2.2.1. Analysis of TOF-SIMS Spectra in the Positive Mode
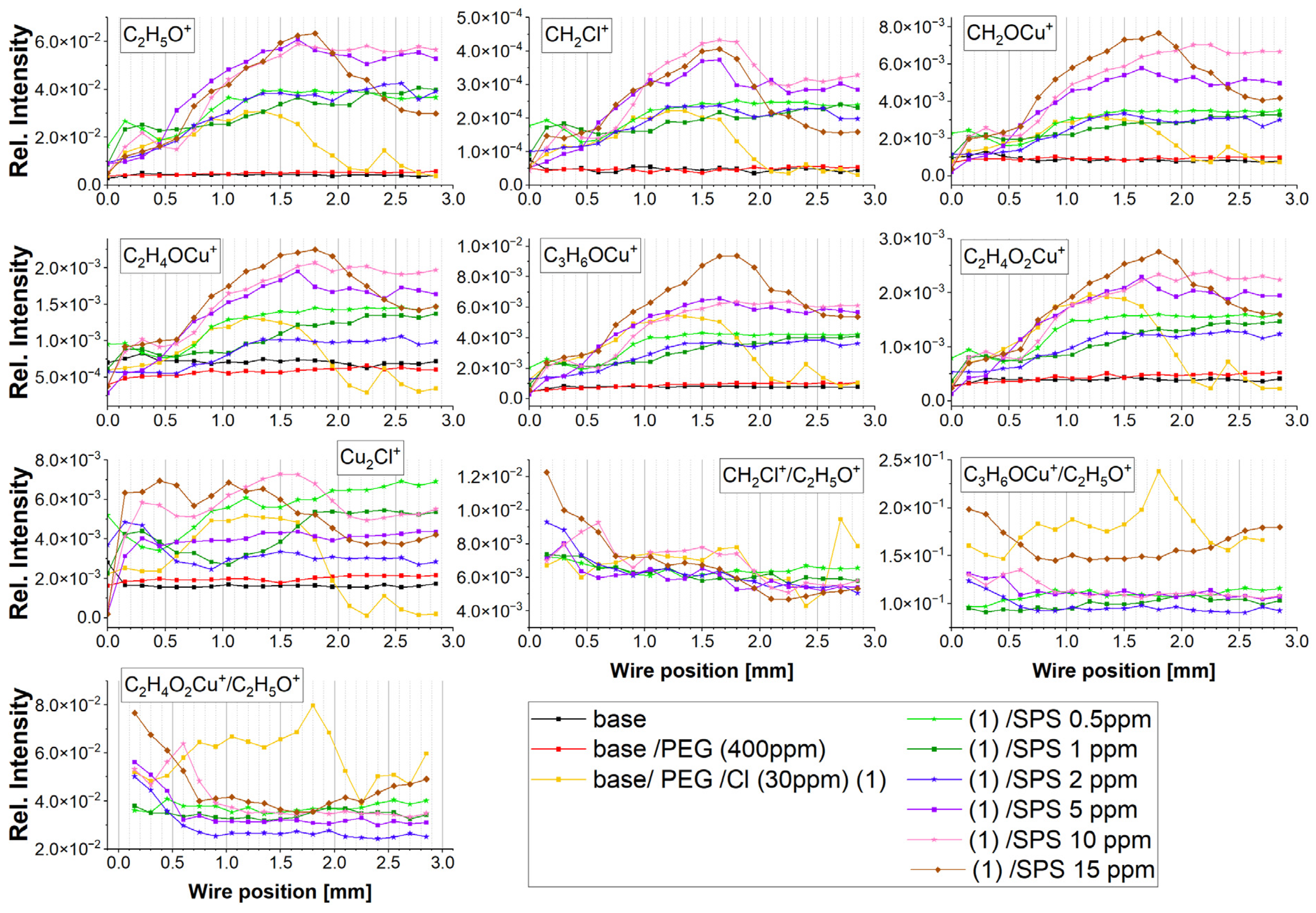
The intensity of the distribution of the most characteristic positive fragments identified in the TOF-SIMS spectra as a function of the wire position is shown in
Figure 3. The assignments of the identified fragments and their
m/
z are listed in
Table 1.
The most prominent fragment, C
2H
5O
+, which overwhelmed the rest of the PEG fragments, corresponded to the adsorbed polyethylene glycol fragments. The intensity of C
2H
5O
+ was approximately six times higher than the intensity of the second most prominent fragment, C
3H
6OCu
+. The intensity of the C
2H
5O
+ fragment can be used for estimating the total PEG surface coverage. Furthermore, the intensity of copper adducts with a PEG molecule decreases in the following order: C
3H
6OCu
+, CH
2OCu
+, C
2H
4O
2Cu
+, C
2H
4OCu
+, C
4H
8O
2Cu
+, and C
3H
6O
2Cu
+. All the copper–PEG adducts were identified in previous studies [
22], and the pathway of the yield of the selected Cu-PEG fragments was proposed. Moreover, the fragment CH
2Cl
+, yielded by a secondary reaction of the
•CH
2• diradical with chloride ions during a bombardment of the copper surface during the TOF-SIMS analysis, was identified [
22]. Accordingly, the reaction that yields CH
2Cl
+ is as follows:
The proposed mechanism for yielding the fragments C
4H
8Cu
+, C
3H
6OCu
+, CH
2OCu
+, C
2H
4O
2Cu
+, and CH
2Cl
+ was provided in the author’s previous paper [
22]. However, the suggested pathways do not consider the influence of the PEG conformation on the yield of the individual Cu-PEG fragments.
Moreover, a more difficult interpretation is related to the Cu-PEG adducts in the presence of MPS molecules adsorbed on the copper surface after the dissociation of SPS. Firstly, it is still unclear whether the copper-PEG adducts correspond to the PEG(Cu
+) complex formed during electrodeposition or are rather yielded from sputtered copper ions from the copper substrate and PEG molecules during the TOF-SIMS measurements. A previous study [
22] proved that Cu-PEG adducts can also be the products of Cu
+ sputtered ions from the copper surface with the layer of PEG deposited from pure 0.1% ethanolic solution during Bi
+ bombardment in the TOF-SIMS instrument. However, if we assume that Cu
+ ions from a Cu
+(PEG) complex that formed during copper electrodeposition can be entirely dissolved through rinsing, we can assume that the PEG conformation on the Cu/Cl adlayer is preserved. Taking into account this assumption, the TOF-SIMS measurements can be used for the reconstruction of the possible conformation of PEG on the copper surface during copper electrodeposition. Contrarily, if the complex Cu
+(PEG) did not exist in a parental form during electrodeposition, as it was postulated in [
20], the TOF-SIMS experiment can reconstruct the competition of the sulfonate ends of the thiolate molecules with ethereal atoms of PEG in capturing the Cu
+ ions sputtered from the copper substrate.
Chloride ions in the positive mode of TOF-SIMS were identified in the forms of CH
2Cl
+ and Cu
2Cl
+. The abundance of the CH
2Cl
+ fragment (Reaction 5) is determined by the proximity of Cl
− ions to the hydrophobic -CH
2CH
2- groups of the PEG skeleton. Under this scenario diradical fragments of
•CH
2• can easily react with Cl
•, resulting in high yields of CH
2Cl
•. Thus, the intensity of the CH
2Cl
+ fragment can be used for estimating the number of CH
2 units attached to the Cl adlayer. A higher yield of CH
2Cl
+ can be expected for gauche conformation around C-C, which increases the probability of the closer proximity of a greater number of hydrogens with underlaid chloride adlayer (
Figure S6b,c, Supplementary Materials). For trans conformation around C-C (
Figure S6a, the dihedral angle along -OCCO- was equal to 180°, which allows only one or two hydrogen atoms to directly contact the chloride adlayer (
Figure S6, Supplementary Materials). Decreasing the dihedral angle to 60° (gauche conformation) favors potentially three hydrogen atoms for hydrophobic interaction, while at 0° we can expect the interaction of four hydrogen atoms with the chloride layer.
