


In Silico Identification of Anti-SARS-CoV-2 Medicinal Plants Using Cheminformatics and Machine Learning

Abstract
:1. Introduction
2. Result
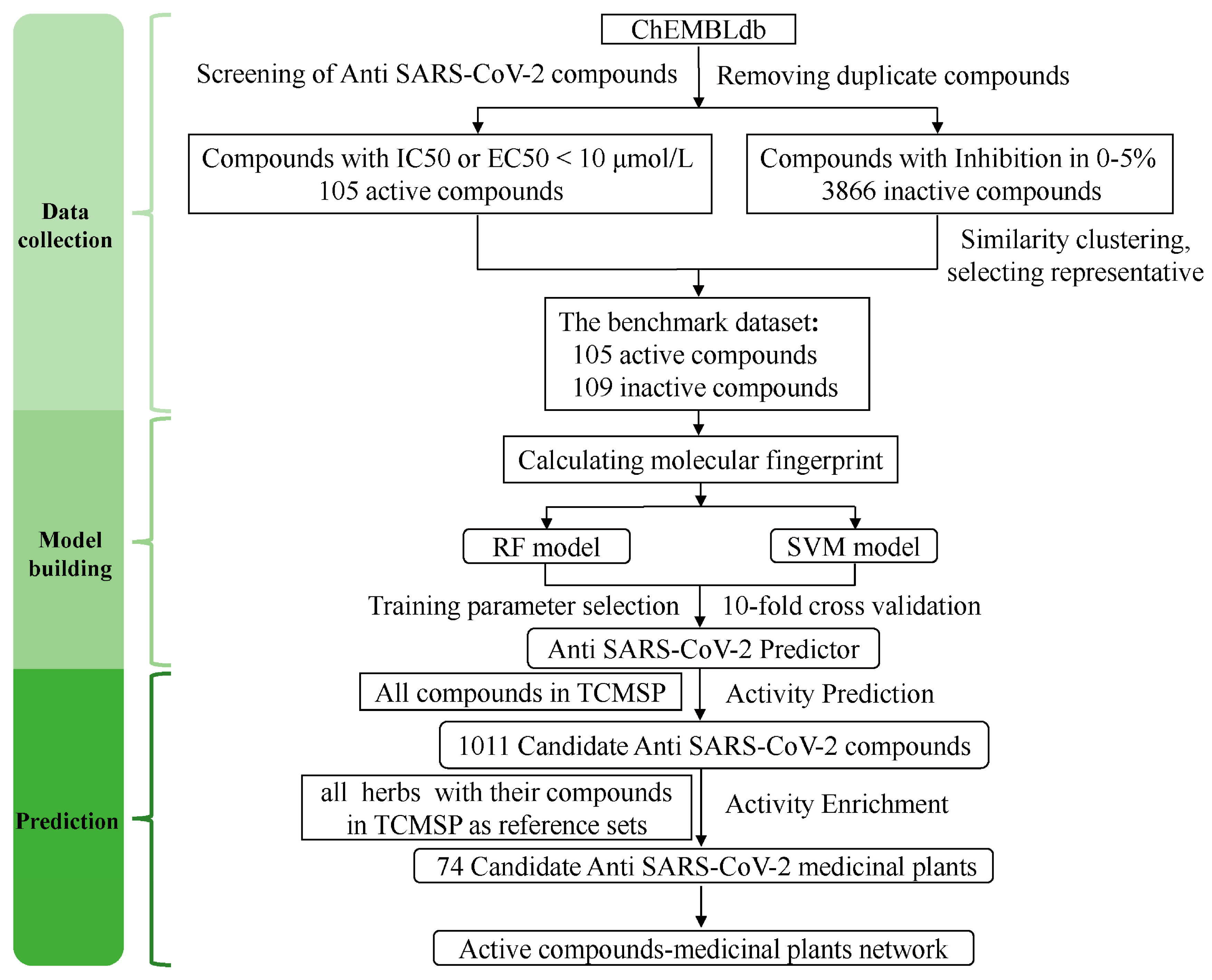
2.1. Development of anti-SARS-CoV-2 Compound Predictor
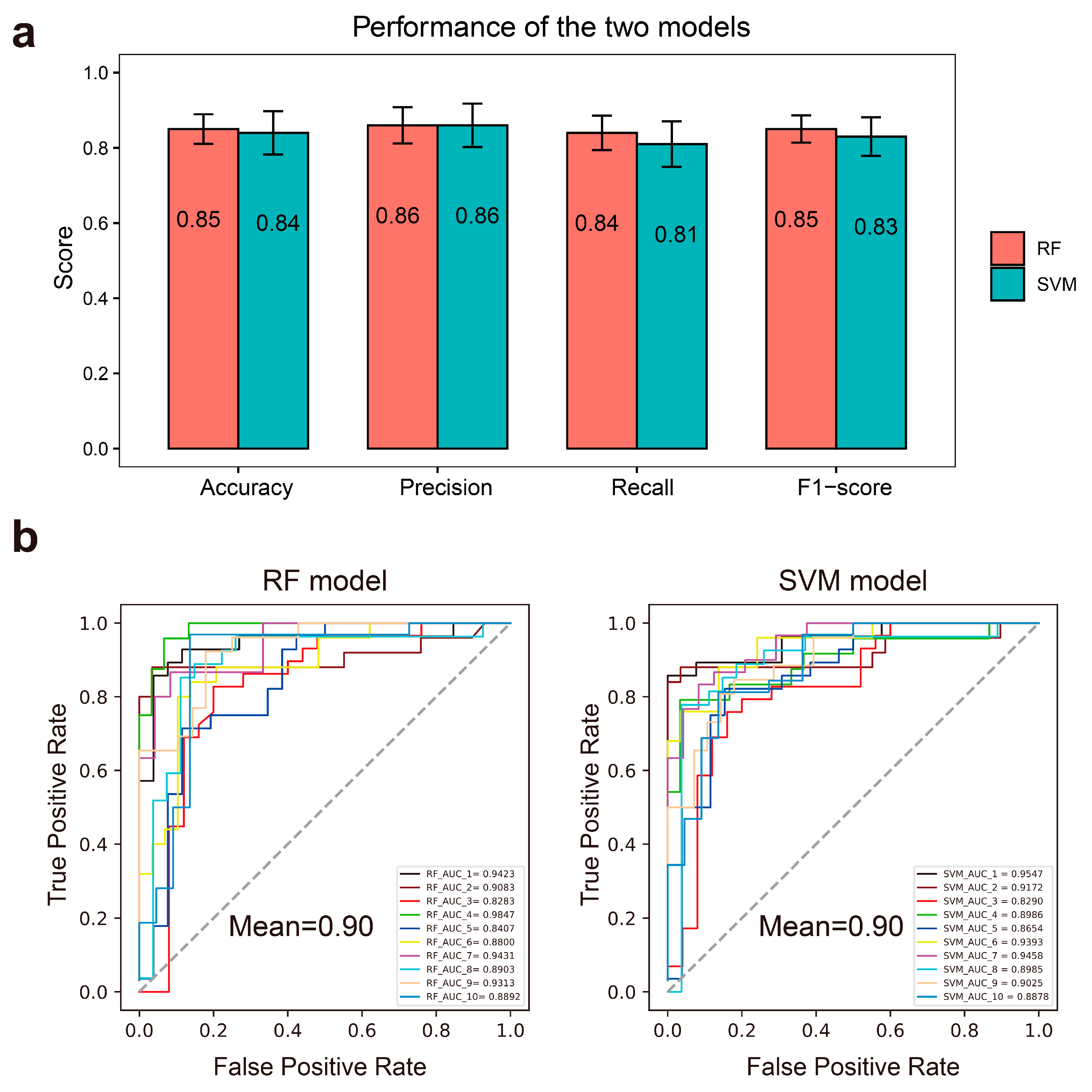
2.2. Performance of the RF and SVM Models
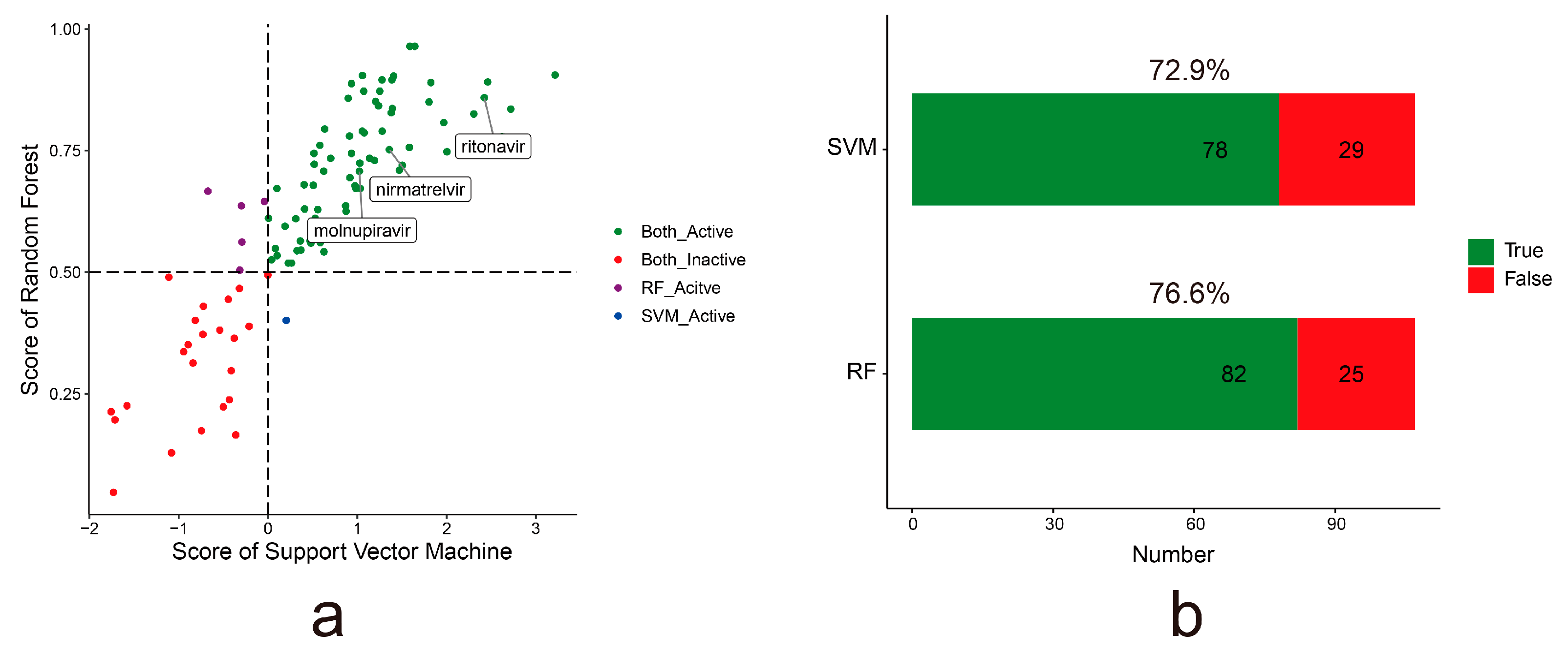
2.3. Accuracy of More Than 72% from the External Dataset
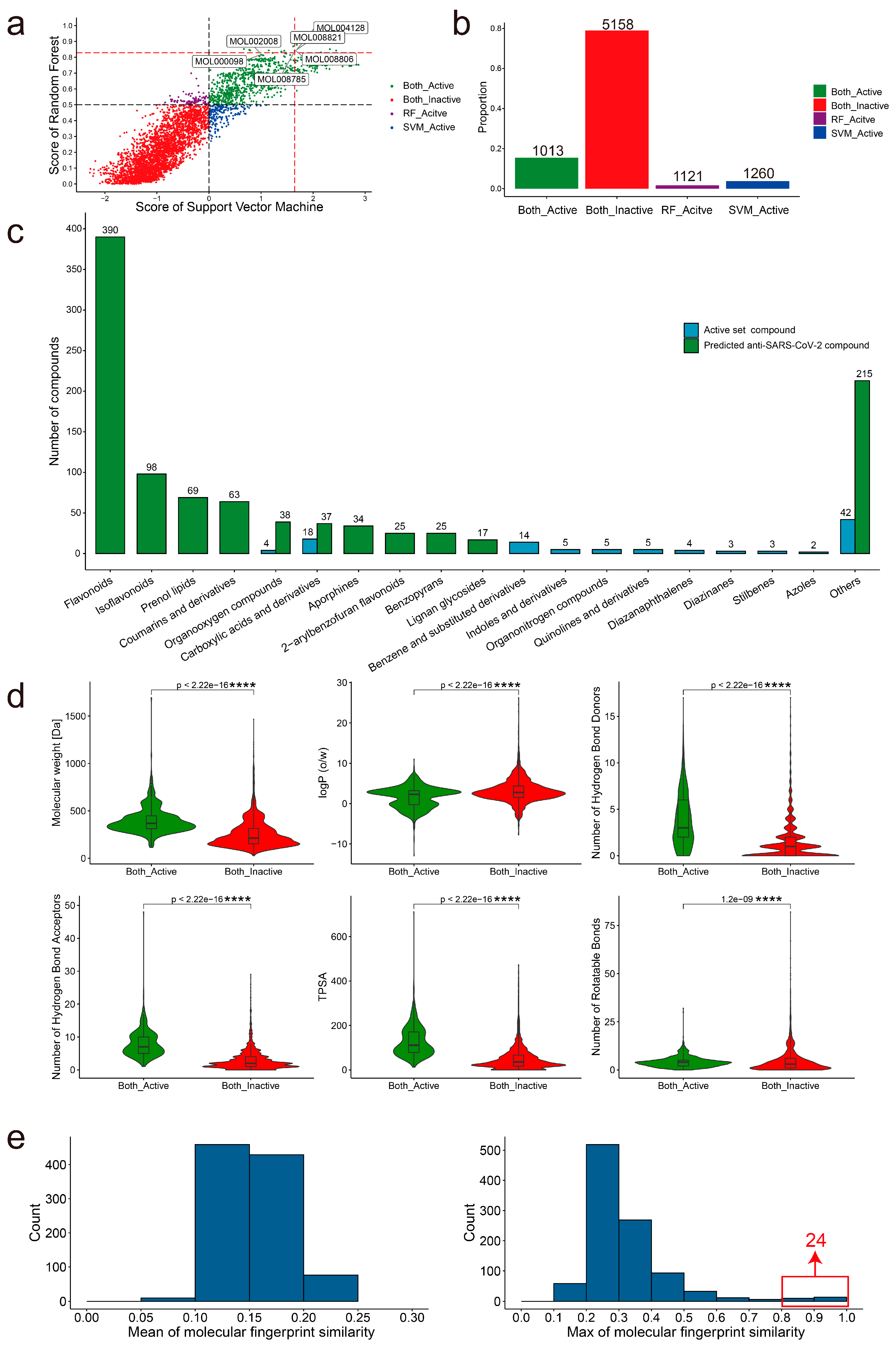
2.4. Identification of Novel Anti-SARS-CoV-2 Compounds by Screening the TCMSP
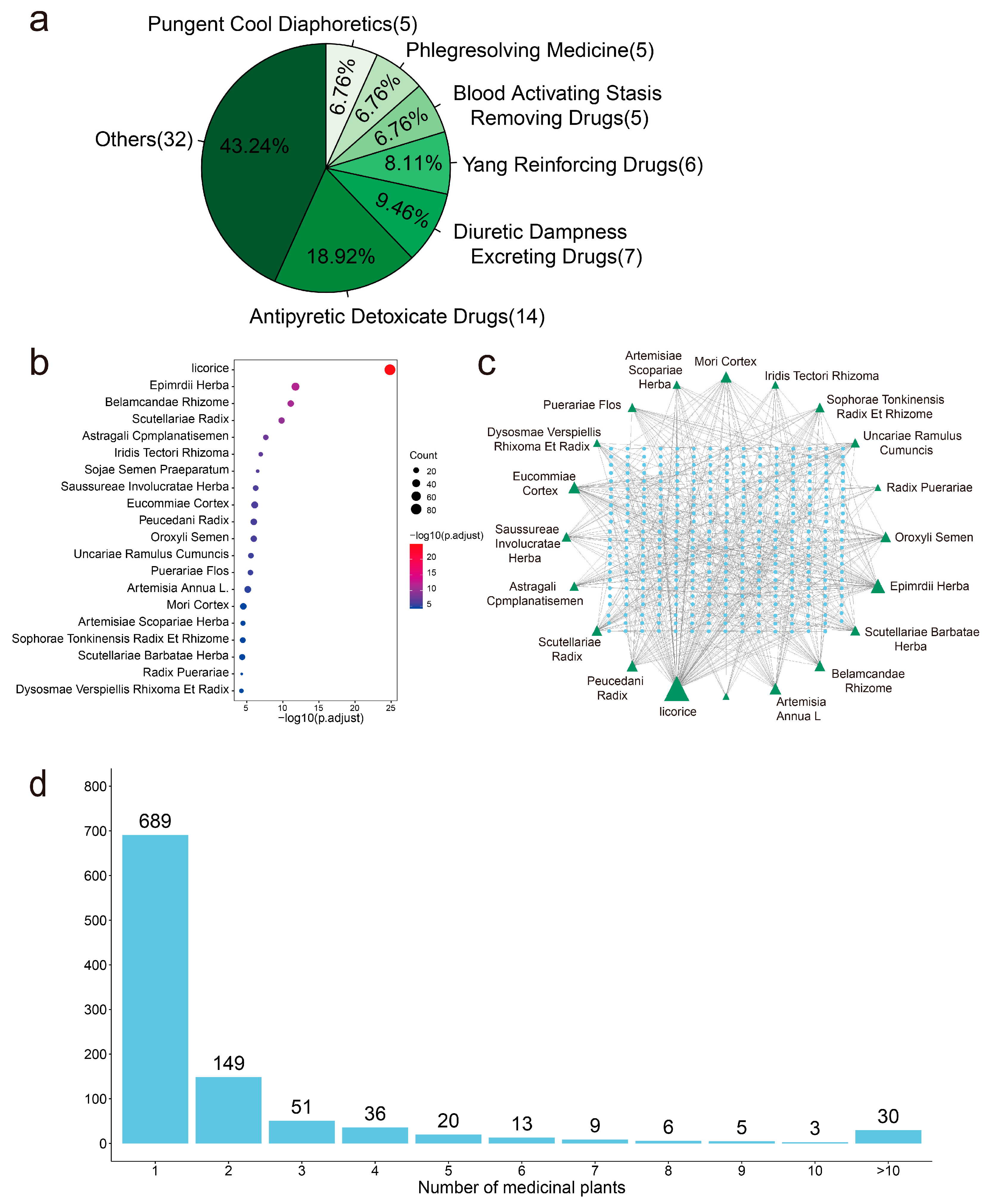
2.5. Identification of 74 Potential Anti-SARS-CoV-2 Medicinal Plants through Enrichment Analysis
3. Discussion
4. Material and methods
4.1. Construction of the Benchmark Dataset
4.2. Machine Learning Analysis of the Benchmark Dataset
4.2.1. Random Forest Model
4.2.2. Support Vector Machine Model
4.2.3. Performance Evaluation
4.3. Anti-SARS-CoV-2 Compound Prediction
4.4. Analysis of Applicability Domain (AD) of Models
4.5. Molecular Docking
4.6. Inferring the Anti-SARS-CoV-2 Medicinal Plants by Enrichment Analysis
4.7. Molecular Descriptors
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ortiz-Prado, E.; Simbana-Rivera, K.; Gomez-Barreno, L.; Rubio-Neira, M.; Guaman, L.P.; Kyriakidis, N.C.; Muslin, C.; Jaramillo, A.M.G.; Barba-Ostria, C.; Cevallos-Robalino, D.; et al. Clinical, molecular, and epidemiological characterization of the SARS-CoV-2 virus and the Coronavirus Disease 2019 (COVID-19), a comprehensive literature review. Diagn. Microbiol. Infect. Dis. 2020, 98, 115094. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.L.; Wang, Y.M.; Li, X.W.; Ren, L.L.; Zhao, J.P.; Hu, Y.; Zhang, L.; Fan, G.H.; Xu, J.Y.; Gu, X.Y.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 5 October 2022).
- Zhang, L.; Zhou, R. Structural Basis of the Potential Binding Mechanism of Remdesivir to SARS-CoV-2 RNA-Dependent RNA Polymerase. J. Phys. Chem. B 2020, 124, 6955–6962. [Google Scholar] [CrossRef] [PubMed]
- Yip, A.J.W.; Low, Z.Y.; Chow, V.T.K.; Lal, S.K. Repurposing Molnupiravir for COVID-19: The Mechanisms of Antiviral Activity. Viruses 2022, 14, 1345. [Google Scholar] [CrossRef] [PubMed]
- Extance, A. COVID-19: What is the evidence for the antiviral Paxlovid? BMJ Br. Med. J. 2022, 377, o1037. [Google Scholar] [CrossRef]
- Tao, K.M.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Pond, S.L.K.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Thomson, E.C.; Willett, B.J. Omicron: A shift in the biology of SARS-CoV-2. Nat. Microbiol. 2022, 7, 1114–1115. [Google Scholar]
- Gandhi, S.; Klein, J.; Robertson, A.; Pena-Hernandez, M.A.; Lin, M.J.; Roychoudhury, P.; Lu, P.W.; Fournier, J.; Ferguson, D.; Bakhash, S.A.M.; et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: A case report. Nat. Commun. 2022, 13, 1547. [Google Scholar] [CrossRef]
