

Challenges for Kinetics Predictions via Neural Network Potentials: A Wilkinson’s Catalyst Case
 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
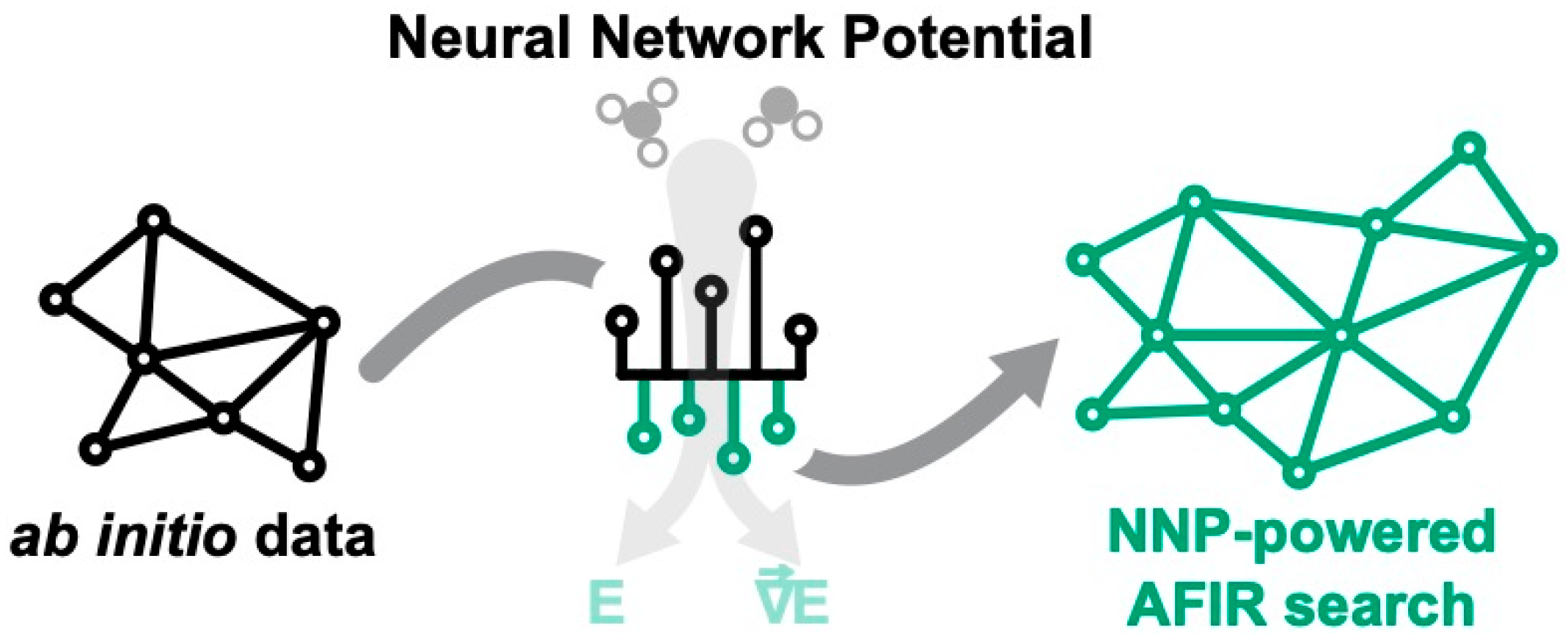
2.1. Reaction Path Search Using the AFIR Method
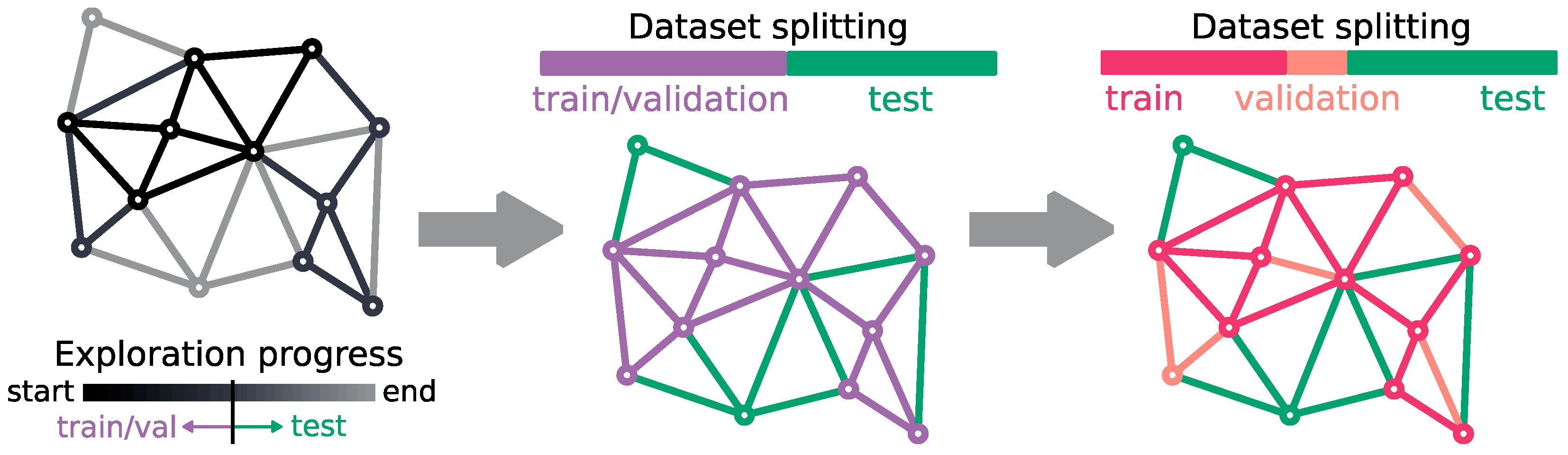
2.2. Dataset Description
2.3. GTM Visualization
2.4. 3D Pairwise-Sorted Distance-Based Descriptors
2.5. Neural Network Potential Architecture
2.6. NNP(+xTB) Models
3. Results
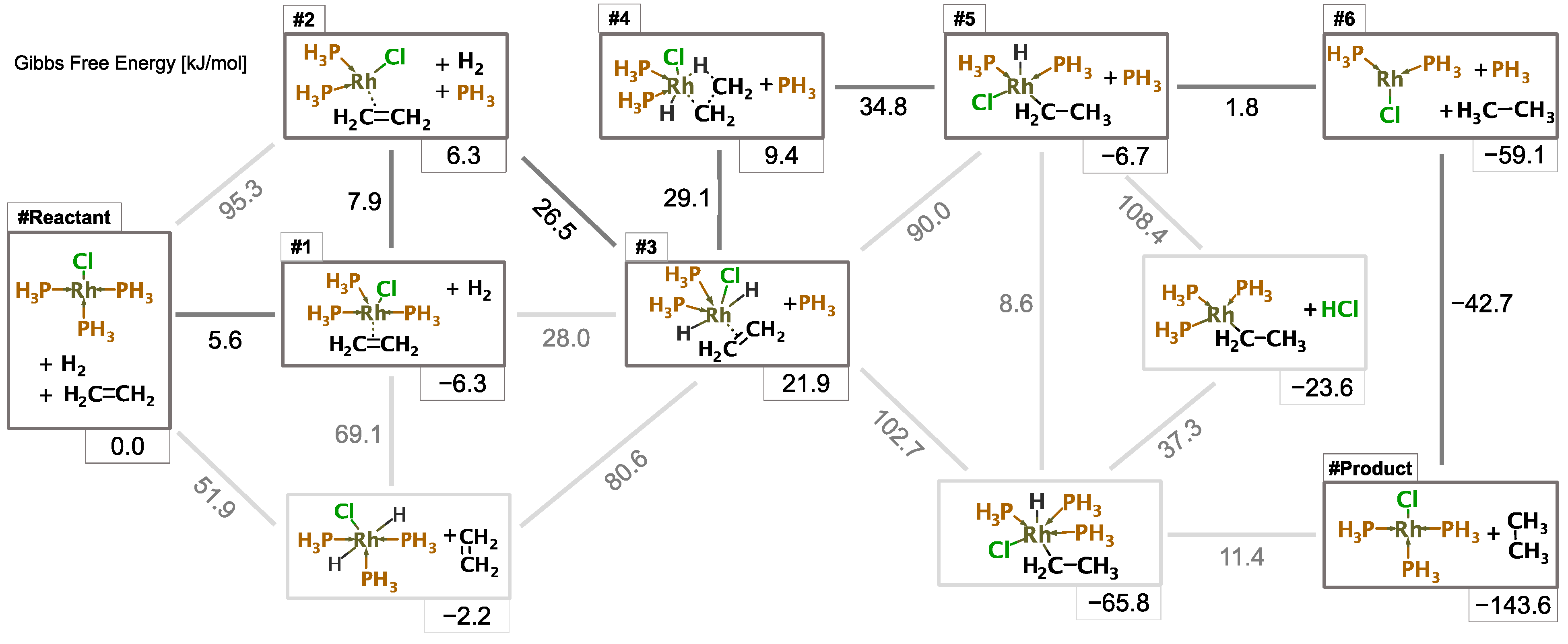
3.1. Reaction Path Network for Hydrogenation Using a Simplified Wilkinson’s Catalyst
3.2. Data Visualization with GTM
3.3. Applicability of Neural Network Potentials to AFIR-Based Reaction Path Search
3.3.1. NNP Performance on Pre-Obtained Geometries
3.3.2. Reaction Path Search Using the NNP Model
- Lack of physics: While general-purpose NNPs, such as SpookyNet, do respect fundamental symmetries (translation, rotation, …), their functional forms (i.e., the mathematical models) are not physics-based. In particular, their asymptotic behavior is not governed by physical principles. Although SpookyNet models already include additional trainable terms that are physics-inspired (EZBL, ED4 and Eelec), these terms do not seem sufficient to ensure physical asymptotic behavior outside the training domain.
