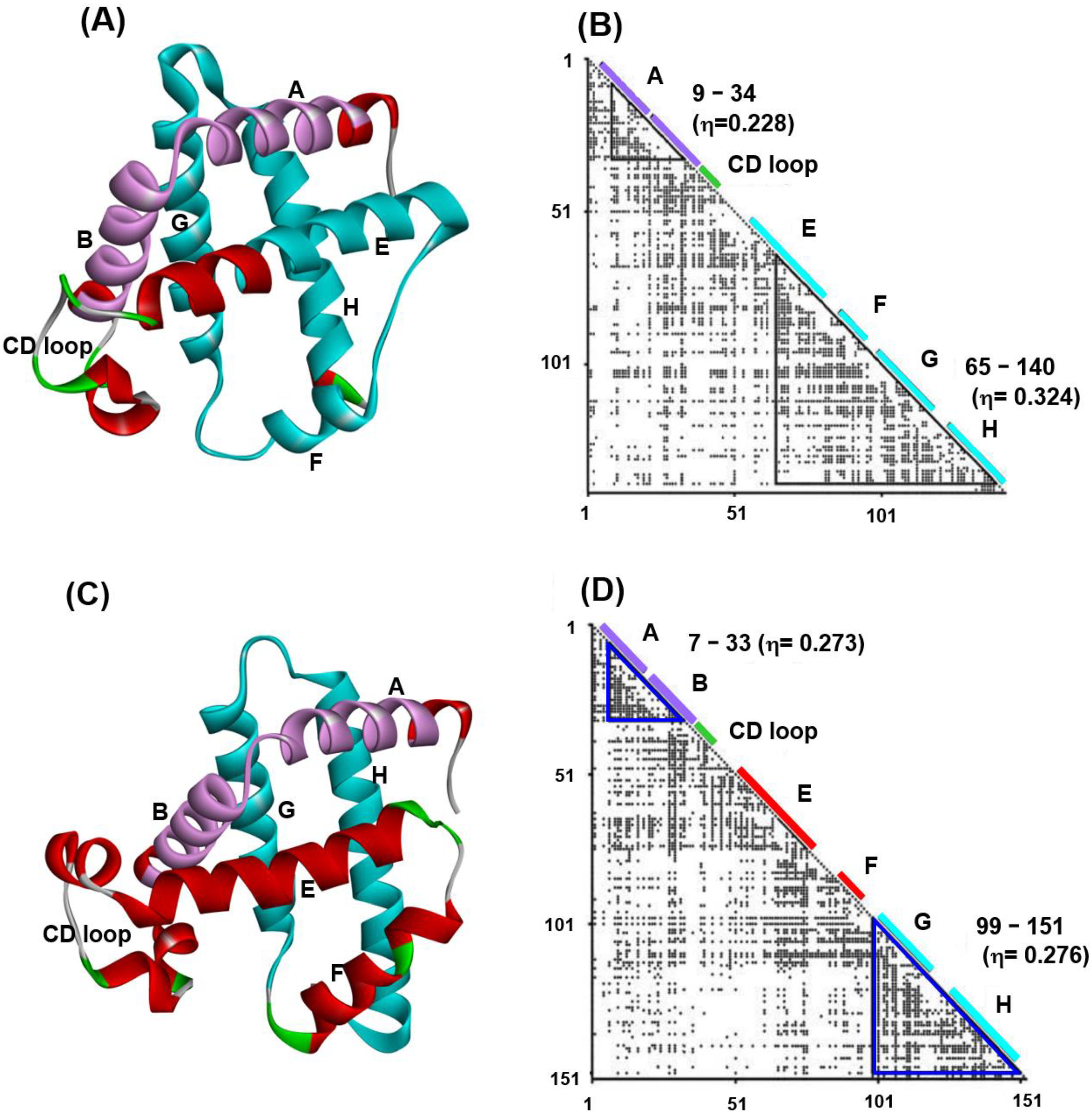
- An area with a local high-density plot region near the diagonal of the obtained map for a sequence is identified and predicted as a compact or structured region. The index of the plot density is called the