The identification of the chloride adlayer was based on the CH2Cl+ as well as the Cu2Cl+ fragments. However, CH2Cl+ unambiguously stands for the chloride ions interacting with PEG, while Cu2Cl+ can also correspond to the chloride adlayer that is not in proximity to the hydrophobic parts of PEG.
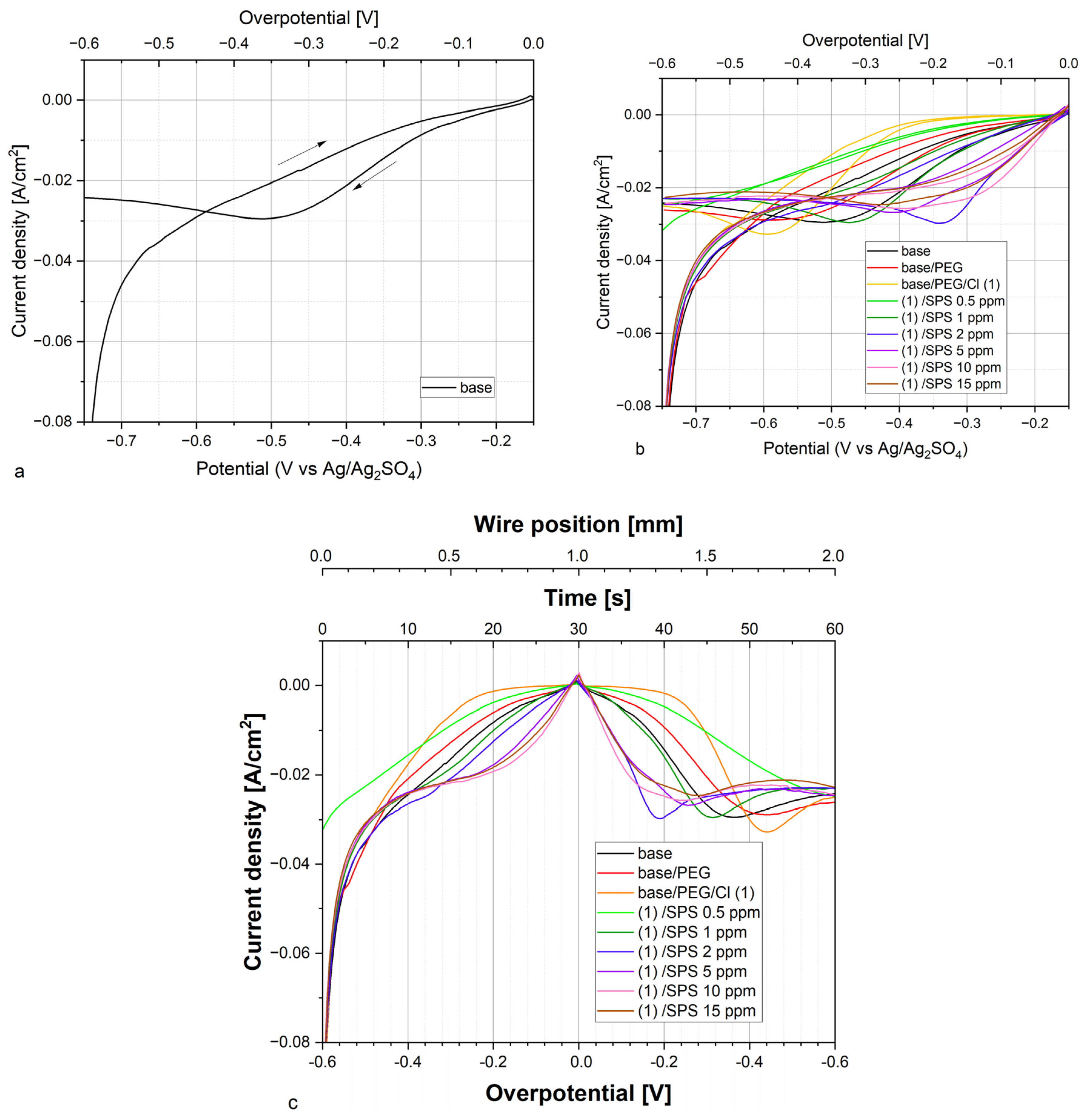
For the base solution and after the addition of PEG, no characteristic fragments for PEG, SPS, or Cl were detected. Very few chloride ions in the form of Cu
2Cl
+ were identified due to the accumulated impurities of chloride on the copper surface [
22,
62,
63], and it was rather constant as a function of the applied overpotential.
After the injection of chloride ions (30 ppm), characteristic fragments for PEG were identified (sample PEG/Cl, yellow colors). The intensity of the C
2H
5O
+ fragment gradually increased, reaching a maximum of approximately 1.2–1.35 mm, which corresponds to the reverse scan and overpotentials −0.12 and −0.18 V, respectively. A very similar distribution was observed for CH
2Cl
+. This strongly suggests that during the increasing adsorption of PEG, when the overpotential was shifting towards the open circuit potential (OCP), the hydrophobic interaction of Cl
− ions with hydrogen atoms of the -CH
2-CH
2- group occurred. The gauche conformation around C-C was favorable for enhancing and maintaining that effect for a wide range of overpotentials, as it was mentioned above. A roughly similar distribution of the intensity of the Cu-PEG fragments could be observed. Moreover, a good correlation between the distribution of intensity of Cu
2Cl
+ and C
2H
5O
+, Cu-PEG adducts, and CH
2Cl
+ proves that the Cu
2Cl
+ fragment corresponded to the Cl adlayer interacting with PEG. During the reverse scan for the higher overpotentials, the co-desorption of chloride ions (the fragment Cu
2Cl
+) and PEG was observed. More detailed information concerning the adsorption/desorption processes can be obtained by examination of the intensity ratio of C
2H
5O
+/Cu
2Cl
+ (
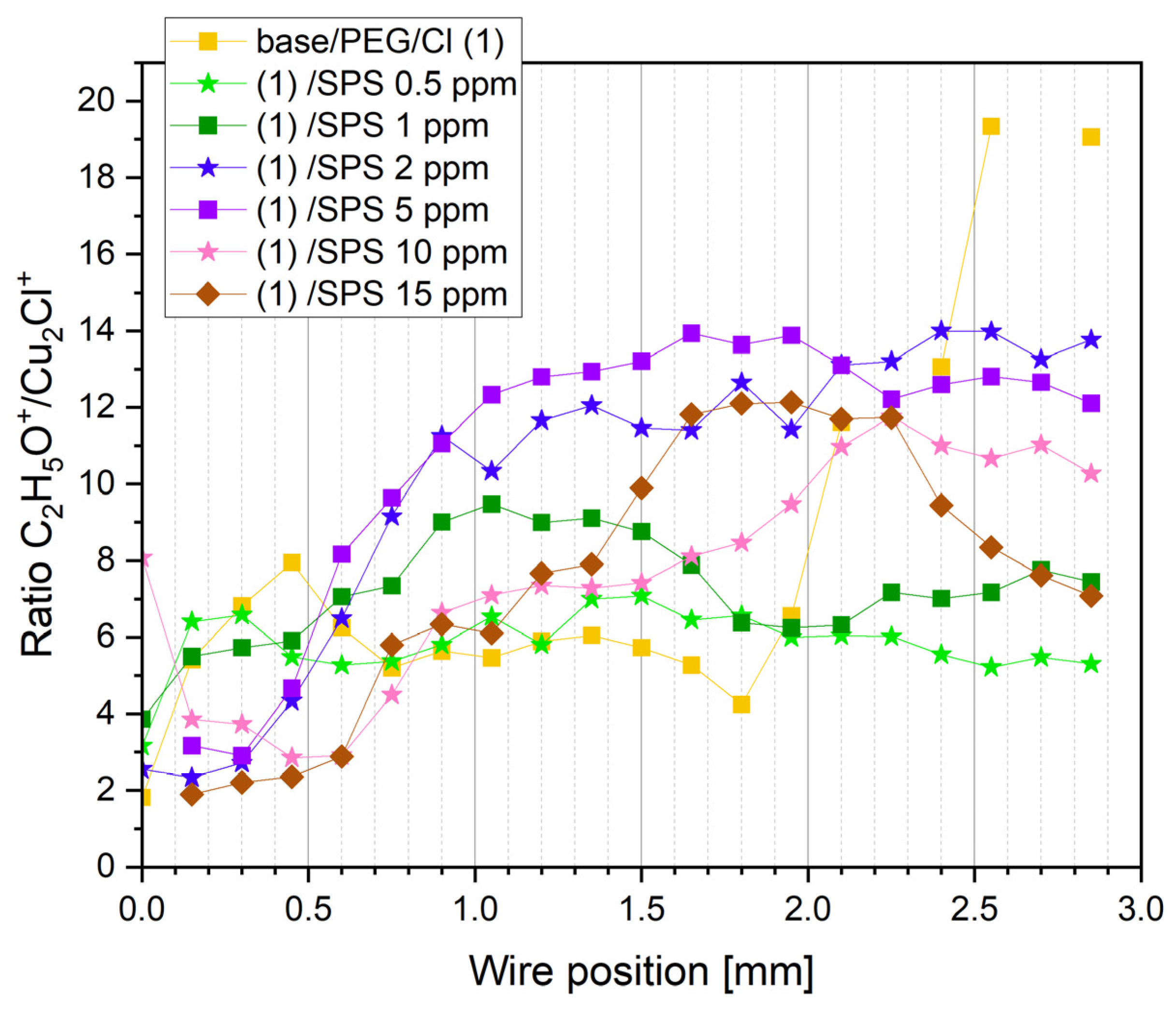
Figure 4).