- Training bias: Due to the aforementioned lack of physics, the NNP considerably relies on the training data, yet the dataset does not contain strongly broken geometries. Indeed, such geometries are not encountered during the DFT-based search, because all paths leading to them would be rightfully assessed as too high in energy for the exploration to continue. Therefore, the trained NNPs cannot properly handle these extreme geometries, leading them to be poorly described.
- Strong exploration forces: Even if sufficient training data is available in the accessible valleys of a potential energy surface (i.e., chemically reasonable geometries), we believe that applying a strong external force can drive a properly described system outside the locally well-defined valleys of the fitted potential.
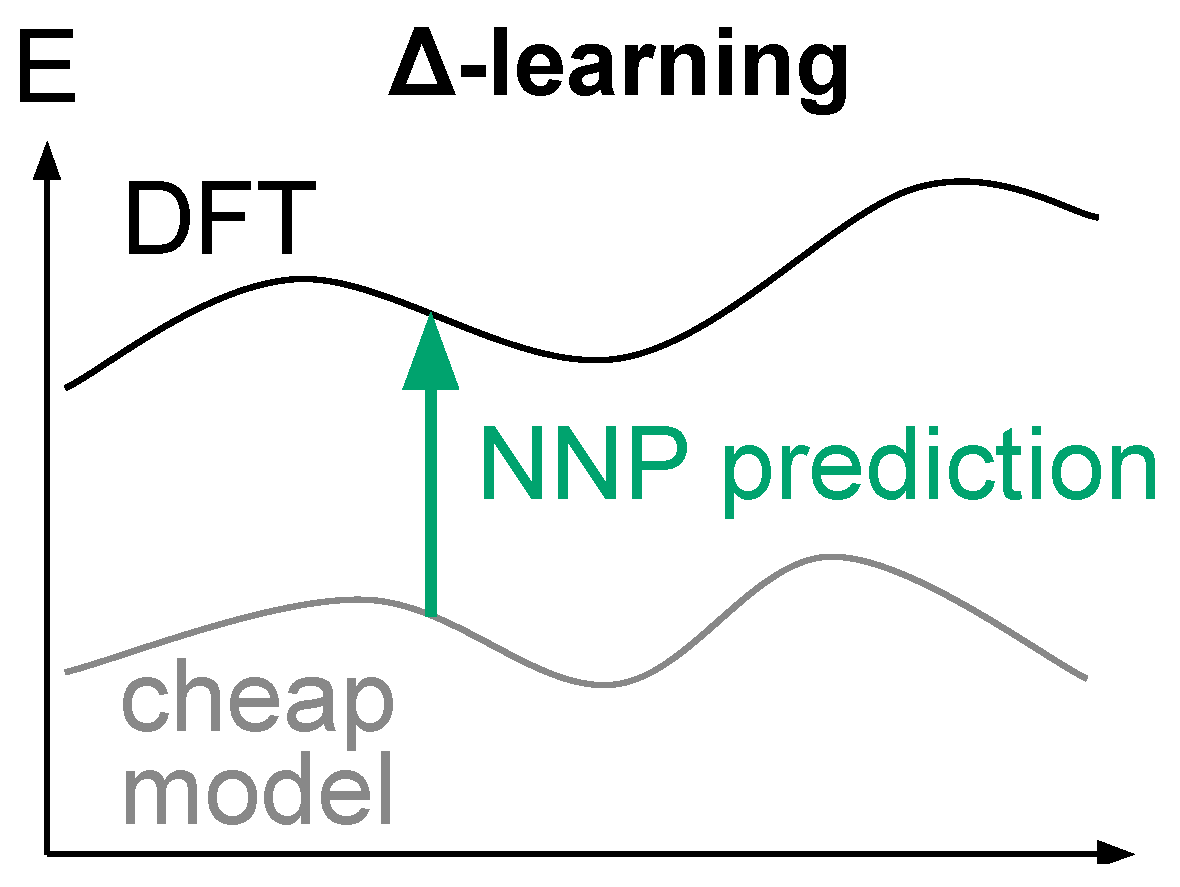
3.3.3. Δ-Learning Solution for Robust NNP-Based Models
- Strong exploration forces are a powerful tool to efficiently sample rare events [76], so we believe that one should focus on designing models that can support them, instead of removing them.
