
Expanded Ligands Based upon Iron(II) Coordination Compounds of Asymmetrical Bis(terpyridine) Domains




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
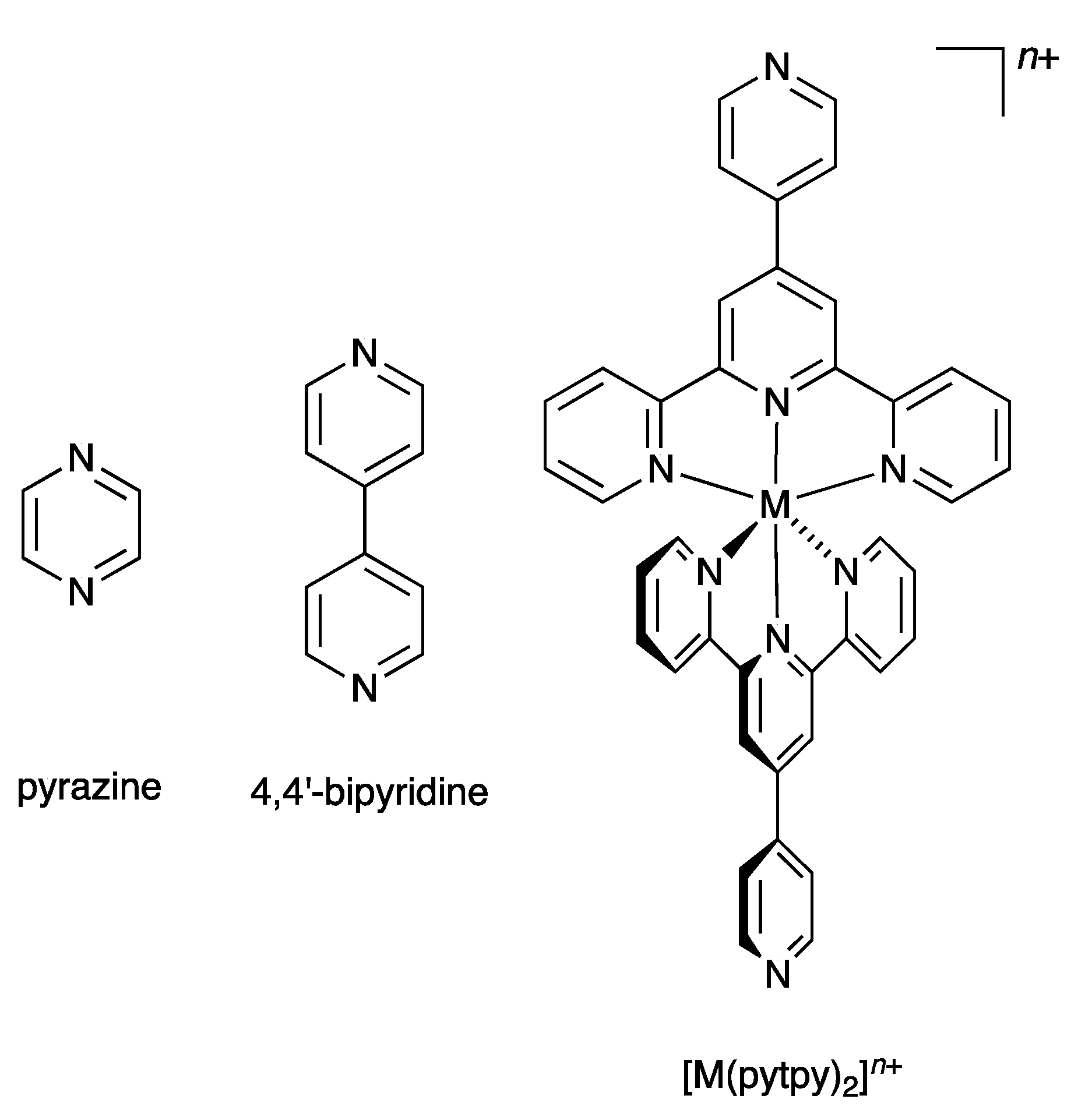
1. Introduction
2. Results and Discussion
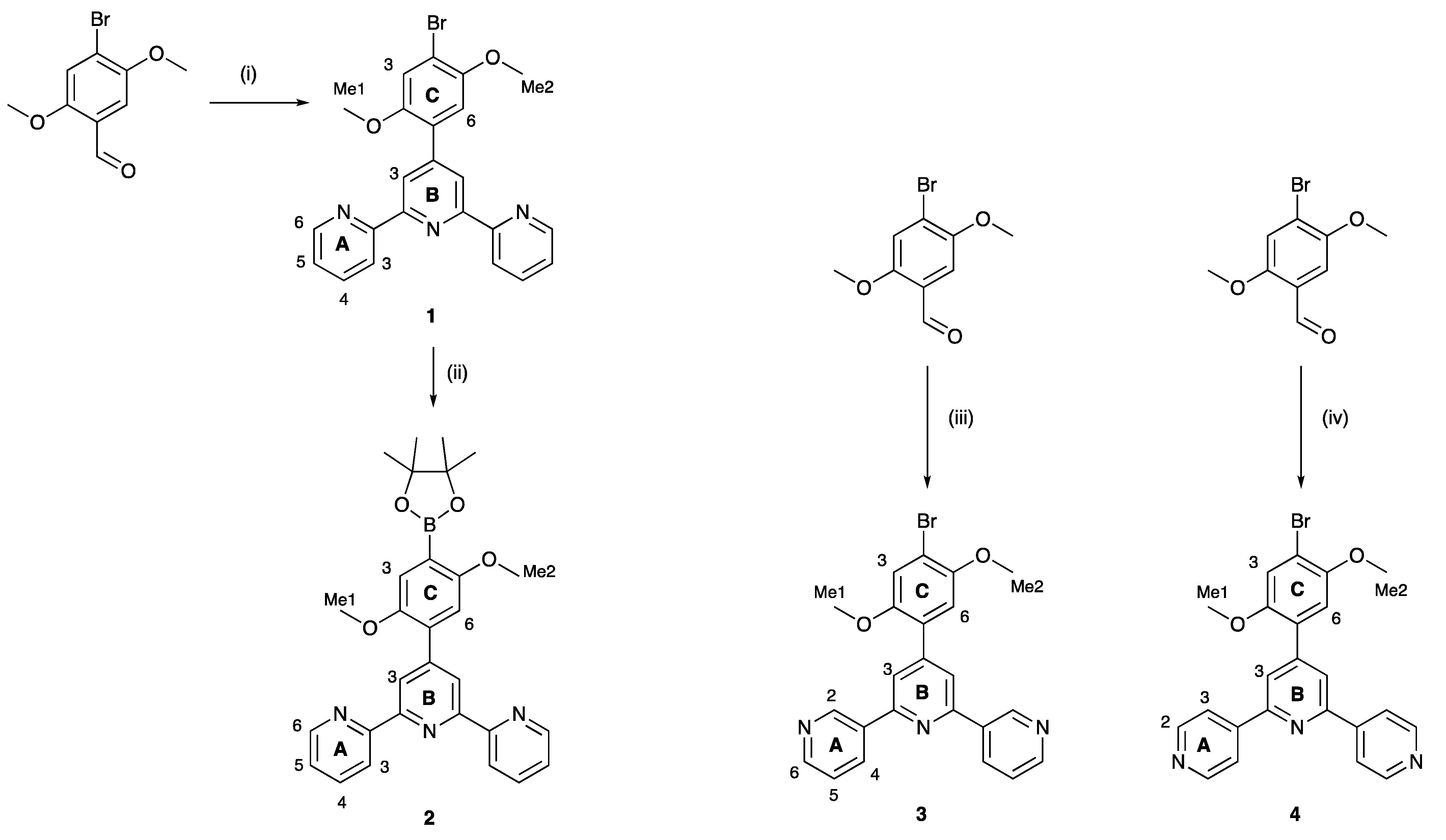
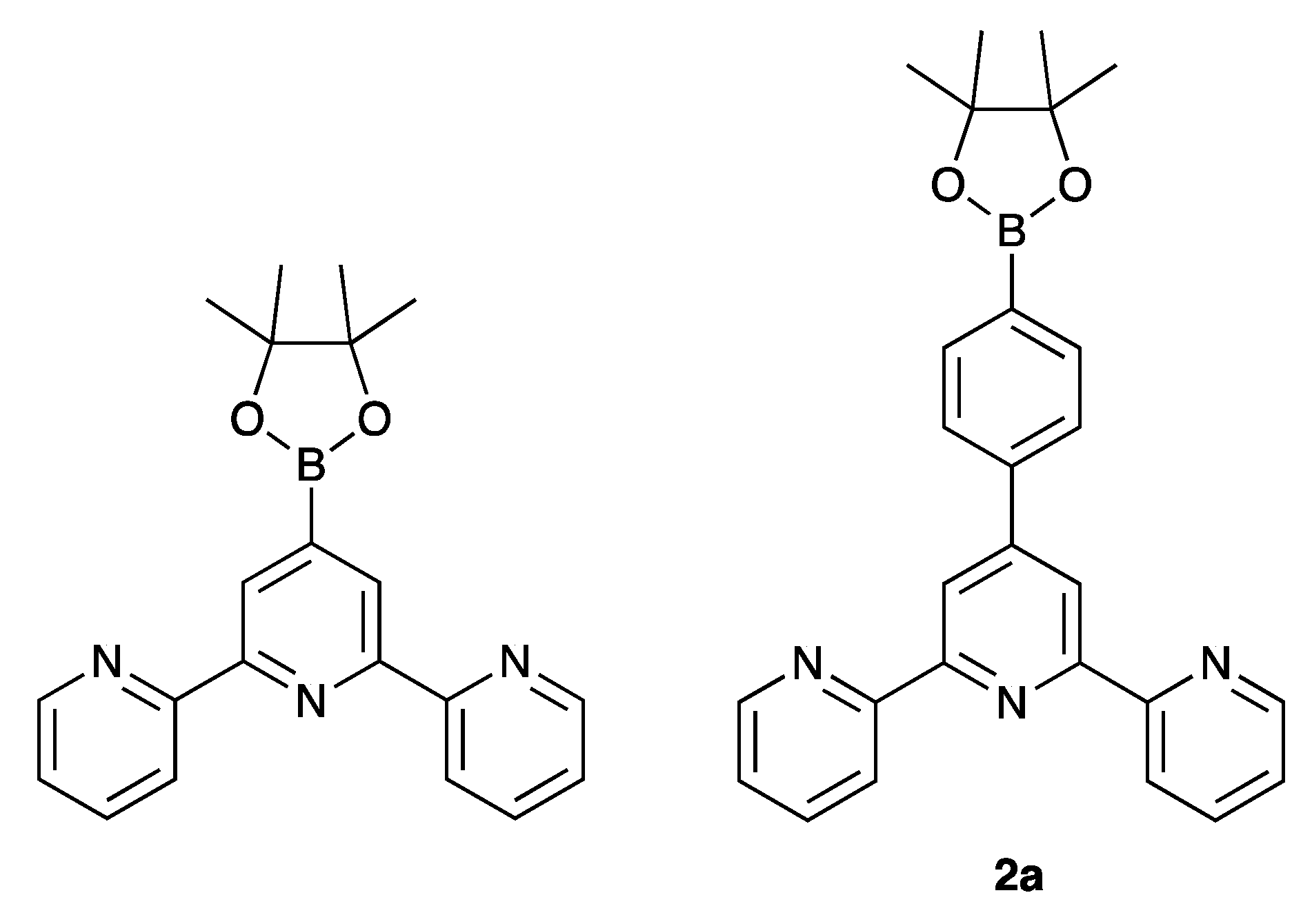
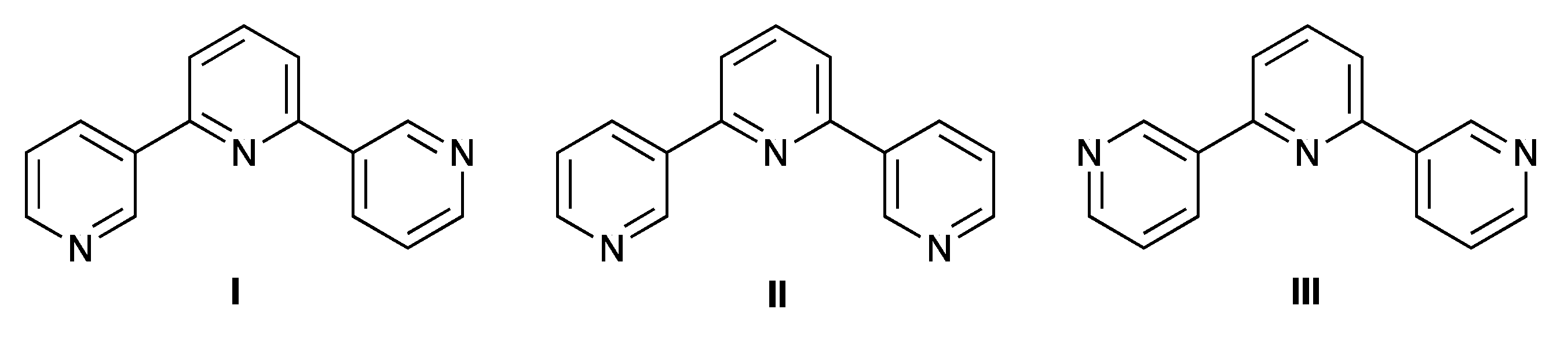
2.1. Synthesis and Characterization of Terpyridine Precursors 1–4
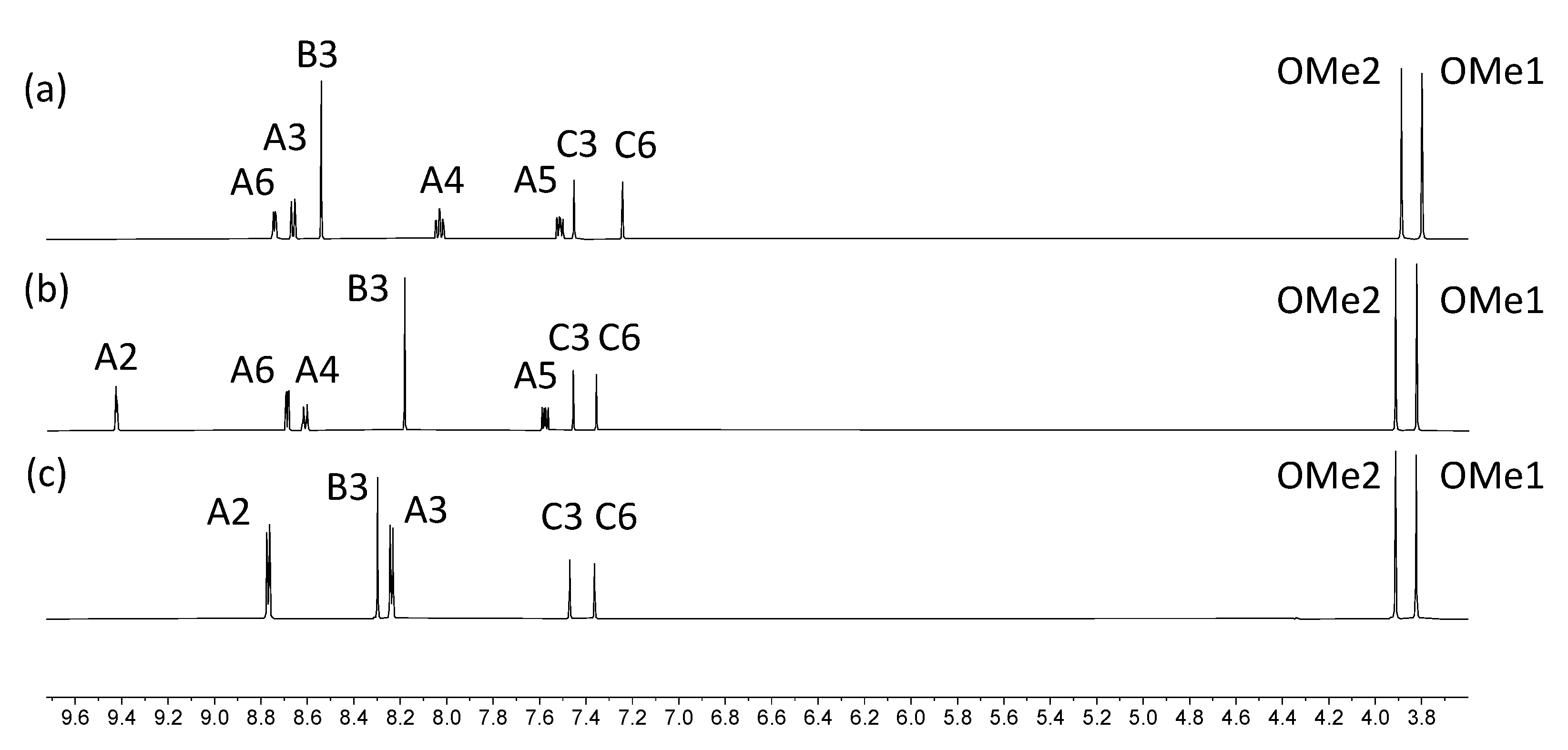
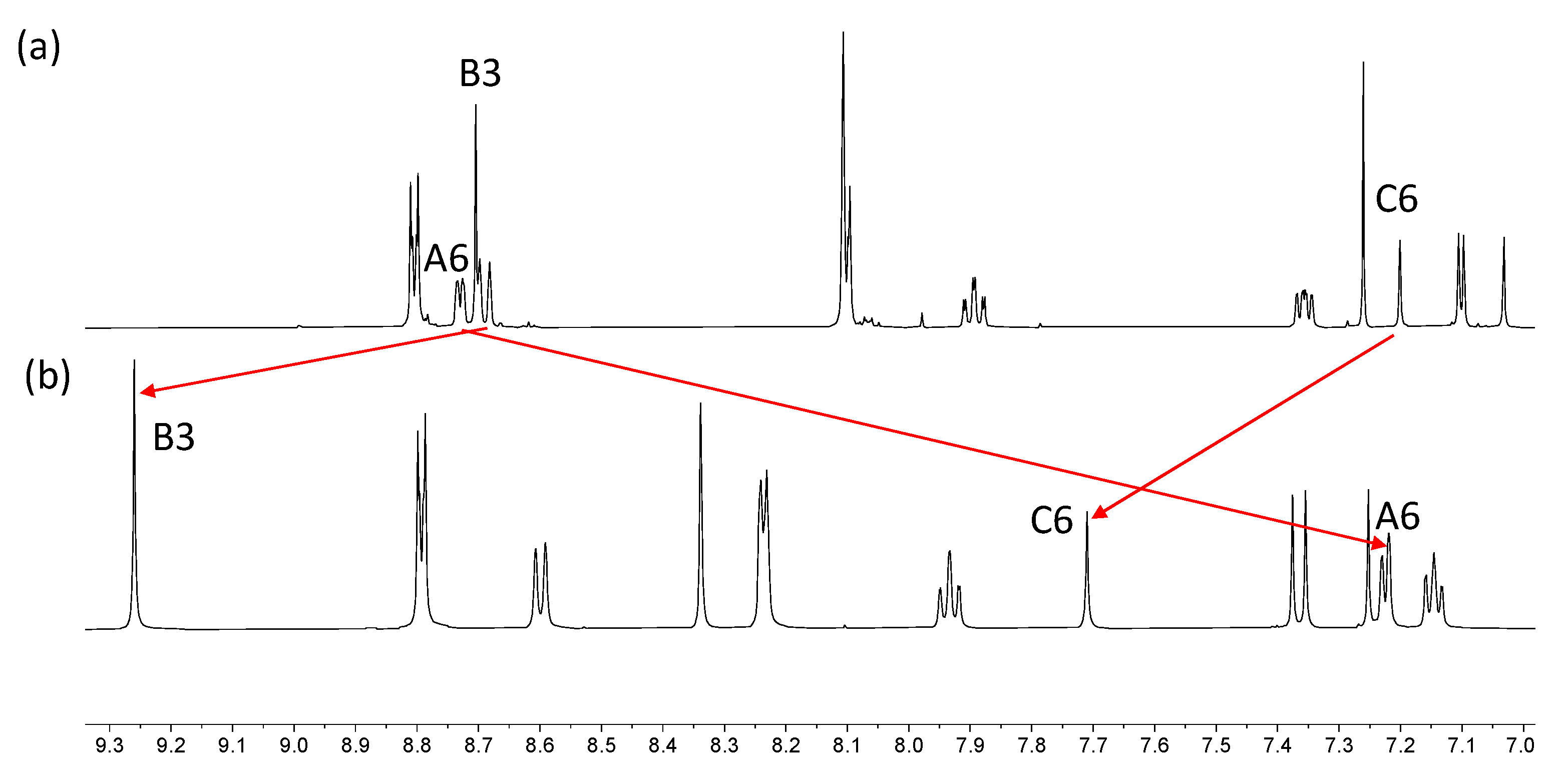
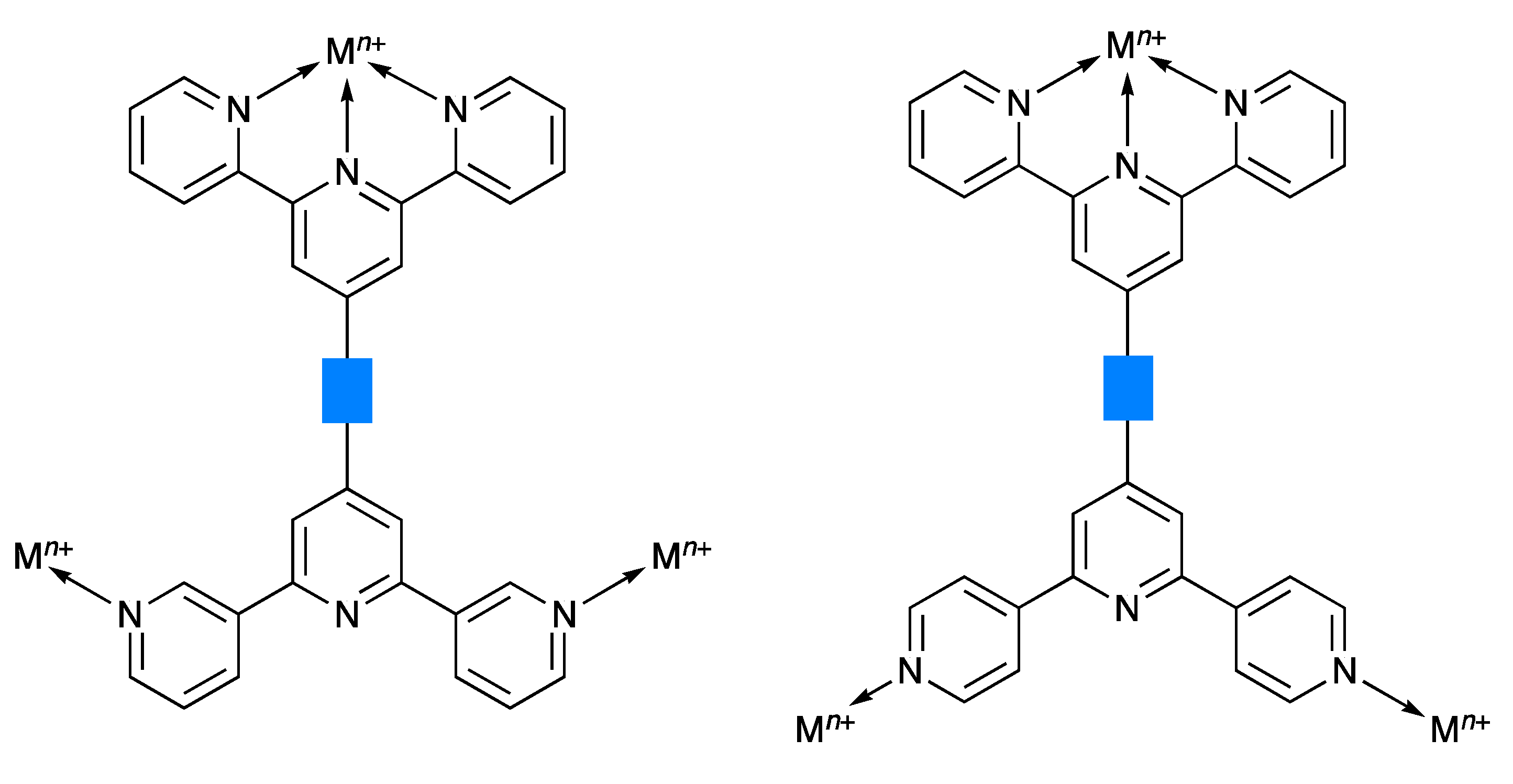
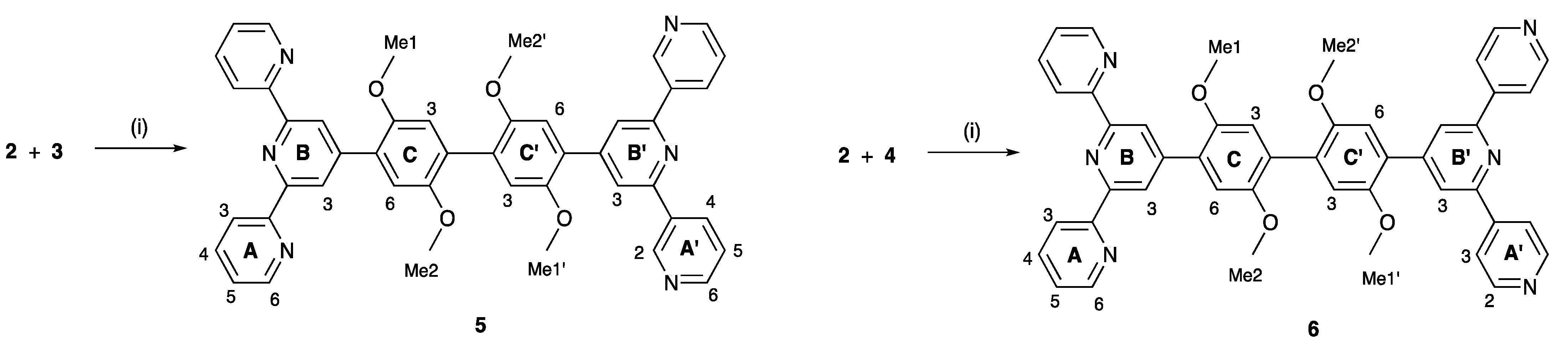
2.2. Synthesis and Characterization of the Asymmetrical Bis(Terpyridine) Ligands 5 and 6
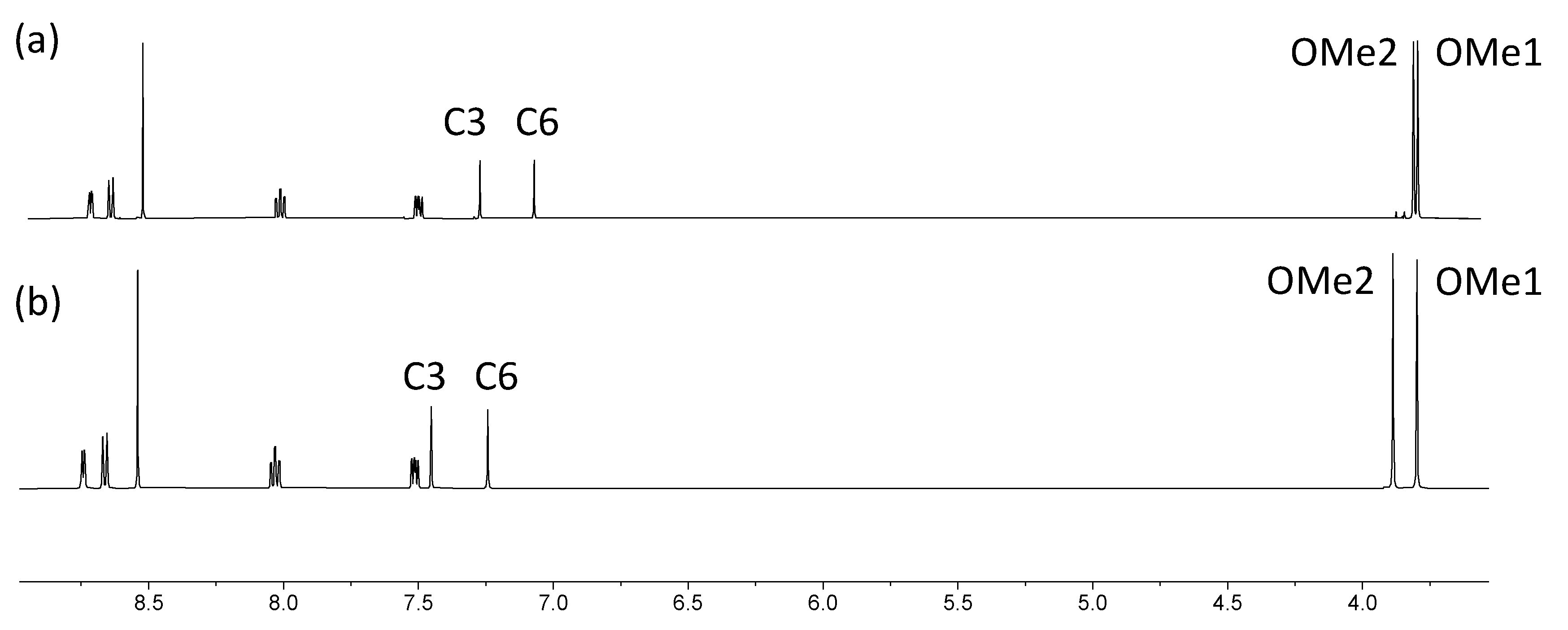
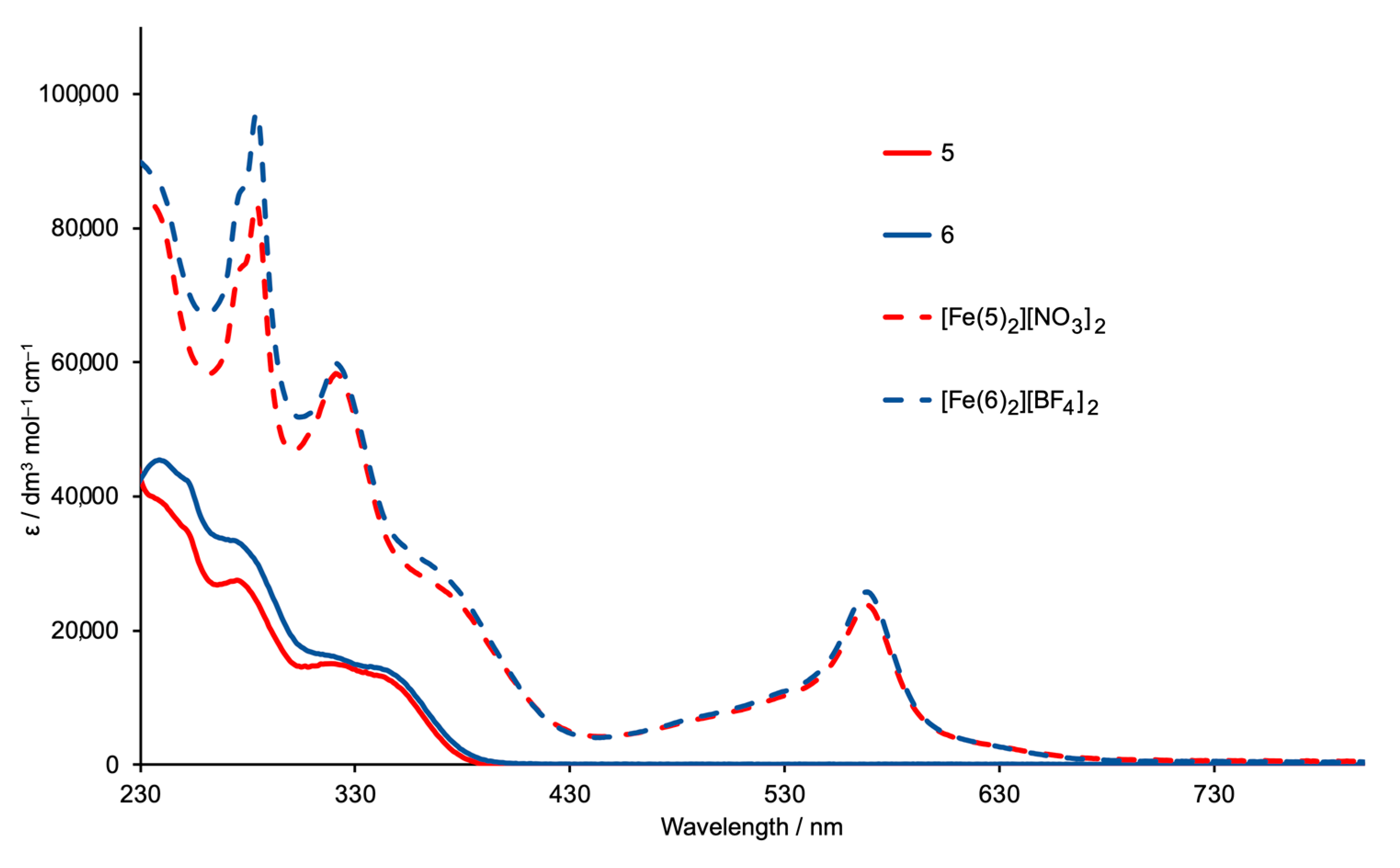
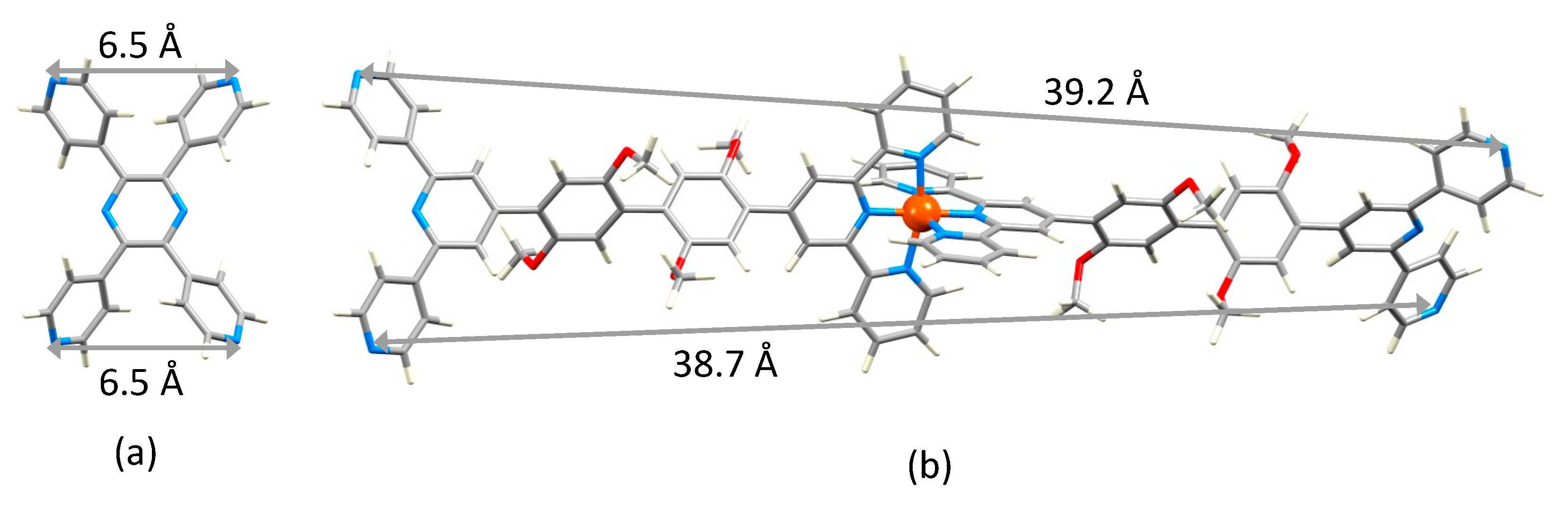
2.3. Expanded Ligands [Fe(5)2]2+ and [Fe(6)2]2+
3. Materials and Methods
3.1. General
3.2. Compound 1
3.3. Compound 2
3.4. Compound 3
3.5. Compound 4
3.6. Compound 5
3.7. Compound 6
3.8. [Fe(5)2][NO3]2
3.9. [Fe(6)2][BF4]2