For a high overpotential, the ratio of C
2H
5O
+/Cu
2Cl
+ increased (positions: 0.0 to 0.45 mm). This was determined by the lower ratio of free chloride ions that did not interact with PEG molecules. Consecutively, for the next examined points (positions: from 0.6 to 0.75 mm), the ratio of C
2H
5O
+/Cu
2Cl
+ decreased, which was accompanied by a higher PEG surface coverage. This means that the higher ratio of free chloride ions was not involved in the interaction with PEG. For the positions from 0.75 to 1.65 mm, we can observe a stable value of theC
2H
5O
+/Cu
2Cl
+ ratio. This was determined by the fact that a strong synergy of the PEG surface coverage with the chloride ion surface coverage occurred involving a greater number of Cl ions with the interaction with PEG. For a very high overpotentials for the reverse scan, a strong increase in the C
2H
5O
+/Cu
2Cl
+ ratio suggests that the chloride layer was desorbed significantly faster than PEG. This observation is supported by the CH
2Cl
+/C
2H
5O
+ ratio (
Figure 3) in this overpotential region (positions: 1.8–2.0 mm). This phenomenon can be explained by the fact that one molecule of PEG with a molar weight of 8000 consists of approximately 20 monomer units that contain approximately 60 or 80 hydrogen atoms that can interact with the chloride adlayer. Even in a situation where the desorption of half of the chloride ions occurred, the remaining chloride ions could still interact with 10 PEG monomer units, allowing for the remaining PEG molecules on the surface. The injection of 0.5 ppm SPS exhibited only a moderate effect on the C
2H
5O
+/Cu
2Cl
+ ratio, while the next portions of SPS (1, 2, and 5 ppm) resulted in an increase in the ratio of the PEG surface coverage to the chloride surface coverage. At the highest concentration of SPS (10 and 15 ppm), the PEG surface coverage, in comparison to the chloride surface coverage, significantly diminished. This behavior was determined by the accumulation of SPS on the chloride adlayer. It is reported that SPS [
24,
28] is very mobile on the chloride adlayer and after dissociation is chemically bonded through the thiolate end to the copper surface. Under these circumstances, the chloride adlayer is partly displaced by MPS adsorbed [
24,
63] units and in that way determines the ratio of PEG/Cl.
The intensity of the Cu-PEG adducts’ distribution was roughly similar (
Figure 3). However, it would be interesting to resolve the mutual correlation of the selected Cu-PEG fragments with conformation around C-C and C-O in the PEG skeleton. It seems that the yield of individual Cu-PEG adducts can be determined by the -O-CH
2-CH
2-O- conformations, and researchers [
19] have distinguished 72 possible conformations of PEG. A recent work [
20] suggested that during the shifting potential towards OCP, gauche conformation around C-O is preferred, which allows for adjusting to the optimal distance of 29 Å between the ethereal oxygen atoms to form hydrogen bonds. Moreover, this conformation is optimal for forming a (Cu
+)PEG complex between two oxygen atoms and Cu
+ [
12,
37]. Furthermore, the conformation TGT in the sequence -O-CH
2-CH
2-O- is favorable for forming PEG complexes with HgCl
2 [
18] in the crystalline state, while it was suggested that in the liquid state, trans conformation around C-C is more energetically stable.
Taking into account the above consideration, it is seen that the estimation of the exact conformation around C-O and C-C is challenging, and very often Raman spectra are applied for this purpose. However, the detection limit of normal Raman for PEG is very low. On the other hand, the SERS variant that requires a specially prepared Cu substrate would significantly improve the detection limit. However, a large inspection area is required to receive an optimal Raman signal intensity at a reasonable time of measurement. In our TOF-SIMS analysis, the inspection area was less than 50 µm, which significantly reduces the application of the Raman method. In combination with thiolate, the Raman measurements are additionally more complicated, since many characteristic peaks for MSA overlap with PEG peaks [
49].
Due to the above reason, we evaluated Cu-PEG adducts in respect to the possibility of estimating feasible conformations (gauche or trans) around C-C and C-O in PEG. For the examination of the contribution of the conformation around C-O, the intensity of the Cu-PEG adducts (C
3H
6OCu
+ and C
2H
4O
2Cu
+) was divided by the C
2H
5O
+ intensity, while for determination of the conformation around C-C bonds, the intensity of the CH
2Cl
+ fragment was divided by C
2H
5O
+. In this way, the higher ratio of C
3H
6OCu
+/C
2H
5O
+ could correspond to the molecular arrangement, as shown in
Figure S6a (Supplementary Materials), while the ratio of C
2H
4O
2Cu
+/C
2H
5O
+ to the complex structure is depicted in
Figure S6b. Likewise, the ratio of CH
2Cl
+/C
2H
5O
+ to the molecular arrangement of the hydrophobic groups -CH
2-CH
2- in gauche conformation around a C-C bond attached to the Cl adlayer is shown in
Figure S6c.