η-value, indicating the strength of the compactness. It would be reasonable to consider the regions with many contacts (a high η-value) as regions structured in the early stage of folding.
3. Predicted Regions by ADM and Relationships to the Folding of a Protein
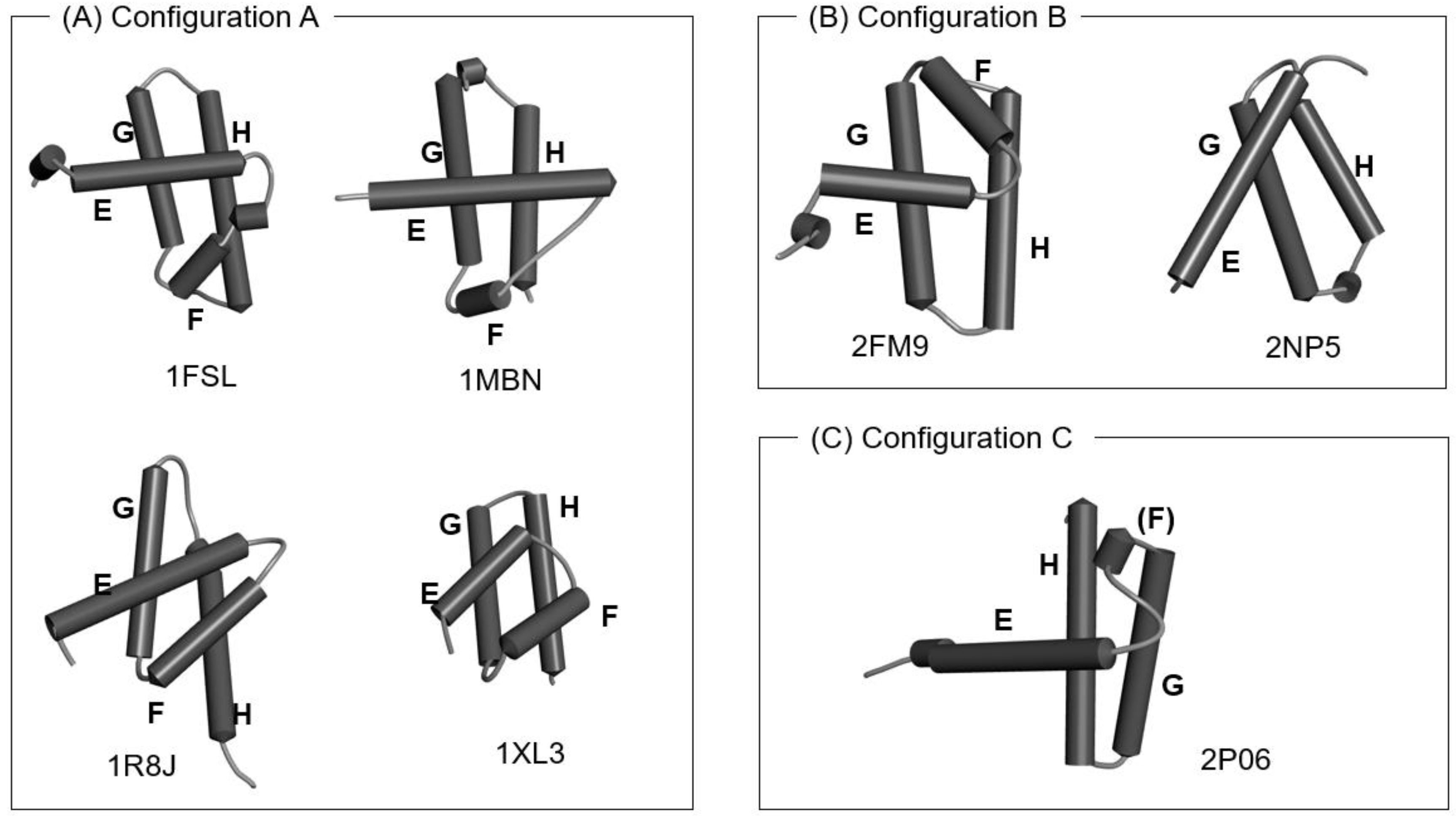
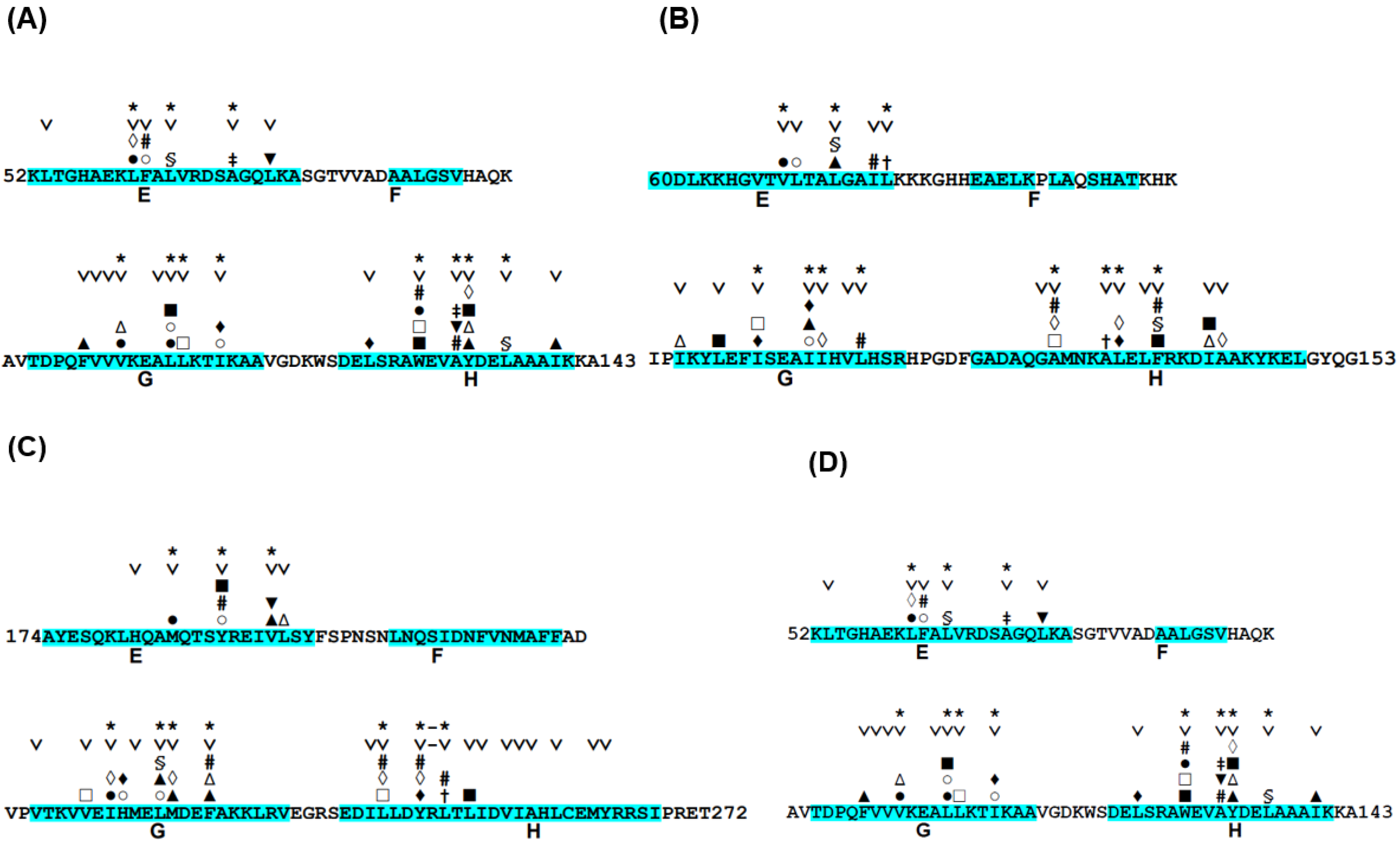
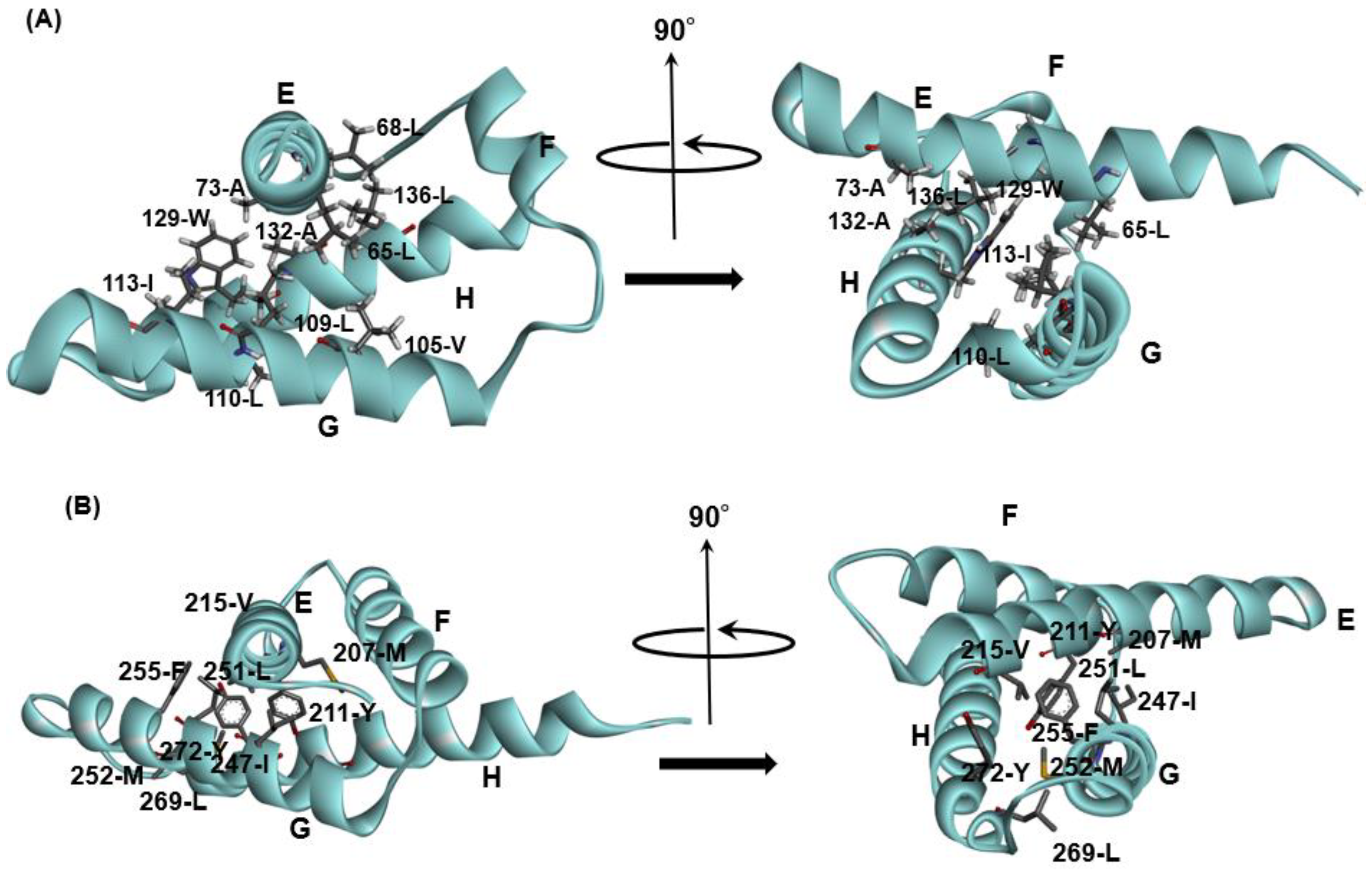
3.1. Hemoglobin E-to-H Helix Unit
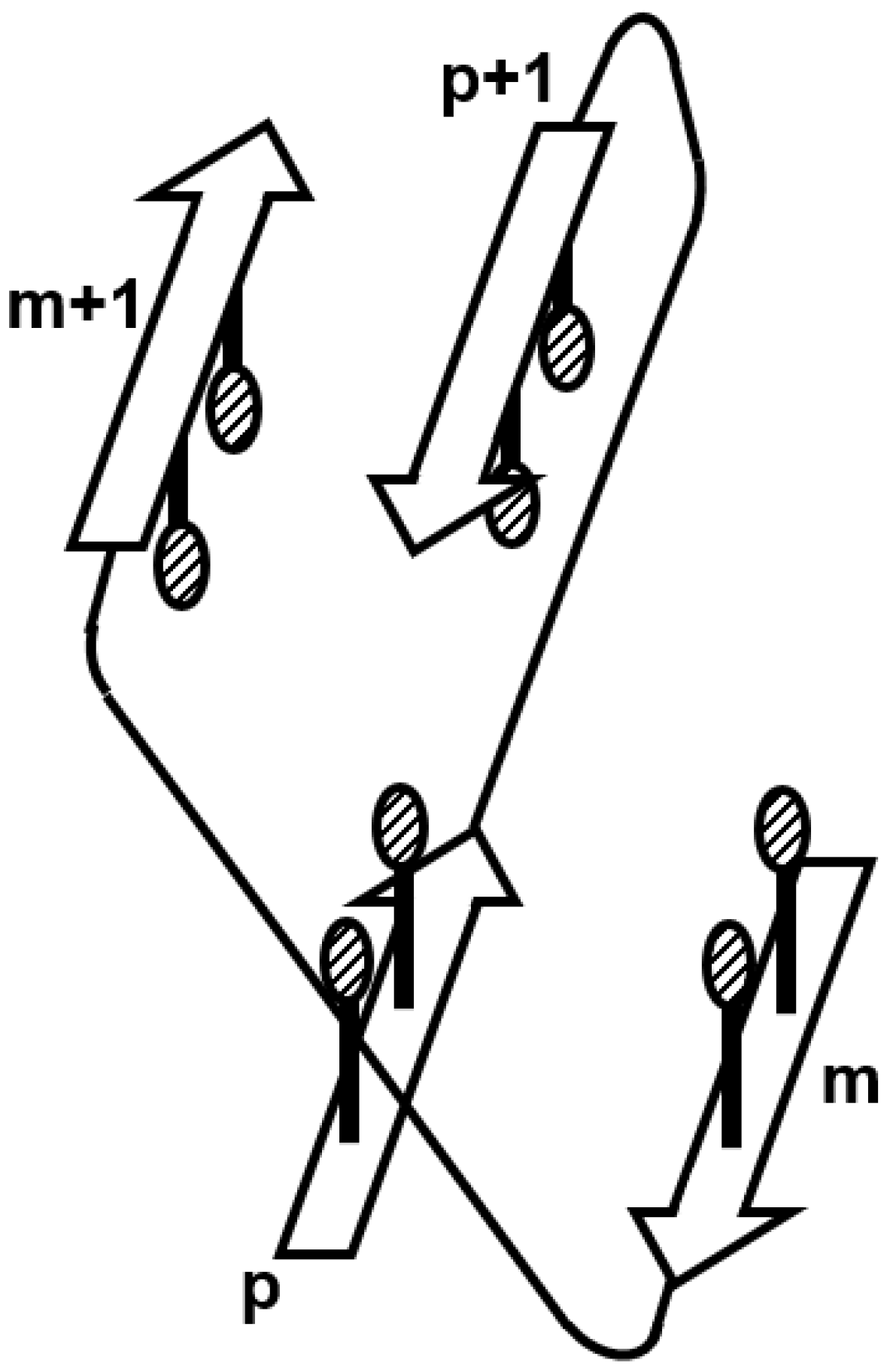
3.2. Ferredoxin-like Fold Proteins