- Chaudhuri, S.; Symons, J.A.; Deval, J. Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antivir. Res. 2018, 155, 76–88. [Google Scholar] [CrossRef]
- Vitiello, A. SARS-CoV-2 and risk of antiviral drug resistance. Ir. J. Med. Sci. 2022, 191, 2367–2368. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Lu, H. Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci. Trends 2020, 14, 69–71. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, Y. Traditional Chinese medicine treatment of COVID-19. Complement. Ther. Clin. Pract. 2020, 39, 101165. [Google Scholar] [CrossRef]
- Du, H.Z.; Hou, X.Y.; Miao, Y.H.; Huang, B.S.; Liu, D.H. Traditional Chinese Medicine: An effective treatment for 2019 novel coronavirus pneumonia (NCP). Chin. J. Nat. Med. 2020, 18, 206–210. [Google Scholar] [CrossRef]
- Huang, Y.F.; Bai, C.; He, F.; Xie, Y.; Zhou, H. Review on the potential action mechanisms of Chinese medicines in treating Coronavirus Disease 2019 (COVID-19). Pharmacol. Res. 2020, 158, 104939. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.F. Diagnosis and Treatment Protocol for Novel Coronavirus Pneumonia (Trial Version 7). Chin. Med. J. Peking 2020, 133, 1087–1095. [Google Scholar]
- Guo, Y.J.; Ma, A.N.; Wang, X.Y.; Yang, C.; Chen, X.; Li, G.; Qiu, F. Research progress on the antiviral activities of natural products and their derivatives: Structure-activity relationships. Front. Chem. 2022, 10, 1005360. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, M.; Arshad, M.; Ahmad, M.; Pomerantz, R.J.; Wigdahl, B.; Parveen, Z. Antiviral potentials of medicinal plants. Virus Res. 2008, 131, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.I.; Sheikh, W.M.; Rather, M.A.; Venkatesalu, V.; Bashir, S.M.; Nabi, S.U. Medicinal plants: Treasure for antiviral drug discovery. Phytother. Res. 2021, 35, 3447–3483. [Google Scholar] [CrossRef]
- Akram, M.; Tahir, I.M.; Shah, S.M.A.; Mahmood, Z.; Altaf, A.; Ahmad, K.; Munir, N.; Daniyal, M.; Nasir, S.; Mehboob, H. Antiviral potential of medicinal plants against HIV, HSV, influenza, hepatitis, and coxsackievirus: A systematic review. Phytother. Res. 2018, 32, 811–822. [Google Scholar] [CrossRef]
- Zandi, K.; Musall, K.; Oo, A.; Cao, D.; Liang, B.; Hassandarvish, P.; Lan, S.; Slack, R.L.; Kirby, K.A.; Bassit, L.; et al. Baicalein and Baicalin Inhibit SARS-CoV-2 RNA-Dependent-RNA Polymerase. Microorganisms 2021, 9, 893. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chan, K.H.; Jiang, Y.; Kao, R.Y.; Lu, H.T.; Fan, K.W.; Cheng, V.C.; Tsui, W.H.; Hung, I.F.; Lee, T.S.; et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J. Clin. Virol. 2004, 31, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Van de Sand, L.; Bormann, M.; Alt, M.; Schipper, L.; Heilingloh, C.S.; Steinmann, E.; Todt, D.; Dittmer, U.; Elsner, C.; Witzke, O.; et al. Glycyrrhizin Effectively Inhibits SARS-CoV-2 Replication by Inhibiting the Viral Main Protease. Viruses 2021, 13, 609. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J.; Morgenstern, B.; Bauer, G.; Chandra, P.; Rabenau, H.; Doerr, H.W. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 2003, 361, 2045–2046. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; He, S.T.; Han, P.; Hong, B.; Liu, K.; Li, M.; Wang, S.; Tong, Y. Cepharanthine: A Promising Old Drug against SARS-CoV-2. Adv. Biol. 2022, 6, e2200148. [Google Scholar] [CrossRef]
- Chen, Y.; de Lomana, M.G.; Friedrich, N.O.; Kirchmair, J. Characterization of the Chemical Space of Known and Readily Obtainable Natural Products. J. Chem. Inf. Model. 2018, 58, 1518–1532. [Google Scholar] [CrossRef]
- Sucher, N.J. The application of Chinese medicine to novel drug discovery. Expert Opin. Drug Discov. 2013, 8, 21–34. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, Z.; Liao, F.; Jiang, T.; Tu, Y. The birth of artemisinin. Pharmacol. Ther. 2020, 216, 107658. [Google Scholar] [CrossRef]
- Torjesen, I. Drug development: The journey of a medicine from lab to shel. Pharm. J. 2015. [Google Scholar] [CrossRef]
- Zhao, Y.; Tian, Y.; Pan, C.; Liang, A.; Zhang, W.; Sheng, Y. Target-Based In Silico Screening for Phytoactive Compounds Targeting SARS-CoV-2. Interdiscip. Sci. 2022, 14, 64–79. [Google Scholar]
- Qi, X.; Li, B.; Omarini, A.B.; Gand, M.; Zhang, X.; Wang, J. Discovery of TCMs and derivatives against the main protease of SARS-CoV-2 via high throughput screening, ADMET analysis, and inhibition assay in vitro. J. Mol. Struct. 2022, 1268, 133709. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, O.V.; Costa, M.C.A.; da Costa, R.M.; Viegas, R.G.; Paluch, A.S.; Ferreira, M.M.C. Traditional herbal compounds as candidates to inhibit the SARS-CoV-2 main protease: An in silico study. J. Biomol. Struct. Dyn. 2021, 1–14. [Google Scholar] [CrossRef]
- Kadioglu, O.; Saeed, M.; Greten, H.J.; Efferth, T. Identification of novel compounds against three targets of SARS CoV-2 coronavirus by combined virtual screening and supervised machine learning. Comput. Biol. Med. 2021, 133, 104359. [Google Scholar] [CrossRef]
- Patel, C.N.; Goswami, D.; Jaiswal, D.G.; Jani, S.P.; Parmar, R.M.; Rawal, R.M.; Pandya, H.A. Excavating phytochemicals from plants possessing antiviral activities for identifying SARS-CoV hemagglutinin-esterase inhibitors by diligent computational workflow. J. Biomol. Struct. Dyn. 2022, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Li, J.H.; Lai, X.Y.; Zhang, M.; Kuang, Y.; Bao, Y.O.; Yu, R.; Hong, W.; Muturi, E.; Xue, H.; et al. Natural triterpenoids from licorice potently inhibit SARS-CoV-2 infection. J. Adv. Res. 2022, 36, 201–210. [Google Scholar] [CrossRef]
