
Polyamine–Drug Conjugates: Do They Boost Drug Activity?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
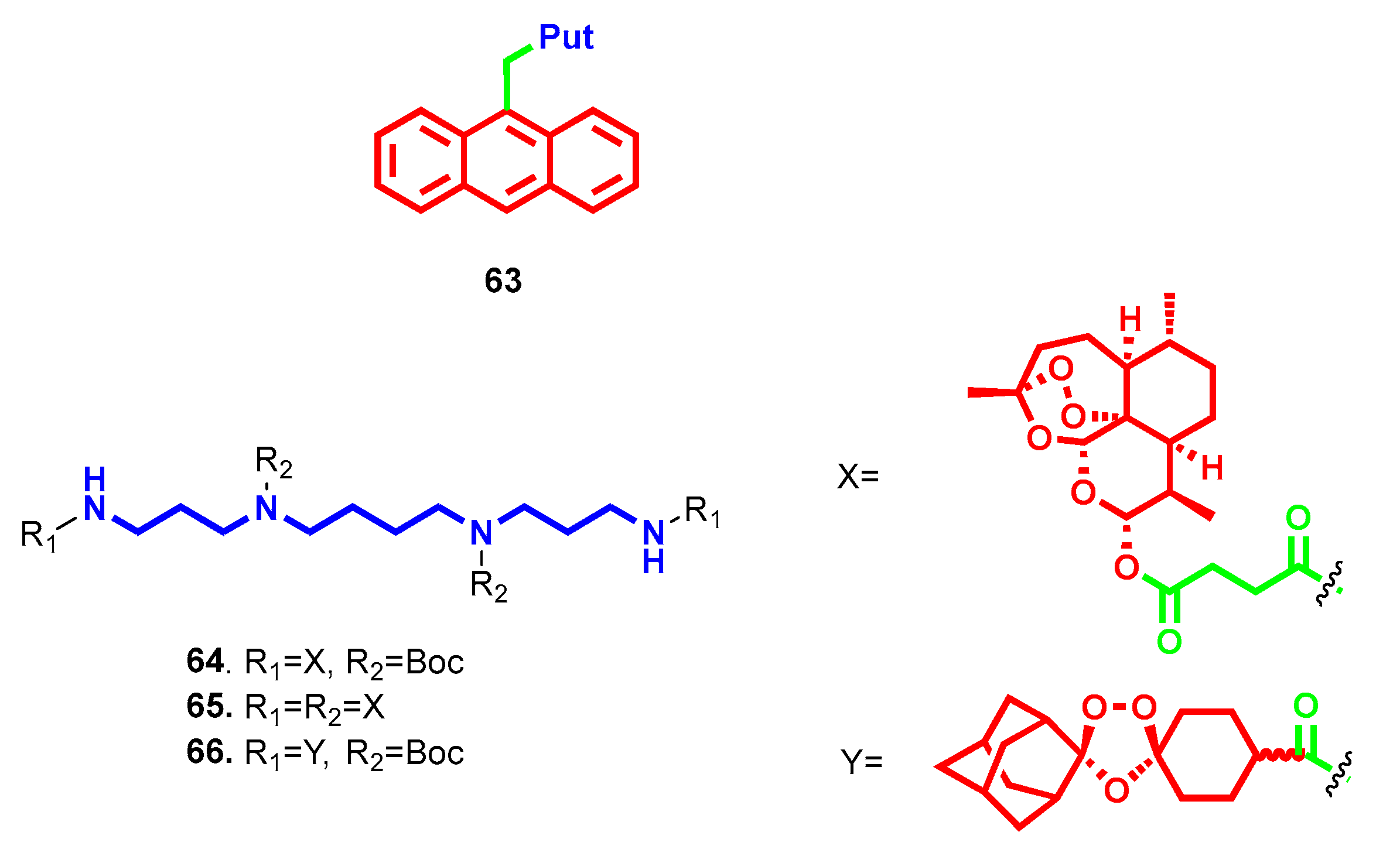{kind=link}
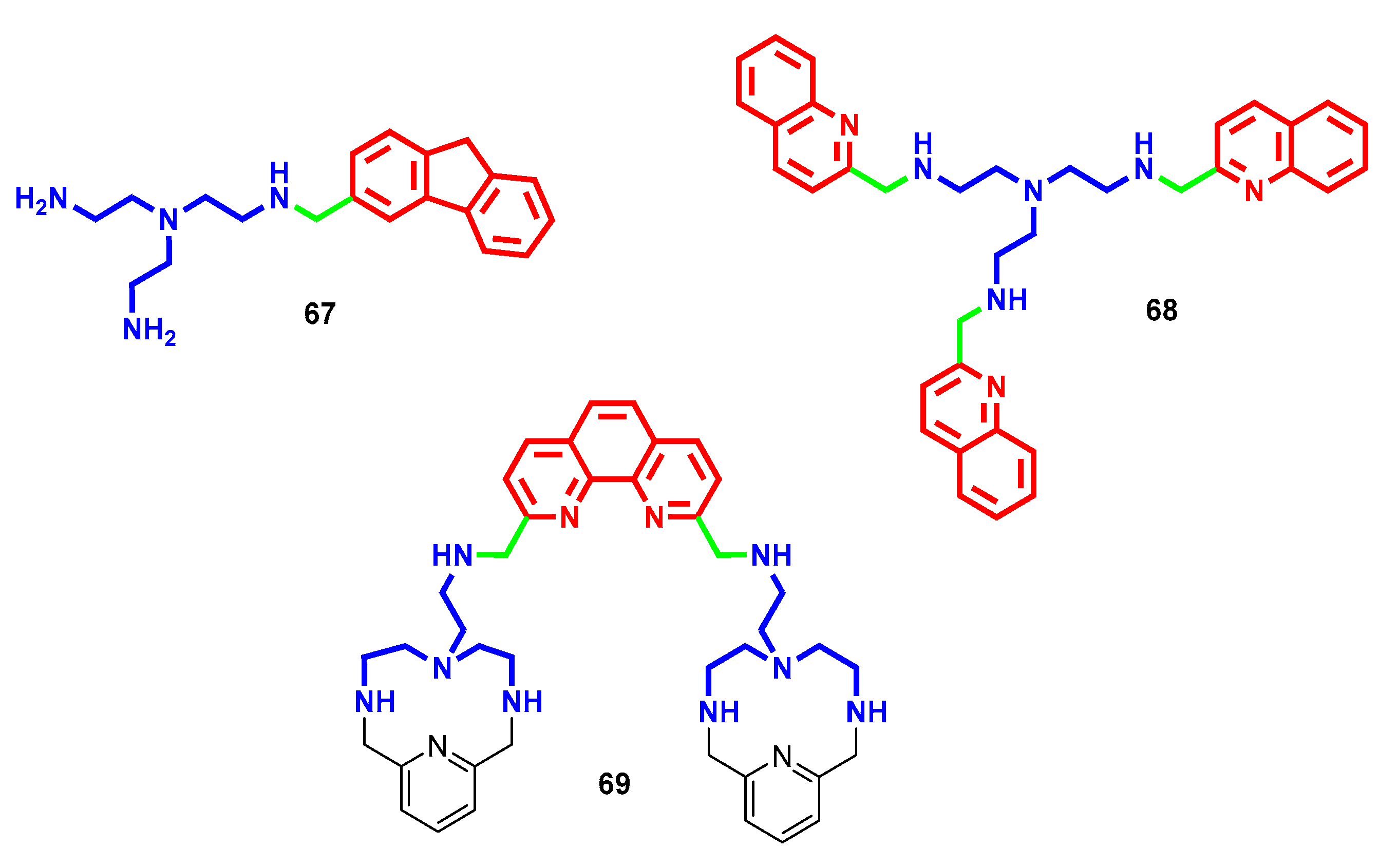{kind=link}
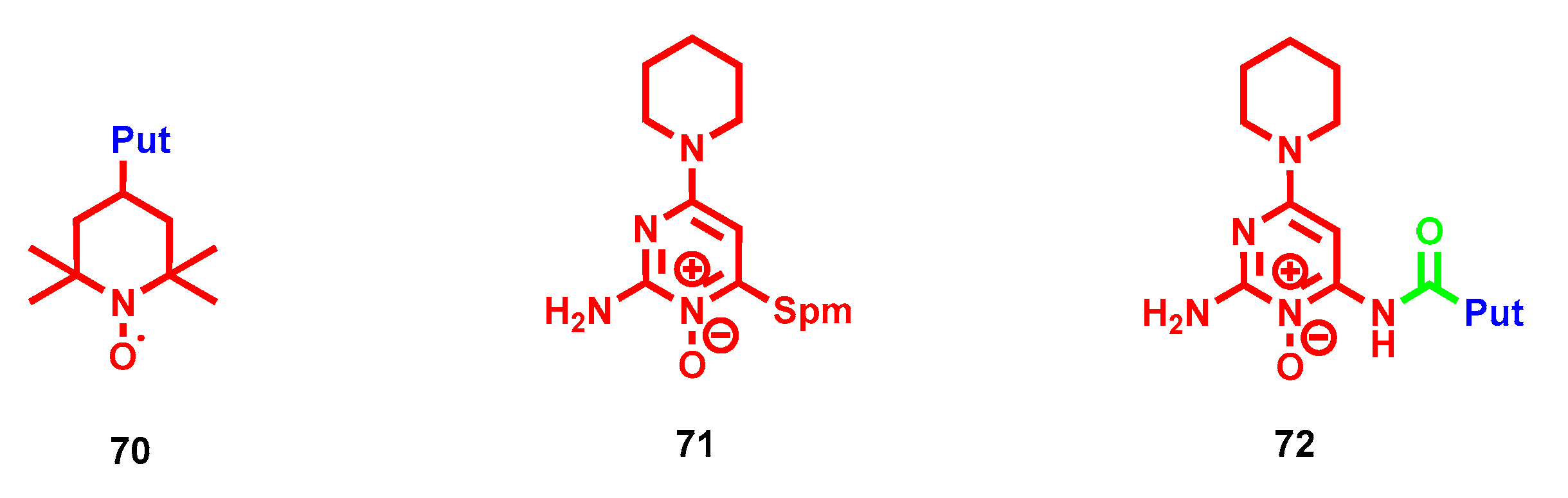{kind=link}
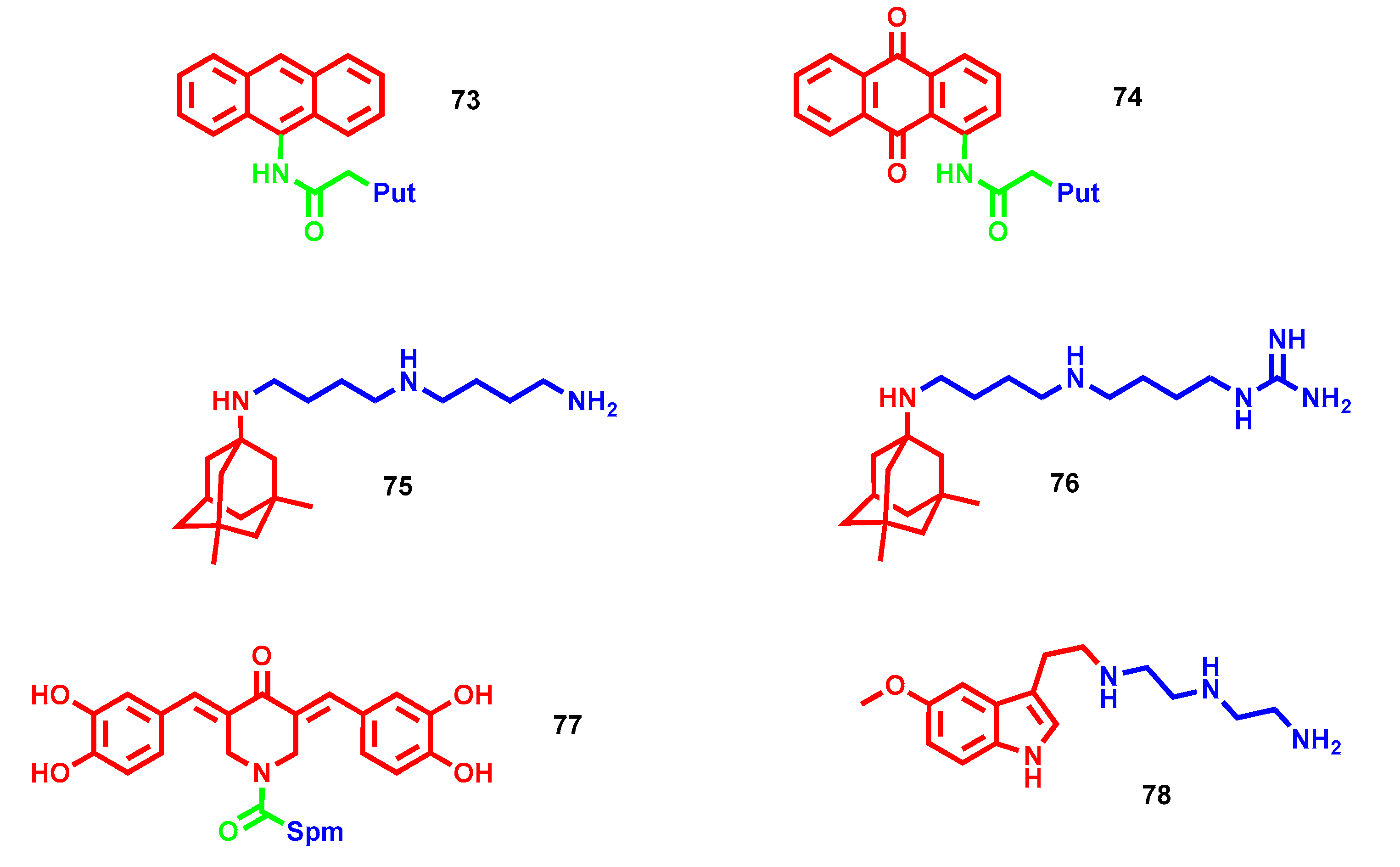{kind=link}
Abstract
:1. Introduction
2. Antitumor Agents
2.1. Natural Scaffolds
2.2. Naphthalimides and Derivatives
2.3. Miscellaneous
3. Antimicrobial Agents
3.1. Antibiotics
3.2. Antiprotozoa
4. Antioxidant
5. Neuroprotective
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chen, D.; Shao, Q.; Yin, L.; Younis, A.; Zheng, B. Polyamine Function in Plants: Metabolism, Regulation on Development, and Roles in Abiotic Stress Responses. Front. Plant Sci. 2019, 9, 1945. [Google Scholar] [CrossRef]
- Michael, A. Polyamine function in archaea and bacteria. J. Biol. Chem. 2018, 293, 18693–18701. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, K.; Kashiwagi, K. The functional role of polyamines in eukaryotic cells. Int. J. Biochem. Cell Biol. 2019, 107, 104–115. [Google Scholar] [CrossRef]
- Pegg, A. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef] [Green Version]
- Casero, R.A.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Soda, K.; Kano, Y.; Chiba, F.; Koizumi, K.; Miyaki, Y. Increased Polyamine Intake Inhibits Age-Associated Alteration in Global DNA Methylation and 1,2-Dimethylhydrazine-Induced Tumorigenesis. PLoS ONE 2013, 8, e64357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casero, R.; Marton, L. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, K.; Barone, S.; Soleimani, M. Polyamines and Their Metabolism: From the Maintenance of Physiological Homeostasis to the Mediation of Disease. Med. Sci. 2022, 10, 38. [Google Scholar] [CrossRef]
- Negrel, S.; Brunel, J. Synthesis and Biological Activities of Naturally Functionalized Polyamines: An Overview. Curr. Med. Chem. 2021, 28, 3406–3448. [Google Scholar] [CrossRef]
- Menzi, M.; Lightfoot, H.L.; Hall, J. Polyamine-oligonucleotide conjugates: A promising direction for nucleic acid tools and therapeutics. Future Med. Chem. 2015, 7, 1733–1749. [Google Scholar] [CrossRef]
- Vanhoutte, R.; Kahler, J.P.; Martin, S.; van Veen, S.; Verhelst, S.H.L. Clickable Polyamine Derivatives as Chemical Probes for the Polyamine Transport System. ChemBioChem 2018, 19, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Novita Sari, I.; Setiawan, T.; Seock Kim, K.; Toni Wijaya, Y.; Won Cho, K.; Young Kwon, H. Metabolism and function of polyamines in cancer progression. Cancer Lett. 2021, 519, 91–104. [Google Scholar] [CrossRef]
- Li, J.; Meng, Y.; Wu, X.; Sun, Y. Polyamines and related signaling pathways in cancer. Cancer Cell Int. 2020, 20, 539. [Google Scholar] [CrossRef] [PubMed]
- Damiani, E.; Wallace, H.M. Polyamines and Cancer. In Polyamines; Alcazar, R., Tiburcio, A.F., Eds.; Springer Protocols: Berlin/Heidelberg, Germany, 2017; pp. 469–488. [Google Scholar]