3.10. Crystallography
3.11. Compound 2
3.12. Compound 3
3.13. Compound 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Damhus, T.; Hartshorn, R.M.; Hutton, A.T. Nomenclature of Inorganic Chemistry, IUPAC Recommendations 2005, IUPAC Red Book; Connelly, N.G., Damhus, T., Hartshorn, R.M., Hutton, A.T., Eds.; RSC Publishing: Cambridge, UK, 2005. [Google Scholar]
- Constable, E.C. A Journey from Solution Self-Assembly to Designed Interfacial Assembly. Adv. Inorg. Chem. 2018, 71, 79–134. [Google Scholar] [CrossRef] [Green Version]
- Constable, E.C.; Housecroft, C.E. More hydra than Janus—Non-classical coordination modes in complexes of oligopyridine ligands. Coord. Chem. Rev. 2017, 350, 84–104. [Google Scholar] [CrossRef] [Green Version]
- Gelmini, L.; Stephan, D.W. The facile preparation of early transition metal/late transition metal heterobimetallic complexes; (η5-C5H5)2Zr(PPh2)2 as a ‘metalloligand’ for Ni, Pd and Pt. Inorg. Chim. Acta 1986, 111, L17–L18. [Google Scholar] [CrossRef]
- Kumar, G.; Kumar, G.; Gupta, R. Effect of pyridyl donors from organic ligands versus metalloligands on material design. Inorg. Chem. Front. 2021, 8, 1334–1373. [Google Scholar] [CrossRef]
- Li, F.; Lindoy, L.F. Metalloligand Strategies for Assembling Heteronuclear Nanocages—Recent Developments. Aust. J. Chem. 2019, 72, 731–741. [Google Scholar] [CrossRef]
- Dzhardimalieva, G.I.; Uflyand, I.E. Design and synthesis of coordination polymers with chelated units and their application in nanomaterials science. RSC Adv. 2017, 7, 42242–42288. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.; Gupta, R. Molecularly designed architectures—The metalloligand way. Chem. Soc. Rev. 2013, 42, 9403–9453. [Google Scholar] [CrossRef]
- Chen, B.; Xiang, S.; Qian, G. Metal—Organic Frameworks with Functional Pores for Recognition of Small Molecules. Acc. Chem. Res. 2010, 43, 1115–1124. [Google Scholar] [CrossRef]
- Constable, E.C. Expanded ligands—An assembly principle for supramolecular chemistry. Coord. Chem. Rev. 2008, 252, 842–855. [Google Scholar] [CrossRef]