- Jan, J.-T.; Cheng, T.-J.R.; Juang, Y.-P.; Ma, H.-H.; Wu, Y.-T.; Yang, W.-B.; Cheng, C.-W.; Chen, X.; Chou, T.-H.; Shie, J.-J. Identification of existing pharmaceuticals and herbal medicines as inhibitors of SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2021579118. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Maldonado, P.; Alvarenga, N.; Burgos-Edwards, A.; Flores-Giubi, M.E.; Barua, J.E.; Romero-Rodriguez, M.C.; Soto-Rifo, R.; Valiente-Echeverria, F.; Langjahr, P.; Cantero-Gonzalez, G.; et al. Screening of Natural Products Inhibitors of SARS-CoV-2 Entry. Molecules 2022, 27, 1743. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Rosado, J.; Pelleau, S.; Cockram, C.; Merkling, S.H.; Nekkab, N.; Demeret, C.; Meola, A.; Kerneis, S.; Terrier, B.; Fafi-Kremer, S.; et al. Multiplex assays for the identification of serological signatures of SARS-CoV-2 infection: An antibody-based diagnostic and machine learning study. Lancet Microbe 2021, 2, e60–e69. [Google Scholar] [CrossRef]
- Zoabi, Y.; Deri-Rozov, S.; Shomron, N. Machine learning-based prediction of COVID-19 diagnosis based on symptoms. NPJ Digit. Med. 2021, 4, 3. [Google Scholar] [CrossRef]
- Jin, W.; Stokes, J.M.; Eastman, R.T.; Itkin, Z.; Zakharov, A.V.; Collins, J.J.; Jaakkola, T.S.; Barzilay, R. Deep learning identifies synergistic drug combinations for treating COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2105070118. [Google Scholar] [CrossRef] [PubMed]
- Lalmuanawma, S.; Hussain, J.; Chhakchhuak, L. Applications of machine learning and artificial intelligence for COVID-19 (SARS-CoV-2) pandemic: A review. Chaos Solitons Fractals 2020, 139, 110059. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Prema, K.V.; Balaji, S. Machine learning models for drug-target interactions: Current knowledge and future directions. Drug Discov. Today 2020, 25, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Nand, M.; Maiti, P.; Joshi, T.; Chandra, S.; Pande, V.; Kuniyal, J.C.; Ramakrishnan, M.A. Virtual screening of anti-HIV1 compounds against SARS-CoV-2: Machine learning modeling, chemoinformatics and molecular dynamics simulation based analysis. Sci. Rep. 2020, 10, 20397. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, J.; Polshakov, D.; Kato-Weinstein, J.; Zhou, Q.Q.; Li, Y.Z.; Granet, R.; Garner, L.; Deng, Y.; Liu, C.; Albaiu, D.; et al. Quantitative Structure-Activity Relationship Machine Learning Models and their Applications for Identifying Viral 3CLpro-and RdRp-Targeting Compounds as Potential Therapeutics for COVID-19 and Related Viral Infections. ACS Omega 2020, 5, 27344–27358. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Zhu, X.T.; Chen, L.L.; Alhussain, L.; Alshiekheid, M.A.; Theyab, A.; Algahtani, M. Target-Specific Machine Learning Scoring Function Improved Structure-Based Virtual Screening Performance for SARS-CoV-2 Drugs Development. Int. J. Mol. Sci. 2022, 23, 11003. [Google Scholar] [CrossRef]