- Casero, R.A.; Woster, P.M. Recent advances in the development of polyamine analogues as antitumor agents. J. Med. Chem. 2009, 52, 4551–4573. [Google Scholar] [CrossRef] [Green Version]
- Barret, J.M.; Kruczynski, A.; Vispé, S.; Annereau, J.P.; Brel, V.; Guminski, Y.; Delcros, J.G.; Lansiaux, A.; Guilbaud, N.; Imbert, T.; et al. F14512, a potent antitumor agent targeting topoisomerase II vectored into cancer cells via the polyamine transport system. Cancer Res. 2008, 68, 9845–9853. [Google Scholar] [CrossRef] [Green Version]
- Tierny, D.; Serres, F.; Segaoula, Z.; Bemelmans, I.; Bouchaert, E.; Pétain, A.; Brel, V.; Couffin, S.; Marchal, T.; Nguyen, L.; et al. Phase I Clinical Pharmacology Study of F14512, a New Polyamine-Vectorized Anticancer Drug, in Naturally Occurring Canine Lymphoma. Clin. Cancer Res. 2015, 21, 5314–5323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leary, A.; Le Tourneau, C.; Varga, A.; Sablin, M.P.; Gomez-Roca, C.; Guilbaud, N.; Petain, A.; Pavlyuk, M.; Delord, J.P. Phase I dose-escalation study of F14512, a polyamine-vectorized topoisomerase II inhibitor, in patients with platinum-refractory or resistant ovarian cancer. Investig. New Drugs 2019, 37, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Kruczynski, A.; Pillon, A.; Créancier, L.; Vandenberghe, I.; Gomes, B.; Brel, V.; Fournier, E.; Annereau, J.P.; Currie, E.; Guminski, Y.; et al. F14512, a polyamine-vectorized anti-cancer drug, currently in clinical trials exhibits a marked preclinical anti-leukemic activity. Leukemia 2013, 27, 2139–2148. [Google Scholar] [CrossRef]