The distribution of the intensity of both the C
3H
6OCu
+/C
2H
5O
+ and C
2H
4O
2Cu
+/C
2H
5O
+ ratios for PEG/Cl was very similar for the region from 0.0 to 2.0 mm and gradually increased for the forward scan simultaneously with the increasing PEG surface coverage exhibited by the C
2H
5O
+ intensity. For the reverse scan, the C
3H
6OCu
+/C
2H
5O
+ and C
2H
4O
2Cu
+/C
2H
5O
+ ratios had relatively stable values in the region from 1.0 to 1.5 mm; then, they gave rise to the position from 1.65 to 1.8 mm. For this region, the PEG surface coverage significantly decreased and correlated with the diminished chloride (Cu
2Cl
+) surface coverage. This means that during the desorption of PEG at overpotentials of −400 and −490 mV, the contribution of the gauche conformation around C-O (see
Table S1, supplement materials) can increase, which is favorable for a higher yield of C
3H
6OCu
+ and C
2H
4O
2Cu
+. On the other hand, the contribution of the gauche conformation around the C-C bond corresponding to the ratio CH
2Cl
+/C
2H
5O
+ was similar at −400 and −480 mV. This means that under the stronger PEG desorption observed at an overpotential of −480 mV, the skeleton of the PEG rotated around the C-C bond due to the desorption of the chloride adlayer, while changes in the C-O rotation were favorable for the stabilization of PEG(Cu+) complexes. The selected conformers of PEG and their influence on the rotation around the C-C bond at the distance of O-O is depicted in
Table S1 and
Figure S7 (Supplementary Materials). It is clearly seen that rotation around the C-O bond neglected to influence the O-O distance, while rotation around C-C resulted in a dihedral angle O-C-C-O higher than 60°, which significantly increased the O-O length. As a consequence, only under a trans conformation around C-C can the formation of Cu-PEG complexes be ruled out. More detailed quantum chemistry calculations and discussions can be found elsewhere [
21]. In this context, the increase in the C
2H
4O
2Cu
+/C
2H
5O
+ ratio can be primarily determined by the free access to Cu
2+ ions by ethereal oxygens rather than gauche conformation around C-O. Moreover, when the ratio of CH
2Cl
+/C
2H
5O
+ was simultaneously reduced, it was expected to be determined by the partial desorption of the Cl adlayer, which reduced the number of monomer units binding to the Cl ions. However, in this molecular arrangement, PEG molecules were identified on the surface, and ethereal oxygen atoms of PEG can form complexes from the other side of the PEG molecule.
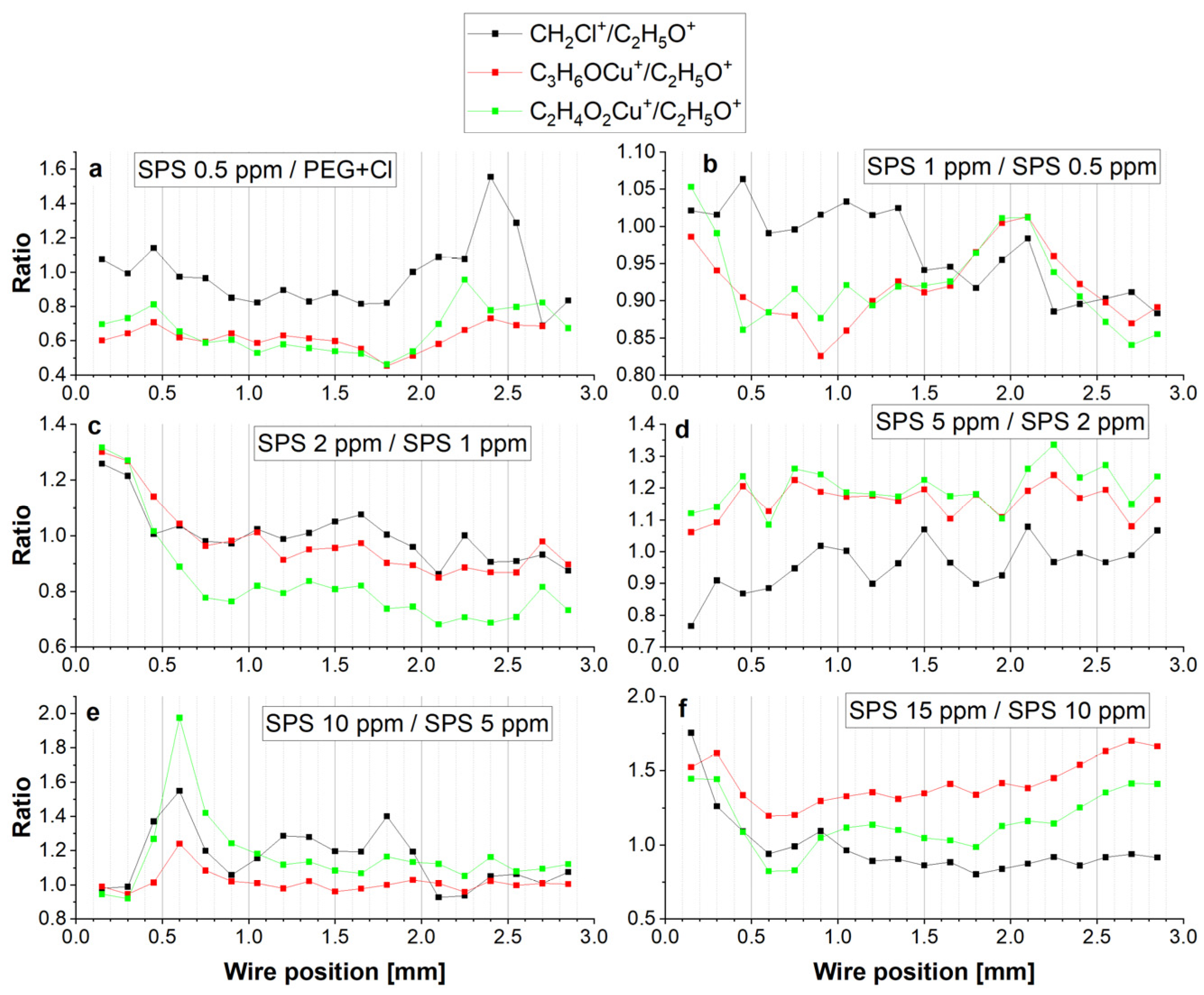
After the injection of 0.5 ppm SPS, the PEG surface coverage (intensity of C
2H
5O
+) slightly increased. This is in contradiction to the model proposed by other researchers [
2,
41,
42,
51], which suggested the partial displacement of PEG by SPS molecules due to the fact of competitive adsorption. The increasing yield of C
2H
5O
+ can be also determined by the existence of MPS molecules on the copper surface due to the matrix effect. However, the higher intensity of all Cu-PEG and CH
2Cl
+ rather suggests that synergetic co-adsorption of SPS and PEG occurred under our experimental condition.
For high overpotentials from −600 to −300 mV (positions: 0.0–0.3 mm) for the forward scan as well as for the reverse scan (1.65–2.0 mm), the PEG surface coverage was significantly greater than for the sample without SPS, while for low overpotentials, it was similar. The distribution of C
2H
5O
+ positively correlated with Cu
2Cl
+ as well as with the Cu-PEG fragments and CH
2Cl. However, closer inspection shows a strong reduction, at approximately40%, of the intensity of the C
3H
6OCu
+/C
2H
5O
+ and C
2H
4O
2Cu
+/C
2H
5O
+ ratios for the sample of PEG/Cl/0.5 ppm SPS in comparison to the sample without SPS (
Figure 5a). On the other hand, the CH
2Cl/C
2H
5O ratio only slightly decreased for the reverse scan. This strongly suggests that the introduction of SPS significantly diminished the yields of the Cu-PEG adducts. The first explanation can combine this fact with the changes in the C-O conformation from gauche to trans for a significant number of monomer units of PEG, while the reorientation of the C-C bond from gauche to trans was less severe. However, the yield of C
3H
6OCu
+ seemed to be rather independent of the conformational changes around C-O, since the distance O-O was constant for the different C-O conformations (
Table S1, Supplementary Materials), and one ion of Cu
+ or Cu
2+ can still easily accommodate in the close proximity of oxygen and form a bond. On the other hand, as it is shown in
Figure 5a, the distribution of C
3H
6OCu
+ and C
2H
4O
2Cu
+ was very similar. This strongly suggests that the primary C
3H
6OCu
+ and C
2H
4O
2Cu
+ contributions after the addition of SPS was the lack of copper ions in the close proximity of oxygen due to the capture of Cu
2+ ions by thiolate molecules. Thiolate molecules in gauche conformation are efficient in the partial dehydration of Cu
2+ and supply Cu
+ ions directly to the chloride adlayer and in that way omit oxygen atoms of PEG. Moreover, thiolate ions in trans conformations that are less efficient in the latter process can also compete with PEG in capturing Cu
2+. In that way, the available Cu
2+ ions for PEG molecules are reduced, diminishing its suppressing role. In this way, the closer chemical environmental surroundings of the ethereal oxygen atom of PEG are diminished in copper ions and metallic copper received after the reduction of Cu
2+. In consequence, due to the lack of copper atoms in close proximity to the ethereal atom of PEG during the sputtering processes in the TOF-SIMS measurements, the yield of Cu-PEG adducts (i.e., C
3H
6OCu
+ and C
2H
4O
2Cu
+) were strongly reduced.