- Xu, T.; Xu, M.; Zhu, W.; Chen, C.Z.; Zhang, Q.; Zheng, W.; Huang, R. Efficient Identification of Anti-SARS-CoV-2 Compounds Using Chemical Structure- and Biological Activity-Based Modeling. J. Med. Chem. 2022, 65, 4590–4599. [Google Scholar] [CrossRef]
- Gawriljuk, V.O.; Zin, P.P.K.; Puhl, A.C.; Zorn, K.M.; Foil, D.H.; Lane, T.R.; Hurst, B.; Tavella, T.A.; Costa, F.T.M.; Lakshmanane, P.; et al. Machine Learning Models Identify Inhibitors of SARS-CoV-2. J. Chem. Inf. Model 2021, 61, 4224–4235. [Google Scholar] [CrossRef]
- Rajput, A.; Kumar, A.; Megha, K.; Thakur, A.; Kumar, M. DrugRepV: A compendium of repurposed drugs and chemicals targeting epidemic and pandemic viruses. Brief Bioinform. 2021, 22, 1076–1084. [Google Scholar] [CrossRef]
- Li, Y.; Tian, Y.; Qin, Z.; Yan, A. Classification of HIV-1 Protease Inhibitors by Machine Learning Methods. ACS Omega 2018, 3, 15837–15849. [Google Scholar] [CrossRef]
- Feunang, Y.D.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Molavi, Z.; Razi, S.; Mirmotalebisohi, S.A.; Adibi, A.; Sameni, M.; Karami, F.; Niazi, V.; Niknam, Z.; Aliashrafi, M.; Taheri, M.; et al. Identification of FDA approved drugs against SARS-CoV-2 RNA dependent RNA polymerase (RdRp) and 3-chymotrypsin-like protease (3CLpro), drug repurposing approach. Biomed. Pharmacother. 2021, 138, 111544. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.N.; Jani, S.P.; Sivakumar, P.K.; Modi, K.M.; Kumar, Y. Medicine, Computational investigation of natural compounds as potential main protease (Mpro) inhibitors for SARS-CoV-2 virus. Comput. Biol. Med. 2022, 151, 106318. [Google Scholar] [CrossRef] [PubMed]
- Mosquera-Yuqui, F.; Lopez-Guerra, N.; Moncayo-Palacio, E.A. Targeting the 3CLpro and RdRp of SARS-CoV-2 with phytochemicals from medicinal plants of the Andean Region: Molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2022, 40, 2010–2023. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Huang, B.; Zhang, Y. Teaching an old dog new tricks: Drug discovery by repositioning natural products and their derivatives. Drug Discov. Today 2022, 27, 1936–1944. [Google Scholar] [CrossRef]
- Song, M.R.; Liu, Y.; Li, T.T.; Liu, X.J.; Hao, Z.H.; Ding, S.Y.; Panichayupakaranant, P.; Zhu, K.; Shen, J.Z. Plant Natural Flavonoids Against Multidrug Resistant Pathogens. Adv. Sci. 2021, 8, e2100749. [Google Scholar] [CrossRef]
- Liskova, A.; Koklesova, L.; Samec, M.; Smejkal, K.; Samuel, S.M.; Varghese, E.; Abotaleb, M.; Biringer, K.; Kudela, E.; Danko, J.; et al. Flavonoids in Cancer Metastasis. Cancers 2020, 12, 1498. [Google Scholar] [CrossRef]
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as Potential Anti-Inflammatory Molecules: A Review. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef]