- SpookyNet models need to be trained on broken geometries to properly describe them. We argue that complementing the training dataset a priori with broken geometries is not reasonable, because one cannot easily predict in advance the pitfalls of a fitted potential, and one cannot reasonably include all possible broken geometries in the training set. A simple argument to convince the reader is to consider N atoms randomly distributed in a box: the probability that the resulting geometry is chemically reasonable is close to zero, therefore illustrating the inconceivably large ratio of broken geometries over reasonable geometries. We further argue that such training bias toward reasonable geometries in available datasets is actually desirable, because we believe it is unreasonable to waste computational resources on unreasonable geometries.
3.3.4. Kinetic Study from Reaction Path Search Using NNP(+xTB)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klippenstein, S.J.; Pande, V.S.; Truhlar, D.G. Chemical Kinetics and Mechanisms of Complex Systems: A Perspective on Recent Theoretical Advances. J. Am. Chem. Soc. 2014, 136, 528–546. [Google Scholar] [CrossRef]
- Maeda, S.; Morokuma, K. Communications: A Systematic Method for Locating Transition Structures of A + B→X Type Reactions. J. Chem. Phys. 2010, 132, 241102. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Taketsugu, T.; Morokuma, K. Exploring Transition State Structures for Intramolecular Pathways by the Artificial Force Induced Reaction Method. J. Comput. Chem. 2014, 35, 166–173. [Google Scholar] [CrossRef]
- Habermann, B.; Villaveces, J.; Koti, P. Tools for Visualization and Analysis of Molecular Networks, Pathways, and -Omics Data. Adv. Appl. Bioinform. Chem. 2015, 2015, 11–22. [Google Scholar] [CrossRef]
- Garay-Ruiz, D.; Álvarez-Moreno, M.; Bo, C.; Martínez-Núñez, E. New Tools for Taming Complex Reaction Networks: The Unimolecular Decomposition of Indole Revisited. ACS Phys. Chem. Au 2022, 2, 225–236. [Google Scholar] [CrossRef]
- Komatsuzaki, T.; Hoshino, K.; Matsunaga, Y.; Rylance, G.J.; Johnston, R.L.; Wales, D.J. How Many Dimensions Are Required to Approximate the Potential Energy Landscape of a Model Protein? J. Chem. Phys. 2005, 122, 84714. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.R.; Bratholm, L.A.; Glowacki, D.R.; Carpenter, B.K. Low Dimensional Representations along Intrinsic Reaction Coordinates and Molecular Dynamics Trajectories Using Interatomic Distance Matrices. Chem. Sci. 2019, 10, 9954–9968. [Google Scholar] [CrossRef]
- Shi, W.; Jia, T.; Li, A. Quasi-Classical Trajectory Analysis with Isometric Feature Mapping and Locally Linear Embedding: Deep Insights into the Multichannel Reaction on an NH3+ (4A) Potential Energy Surface. Phys. Chem. Chem. Phys. 2020, 22, 17460–17471. [Google Scholar] [CrossRef]
- Li, X.; Xie, Y.; Hu, D.; Lan, Z. Analysis of the Geometrical Evolution in On-the-Fly Surface-Hopping Nonadiabatic Dynamics with Machine Learning Dimensionality Reduction Approaches: Classical Multidimensional Scaling and Isometric Feature Mapping. J. Chem. Theory Comput. 2017, 13, 4611–4623. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, T.; Ono, Y.; Taketsugu, T. Visualization of Reaction Route Map and Dynamical Trajectory in Reduced Dimension. Chem. Commun. 2021, 57, 11734–11750. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Ono, Y.; Arai, Z.; Taketsugu, T. Visualization of the Intrinsic Reaction Coordinate and Global Reaction Route Map by Classical Multidimensional Scaling. J. Chem. Theory Comput. 2018, 14, 4263–4270. [Google Scholar] [CrossRef]
- Kireeva, N.; Baskin, I.I.; Gaspar, H.A.; Horvath, D.; Marcou, G.; Varnek, A. Generative Topographic Mapping (GTM): Universal Tool for Data Visualization, Structure-Activity Modeling and Dataset Comparison. Mol. Inform. 2012, 31, 301–312. [Google Scholar] [CrossRef]