- Gentry, A.C.; Pitts, S.L.; Jablonsky, M.J.; Bailly, C.; Graves, D.E.; Osheroff, N. Interactions between the etoposide derivative F14512 and human type II topoisomerases: Implications for the C4 spermine moiety in promoting enzyme-mediated DNA cleavage. Biochemistry 2011, 50, 3240–3249. [Google Scholar] [CrossRef] [Green Version]
- Palermo, G.; Minniti, E.; Greco, M.L.; Riccardi, L.; Simoni, E.; Convertino, M.; Marchetti, C.; Rosini, M.; Sissi, C.; Minarini, A.; et al. An optimized polyamine moiety boosts the potency of human type II topoisomerase poisons as quantified by comparative analysis centered on the clinical candidate F14512. Chem. Commun. 2015, 51, 14310–14313. [Google Scholar] [CrossRef] [Green Version]
- Oviatt, A.A.; Kuriappan, J.A.; Minniti, E.; Vann, K.R.; Onuorah, P.; Minarini, A.; De Vivo, M.; Osheroff, N. Polyamine-containing etoposide derivatives as poisons of human type II topoisomerases: Differential effects on topoisomerase IIα and IIβ. Bioorg. Med. Chem. Lett. 2018, 28, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
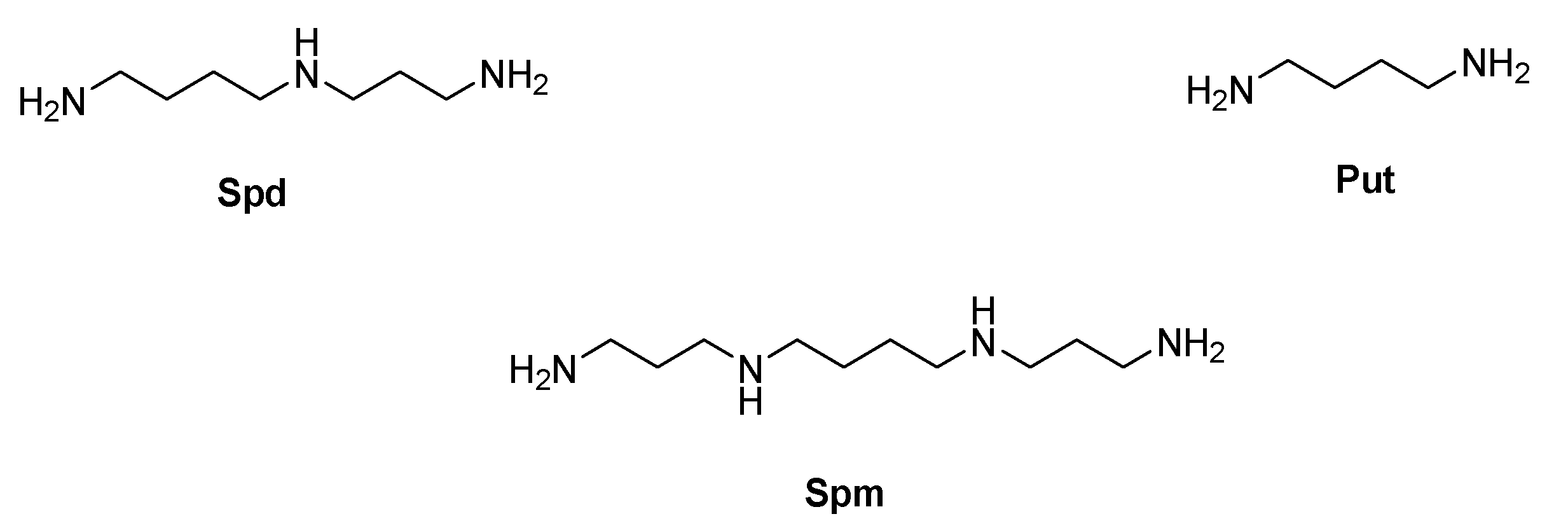
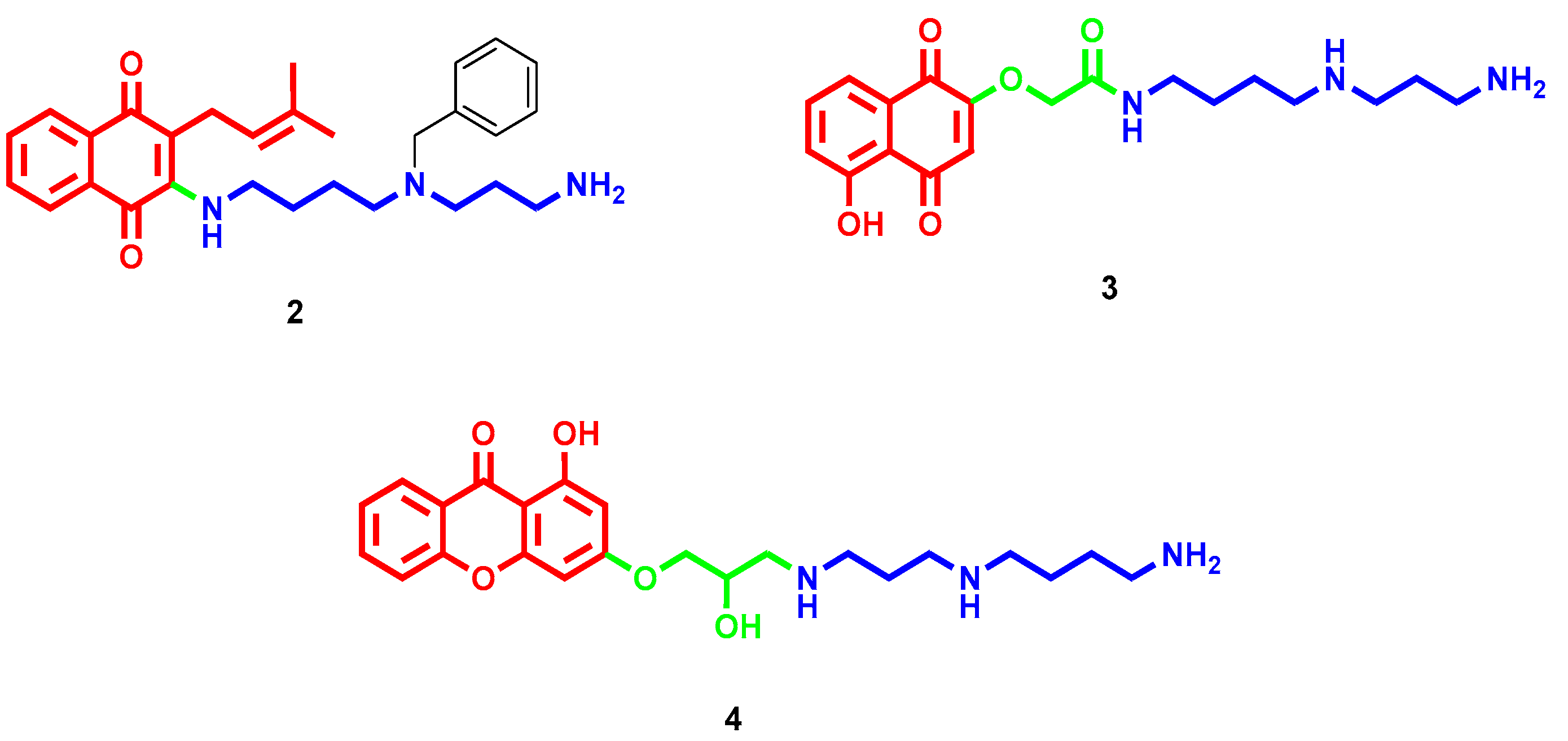