4. ADM Predicted Region and F-Value Analysis
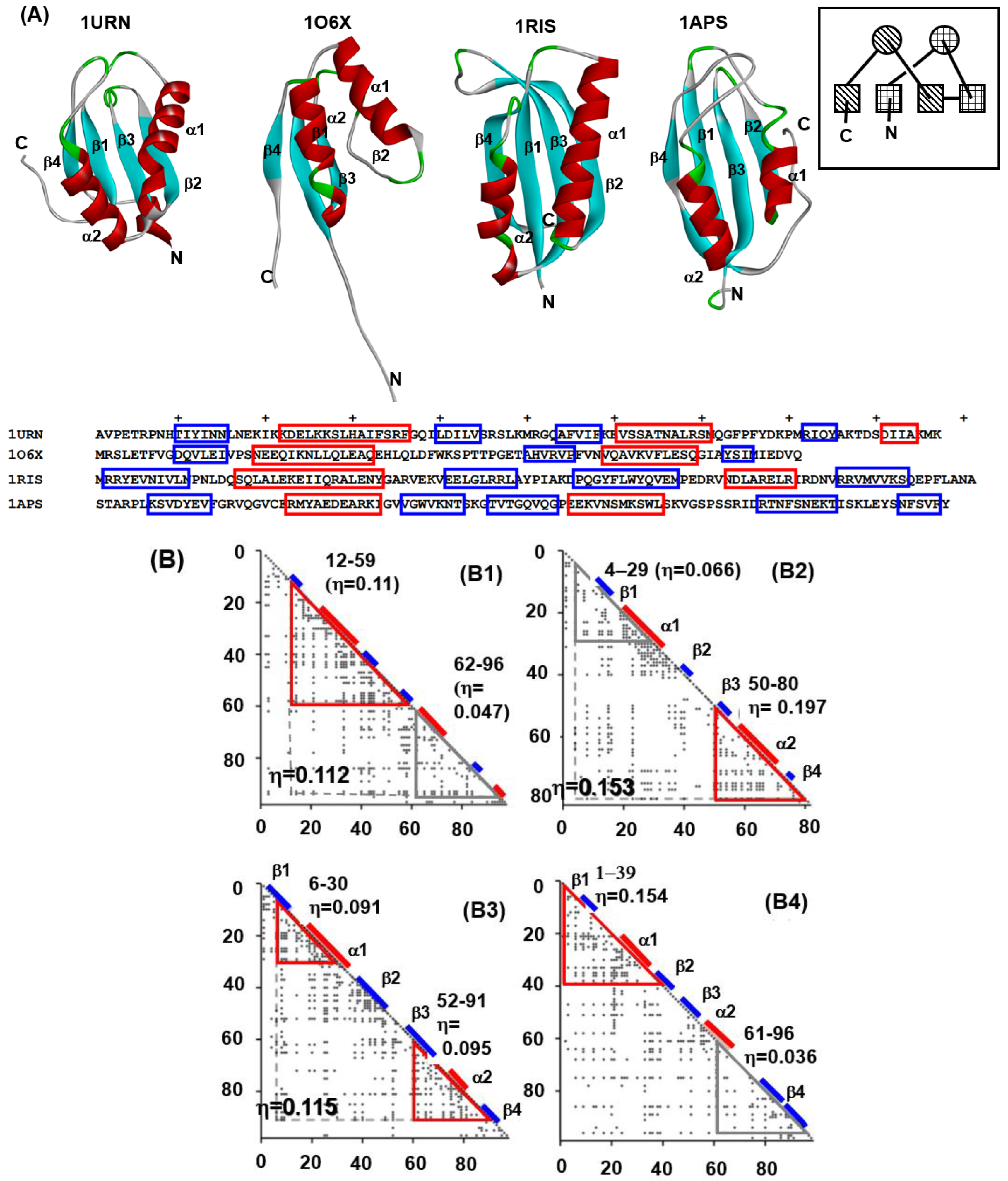
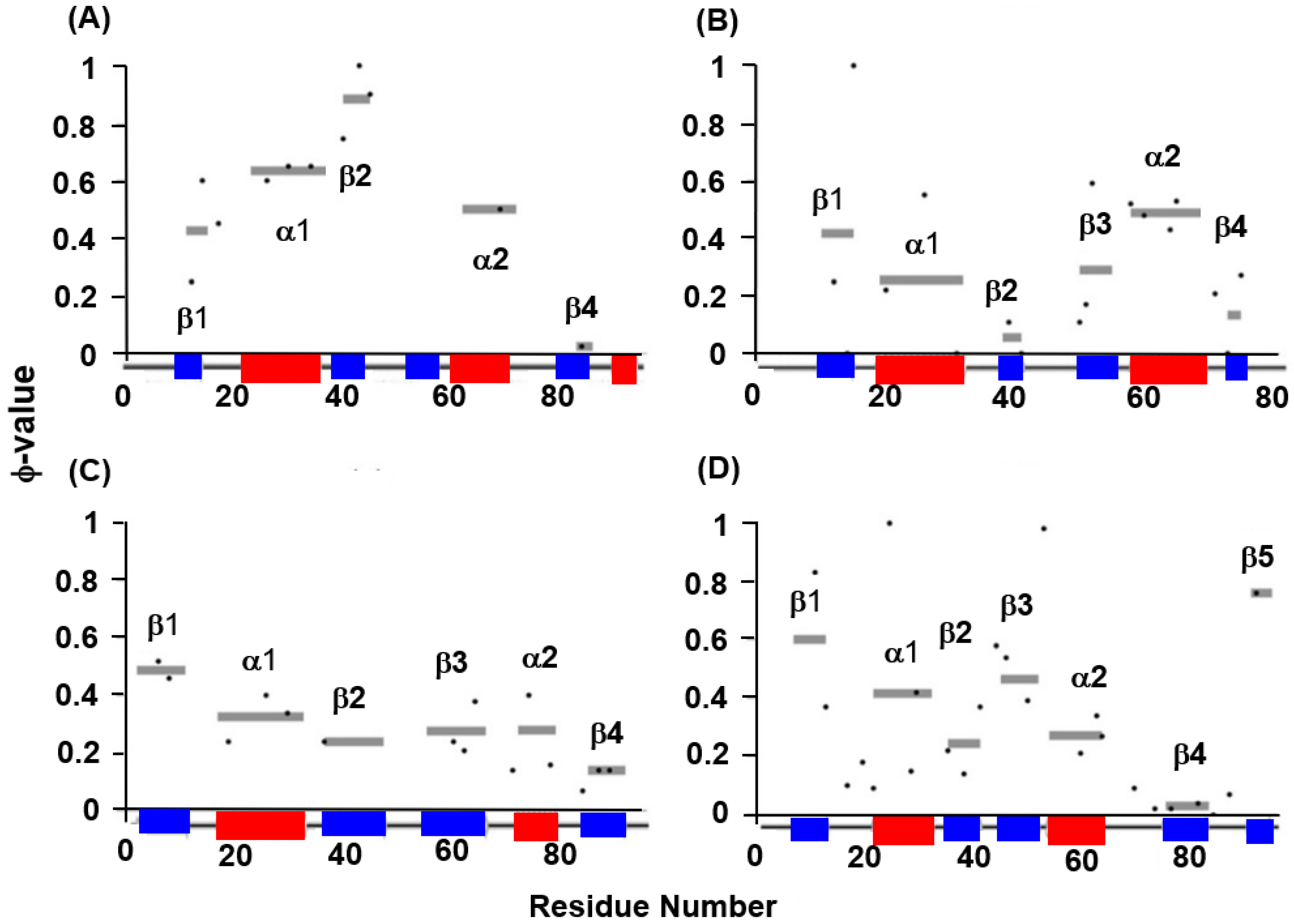
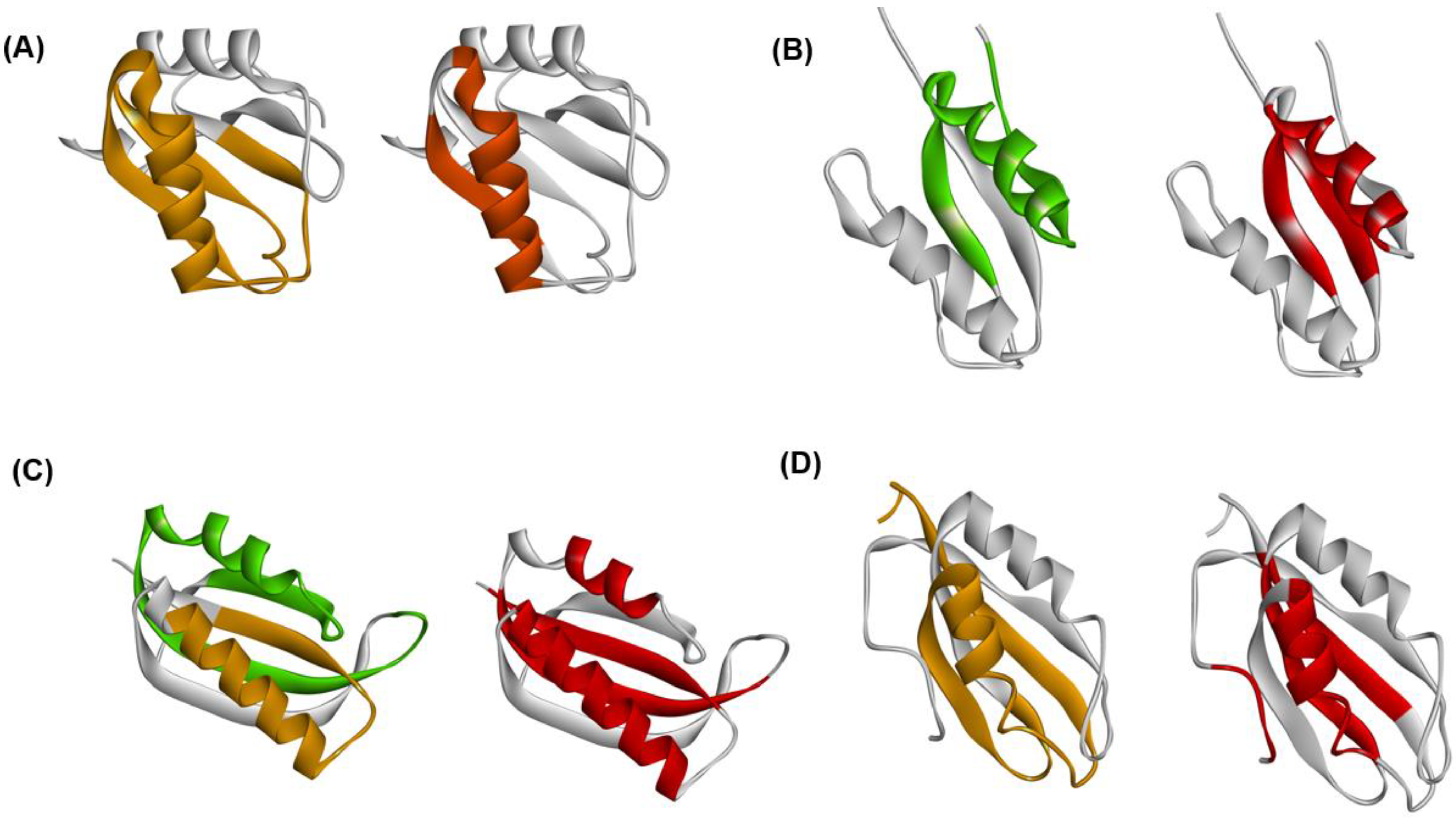
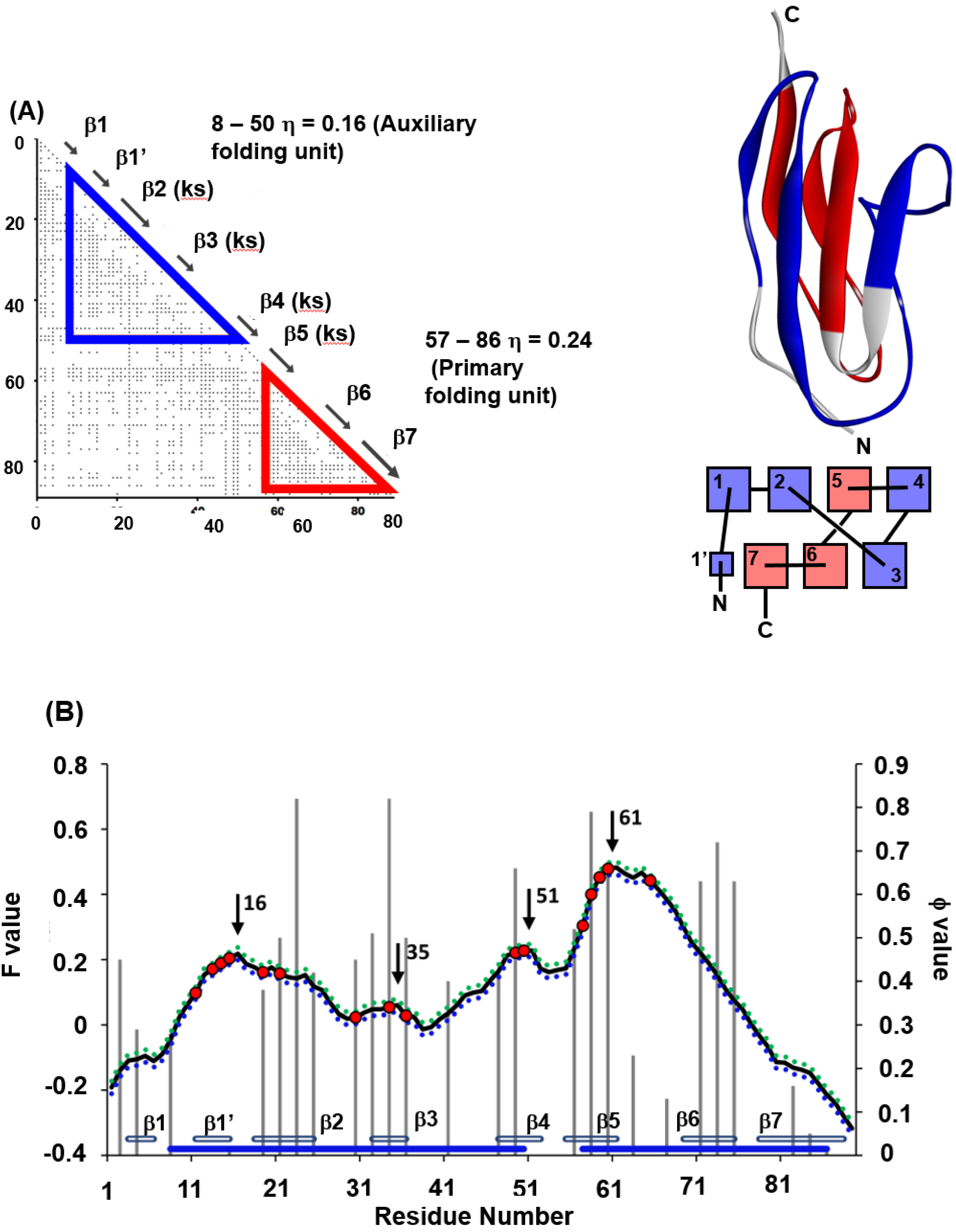
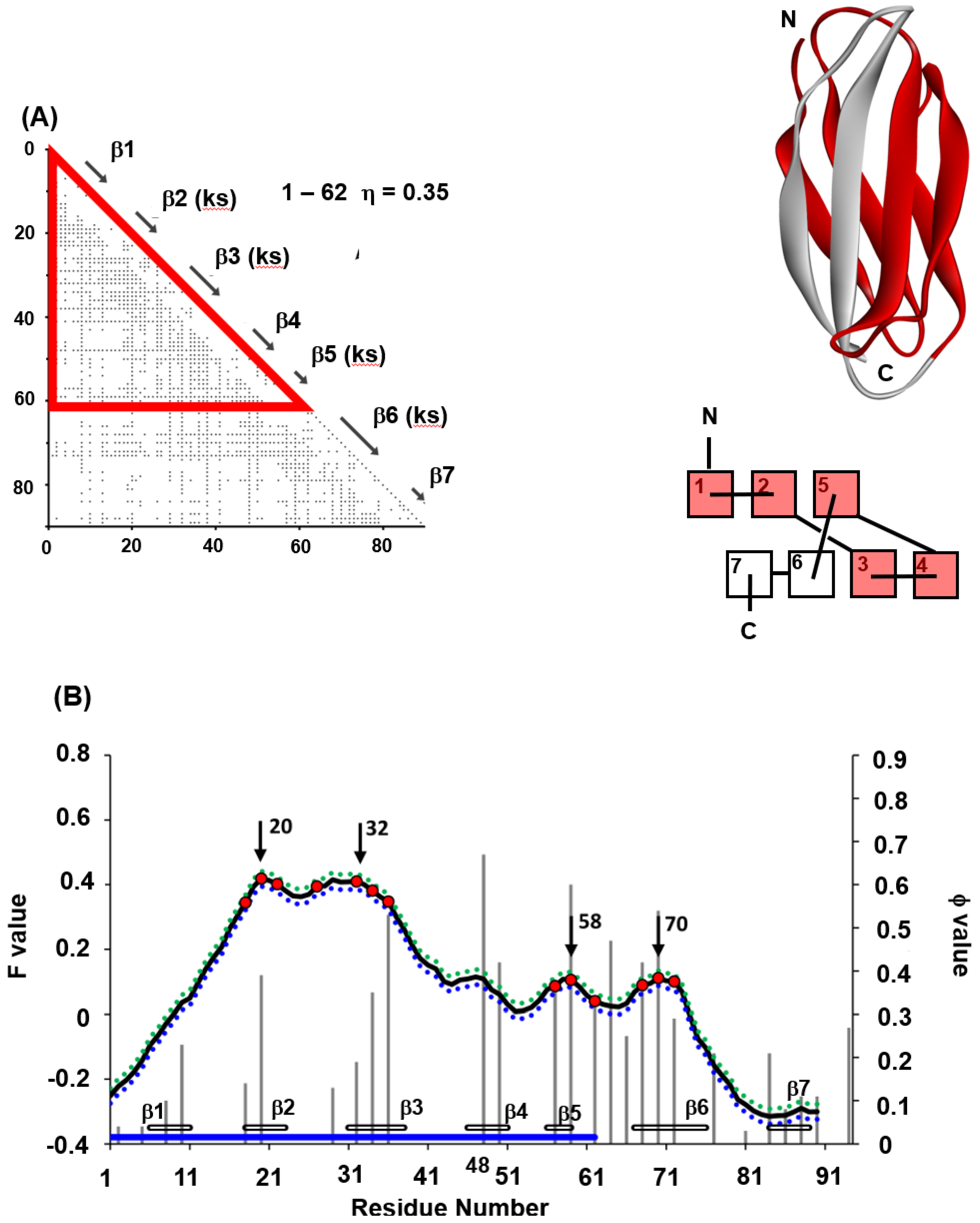