- Sumiya, Y.; Maeda, S. Rate Constant Matrix Contraction Method for Systematic Analysis of Reaction Path Networks. Chem. Lett. 2020, 49, 553–564. [Google Scholar] [CrossRef]
- Gillespie, D.T. Stochastic Simulation of Chemical Kinetics. Annu. Rev. Phys. Chem. 2007, 58, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Sumiya, Y.; Maeda, S. A Reaction Path Network for Wöhler’s Urea Synthesis. Chem. Lett. 2019, 48, 47–50. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and Use of Quantum Mechanical Molecular Models. 76. AM1: A New General Purpose Quantum Mechanical Molecular Model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-Optimization of Parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding Quantum Chemistry Methods. WIREs Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Bursch, M.; Hansen, A.; Pracht, P.; Kohn, J.T.; Grimme, S. Theoretical Study on Conformational Energies of Transition Metal Complexes. Phys. Chem. Chem. Phys. 2021, 23, 287–299. [Google Scholar] [CrossRef]
- Kocer, E.; Ko, T.W.; Behler, J. Neural Network Potentials: A Concise Overview of Methods. Annu. Rev. Phys. Chem. 2022, 73, 163–186. [Google Scholar] [CrossRef]
- Behler, J. Four Generations of High-Dimensional Neural Network Potentials. Chem. Rev. 2021, 121, 10037–10072. [Google Scholar] [CrossRef] [PubMed]
- Unke, O.T.; Chmiela, S.; Gastegger, M.; Schütt, K.T.; Sauceda, H.E.; Müller, K.-R. SpookyNet: Learning Force Fields with Electronic Degrees of Freedom and Nonlocal Effects. Nat. Commun. 2021, 12, 7273. [Google Scholar] [CrossRef] [PubMed]
- Schütt, K.T.; Sauceda, H.E.; Kindermans, P.-J.; Tkatchenko, A.; Müller, K.-R. SchNet—A Deep Learning Architecture for Molecules and Materials. J. Chem. Phys. 2018, 148, 241722. [Google Scholar] [CrossRef] [PubMed]
- Pun, G.P.P.; Batra, R.; Ramprasad, R.; Mishin, Y. Physically Informed Artificial Neural Networks for Atomistic Modeling of Materials. Nat. Commun. 2019, 10, 2339. [Google Scholar] [CrossRef]
- Batzner, S.; Musaelian, A.; Sun, L.; Geiger, M.; Mailoa, J.P.; Kornbluth, M.; Molinari, N.; Smidt, T.E.; Kozinsky, B. E(3)-Equivariant Graph Neural Networks for Data-Efficient and Accurate Interatomic Potentials. Nat. Commun. 2022, 13, 2453. [Google Scholar] [CrossRef]
- Thölke, P.; De Fabritiis, G. TorchMD-NET: Equivariant Transformers for Neural Network Based Molecular Potentials. arXiv 2022, arXiv:2202.02541. [Google Scholar] [CrossRef]
- Schütt, K.; Unke, O.; Gastegger, M. Equivariant Message Passing for the Prediction of Tensorial Properties and Molecular Spectra. In Proceedings of the 38th International Conference on Machine Learning, Virtual, 18–24 July 2021; Meila, M., Zhang, T., Eds.; PMLR Series. MLR Press: Toledo, OH, USA; Volume 139, pp. 9377–9388. [Google Scholar]
- Nandy, A.; Duan, C.; Taylor, M.G.; Liu, F.; Steeves, A.H.; Kulik, H.J. Computational Discovery of Transition-Metal Complexes: From High-Throughput Screening to Machine Learning. Chem. Rev. 2021, 121, 9927–10000. [Google Scholar] [CrossRef]
- Meuwly, M. Machine Learning for Chemical Reactions. Chem. Rev. 2021, 121, 10218–10239. [Google Scholar] [CrossRef]
- Musil, F.; Grisafi, A.; Bartók, A.P.; Ortner, C.; Csányi, G.; Ceriotti, M. Physics-Inspired Structural Representations for Molecules and Materials. Chem. Rev. 2021, 121, 9759–9815. [Google Scholar] [CrossRef]
- Keith, J.A.; Vassilev-Galindo, V.; Cheng, B.; Chmiela, S.; Gastegger, M.; Müller, K.-R.; Tkatchenko, A. Combining Machine Learning and Computational Chemistry for Predictive Insights Into Chemical Systems. Chem. Rev. 2021, 121, 9816–9872. [Google Scholar] [CrossRef]
- Göb, S.; Oliveros, E.; Bossmann, S.H.; Braun, A.M.; Guardani, R.; Nascimento, C.A.O. Modeling the Kinetics of a Photochemical Water Treatment Process by Means of Artificial Neural Networks. Chem. Eng. Process. Process Intensif. 1999, 38, 373–382. [Google Scholar] [CrossRef]