- Liskova, A.; Samec, M.; Koklesova, L.; Samuel, S.M.; Zhai, K.; Al-Ishaq, R.K.; Abotaleb, M.; Nosal, V.; Kajo, K.; Ashrafizadeh, M.; et al. Flavonoids against the SARS-CoV-2 induced inflammatory storm. Biomed. Pharmacother. 2021, 138, 111430. [Google Scholar] [CrossRef]
- Kaul, T.N.; Middleton, E., Jr.; Ogra, P.L. Antiviral effect of flavonoids on human viruses. J. Med. Virol. 1985, 15, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Lani, R.; Hassandarvish, P.; Shu, M.H.; Phoon, W.H.; Chu, J.J.H.; Higgs, S.; Vanlandingham, D.; Abu Bakar, S.; Zandi, K. Antiviral activity of selected flavonoids against Chikungunya virus. Antivir. Res. 2016, 133, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Cataneo, A.H.D.; Avila, E.P.; Mendes, L.A.D.; de Oliveira, V.G.; Ferraz, C.R.; de Almeida, M.V.; Frabasile, S.; dos Santos, C.N.D.; Verri, W.A.; Bordignon, J.; et al. Flavonoids as Molecules with Anti-Zika virus Activity. Front. Microbiol. 2021, 12, 710359. [Google Scholar] [CrossRef] [PubMed]
- Abian, O.; Ortega-Alarcon, D.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Vega, S.; Reyburn, H.T.; Rizzuti, B.; Velazquez-Campoy, A. Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening. Int. J. Biol. Macromol. 2020, 164, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Cui, M.; Zheng, C.; Wang, M.; Sun, R.; Gao, D.; Bao, J.; Ren, S.; Yang, B.; Lin, J.; et al. Myricetin Inhibits SARS-CoV-2 Viral Replication by Targeting M(pro) and Ameliorates Pulmonary Inflammation. Front. Pharmacol. 2021, 12, 669642. [Google Scholar] [CrossRef] [PubMed]
- Al-Maharik, N. Isolation of naturally occurring novel isoflavonoids: An update. Nat. Prod. Rep. 2019, 36, 1156–1195. [Google Scholar] [CrossRef]
- Matos, M.J. Coumarin and Its Derivatives-Editorial. Molecules 2021, 26, 6320. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, M.; Liu, H.; Wei, K.; He, M.; Li, X.; Hu, D.; Yang, S.; Zheng, Y. Antiviral activity of aconite alkaloids from Aconitum carmichaelii Debx. Nat. Prod. Res. 2019, 33, 1486–1490. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef]
- Liuzzo, G.; Patrono, C. COVID 19: In the eye of the cytokine storm. Eur. Heart J. 2021, 42, 150–151. [Google Scholar]
- Li, L.; Wu, Y.; Wang, J.; Yan, H.; Lu, J.; Wan, Y.; Zhang, B.; Zhang, J.; Yang, J.; Wang, X.; et al. Potential treatment of COVID-19 with traditional chinese medicine: What herbs can help win the battle with SARS-CoV-2? Engineering 2021. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.L.; Andurkar, S.V. A review of natural products, their effects on SARS-CoV-2 and their utility as lead compounds in the discovery of drugs for the treatment of COVID-19. Med. Chem. Res. 2022, 31, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Lyu, M.; Fan, G.W.; Xiao, G.X.; Wang, T.Y.; Xu, D.; Gao, J.; Ge, S.Q.; Li, Q.L.; Ma, Y.L.; Zhang, H.; et al. Traditional Chinese medicine in COVID-19. Acta Pharm. Sin. B 2021, 11, 3337–3363. [Google Scholar] [CrossRef]