4.1. Immunoglobulin-like Beta Sandwich Protein
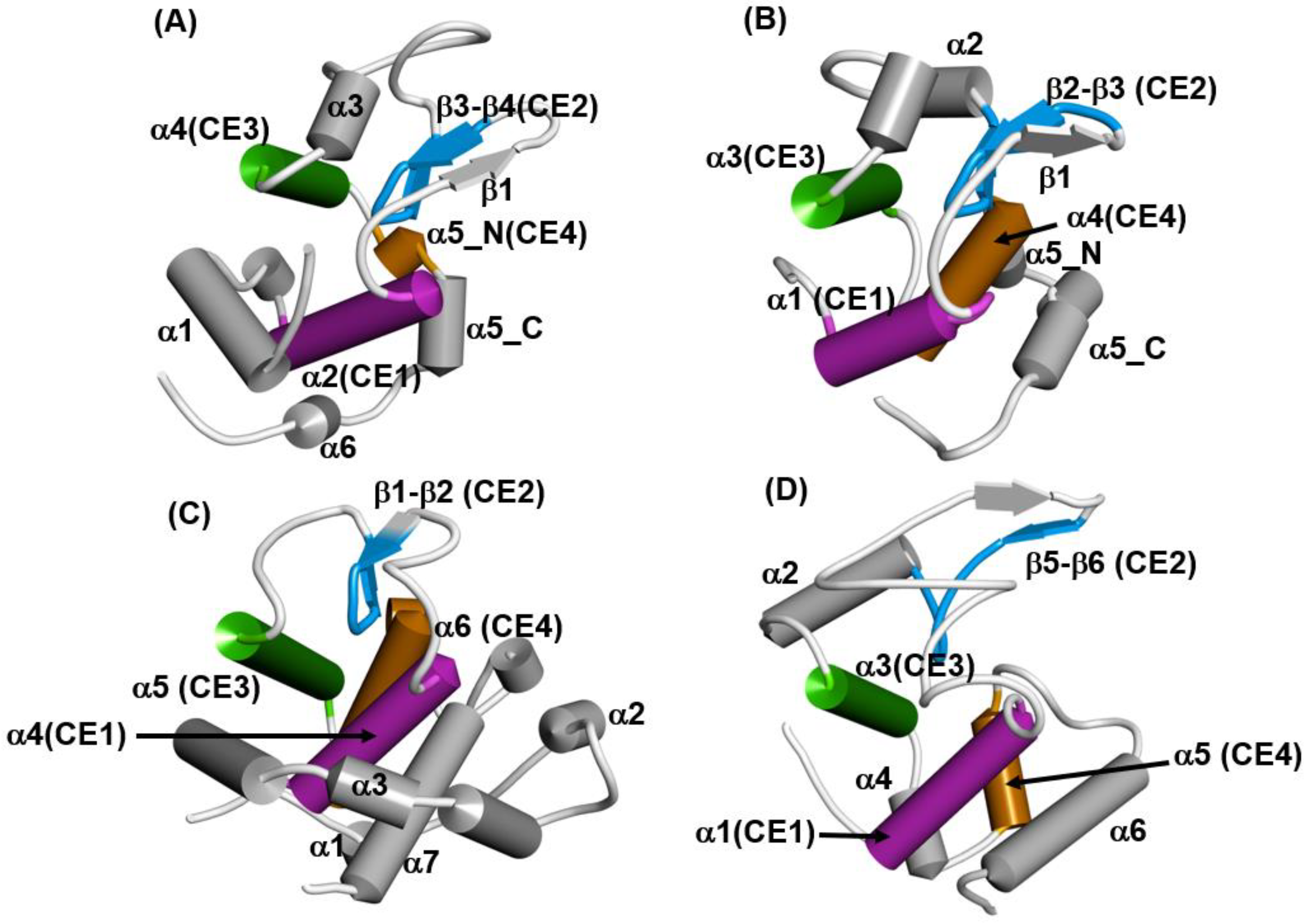
4.2. Lysozyme-like Fold Proteins
- (1)
- In each protein, several regions are predicted by ADM and each predicted region contains one of the common secondary structures.