- Allison, T.C. Application of an Artificial Neural Network to the Prediction of OH Radical Reaction Rate Constants for Evaluating Global Warming Potential. J. Phys. Chem. B 2016, 120, 1854–1863. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, X.; Xu, X.; Zhang, D.H. Communication: An Accurate Global Potential Energy Surface for the OH + CO → H + CO 2 Reaction Using Neural Networks. J. Chem. Phys. 2013, 138, 221104. [Google Scholar] [CrossRef]
- Lu, D.; Behler, J.; Li, J. Accurate Global Potential Energy Surfaces for the H + CH 3 OH Reaction by Neural Network Fitting with Permutation Invariance. J. Phys. Chem. A 2020, 124, 5737–5745. [Google Scholar] [CrossRef]
- Gerrits, N.; Shakouri, K.; Behler, J.; Kroes, G.-J. Accurate Probabilities for Highly Activated Reaction of Polyatomic Molecules on Surfaces Using a High-Dimensional Neural Network Potential: CHD 3 + Cu(111). J. Phys. Chem. Lett. 2019, 10, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef]
- Yang, M.; Bonati, L.; Polino, D.; Parrinello, M. Using Metadynamics to Build Neural Network Potentials for Reactive Events: The Case of Urea Decomposition in Water. Catal. Today 2022, 387, 143–149. [Google Scholar] [CrossRef]
- Yang, X.; Bhowmik, A.; Vegge, T.; Hansen, H.A. Neural Network Potentials for Accelerated Metadynamics of Oxygen Reduction Kinetics at Au–Water Interfaces. Chem. Sci. 2023, 14, 3913–3922. [Google Scholar] [CrossRef]
- Schreiner, M.; Bhowmik, A.; Vegge, T.; Jørgensen, P.B.; Winther, O. NeuralNEB—Neural Networks Can Find Reaction Paths Fast. Mach. Learn. Sci. Technol. 2022, 3, 45022. [Google Scholar] [CrossRef]
- Chu, Q.; Luo, K.H.; Chen, D. Exploring Complex Reaction Networks Using Neural Network-Based Molecular Dynamics Simulation. J. Phys. Chem. Lett. 2022, 13, 4052–4057. [Google Scholar] [CrossRef]
- Osborn, J.A.; Jardine, F.H.; Young, J.F.; Wilkinson, G. The Preparation and Properties of Tris(Triphenylphosphine)Halogenorhodium(I) and Some Reactions Thereof Including Catalytic Homogeneous Hydrogenation of Olefins and Acetylenes and Their Derivatives. J. Chem. Soc. Inorg. Phys. Theor. 1966, 1, 1711–1732. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y. Exploring Paths of Chemical Transformations in Molecular and Periodic Systems: An Approach Utilizing Force. WIREs Comput. Mol. Sci. 2021, 11, e1538. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Hayashi, H.; Mita, T. Toward Ab Initio Reaction Discovery Using the Artificial Force Induced Reaction Method. Annu. Rev. Phys. Chem. 2023, 74, 287–311. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Harabuchi, Y.; Maeda, S.; Tsuda, K. Exploring the Quantum Chemical Energy Landscape with GNN-Guided Artificial Force. J. Chem. Theory Comput. 2023, 19, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Elber, R. Reaction Path Study of Helix Formation in Tetrapeptides: Effect of Side Chains. J. Chem. Phys. 1991, 94, 751–760. [Google Scholar] [CrossRef]
- Maeda, S.; Sugiyama, K.; Sumiya, Y.; Takagi, M.; Saita, K. Global Reaction Route Mapping for Surface Adsorbed Molecules: A Case Study for H2O on Cu(111) Surface. Chem. Lett. 2018, 47, 396–399. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef] [PubMed]
- Hjorth Larsen, A.; Jørgen Mortensen, J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; et al. The Atomic Simulation Environment—A Python Library for Working with Atoms. J. Phys. Condens. Matter 2017, 29, 273002. [Google Scholar] [CrossRef]
- Sumiya, Y.; Harabuchi, Y.; Nagata, Y.; Maeda, S. Quantum Chemical Calculations to Trace Back Reaction Paths for the Prediction of Reactants. JACS Au 2022, 2, 1181–1188. [Google Scholar] [CrossRef]
