
Flavonoid Metabolism in Tetrastigma hemsleyanum Diels et Gilg Based on Metabolome Analysis and Transcriptome Sequencing

Abstract
:1. Introduction
2. Results
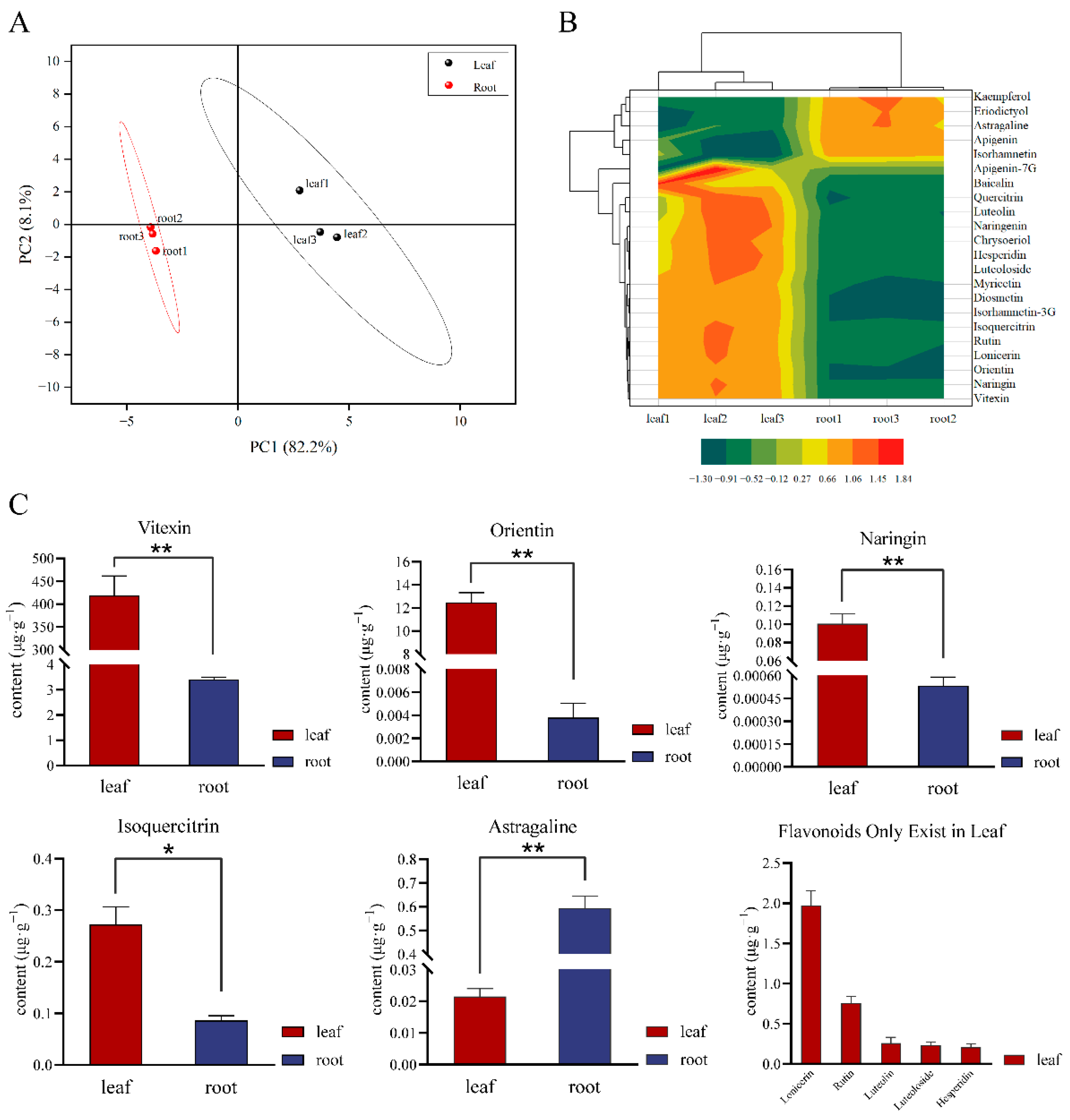
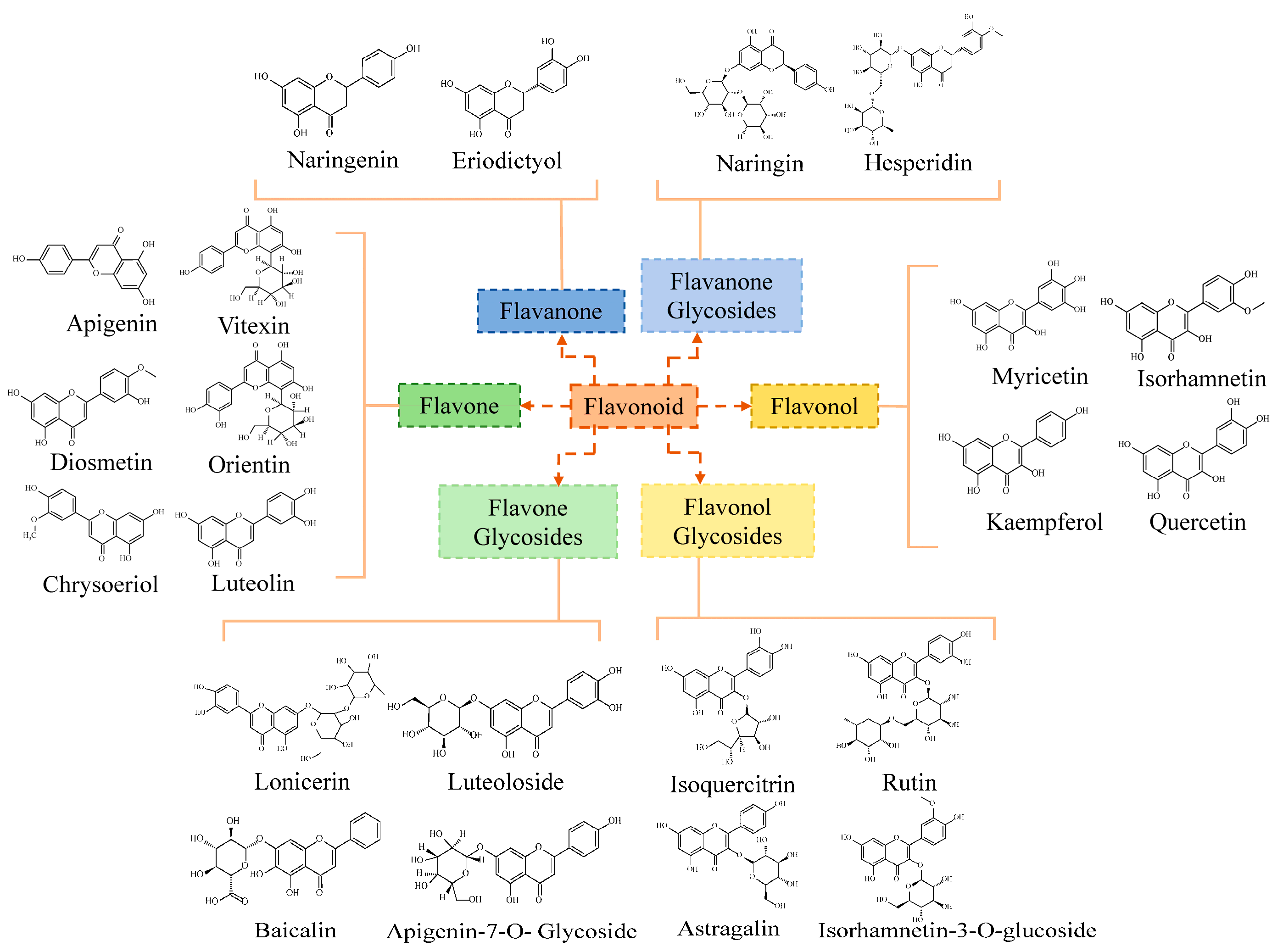
2.1. Flavonoid Components Differ in Leaves and Root Tubers of T. hemsleyanum
2.2. Transcriptome Sequencing of Leaves and Root Tubers of T. hemsleyanum
2.3. Differentially Expressed Genes of Leaves and Root Tubers of T. hemsleyanum
2.4. Analysis Correlation between Flavonoid Biosynthetic Gene Expression and Flavonoid Concentration
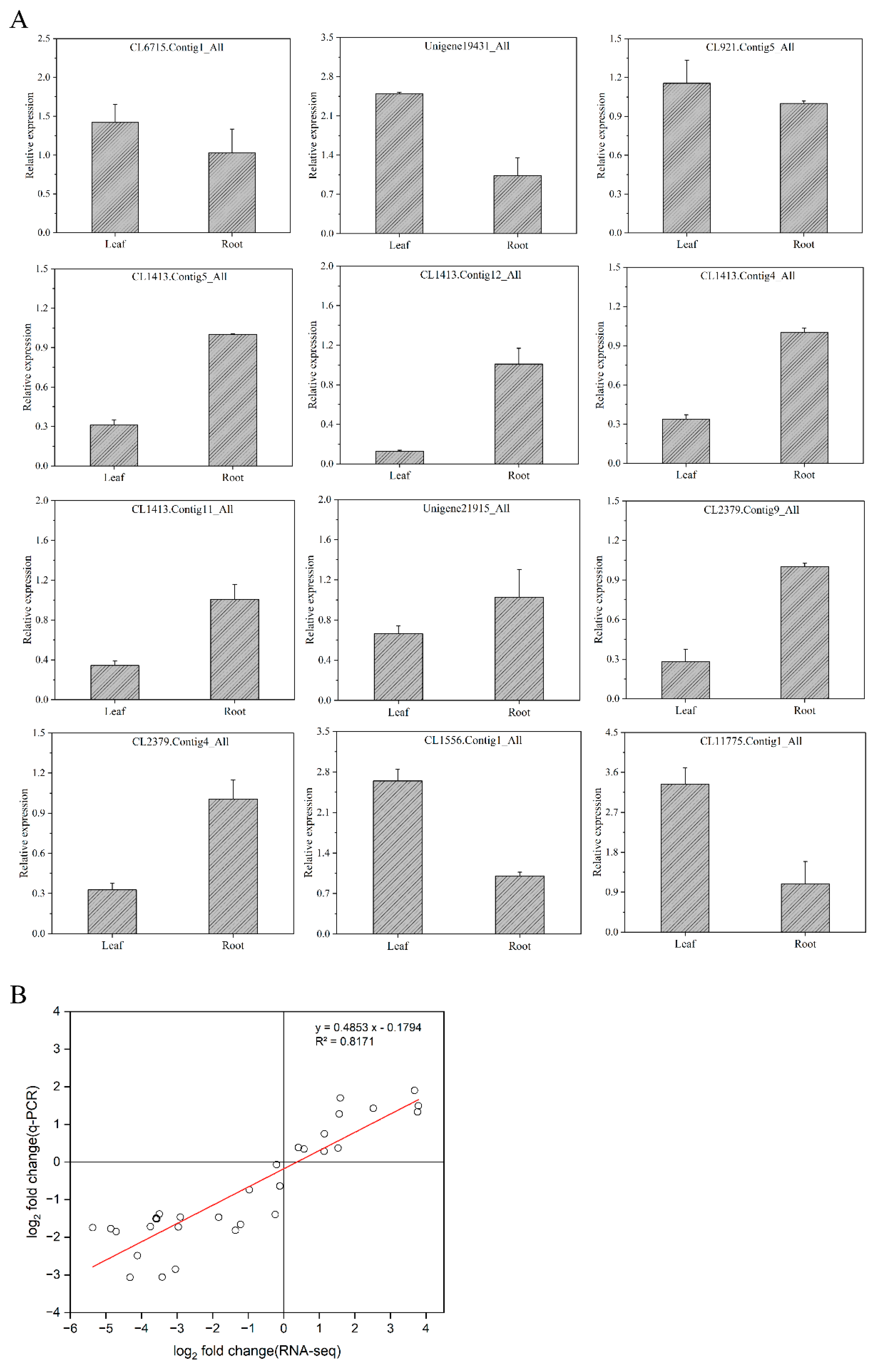
2.5. Validation of the DEGs
2.6. Key Gene Expression and Total Flavonoid Content Trends for T. hemsleyanum from Different Locations
3. Discussion
3.1. Key Role of DEGs in Photosynthesis-Antenna Proteins in Flavonoid Metabolism in Leaves and Root Tubers
3.2. CHI and UFGT Are Critical Genes for Flavonoid Biosynthesis in T. hemsleyanum
4. Materials and Methods
4.1. Plant Materials
4.2. Determination of Flavonoid Metabolomes
4.3. RNA Extraction and Transcriptome Sequencing
4.4. De Novo Assembly and Sequence Clustering
4.5. FPKM Calculation and DEGs Screening
4.6. Functional Annotation and Classification of Genes
4.7. Quantitative Real-Time PCR
4.8. Total Flavonoid Determination
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| T. hemsleyanum | Tetrastigma hemsleyanum Diels et Gilg |
| PAL | Phenylalanine ammonia lyase |
| C4H | Cinnamate 4-hydroxylase |
| 4CL | 4-coumarate-coenzyme A |
| CHI | Chalcone isomerase |
| CHS | Chalcone synthase |
| F3H | Flavanone-3-hydroxylase |
| F3′H | Flavanone-3′-hydroxylase |
| F3′5′H | Flavonoid-3′,5′-hydroxylase |
| FLS | Flavonol synthase |
| FNS | flavone synthase |
| DFR | Dihydroflavonol 4-reductase |
| LDOX | Leucoanthocyanidin dioxygenase |
| 3-Glct | Flavonoid-3-O-glucosyltransferase |
| 7-Glct | Flavonoid-3-O-glucosyltransferase |
| 1,2-Rhat | 1,2-rhamnosyltransferase |
| 1,6-Rhat | 1,6-rhamnosyltransferase |
| UGTs | UDP-Glycosyltransferases |
| OMT | O-methyltransferase |