- Romão, L.; do Canto, V.P.; Netz, P.A.; Moura-Neto, V.; Pinto, Â.; Follmer, C. Conjugation with polyamines enhances the antitumor activity of naphthoquinones against human glioblastoma cells. Anticancer Drugs 2018, 29, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Manickam, M.; Boggu, P.R.; Pillaiyar, T.; Nam, Y.J.; Abdullah, M.; Lee, S.J.; Kang, J.S.; Jung, S.H. Design, synthesis and anticancer activity of 2-amidomethoxy-1,4-naphthoquinones and its conjugates with Biotin/polyamine. Bioorg. Med. Chem. Lett. 2021, 31, 127685. [Google Scholar] [CrossRef]
- Minniti, E.; Byl, J.A.W.; Riccardi, L.; Sissi, C.; Rosini, M.; De Vivo, M.; Minarini, A.; Osheroff, N. Novel xanthone-polyamine conjugates as catalytic inhibitors of human topoisomerase IIα. Bioorg. Med. Chem. Lett. 2017, 27, 4687–4693. [Google Scholar] [CrossRef]
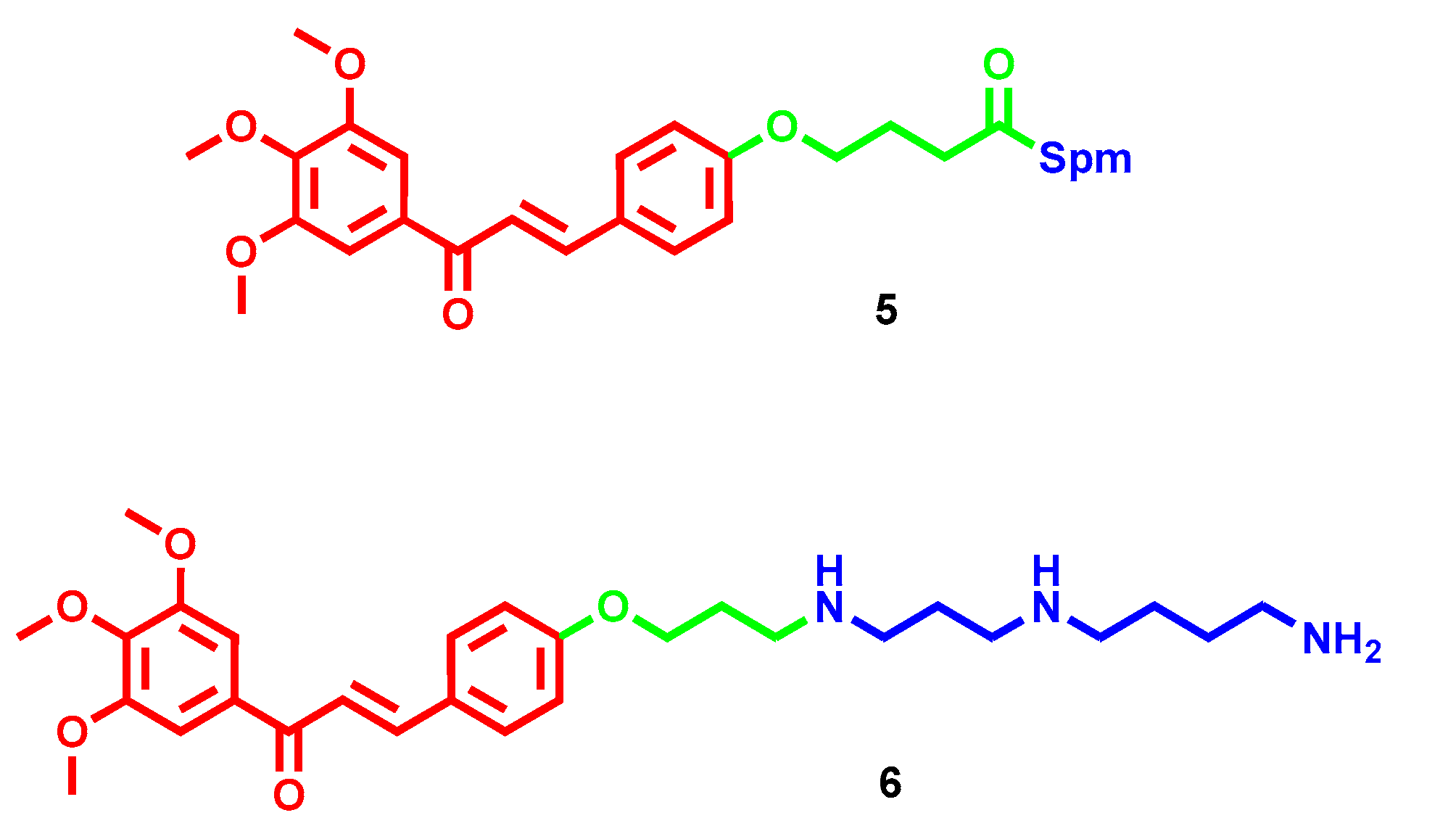
- Rioux, B.; Pouget, C.; Fidanzi-Dugas, C.; Gamond, A.; Laurent, A.; Semaan, J.; Pinon, A.; Champavier, Y.; Léger, D.Y.; Liagre, B.; et al. Design and multi-step synthesis of chalcone-polyamine conjugates as potent antiproliferative agents. Bioorg. Med. Chem. Lett. 2017, 27, 4354–4357. [Google Scholar] [CrossRef]
- Rioux, B.; Pinon, A.; Gamond, A.; Martin, F.; Laurent, A.; Champavier, Y.; Barette, C.; Liagre, B.; Fagnère, C.; Sol, V.; et al. Synthesis and biological evaluation of chalcone-polyamine conjugates as novel vectorized agents in colorectal and prostate cancer chemotherapy. Eur. J. Med. Chem. 2021, 222, 113586. [Google Scholar] [CrossRef]