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Li, S.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzym. Inhib. Med. Chem. 2021, 36, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Crampon, K.; Giorkallos, A.; Deldossi, M.; Baud, S.; Steffenel, L.A. Machine-learning methods for ligand-protein molecular docking. Drug Discov. Today 2022, 27, 151–164. [Google Scholar] [CrossRef]
- Li, W.X.; Tong, X.; Yang, P.P.; Zheng, Y.; Liang, J.H.; Li, G.H.; Liu, D.; Guan, D.G.; Dai, S.X. Screening of antibacterial compounds with novel structure from the FDA approved drugs using machine learning methods. Aging 2022, 14, 1448–1472. [Google Scholar] [CrossRef]
- Batista, G.E.; Prati, R.C.; Monard, M.C. A study of the behavior of several methods for balancing machine learning training data. ACM SIGKDD Explor. Newsl. 2004, 6, 20–29. [Google Scholar] [CrossRef]
- Yang, L.; Shami, A. On hyperparameter optimization of machine learning algorithms: Theory and practice. Neurocomputing 2020, 415, 295–316. [Google Scholar] [CrossRef]
- Branco, P.; Torgo, L.; Ribeiro, R.P. A survey of predictive modeling on imbalanced domains. ACM Comput. Surv. 2016, 49, 1–50. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.B.; Song, Q.B.; Zhu, X.Y.; Sun, H.L.; Xu, B.W.; Zhou, Y.M. A novel ensemble method for classifying imbalanced data. Pattern Recogn. 2015, 48, 1623–1637. [Google Scholar] [CrossRef]
- Yen, S.J.; Lee, Y.S. Cluster-based under-sampling approaches for imbalanced data distributions. Expert Syst. Appl. 2009, 36, 5718–5727. [Google Scholar] [CrossRef]
- Wigh, D.S.; Goodman, J.M.; Lapkin, A.A. A review of molecular representation in the age of machine learning. Wires Comput. Mol. Sci. 2022, 12, e1603. [Google Scholar] [CrossRef]
- Muegge, I.; Mukherjee, P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Felix, E.; Magarinos, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Thakur, A.; Rastogi, A.; Choudhury, S.; Kumar, M. Computational identification of repurposed drugs against viruses causing epidemics and pandemics via drug-target network analysis. Comput. Biol. Med. 2021, 136, 104677. [Google Scholar] [CrossRef]
- Deng, Y.H.; Wang, N.N.; Zou, Z.X.; Zhang, L.; Xu, K.P.; Chen, A.F.; Cao, D.S.; Tan, G.S. Multi-Target Screening and Experimental Validation of Natural Products from Selaginella Plants against Alzheimer’s Disease. Front. Pharmacol. 2017, 8, 539. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, A.; Kaur, G.; Kumar, M. AVCpred: An integrated web server for prediction and design of antiviral compounds. Chem. Biol. Drug Des. 2017, 89, 74–83. [Google Scholar] [CrossRef]
- Sander, J.F.T.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program for Chemistry Aware Data Visualization and Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Morley, C.; Hutchison, G.R. Pybel: A Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [Green Version]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef] [Green Version]