| UFGT | UDP-glycose flavonoid glycosyltransferase |
| MAPK | Mitogen-activated protein kinase. |
| KOG | Clusters of orthologous groups for eukaryotic complete genomes |
| NR | Non-Redundant Protein Sequence Database |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Gong, W.; Liu, T.; Zhou, Z.; Wu, D.; Shu, X.; Xiong, H.; Gong, W.; Liu, T.; Zhou, Z.; Wu, D.; et al. Physicochemical characterizations of starches isolated from Tetrastigma hemsleyanum Diels et Gilg. Int. J. Biol. Macromol. 2021, 183, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Ji, W.W.; Wang, J.; Chen, H.J.; Peng, X.; Cheng, K.J.; Qiu, D.; Yang, W.J. A comprehensive review on traditional uses, chemical compositions, pharmacology properties and toxicology of Tetrastigma hemsleyanum. J. Ethnopharmacol. 2021, 264, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Liu, Y.X.; Fanady, B.; Han, Y.F.; Xie, L.; Chen, Z.Y.; Yu, K.B.; Peng, X.; Zhang, X.L.; Ge, Z.Y. Ultra-flexible light-permeable organic solar cells for the herbal photosynthetic growth. Nano Energy 2021, 86, 1–11. [Google Scholar] [CrossRef]
- Xiang, Q.; Hu, S.; Ligaba-Osena, A.; Yang, J.; Tong, F.; Guo, W. Seasonal variation in transcriptomic profiling of Tetrastigma hemsleyanum fully developed tuberous roots enriches candidate genes in essential metabolic pathways and phytohormone signaling. Front. Plant Sci. 2021, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.L.; Xu, Z.J.; Cao, Y.F.; Wang, F.; Chu, C.; Zhang, C.; Tao, Y.; Wang, P. HPLC fingerprinting-based multivariate analysis of chemical components in Tetrastigma Hemsleyanum Diels et Gilg: Correlation to their antioxidant and neuraminidase inhibition activities. J. Pharm. Biomed. Anal. 2021, 205, 1–11. [Google Scholar] [CrossRef]
- Han, M.Q.; Xu, L.Y.; Bai, Y.; Hu, X.T.; Wu, X.Q.; Zheng, B.S. Effect of light quality on photosynthetic characteristics and total flavonoid content in Tetrastigma hemsleyanum. J. Plant Physiol. 2019, 55, 883–890. [Google Scholar]