- Li, Q.; Zhai, Y.; Luo, W.; Zhu, Z.; Zhang, X.; Xie, S.; Hong, C.; Wang, Y.; Su, Y.; Zhao, J.; et al. Synthesis and biological properties of polyamine modified flavonoids as hepatocellular carcinoma inhibitors. Eur. J. Med. Chem. 2016, 121, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhu, Z.X.; Zhang, X.; Luo, W.; Chang, L.P.; Chen, S.; Wang, Y.X.; Xie, S.Q.; Chang, C.C.; Wang, C.J. The lead optimization of the polyamine conjugate of flavonoid with a naphthalene motif: Synthesis and biological evaluation. Eur. J. Med. Chem. 2018, 146, 564–576. [Google Scholar] [CrossRef]
- Dai, F.; Li, Q.; Wang, Y.; Ge, C.; Feng, C.; Xie, S.; He, H.; Xu, X.; Wang, C. Design, Synthesis, and Biological Evaluation of Mitochondria-Targeted Flavone-Naphthalimide-Polyamine Conjugates with Antimetastatic Activity. J. Med. Chem. 2017, 60, 2071–2083. [Google Scholar] [CrossRef]
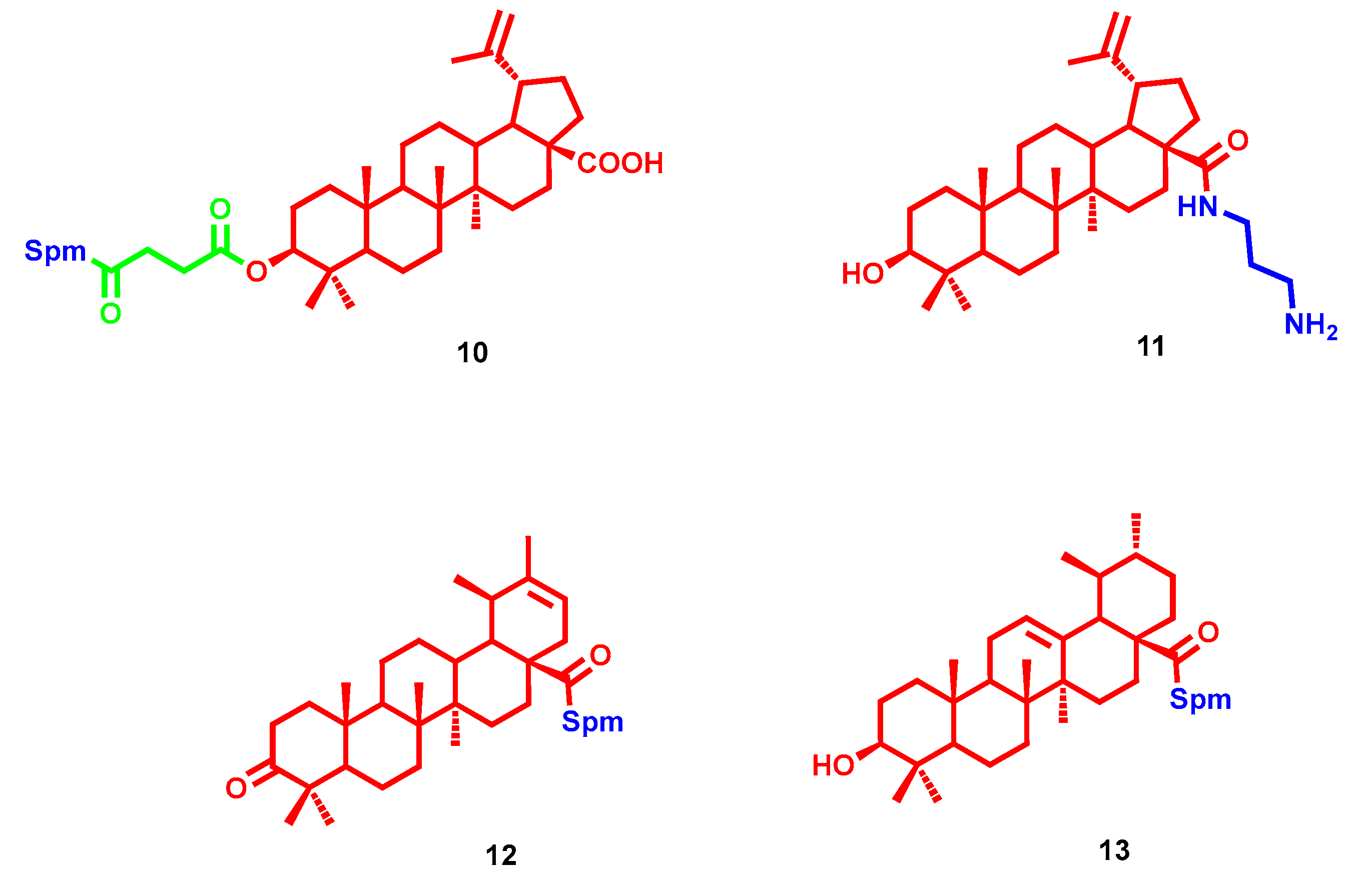
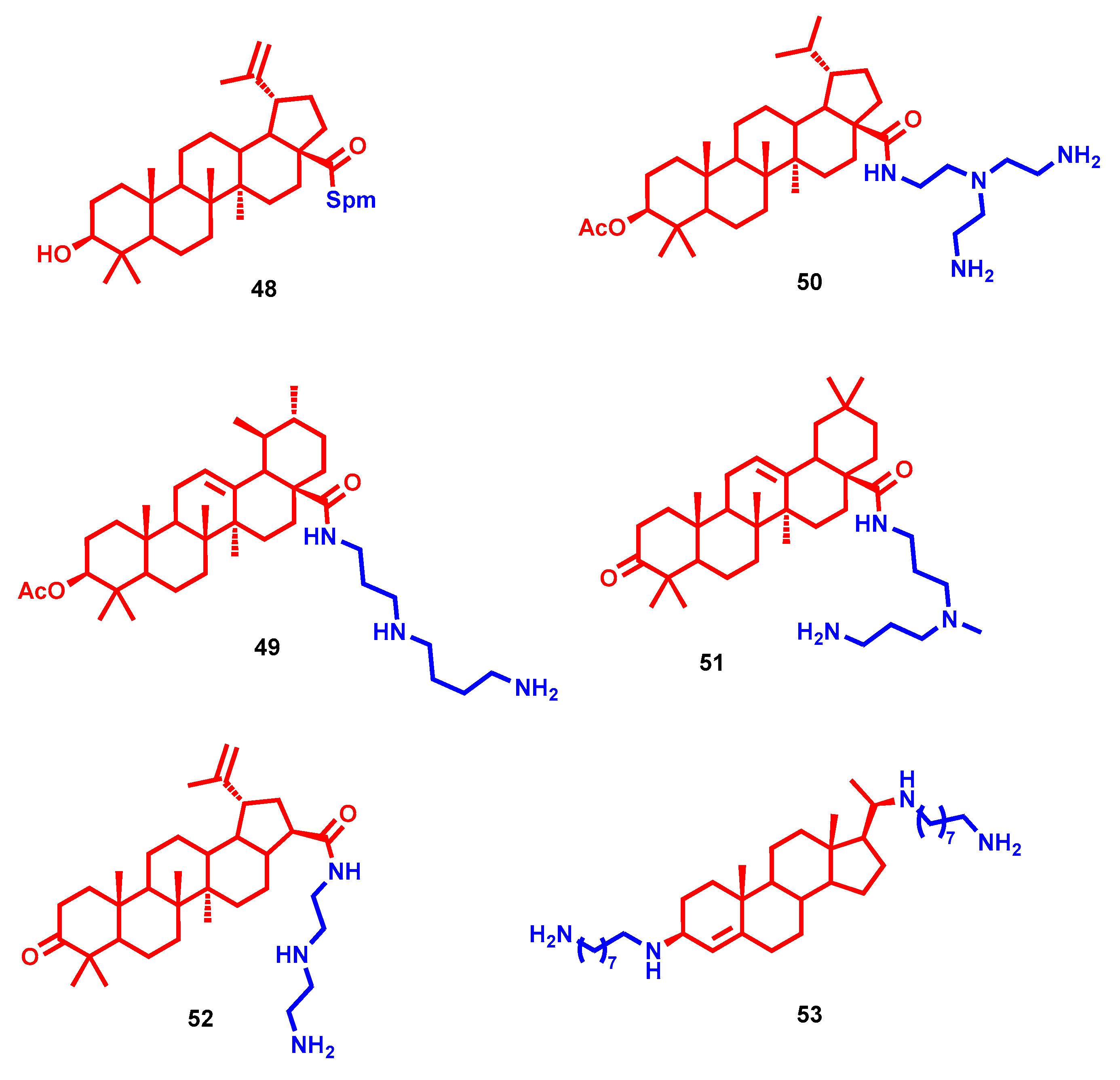
- Bildziukevich, U.; Vida, N.; Rárová, L.; Kolář, M.; Šaman, D.; Havlíček, L.; Drašar, P.; Wimmer, Z. Polyamine derivatives of betulinic acid and β-sitosterol: A comparative investigation. Steroids 2015, 100, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, O.B.; Giniyatullina, G.V.; Mustafin, A.G.; Babkov, D.A.; Sokolova, E.V.; Spasov, A.A. Evaluation of Cytotoxicity and α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids. Molecules 2020, 25, 4833. [Google Scholar] [CrossRef]