The injection of the next portion of SPS (PEG/Cl/SPS1ppm) caused a decrease of both of the C
3H
6OCu
+/C
2H
5O
+ and C
2H
4O
2Cu
+/C
2H
5O
+ ratios again, similarly as it was observed for PEG/Cl/SPS0.5ppm (
Figure 5b), while the ratio CH
2Cl/C
2H
5O was maintained. This means that additional amounts of SPS enhanced the effect of capturing Cu
2+ ions and, therefore, reduced the number of these ions, so they were not available for PEG.
For greater concentrations of SPS (2 ppm) for higher overpotentials (positions: 0.0–0.6 mm), the PEG surface coverage was reduced (
Figure 3), while for the rest of the potential range (positions: 0.75–2.0 mm), it slightly increased in comparison to the PEG/Cl/SPS1ppm. However, the ratio of C
2H
4O
2Cu
+/C
2H
5O
+ (
Figure 5c) was significantly reduced for the range from 0.6 to 2.0 mm and after the CV measurements when switching off of the current occurred (positions: 2.0–2.8 mm). Furthermore, this C
3H
6OCu
+/C
2H
5O
+ ratio was very comparable to that observed for SPS 1 ppm (
Figure 3 and
Figure 5c). This strongly suggests that at similar surface coverage, expressed by the intensity of the C
2H
5O
+ fragment, the intensity ratio of C
2H
4O
2Cu
+/C
2H
5O
+ was the main factor that determined the suppressing activities of PEG. This strongly implies that the copper ions that can be captured by two ethereal oxygen atoms (C
2H
4O
2Cu
+) can form a more stable complex than C
3H
6OCu
+ and, in effect, enhance the suppressing abilities of PEG. In consequence, the lower ratio of C
2H
4O
2Cu
+/C
2H
5O
+ determines the lower suppressing abilities of PEG. Moreover, the ratio of C
2H
5O
+/Cu
2Cl
+ expresses the ratio of the PEG surface coverage to the chloride adlayer surface coverage.
Figure 4 depicts that for the samples containing SPS (from 0.5 to 5 ppm), an increase in the PEG surface coverage to chloride surface coverage as a function of the SPS concentration took place, and it demonstrates a similar distribution as the intensity of C
2H
5O
+, while at 10 and 15 ppm of SPS, it was significantly reduced.
The injection of the subsequent portion of SPS (PEG/Cl/SPS5ppm) strongly increased the amount of PEG for low overpotentials for the forward scan (positions: 0.6–1.0 mm) and for the whole reverse scan. Moreover, the ratio of C
2H
4O
2Cu
+/C
2H
5O
+ increased for the forward as well for the reverse scans in comparison to PEG/Cl/SPS2ppm (
Figure 3 and
Figure 5d). This means that the suppressing abilities should increase, while the CV curve (
Figure 1) demonstrates that the suppressing abilities were still diminished. This inconsistency can be explained by the significantly higher surface coverage of the thiolate observed than that at the SPS concentration. A detailed discussion is provided in the next section.
Similarly, for SPS 10 ppm (
Figure 3 and
Figure 5e), we can observe the increase in the C
2H
4O
2Cu
+/C
2H
5O
+ ratio in comparison to SPS 5 ppm. However, the C
2H
4O
2Cu
+/C
2H
5O
+ and CH
2Cl
+/C
2H
5O
+ ratios were comparable for SPS 10 and 15 ppm and demonstrate slightly higher values than for SPS 5 ppm. Moreover, the C
3H
6OCu
+/C
2H
5O
+ ratio was significantly higher for the sample with a greater concentration of SPS (15 ppm).
2.2.2. Analysis of TOF-SIMS Spectra in the Negative Mode
The most prominent ions identified in the TOF-SIMS spectra are listed in
Table 2.
Figure 6 depicts the intensity distribution of the selected negative fragments: CH
2SO
3−, CuS
−, SO
4−, C
2H
3SO
3−, CuSO
−, C
3H
5SO
3−, CuSC
3H
6SO
3−, Cu
2Cl
3−, the ratio of CH
2SO
3−/C
3H
5SO
3−, and the total thiols. The total thiols were obtained by summation of the intensities: CH
2SO
3−, CuS
−, C
2H
3SO
3−, C
3H
5SO
3−, and CuSC
3H
6SO
3−.
The sulfate ions were identified in the form of SO4− and HSO4− while the Cl adlayer was in the form Cu2Cl3−.
For the base solution, the sulfate ions were adsorbed as the potential increased towards OCP, while after switching to the reverse scan, the sulfate surface coverage gradually decreased (
Figure 6). A similar distribution was observed for HSO
4− (
Figure S8, Supplementary Materials). However, the ratio of HSO
4−/SO
4− demonstrated approximately three times higher values than SO
4−. This ratio is in contradiction to the results reported in [
20] that provide evidence suggesting that the SO
4− ion rather than the HSO
4− ion dominates on a copper surface immersed in sulfuric acid solution. However, the existence of HSO
4− ions in the TOF-SIMS mass spectra can be determined by the hydrogen radical ions’ transfer to sulfate ions, which can give rise to the abundance of HSO
4−. This needs a separate study. After the addition of PEG, a minor lower sulfate surface coverage was observed, while after the injection of chloride ions, the significant replacement of sulfate ions by chloride ions was observed. This is consistent with previous studies [
63]. The addition of SPS reduced the amounts of sulfate for the whole overpotential range and was roughly similar for all samples at different SPS concentrations. On the other hand, after the addition of SPS (0.5 and 1 ppm), the ratio HSO
4−/SO
4− was reduced (
Figure S8, Supplementary Materials).