- (2)
- In each protein, each ADMpr contains one peak of the F-value plot, and the CHR close to the peak is within ±5 residues.
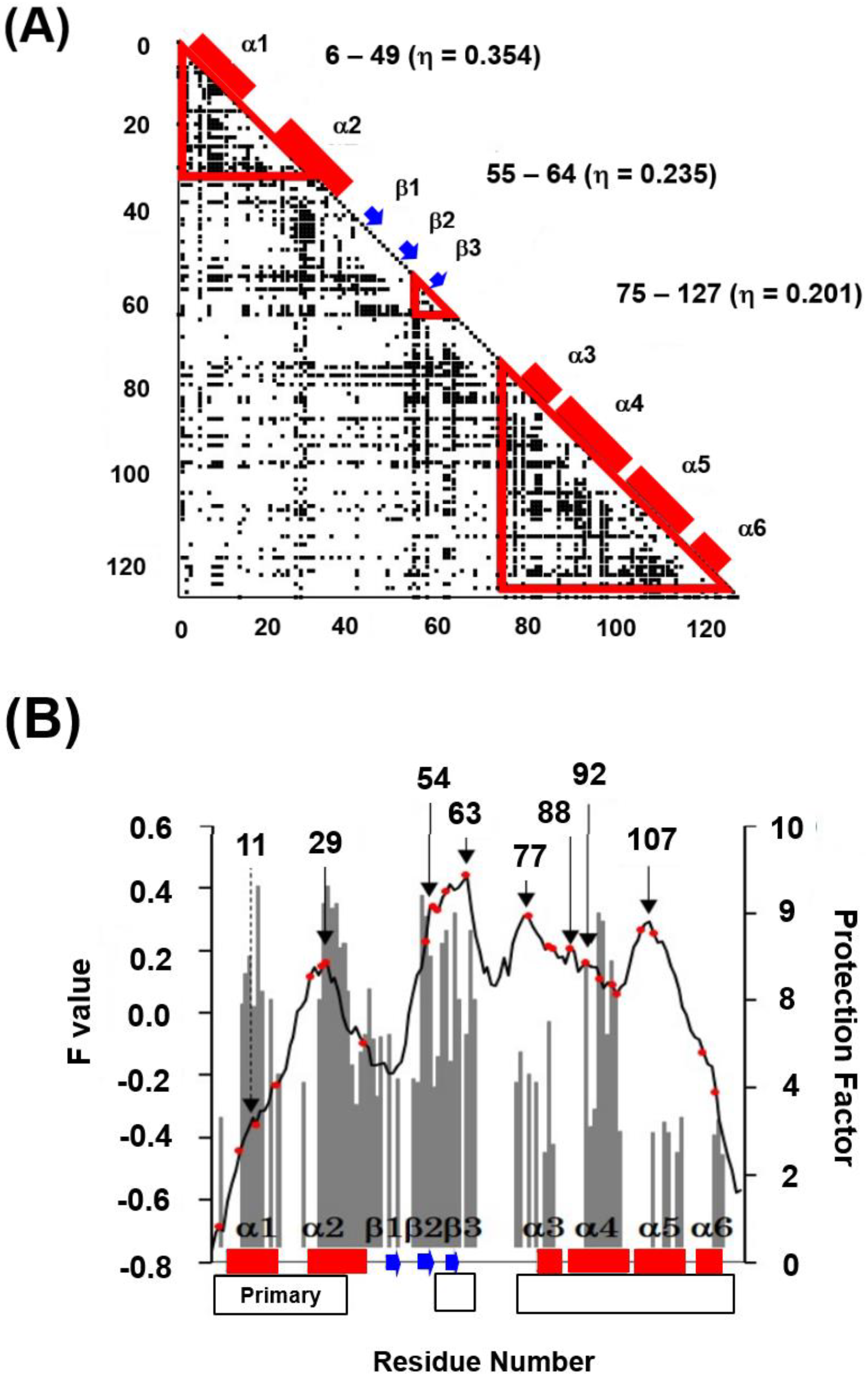
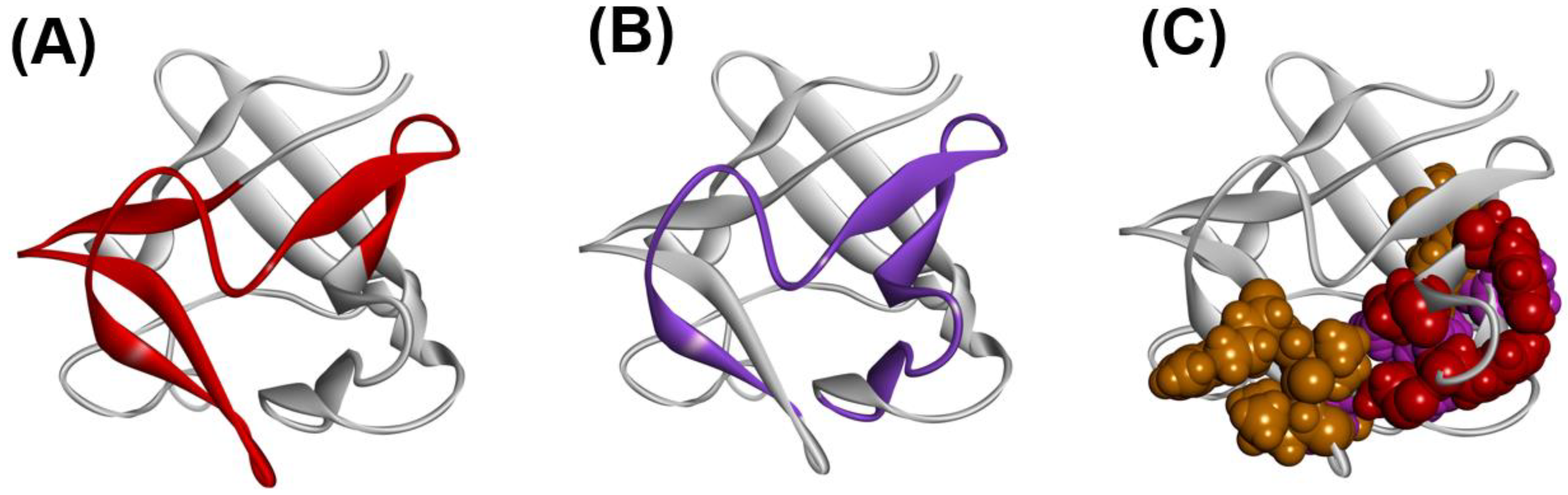
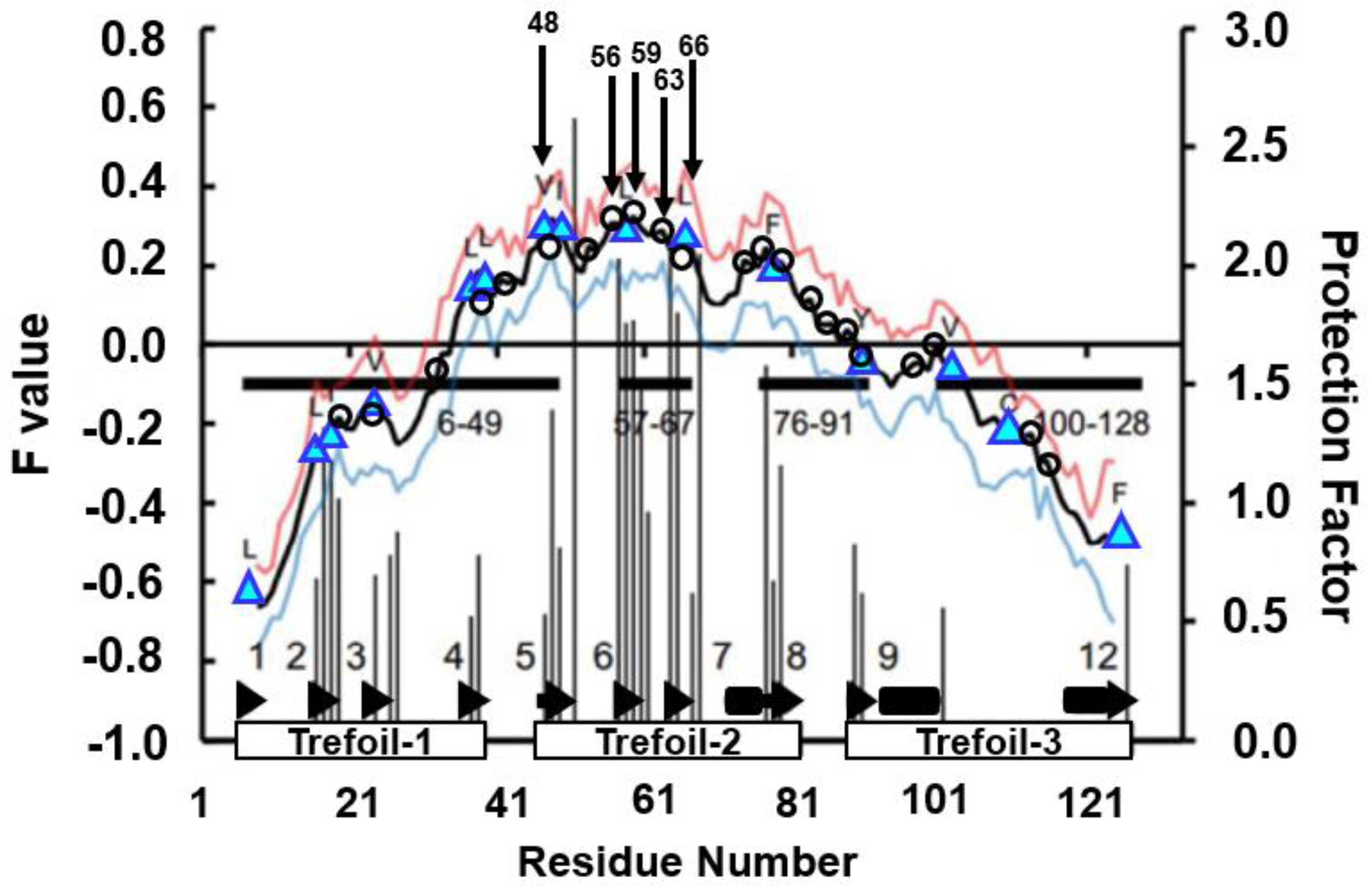
4.3. Trefoil Protein
5. Perspective
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anfinsen, C.B.; Scheraga, H.A. Experimental and theoretical aspects of protein folding. Adv. Protein Chem. 1975, 29, 205–300. [Google Scholar] [PubMed]
- Anfinsen, C.B.; Haber, E.; Sela, M.; White, F.H., Jr. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc. Natl. Acad. Sci. USA 1962, 47, 1309–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Potapenko, A.; Bridgrand, A.; Meyer, C.; Kohl, S.A.A.; Ballard, A.; et al. Highly acculate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Alexander Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.K.; Udgaonkar, J.B. Early events in protein folding. Curr. Sci. 2009, 96, 1053–1070. [Google Scholar]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [Green Version]
- Englander, S.W.; Mayne, L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. USA 2014, 111, 15873–15880. [Google Scholar] [CrossRef] [Green Version]
- Rose, G.D. Protein folding—Seeing is deceiving. Protein Sci. 2021, 30, 1606–1616. [Google Scholar] [CrossRef]
- Jennings, P.; Write, P.E. Formation of a Molten Globule Intermediate Early in the Kinetic Folding Pathway of Apomyoglobin. Science 1993, 262, 892–896. [Google Scholar] [CrossRef]
- Nishimura, C.; Prytulla, S.; Dyson, H.J.; Wright, P.E. Conservation of folding pathways in evolutionarily distant globin sequences. Nat. Struct. Biol. 2000, 7, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Daggett, V.; Fersht, A.R. Transition states in protein folding. In Mechanisms of Protein Folding, 2nd ed.; Pain, R.H., Ed.; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Haglund, E.; Lindberg, M.O.; Oliveberg, M. Changes of protein folding pathways by circular permutation: Overlapping nuclei promote global cooperativity. J. Biol. Chem. 2008, 283, 7904–27915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Ron, O.; Dror, R.O.; Mildorf, T.J.; Stefano Piana, S.; Shaw, D.E. Identifying localized changes in large systems: Change-point detection for biomolecular simulations. Proc. Natl. Acad. Sci. USA 2015, 112, 7454–7459. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How fastfolding proteins fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef]
- Paci, E.; Clarke, J.; Steward, A.; Vendruscolo, M.; Karplus, M. Self-consistent determination of the transition state for protein folding: Application to a fibronectin type III domain. Proc. Nat. Acad. Sci. USA 2003, 100, 394–399. [Google Scholar] [CrossRef] [Green Version]
- Garbuzynsliy, S.O.; Ivankov, D.N.; Bogatyreva, N.S.; Finkelstein, A.V. Golden triangle for folding rates of globular proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Rao, N.; Liu, G.; Yang, Y.; Wang, G. Predicting Protein Folding Rates Using the Concept of Chou’s Pseudo Amino Acid Composition. J. Comput. Chem. 2011, 32, 1612–1617. [Google Scholar] [CrossRef]
- Baker, D. A surprising simplicity to protein folding. Nature 2000, 405, 39–42. [Google Scholar] [CrossRef]
- Raimondi, D.; Orlando, G.; Pancsa, R.; Khan, T.; Vranken, W.F. Exploring the Sequence-based Prediction of Folding Initiation Sites in Proteins. Sci. Rep. 2017, 7, 8826. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, T.; Némethy, G.; Scheraga, H.A. Prediction of the location of structural domains in globular proteins. J. Protein Chem. 1988, 7, 427–471. [Google Scholar] [CrossRef]
- Aumpuchin, P.; Kikuchi, T. Prediction of folding mechanisms for Ig-like beta sandwich proteins based on inter-residue average distance statistics methods. Proteins 2019, 87, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T. Analysis of 3D structural differences in the IgG-binding domains based on the interresidue average-distance statistics. Amino Acids. 2008, 35, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Fujita, A.; Kawai, Y.; Kikuchi, T. Similar structures to the E-to-H helix unit in the globin-like fold are found in other helical folds. Biomolecules 2014, 4, 268–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, L.; Park, J. DaliLite workbench for protein structure comparison. Bioinformatics 2000, 16, 566–567. [Google Scholar] [CrossRef] [PubMed]
- Shrake, A.; Rupley, J.A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef]