- Li, Z.; Zhou, C.H.; Du, X.B.; Liu, S.Z.; Bai, Y.; Jiang, L.T.; Wang, J.Y.; Pan, J.Y.; Lou, Y.Y.; Zheng, B.S. Current situation and countermeasures on research and development of Tetrastigma hemsleyanum. J. Zhejiang Sci. Tech. 2020, 40, 87–92. [Google Scholar] [CrossRef]
- Williams, J.S.; Thomas, M.; Clarke, D.J. The gene stlA encodes a phenylalanine ammonia-lyase that is involved in the production of a stilbene antibiotic in Photorhabdus luminescens TT01. Microbiology 2005, 151, 2543–2550. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.X.; Zhang, X.; Han, X.J.; Zhang, Y.Y.; Gao, S.; Liu, C.J.; Lou, H.X. Identification of chalcone isomerase in the basal land plants reveals an ancient evolution of enzymatic cyclization activity for synthesis of flavonoids. New Phytol. 2018, 217, 909–924. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The Flavonoid Biosynthesis Network in Plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef]
- Sun, W.; Shen, H.; Xu, H.; Tang, X.; Tang, M.; Ju, Z.; Yi, Y. Chalcone Isomerase a Key Enzyme for Anthocyanin Biosynthesis in Ophiorrhiza japonica. Front. Plant Sci. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.K.; Alerding, A.B.; Crosby, K.C.; Bandara, A.B.; Westwood, J.H.; Winkel, B.S. Functional analysis of a predicted flavonol synthase gene family in Arabidopsis. Plant Physiol. 2008, 147, 1046–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Li, C.; Li, H.; Lu, S. Identification and characterization of flavonoid biosynthetic enzyme genes in Salvia miltiorrhiza (Lamiaceae). Molecules 2018, 23, 1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.C.; Hu, G.B.; Hu, F.C.; Wang, H.C.; Yang, Z.Y.; Lai, B. The UDP glucose: Flavonoid-3-O-glucosyltransferase (UFGT) gene regulates anthocyanin biosynthesis in litchi (Litchi chinesis Sonn.) during fruit coloration. Mol. Biol. Rep. 2012, 39, 6409–6415. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, H.; Zhao, J.; Yang, Y.; Yang, B.; Feng, L.; Zhang, Y.; Wei, P.; Hou, D.; Zhao, J.; et al. Transcriptome and metabolome analysis to reveal major genes of saikosaponin biosynthesis in Bupleurum chinense. BMC Genom. 2021, 22, 1–14. [Google Scholar] [CrossRef]