The chloride surface coverage significantly increased after adding 30 ppm Cl ions into the base/PEG solution. The distribution of chloride ions was inversely correlated with chloride ions. This means that chloride ions replaced sulfate ions on the copper surface and vice versa at the region from 1.6 to 2.0 mm, where the strong replacement of chloride ions by sulfate ions was observed. This observation is congruent with previous reports [
20,
22,
63]. After the addition of SPS, the sulfate surface coverage was maintained at the minimum level. This can be determined by the possible impurities adsorbed on the copper surface during a sample’s transfer to the TOF-SIMS instrument. We can assume that sulfate ions are not present on the surface when PEG, Cl, and SPS are adsorbed. The chloride impurities observed for the base and base/PEG solutions were very low and did not demonstrate significant variation as a function of the applied overpotential. After the addition of SPS up to 2 ppm, the Cu
2Cl
3− intensity increased, while for the samples with higher concentrations of SPS (5, 10, and 15 ppm), it decreased. This shows that at lower SPS concentrations (up to 2 ppm), chloride ions were co-adsorbed with thiolate ions, while for concentrations of SPS of 5, 10, and 15 ppm, the chloride ions were partly replaced by thiolate ions. The proportion of thiolate/chloride ions is shown in
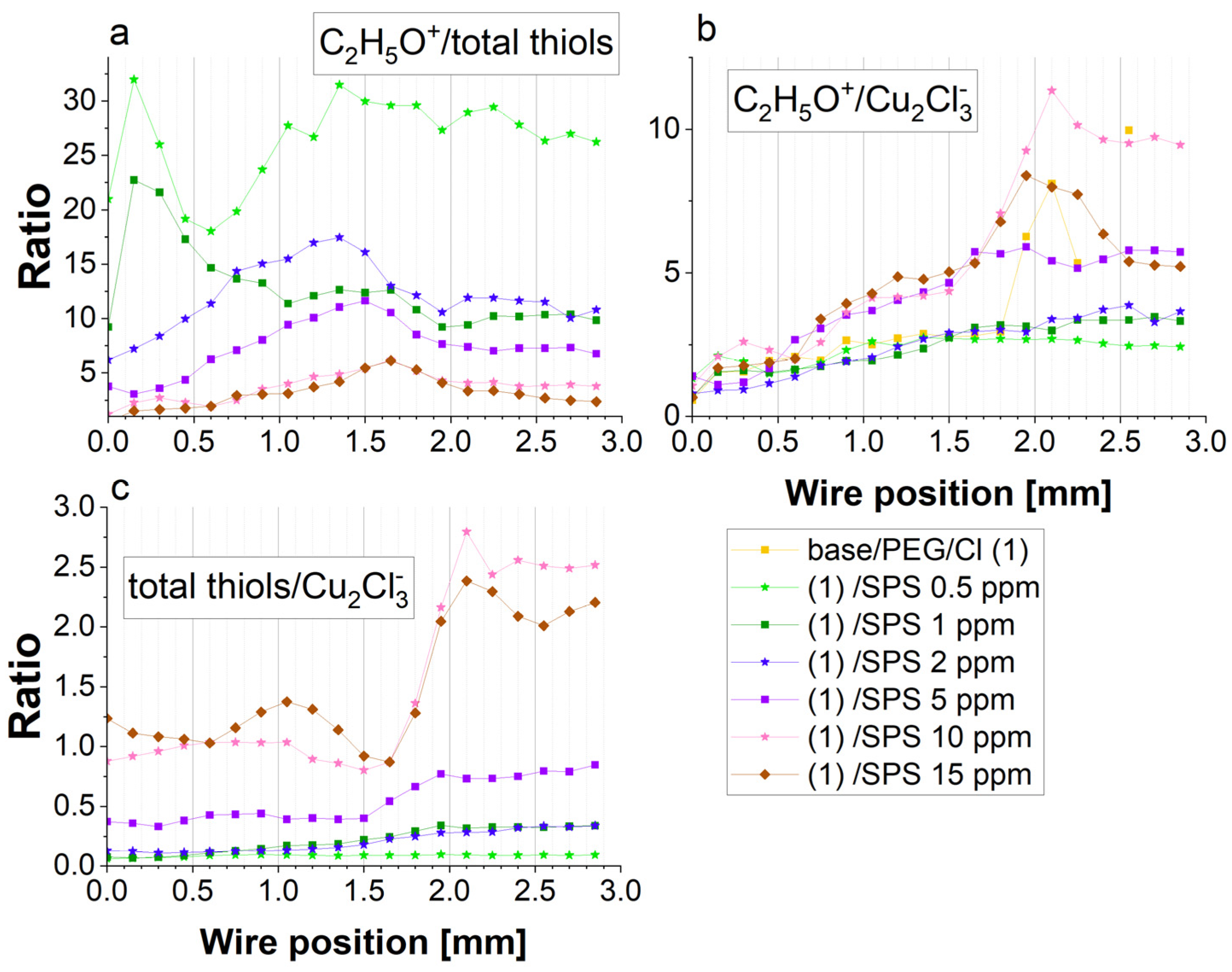
Figure 7. Indeed, the thiolate/chloride ions greatly increased for 5, 10, and 15 ppm of SPS. Accordingly, for 10 and 15 ppm SPS, this ratio was roughly similar, with some visible deviations for 15 ppm, especially for the lower overpotentials for the forward and reverse scans, when the thiolate surface coverage dominated over the chloride surface coverage.
The distribution of characteristic thiolate ions is shown in
Figure 6. More general discussion about the mechanism of yielding both Cu
2Cl
3− and thiolate fragments (CH
2SO
3−, C
3H
5SO
3−, and CuSC
3H
6SO
3−) can be found elsewhere [
22].
For the sample PEG/Cl/SPS0.5ppm, we can observe the constant intensity of CH
2SO
3 for the positions from 0.0 to 0.5 mm and then the subsequently slight increases for the lower overpotential (0.5–1.0 mm). For the reverse scan, the intensity was constant. Roughly similar behavior can be observed for C
3H
5SO
3−. In turn, the CH
2SO
3− intensity increased linearly for SPS 1 ppm. While for SPS 2 ppm, for the positions 0.0–0.5 mm, it was unchanged, and for the rest of the overpotentials (forward and reverse scan), it increased linearly, demonstrating similar values to that of SPS 1 ppm. A significantly higher thiolate surface coverage was observed at 5, 10, and 15 ppm of SPS. The intensity of the fragment C
3H
5SO
3− for 0.5, 1, 2, and 5 ppm of SPS in comparison to CH
2SO
3− slightly increased in the range of 0.65 to 2.0 mm (
Figure 6), since the ratio CH
2SO
3−/C
3H
5SO
3− slightly decreased. Recently [
63], we showed that the ratio of CH
2SO
3−/C
3H
5SO
3− can be assigned to the gauche/trans conformation of MPS molecules adsorbed on the copper surface. On the other hand, at 10 and 15 ppm SPS, we could observe a great variation in the ratio of CH
2SO
3−/C
3H
5SO
3−. For 10 ppm, in the range of 0.64 to 1.5 mm, the CH
2SO
3−/C
3H
5SO
3− ratio strongly increased, while at 15 ppm of SPS, it strongly decreased. This previously suggests [
37,
63] that the gauche conformation of thiolate molecules is favorable to the partial dehydration of Cu
2+ ions and after the reduction transfer of Cu
+ ion to the chloride adlayer. In terms of functionality, we can expect significantly higher accelerating abilities for 10 ppm than for 15 ppm for the reverse scan. This is consistent with the CV curves for 10 and 15 ppm of SPS (see
Figure 1).
The scale of acceleration should be estimated based on the total thiolate surface coverage (
Figure 6), the gauche/trans ratio (
Figure 6), the thiolate/chloride ratio (
Figure 7c), and the ratio of PEG/thiolate expressed as C
2H
5O
−/thiolate (
Figure 7a) as well as PEG/chloride expressed by C
2H
5O
−/Cu
2Cl
3− (
Figure 7b). As it is depicted in
Figure 6, the thiolate surface coverage, calculated as the sum of the intensity of CH
2SO
3−, C
2H
3SO
3−, C
3H
5SO
3−, and CuSC
3H
6SO
3−, was slightly higher at 15 ppm than at 10 ppm of SPS. However, as was shown above, significantly higher thiolate molecules remained in the trans conformation for 15 ppm of SPS. Even though the total number of MPS molecules in the gauche conformation was still slightly greater at 10 ppm than for 15 ppm for the forward scan, a greater contribution of the trans conformation (C
3H
5SO
3−) may decrease the accelerating effect of thiolate in the gauche conformation for PEG/Cl/SPS15ppm.
For 5 ppm SPS, the thiolate total surface coverage was moderate, while for 0.5, 1, and 2 ppm, it was significantly lower. An interesting situation was observed at 1 and 2 ppm, since we observed a very similar distribution of thiolate surface coverage and a slightly lower CH2SO3−/C3H5SO3− ratio for 2 ppm SPS. Since the CV curves showed that for 2 ppm, the accelerating effect was stronger than for 1 ppm, the results based on the negative ions cannot supply us with the relevant answer for why that effect was not significantly observed in the distribution of the negative ions. Additional insight can be received when we compare the distribution of the thiolate surface coverage with the PEG surface coverage.