- Matsuoka, M.; Kikuchi, T. Sequence analysis on the information of folding initiation segments in ferredoxin-like fold proteins. BMC Struct. Biol. 2014, 14, 15. [Google Scholar] [CrossRef] [Green Version]
- Ternström, T.; Mayor, U.; Akke, M.; Oliveberg, M. From snapshot to movie: Phi analysis of protein folding transition states taken one step further. Proc. Natl. Acad. Sci. USA 1999, 96, 14854–14859. [Google Scholar] [CrossRef] [Green Version]
- Villegas, V.V.; Martínez, J.C.J.; Avilés, F.X.F.; Serrano, L.L. Structure of the transition state in the folding process of human procarboxypeptidase A2 activation domain. J. Mol. Biol. 1998, 283, 1027–1036. [Google Scholar] [CrossRef]
- Chiti, F.; Taddei, N.; White, P.M.; Bucciantini, M.; Magherini, F.; Stefani, M.; Dobson, C.M. Mutational analysis of acylphosphatase suggests the importance of topology and contact order in protein folding. Nat. Struct. Biol. 1999, 6, 1005–1009. [Google Scholar]
- Fowler, S.B.; Clarke, J. Mapping the folding pathway of an immunoglobulin domain: Structural detail from phi value analysis and movement of the transition state. Structure 2001, 9, 355–366. [Google Scholar] [CrossRef]
- Hamill, S.J.; Steward, A.; Clarke, J. The folding of an immunoglobulin-like Greek key protein is defined by a common-core nucleus and regions constrained by topology. J. Mol. Biol. 2000, 297, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Radford, S.E.; Buck, M.; Topping, D.K.; Dobson, C.M.; Evans, P.A. Hydrogen exchange in native and denatured states of hen egg-white lysozyme. Proteins 1992, 14, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Kister, A.E.; Finkelstein, A.V.; Gelfand, I.M. Common features in structures and sequences of sandwich-like proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 14137–14141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, T.; Kabata, M.; Kikuchi, T. Properties of amino acid sequences of lysozyme-like superfamily proteins relating to their folding mechanisms. J. Proteom. Bioinform. 2017, 10, 94–107. [Google Scholar] [CrossRef]
- Callewaert, L.; Michiels, C.W. Lysozyme in the animal kingdom. J. Biosci. 2010, 3, 127–160. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, A.D. Three-fold structural pattern in the soybean trypsin inhibitor (Kunitz). J. Mol. Biol. 1979, 133, 557–563. [Google Scholar] [CrossRef]
- Murzin, A.G.; Lesk, A.M.; Chothia, C. β-Trefoil fold. Patterns of structure and sequence in the Kunitz inhibitors interleukins-1β and 1α and fibroblast growth factors. J. Mol. Biol. 1992, 223, 531–543. [Google Scholar] [CrossRef]
- Ponting, C.P.; Russell, R.B. Identification of distant homologues of fibroblast growth factors suggests a common ancestor for all β-trefoil proteins. J. Mol. Biol. 2000, 302, 1041–1047. [Google Scholar] [CrossRef]
- Lee, J.; Blaber, S.I.; Dubey, V.K.; Blaber, M. A polypeptide “building block” for the β-trefoil fold identified by “top-down symmetric deconstruction”. J. Mol. Biol. 2011, 407, 744–763. [Google Scholar] [CrossRef]
- Broom, A.; Doxey, A.C.; Lobsanov, Y.D.; Berthin, L.G.; Rose, D.R.; Howell, L.; McConley, B.J.; Meiering, E.M. Modular evolution and the origins of symmetry: Reconstruction of a three-fold symmetric globular protein. Structure 2012, 20, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Longo, L.; Lee, J.; Blaber, M. Experimental support for the foldabilityfunction tradeoff hypothesis: Segregation of the folding nucleus and functional regions in fibroblast growth factor-1. Protein Sci. 2012, 21, 1911–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, L.M.; Kumru, O.S.; Middaugh, C.R.; Blaber, M. Evolution and design of protein structure by folding nucleus symmetric expansion. Structure 2014, 22, 1377–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Longo, L.M.; Sutherland, M.A.; Blaber, M. Evolution of a protein folding nucleus. Protein Sci. 2016, 25, 1227–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirioka, T.; Aumpuchin, P.; Kikuchi, T. Detection of folding sites of β-trefoil fold proteins based on amino acid sequence analyses and structure-based sequence alignment. J. Proteom. Bioinform. 2017, 10, 10222–10235. [Google Scholar] [CrossRef]
- Wang, H.M.; Yu, C. Investigating the refolding pathway of human acidic fibroblast growth factor (hFGF-1) from the residual structure(s) obtained by denatured-state hydrogen/deuterium exchange. Biophys. J. 2011, 100, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Agashe, V.R.; Shastry, M.C.; Udgaonkar, J.B. Initial hydrophobic collapse in the folding of barstar. Nature 1995, 377, 754–757. [Google Scholar] [CrossRef]