- Dragos, H.; Gilles, M.; Alexandre, V. Predicting the predictability: A unified approach to the applicability domain problem of QSAR models. J. Chem. Inf. Model 2009, 49, 1762–1776. [Google Scholar] [CrossRef] [PubMed]
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of different approaches to define the applicability domain of QSAR models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef]
- Sun, L.X.; Yang, H.B.; Li, J.; Wang, T.D.Y.; Li, W.H.; Liu, G.X.; Tang, Y. In Silico Prediction of Compounds Binding to Human Plasma Proteins by QSAR Models. ChemMedChem 2018, 13, 572–581. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, H.B.; Wu, Z.R.; Liu, G.X.; Tang, Y.; Li, W.H. Prediction of Farnesoid X Receptor Disruptors with Machine Learning Methods. Chem. Res. Toxicol. 2018, 31, 1128–1137. [Google Scholar] [CrossRef]
- Tian, Y.J.; Zhang, S.D.; Yin, H.Y.; Yan, A.X. Quantitative structure-activity relationship (QSAR) models and their applicability domain analysis on HIV-1 protease inhibitors by machine learning methods. Chemometr. Intell. Lab. 2020, 196, 103888. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.D.; Lee, K.H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Qurey Compound 1 | Match Compound 2 | Max TC 3 | IC50 4 (μmol/L) | SVM Probability | SVM Label | RF Probability | RF Label |
|---|---|---|---|---|---|---|---|
| MOL000098 | CHEMBL50 | 1 | 2.3 | 1.00 | 1 | 0.82 | 1 |
| MOL002008 | CHEMBL50 | 1 | 2.3 | 1.00 | 1 | 0.82 | 1 |
| MOL004128 | CHEMBL507100 | 1 | 3.9 | 1.65 | 1 | 0.84 | 1 |
| MOL008785 | CHEMBL507100 | 1 | 3.9 | 1.65 | 1 | 0.84 | 1 |
| MOL008806 | CHEMBL507100 | 1 | 3.9 | 1.65 | 1 | 0.84 | 1 |
| MOL008821 | CHEMBL507100 | 1 | 3.9 | 1.65 | 1 | 0.84 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.; Zheng, Y.; Tong, X.; Yang, N.; Dai, S. In Silico Identification of Anti-SARS-CoV-2 Medicinal Plants Using Cheminformatics and Machine Learning. Molecules 2023, 28, 208. https://doi.org/10.3390/molecules28010208
Liang J, Zheng Y, Tong X, Yang N, Dai S. In Silico Identification of Anti-SARS-CoV-2 Medicinal Plants Using Cheminformatics and Machine Learning. Molecules. 2023; 28(1):208. https://doi.org/10.3390/molecules28010208
Chicago/Turabian StyleLiang, Jihao, Yang Zheng, Xin Tong, Naixue Yang, and Shaoxing Dai. 2023. "In Silico Identification of Anti-SARS-CoV-2 Medicinal Plants Using Cheminformatics and Machine Learning" Molecules 28, no. 1: 208. https://doi.org/10.3390/molecules28010208
APA StyleLiang, J., Zheng, Y., Tong, X., Yang, N., & Dai, S. (2023). In Silico Identification of Anti-SARS-CoV-2 Medicinal Plants Using Cheminformatics and Machine Learning. Molecules, 28(1), 208. https://doi.org/10.3390/molecules28010208