Figure 7a depicts the distribution of the C
2H
5O
+/thiolate ratio. For 1 ppm SPS, the C
2H
5O
+/thiolate ratio decreased for the forward scan, while for 2 ppm, the C
2H
5O
+/thiolate ratio increased. The crossing point was observed at 0.75 mm, while for low overpotentials for the forward scan for 2 ppm SPS, the relative PEG surface coverage in comparison to thiolate was slightly greater than for 1 ppm. The latter fact suggests that the suppressing abilities can slightly dominate for 2 ppm SPS over 1 ppm SPS for the reverse scan, which is in contradiction with the CV curve (
Figure 1). It seems that the resultant electrochemical effect was determined not only by the ratio of C
2H
5O
+/total thiols through the effective capturing of Cu
2+. More precisely, the latter effect could be related to the interactions with hydrated Cu
2+ ions, as it was supposed by Liu et al. [
20]. It was reported [
37] that sulfonate ions in the gauche conformation can efficiently increase the velocity of the partial dehydration of Cu
2+. On the contrary, it is likely that PEG molecules can interact with hydrated Cu
2+ by two ethereal oxygen atoms due to the appropriate fit of the O-O distance (2.9 Å for a dihedral angle -O-C-C-O- at 60 degrees; see
Table S1 and Figure S7, Supplementary Materials) in PEG and the O-O distance in hydrated water of Cu
2+. After capturing the Cu
2+ by MPS units by ion–pair interaction, the Cu
2+ ion was not available for PEG and vice versa. For the examination of the Cu
2+ ions capturing scale by PEG molecules, the C
2H
4O
2Cu
+/C
2H
5O
+ ratio can be used (
Figure 3). It was clearly seen that for 2 ppm of SPS, the C
2H
4O
2Cu
+/C
2H
5O
+ ratio strongly decreased in the range of 0.0–0.75 mm and demonstrated a significantly lower value than for 1 ppm SPS (
Figure 5c) for the region 0.6–2.0 mm and under OCP after the CV experiment (positions: 2.0–3.0 mm). The latter fact can explain the stronger accelerating abilities for 2 ppm SPS, since the accelerating role of thiolate contribution was not compensated for by the suppressing abilities of PEG. Thus, the resultant electrochemical abilities were determined by the influence of the suppressing properties of PEG based on the interaction of ethereal oxygen atoms of PEG with hydrated Cu
2+ and the accelerating abilities of the MPS that was connected with the gauche conformation of MPS, favored for trapping Cu
2+. The latter hypothesis was proposed by spectroscopic studies on the electrochemistry of PEG/Cl [
20], while the role of the gauche conformation of thiolate molecules was examined by SHINERS elsewhere [
37]. On the other hand, similar spectroscopic studies devoted to the electrochemical properties of PEG/SPS/Cl were not provided. Here, we have to point out that the TOF-SIMS data do not supply information regarding possible changes in the “structural water” observed at the interface of Cu/Cl/PEG, which were considered and examined by Liu et al. [
20] and Schmitt et al. [
37].
An analogous interpretation can be provided for the samples of PEG/Cl/SPS10ppm and PEG/Cl/SPS15ppm. For the forward scan, the CV curves were very similar for 10 and 15 ppm of SPS, while for the reverse scan, a significant increase in the accelerating abilities was observed. For 10 ppm SPS, at the region from 0.0 to 0.6 mm, the contribution of the gauche conformation around C-C (CH2Cl+/C2H5O+) and the C2H4O2Cu+/C2H5O+ ratio increased, while for the low overpotentials, less than −0.25 V, it significantly decreased, which was accompanied by a significant increase in the PEG surface coverage for that region. For the reverse scan, the contribution of the gauche conformations about C-C and the C2H4O2Cu+/C2H5O+ ratio was rather constant. Different behavior was observed at 15 ppm SPS, when a significant decrease in the gauche conformation was observed for the higher overpotentials (positions: 0.0–0.75 mm) and, subsequently, remained rather constant for the rest of the CV curve. However, for the reverse scan, the contribution of the C2H4O2Cu+/C2H5O+ ratio for 15 ppm SPS was higher than at 10 ppm of SPS, while the gauche conformation about C-C was slightly more dominant at 10 ppm SPS than for 15 ppm SPS.
Moreover, for these samples (10 and 15 ppm SPS), both the total PEG surface coverage (C
2H
5O
+) as well as the ratio of C
2H
5O
+/thiolate was very similar. However, we can observe a significant increase in the ratio of C
3H
6OCu
+/C
2H
5O
+. The fragment of C
3H
6OCu
+ corresponded to a stable complex, while for the forward scan, we did not see any differences between 10 and 15 ppm SPS. Due to the fact of this reason, the conformation that was favorable for a higher yield of C
3H
6OCu did not have a major impact on the potentially greater suppressing properties of PEG. Careful inspection of the CH
2SO
3− fragment clearly shows that the switching of the CV scan from forward to reverse determined the changes in the gauche conformation for 15 ppm SPS, while this effect was not observed for 10 ppm. For the forward scan, the gauche conformation increased, while for the reverse scan, it diminished. As a consequence, the ratio of the gauche/trans of the thiolate molecules strongly dominated for the reverse scan for 10 ppm over 15 ppm SPS (
Figure 6), determining the higher accelerating abilities.
Interestingly, the variation in the CH2SO3−/C3H5SO3− ratio at 15 ppm SPS correlated quite well with the surface coverage of PEG. The reduction of the PEG surface coverage observed around position 0.5 mm (overpotential: −0.3 V) and the subsequent increase determined the significant rotation of the PEG skeleton around the C-C and C-O bonds. Moreover, the strong correlation of CH2Cl+ with both Cu2Cl and Cu2Cl3− for the entire range of wire strongly suggests the hydrophobic interaction of the CH2-CH2 group of PEG with the Cl adlayer, while the rotations around C-O were relatively independent of the Cl adlayer. Interestingly, for very high overpotentials for the reverse scan, the amounts of Cu-PEG were still maintained, while the hydrophobic interaction diminished due to the reduction of the Cl surface coverage. This means that under a high thiolate surface coverage (15 ppm), the PEG can remain on the surface even if the hydrophobic interaction of the CH2CH2 groups was significantly reduced due to the decreasing availability of Cl sites on the copper. This suggests that ethereal atoms of PEG can efficiently interact with the copper surface when PEG molecules are stabilized by the gauche conformation of thiolate molecules under a desorption regime.
However, the primary reason for the conformal reorientation of both PEG and thiolate was the higher accumulation of PEG and thiolate monolayers on the copper surface. As a consequence, we could observe higher numbers of Cu-PEG adducts at 15 ppm SPS than for 10 ppm SPS, while CH2Cl slightly diminished. Moreover, significantly higher thiolate surface coverage in the trans conformation was responsible for reducing the accelerating abilities.
The CuS
− fragment corresponded to the thiolate ends of the MPS molecules after dissociative adsorption of SPS on the copper surface. A careful inspection of
Figure 4 shows that Cu-S positively correlated with the CH
2SO
3− fragment, while C
3H
5SO
3− demonstrated a slightly different distribution. On the other hand, the distribution of CuSC
3H
6SO
3− was more positively correlated with the C
3H
5SO
3− fragment than with CH
2SO
3−. These differences can be determined by the fact that thiolate molecules in the gauche conformation (CH
2SO
3−) can be positioned more perpendicular to the Bi primary during the TOF-SIMS measurements, and in this way, the yield of CuSC
3H
6SO
3− is reduced for this conformation due to the higher fragmentation of molecule. On the contrary, under the trans conformation, MPS molecules are tilted more parallel to the primary Bi beam, preserving the MPS molecules bonded to the copper surface against higher fragmentation. As a consequence, a higher yield of CuSC
3H
6SO
3− was observed. At this point, it should be expressed that the primary Bi beam strikes the sample at an angle of 45°.
The assignment of the CH
2SO
3− to the gauche and C
3H
5SO
3− to the trans conformation was recently reported [
63].
For the sample PEG/Cl/SPS2ppm, we can observe a reduced ratio of CH2SO3−/C3H5SO3− that corresponded to the gauche/trans ratio. The decreased value of the gauche conformer (CH2SO3−) was accompanied by a reduced intensity of Cu-S.