- Bar-Peled, M.; Fluhr, R.; Gressel, J. Juvenile-specific localization and accumulation of a Rhamnosyltransferase and its bitter flavonoid in foliage, flowers, and young Citrus fruits. Plant Physiol. 1993, 103, 1377–1384. [Google Scholar] [CrossRef] [Green Version]
- Gou, J.Y.; Felippes, F.F.; Liu, C.J.; Weigel, D.; Wang, J.W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef] [Green Version]
- Koja, E.; Ohata, S.; Maruyama, Y.; Suzuki, H.; Shimosaka, M.; Taguchi, G. Identification and characterization of a rhamnosyltransferase involved in rutin biosynthesis in Fagopyrum esculentum (common buckwheat). Biosci. Biotechnol. Biochem. 2018, 82, 1790–1802. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Wu, H.; Chen, H.; Zhang, Y.; Qiu, D.; Zhang, Z. Transcriptome profiling reveals candidate flavonol-related genes of Tetrastigma hemsleyanum under cold stress. BMC Genom. 2019, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.S. Medicinal Plant Cultivation, 1st ed.; Higher Education Press: Beijing, China, 2009; p. 47. [Google Scholar]
- Benavente-Garcia, O.; Castillo, J.; del Rio Conesa, J.A. Changes in neodiosmin levels during the development of Citrus aurantium leaves and fruits. postulation of a neodiosmin biosynthetic pathway. J. Agric. Food Chem. 1993, 41, 1916–1919. [Google Scholar] [CrossRef]
- Flora of China Editorial Committee. Flora of China; Science Press: Beijing, China, 1998; p. 120. [Google Scholar]
- Zhejiang Food and Medical Products Administration. Zhejiang Provincial Standards of Processing Chinese Crud Drugs, 7th ed.; China Medical Science Press: Beijing, China, 2015; pp. 5–6. [Google Scholar]
- Zhang, J.J.; Zhang, Z.F.; Lv, Y.G.; Lu, M.D.; Fang, L.L. Comparison of antioxidant activities of aerial part and underground part of T. hemsleyanum from Zhejiang Province. J. Zhejiang Agric. Sci. 2021, 62, 709–711. [Google Scholar]