- Constable, E.C.; Schofield, E. Metal-directed assembly of a box-like structure. Chem. Commun. 1998, 403–404. [Google Scholar] [CrossRef]
- Constable, E.C.; Dunphy, E.L.; Housecroft, C.E.; Kylberg, W.; Neuburger, M.; Schaffner, S.; Schofield, E.R.; Smith, C.B. Structural development of free or coordinated 4′-(4-pyridyl)-2,2′:6′,2″-terpyridine ligands through N-alkylation: New strategies for metallomacrocycle formation. Chem. Eur. J. 2006, 12, 4600–4610. [Google Scholar] [CrossRef] [PubMed]
- Beves, J.E.; Constable, E.C.; Housecroft, C.E.; Kepert, C.J.; Price, D.J. The first example of a coordination polymer from the expanded 4,4′-bipyridine ligand [Ru(pytpy)2]2+ (pytpy = 4′-(4-pyridyl)-2,2′:6′,2″-terpyridine). CrystEngCommun 2007, 9, 456–459. [Google Scholar] [CrossRef]
- Beves, J.E.; Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Schaffner, S. A palladium (II) complex of 4′-(4-pyridyl)-2,2′:6′,2″-terpyridine: Lattice control through an interplay of stacking and hydrogen bonding effects. Inorg. Chem. Commun. 2007, 10, 1185–1188. [Google Scholar] [CrossRef]