- Bildziukevich, U.; Malík, M.; Özdemir, Z.; Rárová, L.; Janovská, L.; Šlouf, M.; Šaman, D.; Šarek, J.; Nonappa; Wimmer, Z. Spermine amides of selected triterpenoid acids: Dynamic supramolecular system formation influences the cytotoxicity of the drugs. J. Mater. Chem. B 2020, 8, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.; Knorrscheidt, A.; Flemming, F.; Wiemann, J.; Sommerwerk, S.; Pavel, I.Z.; Al-Harrasi, A.; Csuk, R. Synthesis and proapoptotic activity of oleanolic acid derived amides. Bioorg. Chem. 2016, 68, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Kahnt, M.; Fischer Née Heller, L.; Al-Harrasi, A.; Csuk, R. Ethylenediamine Derived Carboxamides of Betulinic and Ursolic Acid as Potential Cytotoxic Agents. Molecules 2018, 23, 2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahnt, M.; Loesche, A.; Serbian, I.; Hoenke, S.; Fischer, L.; Al-Harrasi, A.; Csuk, R. The cytotoxicity of oleanane derived aminocarboxamides depends on their aminoalkyl substituents. Steroids 2019, 149, 108422. [Google Scholar] [CrossRef]
- Vida, N.; Svobodová, H.; Rárová, L.; Drašar, P.; Saman, D.; Cvačka, J.; Wimmer, Z. Polyamine conjugates of stigmasterol. Steroids 2012, 77, 1212–1218. [Google Scholar] [CrossRef]
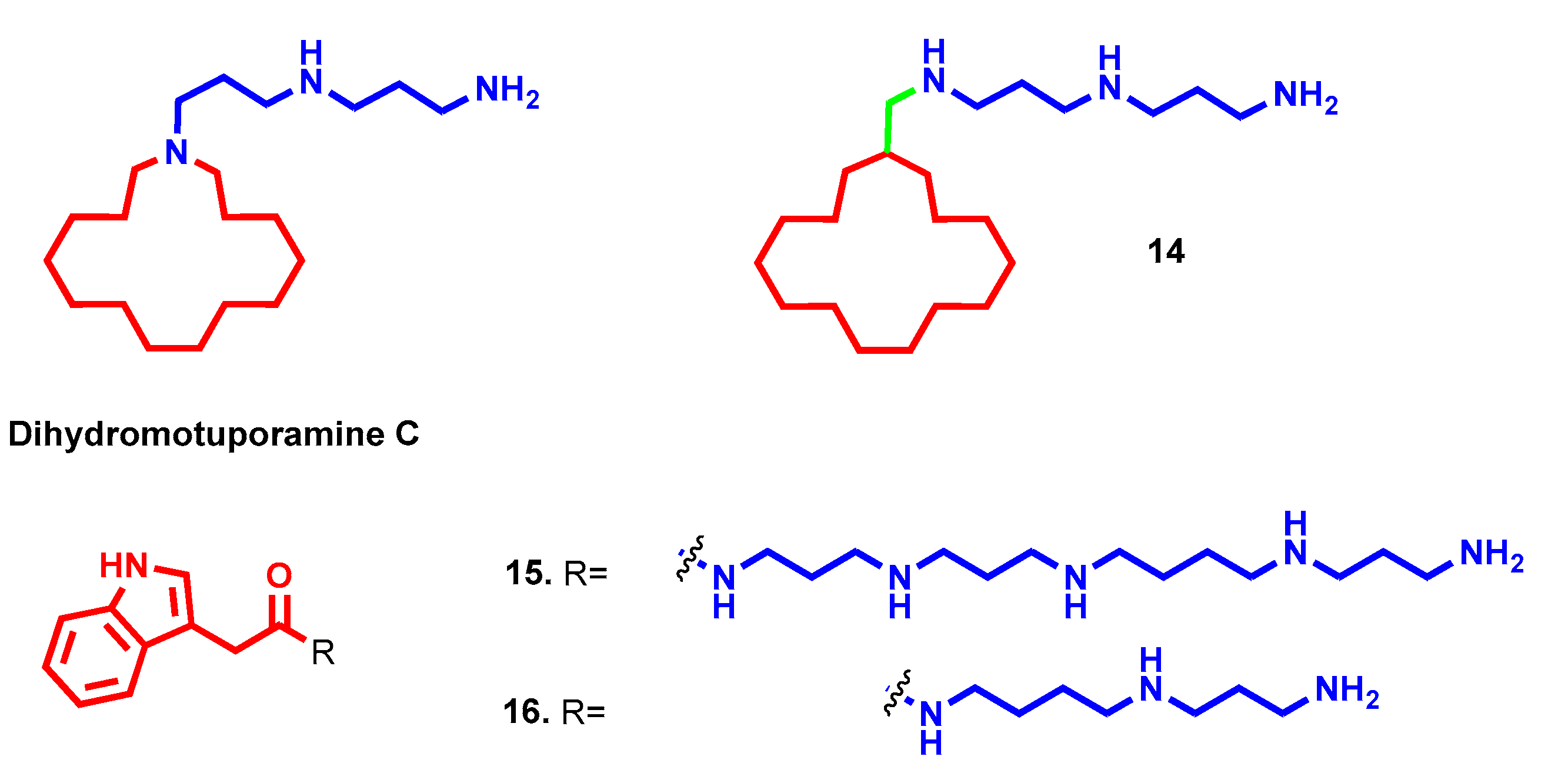
- Muth, A.; Pandey, V.; Kaur, N.; Wason, M.; Baker, C.; Han, X.; Johnson, T.R.; Altomare, D.A.; Phanstiel, O. Synthesis and biological evaluation of antimetastatic agents predicated upon dihydromotuporamine C and its carbocyclic derivatives. J. Med. Chem. 2014, 57, 4023–4034. [Google Scholar] [CrossRef]