- Su, Q.; Sun, Z.; Liu, Y.; Lei, J.; Zhu, W.; Nanyan, L. Physiological and comparative transcriptome analysis of the response and adaptation mechanism of the photosynthetic function of mulberry (Morus alba L.) leaves to flooding stress. Plant Signal. 2022, 17, e2094619. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, Y.; Sun, S.; Li, L.; Wang, K. Expression profiles and characteristics of apple lncRNAs in roots, phloem, leaves, flowers, and fruit. Int. J. Mol. Sci 2022, 23, 5931. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, H.; Zhang, X. Integrative transcriptomic and metabolomic analysis of D-leaf of seven pineapple varieties differing in N-P-K% contents. BMC Plant Biol. 2021, 21, 1–19. [Google Scholar] [CrossRef]
- Wang, F.; Wu, Y.; Wu, W.; Huang, Y.; Zhu, C.; Zhang, R.; Chen, J.; Zeng, J. Integrative analysis of metabolome and transcriptome profiles provides insight into the fruit pericarp pigmentation disorder caused by ’Candidatus Liberibacter asiaticus’ infection. BMC Plant Biol. 2021, 21, 1–16. [Google Scholar] [CrossRef]
- Hong, Y.; Li, M.; Dai, S. iTRAQ-Based protein profiling provides insights into the mechanism of light-induced anthocyanin biosynthesis in Chrysanthemum (Chrysanthemum × morifolium). Genes 2019, 10, 1024. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Gu, Y.W.; Liu, Z.S.; Jiang, L.T.; Han, M.Q.; Geng, D.J. Flavonoids metabolism and physiological response to ultraviolet treatments in Tetrastigma hemsleyanum Diels et Gilg. Front. Plant Sci. 2022, 13, 926197. [Google Scholar] [CrossRef]
- Ma, H.; Chen, J.; Zhang, Z.; Ma, L.; Yang, Z.; Zhang, Q.; Li, X.; Xiao, J.; Wang, S. MAPK kinase 10.2 promotes disease resistance and drought tolerance by activating different MAPKs in rice. Plant J. 2017, 92, 557–570. [Google Scholar] [CrossRef] [Green Version]
- Ryu, T.K.; Roh, E.; Shin, H.S.; Kim, J.E. Inhibitory effect of lotusine on solar UV-induced matrix metalloproteinase-1 expression. Plants 2022, 11, 773. [Google Scholar] [CrossRef]