- Beves, J.E.; Constable, E.C.; Housecroft, C.E.; Kepert, C.J.; Price, D.J.; Schaffner, S. The conjugate acid of bis{4′-(4-pyridyl)-2,2′:6′,2″-terpyridine}iron(II) as a self-complementary hydrogen-bonded building block. CrystEngCommun 2007, 9, 1073–1077. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Schaffner, S.; Schaper, F. Preparation and structural characterisation of bis(4′-(3-pyridyl)-2,2′:6′,2″-terpyridine)ruthenium(II) hexafluorophosphate. Inorg. Chem. Commun. 2006, 9, 433–436. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Schaffner, S.; Schaper, F. The solid-state structure of bis(4′-(4-pyridyl)-2,2′:6′,2″-terpyridine)ruthenium hexafluorophosphate nitrate—an expanded 4,4′-bipyridine. Inorg. Chem. Commun. 2006, 9, 616–619. [Google Scholar] [CrossRef]
- Constable, E.C.; Dunphy, E.L.; Housecroft, C.E.; Neuburger, M.; Schaffner, S.; Schaper, F.; Batten, S.R. Expanded ligands: Bis(2,2′:6′,2″-terpyridine carboxylic acid)ruthenium(II) complexes as metallosupramolecular analogues of dicarboxylic acids. Dalton Trans. 2007, 4323–4332. [Google Scholar] [CrossRef]