This hypothesis is supported by the intensity distribution of the CuSO− fragment. The latter fragment can be assigned to the sulfonate ends and is formed by the reaction of the SO3 group with sputtered copper during Bi bombardment in the TOF-SIMS instrument. The distribution of CuS− demonstrated generally good agreement with CuS− while exhibiting more distinct differences between samples. However, some inconsistency was observed for 5 and 15 ppm SPS, when a very similar distribution of CuSO− was observed, while for CuS− it was significantly different.
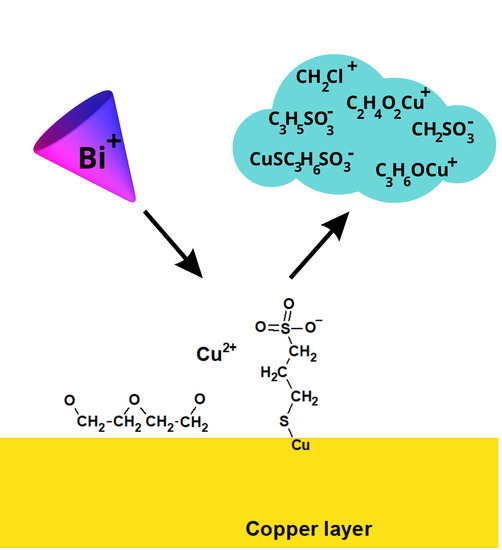
Overall, the interaction between PEG and SPS on the copper surface under reactive conditions (copper electrodeposition) is illustrated in
Figure 8. The chloride adlayer was omitted for clarity. SPS molecules after dissociative adsorption on the copper surfaces reduce into two MPS units, as postulated by Hai et al. and proven by other investigations [
22,
23,
28,
66]. However, the model postulated by Hai et al. does not take into account the incorporation of thiolate molecules into the copper deposit. The latter fact was proved recently [
63] and suggested previously [
50,
67]. On the contrary, PEG molecules are not able to incorporate into a copper deposit, as suggested previously [
22,
50]. PEG molecules are bonded to the chloride adlayer through the hydrophobic -CH
2CH
2- part of PEG. This fact was suggested previously for PEG/Cl [
20,
21,
22], and it was proven in that work for the PEG/SPS/Cl component of an electroplating bath. Contrary to previous studies, PEG was co-adsorbed with SPS under our experimental conditions in an SPS concentration range from 0.5 to 15 ppm. However, the ratio of MPS/PEG increased as a function of the SPS concentration. The first function of MPS in decreasing the PEG-suppressing role relies on the limited access of hydrophobic parts to the chloride adlayer due to the partial displacement of chloride ions by MPS molecules. In this way, ethereal oxygen atoms are pointed out toward the solution at a greater distance from the electrode surface. This latter fact minimized their effectiveness in capturing Cu
2+. On the other hand, MPS molecules are very efficient at capturing Cu
2+, especially in gauche conformations, as is schematically shown in
Figure 8. The formation of Cu-MPS bonds can be found in our previous paper (Figure 11) [
63]. The main idea depicted in
Figure 8 is the demonstration that PEG exists only on the copper surface, while MPS molecules can also be incorporated into the copper deposit and exhibit competition in catching Cu
2+ ions.
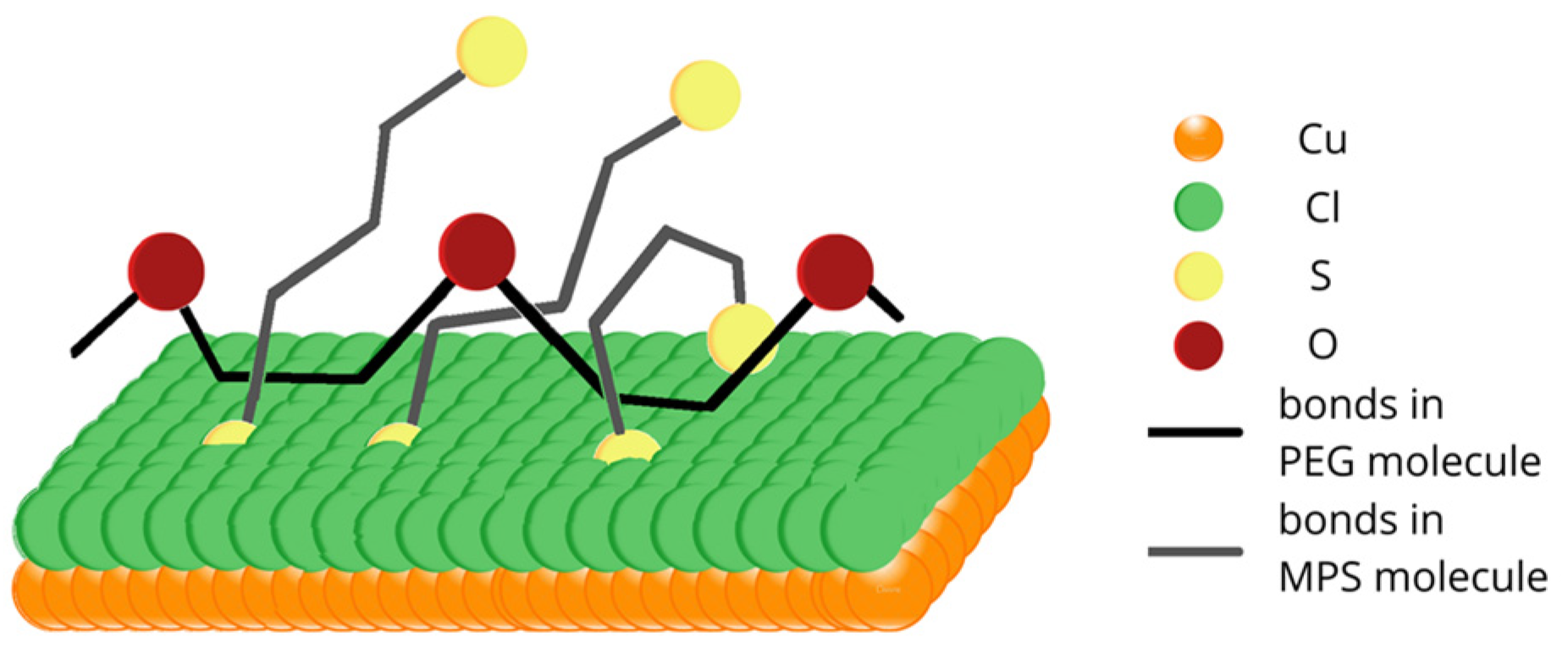
One of the possible molecular arrangements of MPS thiolate molecules and PEG in 3D projection is shown in
Figure 9. All hydrogen atoms and oxygen atoms located around sulfur pointed towards the top were omitted for clarity. MPS molecules (sulfur atoms indicated by the yellow color and bonds by grey) penetrate and disrupt the chloride adlayer by forming chemical bonds with the copper substrate. On the contrary, PEG molecules (oxygen atoms in PEG are indicated by the brown color and bonds by black) can interact with the chloride adlayer through hydrophobic interactions. Due to the high mobility of MPS and the high length of PEG 8000, MPS and PEG molecules can be wrapped around each other. In this model, sulfonate groups efficiently capture Cu
2+ ions and, in this way, reduce similar abilities for the ethereal oxygen of PEG. The proposed mechanism is in contradiction to the model postulated by Hai et al. [
23], where the primary antisuppressing effect of MPS relies on the partial rupturing of coordination bonds of Cu(+)-Cl- in the PEG(Cu
+)Cl
− complexes by displacement of Cl ions from the copper surface. The second antisuppressing effect of MPS molecules is connected with the coordinative dissolution of the PEG-(Cu+)Cl- complex by desorbed MPS units. However, the existence of PEG(Cu+)Cl- complexes that play a central role in the suppressing abilities of PEG was undermined recently [
20].












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}