- Zhou, H.; Ren, S.; Han, Y.; Zhang, Q.; Qin, L.; Xing, Y. Identification and analysis of mitogen-activated protein kinase (MAPK) cascades in Fragaria vesca. Int J Mol Sci 2017, 18, 1766. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Zhao, H.; Sun, L.; Wu, W.; Li, C.; Wu, Q. Genome-wide identification of MAPK gene family members in Fagopyrum tataricum and their expression during development and stress responses. BMC Genom. 2022, 23, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, L.P.; Zhang, L.; Yan, P.; Ahammed, G.J.; Han, W.Y. Methyl salicylate enhances flavonoid biosynthesis in tea leaves by stimulating the Phenylpropanoid Pathway. Molecules 2019, 24, 362. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Shen, Y.; Ni, Z.; Wang, Q.; Lei, Z.; Xu, N.; Deng, Q.; Lin, L.; Wang, J.; Lv, X.; et al. Exogenous melatonin application delays senescence of kiwifruit leaves by regulating the antioxidant capacity and biosynthesis of flavonoids. Front. Plant Sci. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Isah, T. Stress and defense responses in plant secondary metabolites production. Biol. Res. 2019, 52, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, N.I.; Xu, H.; Li, X.; Kim, S.J.; Park, S.U. Enhancement of flavone levels through overexpression of chalcone isomerase in hairy root cultures of Scutellaria baicalensis. Funct. Integr. Genom. 2011, 11, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, G.; Tang, N.; Li, Z. Transcriptome analysis reveals molecular signatures of luteoloside accumulation in senescing leaves of Lonicera macranthoides. Int. J. Mol. Sci. 2018, 19, 1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Zhu, Y.; Fan, J.; Wang, D.; Gong, X.; Ouyang, Z. Accumulation of flavonoid glycosides and UFGT gene expression in Mulberry Leaves (Morus alba L.) before and after frost. Chem Biodivers 2017, 14, e1600496. [Google Scholar] [CrossRef]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, J.; Ni, S.; Xing, B.; Cheng, K.; Peng, X. Transcriptome and Metabonomics Combined Analysis Revealed the Defense Mechanism Involved in Hydrogen-Rich Water-Regulated Cold Stress Response of Tetrastigma hemsleyanum. Front. Plant Sci. 2022, 13, 1–17. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | All > 300 bp | ≥500 bp | ≥1000 bp | N50 | Total Length | Max Length | Min Length | Average Length |