- Wang, J.; Hanan, G.S. A Facile Route to Sterically Hindered and Non-hindered 4′-Aryl-2,2′:6′,2″-terpyridines. Synlett 2005, 1251–1254. [Google Scholar] [CrossRef]
- Sun, Q.; Tang, L.; Zhang, Z.; Zhang, K.; Xie, Z.; Chi, Z.; Zhang, H.; Yang, W. Bright NUV mechanofluorescence from a terpyridine-based pure organic crystal. Chem. Commun. 2018, 54, 94–97. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 5th ed.; Pearson: Harlow, UK, 2018; p. 413. ISBN 978-1-292-13414-7. [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Schwalbe, M.; Metzinger, R.; Teets, T.S.; Nocera, D.G. Terpyridine–Porphyrin Hetero-Pacman Compounds. Chem. Eur. J. 2012, 18, 15449–15458. [Google Scholar] [CrossRef] [PubMed]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualising crystal structures. Acta Cryst. 2002, B58, 389–397. [Google Scholar] [CrossRef]
- Spartan; Version 18; Wavefunction Inc.: Irvine, CA, USA, 2020.
- Chen, Y.; Yang, T.; Huang, J.; Yong, H.-Y. Two Zn(II)-based coordination polymers: Treatment effect on the cardiac arrest induced by anesthesia by regulating Sirt1 expression. Inorg. Nano-Metal Chem. 2020, 51, 1471–1476. [Google Scholar] [CrossRef]
- Software for the Integration of CCD Detector System Bruker Analytical X-ray Systems; Bruker axs: Madison, WI, USA, 2013.
- Sheldrick, G.M. ShelXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Clegg, J.K.; Jiang, Q.; Lui, X.; Yan, H.; Zhong, W.; Beves, J.E. Multi-pyridine decorated Fe(ii) and Ru(ii) complexes by Pd(0)-catalysed cross couplings: New building blocks for metallosupramolecular assemblies. Dalton Trans. 2013, 42, 15625–15636. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-L.; Chen, Q.-W.; Zhang, Z.-W.; Chen, Q.; Wei, L.-Q.; Lin, N. Efficient heterogeneous catalyst of Fe(II)-based coordination complexes for Friedel-Crafts alkylation reaction. J. Solid State Chem. 2022, 310, 123045. [Google Scholar] [CrossRef]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocco, D.; Prescimone, A.; Housecroft, C.E.; Constable, E.C. Expanded Ligands Based upon Iron(II) Coordination Compounds of Asymmetrical Bis(terpyridine) Domains. Molecules 2023, 28, 82. https://doi.org/10.3390/molecules28010082
Rocco D, Prescimone A, Housecroft CE, Constable EC. Expanded Ligands Based upon Iron(II) Coordination Compounds of Asymmetrical Bis(terpyridine) Domains. Molecules. 2023; 28(1):82. https://doi.org/10.3390/molecules28010082
Chicago/Turabian StyleRocco, Dalila, Alessandro Prescimone, Catherine E. Housecroft, and Edwin C. Constable. 2023. "Expanded Ligands Based upon Iron(II) Coordination Compounds of Asymmetrical Bis(terpyridine) Domains" Molecules 28, no. 1: 82. https://doi.org/10.3390/molecules28010082
APA StyleRocco, D., Prescimone, A., Housecroft, C. E., & Constable, E. C. (2023). Expanded Ligands Based upon Iron(II) Coordination Compounds of Asymmetrical Bis(terpyridine) Domains. Molecules, 28(1), 82. https://doi.org/10.3390/molecules28010082