- Harabuchi, Y.; Maeda, S. Theoretical Chemical Reaction Database Construction Based on Quantum Chemistry-Aided Retrosynthetic Analysis. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Zabolotna, Y.; Bonachera, F.; Horvath, D.; Lin, A.; Marcou, G.; Klimchuk, O.; Varnek, A. Chemspace Atlas: Multiscale Chemography of Ultralarge Libraries for Drug Discovery. J. Chem. Inf. Model. 2022, 62, 4537–4548. [Google Scholar] [CrossRef] [PubMed]
- Horvath, D.; Brown, J.; Marcou, G.; Varnek, A. An Evolutionary Optimizer of Libsvm Models. Challenges 2014, 5, 450–472. [Google Scholar] [CrossRef]
- Sengupta, U.; Carballo-Pacheco, M.; Strodel, B. Automated Markov State Models for Molecular Dynamics Simulations of Aggregation and Self-Assembly. J. Chem. Phys. 2019, 150, 115101. [Google Scholar] [CrossRef] [PubMed]
- Roet, S.; Daub, C.D.; Riccardi, E. Chemistrees: Data-Driven Identification of Reaction Pathways via Machine Learning. J. Chem. Theory Comput. 2021, 17, 6193–6202. [Google Scholar] [CrossRef]
- Hansen, K.; Biegler, F.; Ramakrishnan, R.; Pronobis, W.; von Lilienfeld, O.A.; Müller, K.-R.; Tkatchenko, A. Machine Learning Predictions of Molecular Properties: Accurate Many-Body Potentials and Nonlocality in Chemical Space. J. Phys. Chem. Lett. 2015, 6, 2326–2331. [Google Scholar] [CrossRef]
- Chen, X.; Jørgensen, M.S.; Li, J.; Hammer, B. Atomic Energies from a Convolutional Neural Network. J. Chem. Theory Comput. 2018, 14, 3933–3942. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Han, J.; Weinan, E. DeePMD-Kit: A Deep Learning Package for Many-Body Potential Energy Representation and Molecular Dynamics. Comput. Phys. Commun. 2018, 228, 178–184. [Google Scholar] [CrossRef]
- Bose, S.; Dhawan, D.; Nandi, S.; Sarkar, R.R.; Ghosh, D. Machine Learning Prediction of Interaction Energies in Rigid Water Clusters. Phys. Chem. Chem. Phys. 2018, 20, 22987–22996. [Google Scholar] [CrossRef]
- Pinheiro, M.; Ge, F.; Ferré, N.; Dral, P.O.; Barbatti, M. Choosing the Right Molecular Machine Learning Potential. Chem. Sci. 2021, 12, 14396–14413. [Google Scholar] [CrossRef]
- Unke, O.T.; Chmiela, S.; Sauceda, H.E.; Gastegger, M.; Poltavsky, I.; Schütt, K.T.; Tkatchenko, A.; Müller, K.-R. Machine Learning Force Fields. Chem. Rev. 2021, 121, 10142–10186. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention Is All You Need. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; Guyon, I., Luxburg, U.V., Bengio, S., Wallach, H., Fergus, R., Vishwanathan, S., Garnett, R., Eds.; Curran Associates, Inc.: Red Hook, NY, USA, 2017; Volume 30. [Google Scholar]
- Ziegler, J.F.; Biersack, J.P. The Stopping and Range of Ions in Matter. In Treatise on Heavy-Ion Science; Bromley, D.A., Ed.; Springer: Boston, MA, USA, 1985; pp. 93–129. ISBN 978-1-4615-8105-5. [Google Scholar]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A Generally Applicable Atomic-Charge Dependent London Dispersion Correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Dral, P.O.; Rupp, M.; von Lilienfeld, O.A. Big Data Meets Quantum Chemistry Approximations: The Δ-Machine Learning Approach. J. Chem. Theory Comput. 2015, 11, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-XTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Birch, A.J.; Williamson, D.H. Homogeneous Hydrogenation Catalysts in Organic Synthesis. In Organic Reactions; Denmark, S.E., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 1–186. ISBN 978-0-471-26418-7. [Google Scholar]
- Koga, N.; Daniel, C.; Han, J.; Fu, X.Y.; Morokuma, K. Potential Energy Profile of a Full Catalytic Cycle of Olefin Hydrogenation by the Wilkinson Catalyst. J. Am. Chem. Soc. 1987, 109, 3455–3456. [Google Scholar] [CrossRef]
- Orpen, A.G.; Connelly, N.G. Structural Systematics: The Role of P-A .Sigma.* Orbitals in Metal-Phosphorus.Pi.-Bonding in Redox-Related Pairs of M-PA3 Complexes (A = R, Ar, OR; R = Alkyl). Organometallics 1990, 9, 1206–1210. [Google Scholar] [CrossRef]
- Pacchioni, G.; Bagus, P.S. Metal-Phosphine Bonding Revisited. .Sigma.-Basicity, .Pi.-Acidity, and the Role of Phosphorus d Orbitals in Zerovalent Metal-Phospine Complexes. Inorg. Chem. 1992, 31, 4391–4398. [Google Scholar] [CrossRef]