|---|---|---|---|---|---|---|---|---|
| Y-Unigene | 53,039 | 29,973 | 14,690 | 1275 | 47,471,677 | 15,654 | 301 | 895.03 |
| G-Unigene | 60,953 | 33,607 | 16,023 | 1236 | 53,338,272 | 15,654 | 301 | 875.07 |
| Compound | Regression Equation | r | Linear Range |
|---|---|---|---|
| Vitexin | Y = 8,542,235.9 X + 18,454,664.6 | 0.9987 | 20.000–2000.000 |
| Orientin | Y = 9,071,337.1 X + 33,093.6 | 0.9996 | 1.000–100.000 |
| Afzelin | Y = 9,658,807.6 X + 92,259.2 | 0.9993 | 0.001–0.050 |
| Rutin | Y = 4,950,860.8 X + 231,551.2 | 0.9994 | 0.010–0.500 |
| Isoquercitrin | Y = 3,356,305.1 X + 593,847.8 | 0.9989 | 0.020–1.000 |
| Luteolin | Y = 9,109,434.3 X + 35,359.9 | 0.9998 | 0.020–1.000 |
| Luteoloside | Y = 3,168,802.6 X + 166,068.3 | 0.9994 | 0.100–5.000 |
| Isoorientin | Y = 4,845,698.3 X + 4,222,807.8 | 0.9987 | 1.000–100.000 |
| Naringin | Y = 3,972,039.5 X + 64,183.6 | 0.9976 | 0.010–0.500 |
| Isorhamnetin-3-O-Glucoside | Y = 8,988,517.2 X + 5,992,231.1 | 0.9982 | 0.020–1.000 |
| Isorhamnetin | Y = 1,476,424.2 X + 2,111,521.6 | 0.9986 | 0.001–0.050 |
| Astragaline | Y = 2,210,508.5 X + 2,507,321.4 | 0.9986 | 0.010–0.500 |
| Eriodictyol | Y = 691,932.1 X + 84659.5 | 0.9991 | 0.010–0.500 |
| Taxifolin | Y = 3,575,802.9 X + 284,658.7 | 0.9992 | 0.001–0.050 |
| Isovitexin | Y = 8,542,235.9 X + 18,454,664.6 | 0.9987 | 20.000–2000.000 |
| Calycosin-7-O-glucoside | Y = 22,042.7 X + 2463.8 | 0.9986 | 0.020–1.000 |
| Hispidulin | Y = 207,676.0 X + 105,248.9 | 0.9993 | 0.020–1.000 |
| Naringenin | Y = 12,452,147.9 X +74,631.9 | 0.9992 | 0.001–0.050 |
| Apigenin | Y = 2,885,631.5 X + 238,287.7 | 0.9989 | 0.001–0.050 |
| Quercitrin | Y = 12,206,204.2 X + 1,425,428.8 | 0.9992 | 0.001–0.050 |
| Apigenin-7-O-Glycoside | Y = 1,806,515.0 X + 16,658.0 | 0.9996 | 0.010–0.500 |
| Gene | Gene ID | Tm (°C) | Primer F (5′-3′) | Primer R (5′-3′) |
|---|---|---|---|---|
| MDH | \ | 60 | TGTTGCTACGACTGATGT | CCTGAGACTTGTAGATGGAA |