- Elove, G.A.; Chaffotte, A.F.; Roder, H.; Goldberg, M.E. Early steps in cytochrome c folding probed by time-resolved circular dichroism and fluorescence spectroscopy. Biochemistry 1992, 31, 6876–6883. [Google Scholar] [CrossRef]
- Houry, W.A.; Rothwarf, D.M.; Scheraga, H.A. Circular dichroism evidence for the presence of burst-phase intermediates on the conformational folding pathway of ribonuclease A. Biochemistry 1996, 35, 10125–10133. [Google Scholar] [CrossRef]
- Bryngelson, J.D.; Wolynes, P.G. Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. USA 1987, 84, 7524–7528. [Google Scholar] [CrossRef] [Green Version]
- Onuchic, J.N.; Wolynes, P.G.; Luthey-Schulten, Z.; Socci, N.D. Towards an outline of the topography of a realistic protein folding funnel. Proc. Natl. Acad. Sci. USA 1995, 92, 3626–3630. [Google Scholar] [CrossRef] [Green Version]
- José Nelson Onuchic, J.N.; Socci, N.D.; Luthey-Schulten, Z.; Wolynes, P.G. Protein folding funnels: The nature of the transition state ensemble. Fold. Des. 1996, 1, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Pletneva, E.V.; Gray, H.B.; Winkler, J.R. Snapshots of cytochrome c folding. Proc. Natl. Acad. Sci. USA 2005, 102, 18397–18402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradeep, L.; Udgaonkar, J.B. Osmolytes induce structure in an early intermediate on the folding pathway of barstar. J. Biol. Chem. 2004, 279, 40303–40313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgescu, R.E.; Li, J.H.; Goldberg, M.E.; Tasayco, M.L.; Chaffotte, A.F. Proline isomerization-independent accumulation of an early intermediate and heterogeneity of the folding pathways of a mixed alpha/beta protein, Escherichia coli thioredoxin. Biochemistry 1998, 37, 10286–10297. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kondrashkina, E.; Kayatekin, C.; Matthews, C.R.; Bilsel, O. Microsecond acquisition of heterogeneous structure in the folding of a TIM barrel protein. Proc. Natl. Acad. Sci. USA 2008, 105, 13367–13372. [Google Scholar] [CrossRef] [Green Version]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein (Source, PDB ID) |
|---|
| Leghemoglobin (soybean, 1FSL) |
| Myoglobin (sperm whale, 1MBN) |
| Circadian clock protein KaiA (Synechococcus, 1R8J) |
| Secretion control protein SipA (Yersinia, 1XL3) |
| Cell invasion protein SipA (Salmomella, 2FM9) |
| Transcriptional regulator RHA1_ro04179 (Rodococcus, 2NP5) |
| Hypothetical protein AF0060 (E. coli, 2P06) |
| Protein | E Helix | G Helix | H Helix |
|---|---|---|---|
| 1FSL | φxxφxxxxφ | φxxxφφxxφ | φxxφφxxφ |
| 1MBN | φxxxφxxxφ | φxxxφφxxφ | φxxφφxxφ |
| 1R8J | φxxxφxxxφ | φxxxφφxxφ | φxxφxxxφ |
| 1XL3 | φxxxφxxxφ | φxxφφxxxφ | φxxφφxxxφ |
| Interaction between CEs | Hen Egg White Lysozyme (PDB ID: 2VB1) | Tapes japonica Lysozyme (PDB ID: 2DQA) |
|---|---|---|
| CE1⇔CE2 | Trp28-Leu56 | Met14-Phe39 |
| CE1⇔CE3 | Trp28-Ala95 | Met14-Val70 |
| CE1⇔CE4 | Trp28-Met105 | Met14-Phe90 |
| CE2⇔CE3 | Ile58-Ala95 | Ile41-Val70 |
| CE3⇔CE4 | Ala95-Trp108 | Met74-Phe90 |
| Goose lysozyme (PDB ID: 153L) | λ phage lysozyme (PDB ID: 1AM7) | |
| CE1⇔CE2 | Ile69-Leu93 | Leu12-Tyr67 |
| CE1⇔CE3 | Ile69-Leu120 | Leu12-Ala95, |
| CE1⇔CE4 | Val65-Ile144, | Phe11-Ile113, |
| CE2⇔CE3 | Leu93-Ile113 | Tyr67-Ala95 |
| CE3⇔CE4 | Leu120-Ile144, | (Ile99-Ile108) |
| Highly Protected Residues in the H/D Exchange Experiment | Residues at the Highest Peaks in the F-Value Plot | Difference in the Sequence from Highly Protected Residues | Conserved Hydrophobic Residues near a Peak in the F-Value Plot | Difference in the Sequence from Highly Protected Residues |
|---|---|---|---|---|
| 51-Ser | 48-Tyr | three residues | 47-Val | four residues |
| 49-Ile | two residues | |||
| 57-Tyr | 56-Gln | one residue | 58-Leu | one residue |
| 59-Ala | two residues | |||
| 64-Gly | 63-Asp | one residue | 66-Leu | two residues |
| 68-Gly | 66-Leu | two residues | 66-Leu | two residues |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kikuchi, T. Decoding an Amino Acid Sequence to Extract Information on Protein Folding. Molecules 2022, 27, 3020. https://doi.org/10.3390/molecules27093020
Kikuchi T. Decoding an Amino Acid Sequence to Extract Information on Protein Folding. Molecules. 2022; 27(9):3020. https://doi.org/10.3390/molecules27093020
Chicago/Turabian StyleKikuchi, Takeshi. 2022. "Decoding an Amino Acid Sequence to Extract Information on Protein Folding" Molecules 27, no. 9: 3020. https://doi.org/10.3390/molecules27093020
APA StyleKikuchi, T. (2022). Decoding an Amino Acid Sequence to Extract Information on Protein Folding. Molecules, 27(9), 3020. https://doi.org/10.3390/molecules27093020