- Tew, D.P.; Klopper, W.; Neiss, C.; Hättig, C. Quintuple-ζ Quality Coupled-Cluster Correlation Energies with Triple-ζ Basis Sets. Phys. Chem. Chem. Phys. 2007, 9, 1921–1930. [Google Scholar] [CrossRef]
- Knizia, G.; Adler, T.B.; Werner, H.-J. Simplified CCSD(T)-F12 Methods: Theory and Benchmarks. J. Chem. Phys. 2009, 130, 54104. [Google Scholar] [CrossRef]
- Krogh, A.; Vedelsby, J. Neural Network Ensembles, Cross Validation, and Active Learning. In Proceedings of the Advances in Neural Information Processing Systems, Denver, CO, USA, 28 November–1 December 1994; Tesauro, G., Touretzky, D., Leen, T., Eds.; MIT Press: Cambridge, MA, USA, 1994; Volume 7. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Maeda, S.; Ohno, K.; Morokuma, K. Systematic Exploration of the Mechanism of Chemical Reactions: The Global Reaction Route Mapping (GRRM) Strategy Using the ADDF and AFIR Methods. Phys. Chem. Chem. Phys. 2013, 15, 3683. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Nebgen, B.T.; Zubatyuk, R.; Lubbers, N.; Devereux, C.; Barros, K.; Tretiak, S.; Isayev, O.; Roitberg, A.E. Approaching Coupled Cluster Accuracy with a General-Purpose Neural Network Potential through Transfer Learning. Nat. Commun. 2019, 10, 2903. [Google Scholar] [CrossRef]
- Nandi, A.; Qu, C.; Houston, P.L.; Conte, R.; Bowman, J.M. Δ-Machine Learning for Potential Energy Surfaces: A PIP Approach to Bring a DFT-Based PES to CCSD(T) Level of Theory. J. Chem. Phys. 2021, 154, 51102. [Google Scholar] [CrossRef]
- Bowman, J.M.; Qu, C.; Conte, R.; Nandi, A.; Houston, P.L.; Yu, Q. Δ-Machine Learned Potential Energy Surfaces and Force Fields. J. Chem. Theory Comput. 2023, 19, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Poirier, B. An Algorithm to Find (and Plug) “Holes” in Multi-Dimensional Surfaces. J. Chem. Phys. 2020, 152, 214102. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, T. Application of Reaction Path Search Calculations to Potential Energy Surface Fits. J. Phys. Chem. A 2021, 125, 3994–4002. [Google Scholar] [CrossRef] [PubMed]
- Käser, S.; Vazquez-Salazar, L.I.; Meuwly, M.; Töpfer, K. Neural Network Potentials for Chemistry: Concepts, Applications and Prospects. Digit. Discov. 2023, 2, 28–58. [Google Scholar] [CrossRef]
- Marvin Version 23.2, ChemAxon. Available online: https://www.chemaxon.com (accessed on 25 May 2023).
- Rego, N.; Koes, D. 3Dmol.Js: Molecular Visualization with WebGL. Bioinformatics 2015, 31, 1322–1324. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted Yield | GFN2-xTB | NNP(+xTB) 20% Training | NNP(+xTB) 50% Training | NNP(+xTB) 80% Training | DFT |
|---|---|---|---|---|---|
| 250 K | 0.50% | 0.00% | 2.09% | 31.42% | 98.47% |
| 300 K | 1.42% | 0.00% | 96.47% | 100% | 100% |
| 350 K | 2.79% | 0.00% | 99.95% | 99.98% | 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staub, R.; Gantzer, P.; Harabuchi, Y.; Maeda, S.; Varnek, A. Challenges for Kinetics Predictions via Neural Network Potentials: A Wilkinson’s Catalyst Case. Molecules 2023, 28, 4477. https://doi.org/10.3390/molecules28114477
Staub R, Gantzer P, Harabuchi Y, Maeda S, Varnek A. Challenges for Kinetics Predictions via Neural Network Potentials: A Wilkinson’s Catalyst Case. Molecules. 2023; 28(11):4477. https://doi.org/10.3390/molecules28114477
Chicago/Turabian StyleStaub, Ruben, Philippe Gantzer, Yu Harabuchi, Satoshi Maeda, and Alexandre Varnek. 2023. "Challenges for Kinetics Predictions via Neural Network Potentials: A Wilkinson’s Catalyst Case" Molecules 28, no. 11: 4477. https://doi.org/10.3390/molecules28114477
APA StyleStaub, R., Gantzer, P., Harabuchi, Y., Maeda, S., & Varnek, A. (2023). Challenges for Kinetics Predictions via Neural Network Potentials: A Wilkinson’s Catalyst Case. Molecules, 28(11), 4477. https://doi.org/10.3390/molecules28114477