| PAL | CL2379.Contig9_All | 56.02 | ACCAACCATGTCCAAAGTGC | CGCCACAAGGTATGTGGAAG |
| CL2379.Contig4_All | 56.06 | TGGTGACCTCACCTTCTCAC | GAAGCCCATCCATTCCGAAC | |
| CHS | CL1413.Contig5_All | 55.9 | GGAACTGTCCTTCGAACTGC | GTCCGCGGAATGTAACAACA |
| CL1413.Contig12_All | 55.99 | TAGCACGTTGAGCGTTTCTG | AGAGTTGGTGGCATAAGGCT | |
| CL1413.Contig4_All | 55.71 | CCAACTTGTCTCAGCAGCTC | AATCAAAGTGGGCACAGTGG | |
| CL1413.Contig11_All | 55.89 | TGTACCATCAAGGGTGCCAT | GGGCTTGGCCAACTAAAGAG | |
| CHI | CL6715.Contig1_All | 56.01 | GTGCAGGGTGTGAAGTTTGT | TTTGGCTTCCTTCCAACAGC |
| Unigene19431_All | 56.47 | ACGCCATGGATAGAGAGCAA | CCCATGGTTGAGGATTCGGA | |
| CL921.Contig4_All | 55.96 | ACTGAACACCATCCACGACT | GACCAACGAATGCCTCGAAA | |
| FNS | Unigene21915_All | 55.99 | AAAGACGAACTCTCCACCGT | GATGTTCGCAACTCCGTTCA |
| UFGT | CL11556.Contig3_All | 56.05 | TTGACTTGCCTGAGTGTCCT | AACTCGGATGCTGAGTTGGA |
| CL11775.Contig1_All | 55.90 | CAACGGCGGAATGAGCTAAA | TCTTGTGGTCCCTTCGTCAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Y.; Jiang, L.; Li, Z.; Liu, S.; Hu, X.; Gao, F. Flavonoid Metabolism in Tetrastigma hemsleyanum Diels et Gilg Based on Metabolome Analysis and Transcriptome Sequencing. Molecules 2023, 28, 83. https://doi.org/10.3390/molecules28010083
Bai Y, Jiang L, Li Z, Liu S, Hu X, Gao F. Flavonoid Metabolism in Tetrastigma hemsleyanum Diels et Gilg Based on Metabolome Analysis and Transcriptome Sequencing. Molecules. 2023; 28(1):83. https://doi.org/10.3390/molecules28010083
Chicago/Turabian StyleBai, Yan, Lingtai Jiang, Zhe Li, Shouzan Liu, Xiaotian Hu, and Fei Gao. 2023. "Flavonoid Metabolism in Tetrastigma hemsleyanum Diels et Gilg Based on Metabolome Analysis and Transcriptome Sequencing" Molecules 28, no. 1: 83. https://doi.org/10.3390/molecules28010083
APA StyleBai, Y., Jiang, L., Li, Z., Liu, S., Hu, X., & Gao, F. (2023). Flavonoid Metabolism in Tetrastigma hemsleyanum Diels et Gilg Based on Metabolome Analysis and Transcriptome Sequencing. Molecules, 28(1), 83. https://doi.org/10.3390/molecules28010083