- Skruber, K.; Chaplin, K.J.; Phanstiel, O. Synthesis and Bioevaluation of Macrocycle-Polyamine Conjugates as Cell Migration Inhibitors. J. Med. Chem. 2017, 60, 8606–8619. [Google Scholar] [CrossRef]
- Borselli, D.; Blanchet, M.; Bolla, J.M.; Muth, A.; Skruber, K.; Phanstiel, O.; Brunel, J.M. Motuporamine Derivatives as Antimicrobial Agents and Antibiotic Enhancers against Resistant Gram-Negative Bacteria. ChemBioChem 2017, 18, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Vassileiou, C.; Kalantzi, S.; Vachlioti, E.; Athanassopoulos, C.M.; Koutsakis, C.; Piperigkou, Z.; Karamanos, N.; Stivarou, T.; Lymberi, P.; Avgoustakis, K.; et al. New Analogs of Polyamine Toxins from Spiders and Wasps: Liquid Phase Fragment Synthesis and Evaluation of Antiproliferative Activity. Molecules 2022, 27, 447. [Google Scholar] [CrossRef]
- Kamal, A.; Bolla, N.R.; Srikanth, P.S.; Srivastava, A.K. Naphthalimide derivatives with therapeutic characteristics: A patent review. Expert Opin. Ther. Pat. 2013, 23, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Veale, E.B.; Phelan, C.M.; Murphy, S.A.; Tocci, G.M.; Gillespie, L.J.; Frimannsson, D.O.; Kelly, J.M.; Gunnlaugsson, T. Recent advances in the development of 1,8-naphthalimide based DNA targeting binders, anticancer and fluorescent cellular imaging agents. Chem. Soc. Rev. 2013, 42, 1601–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
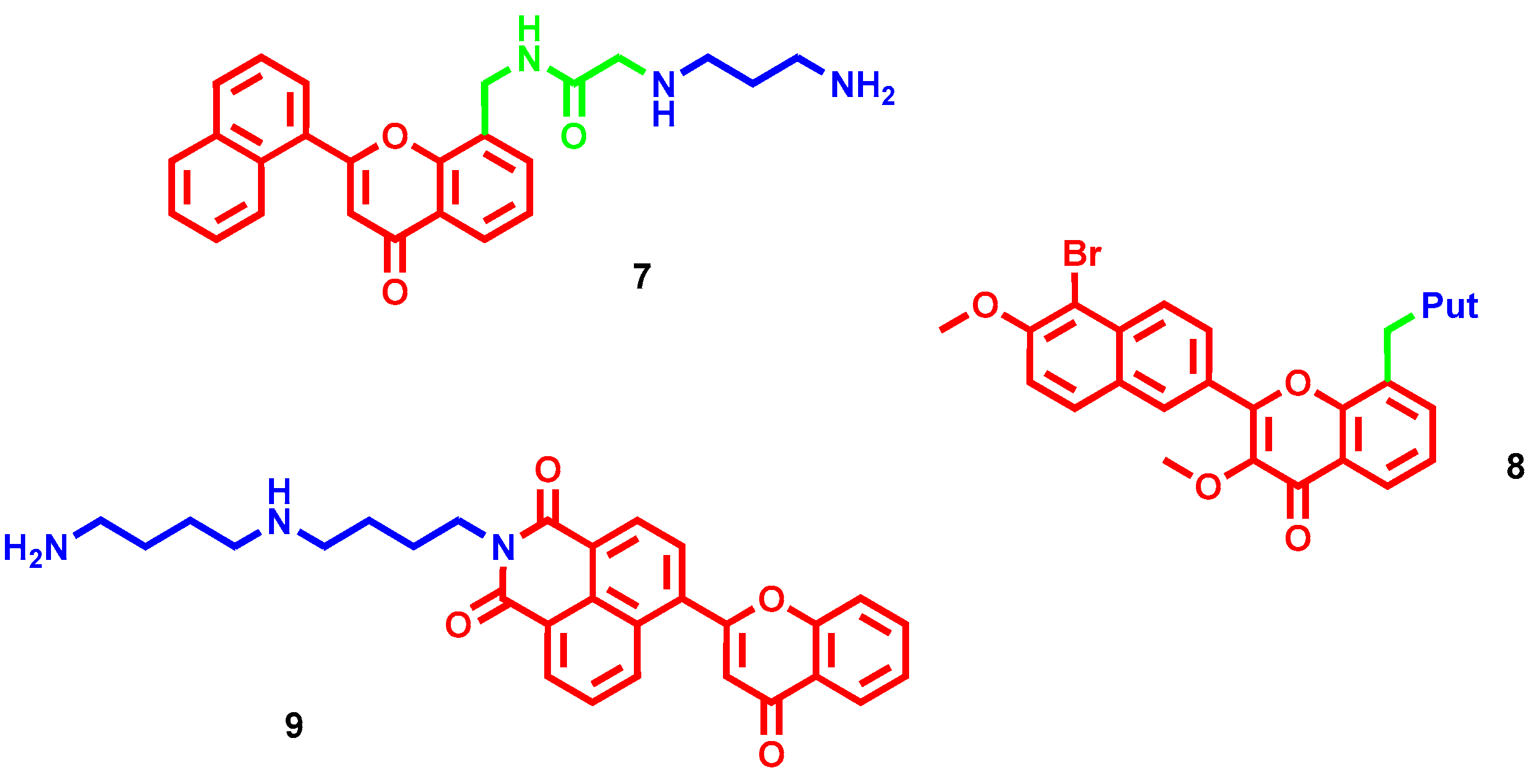
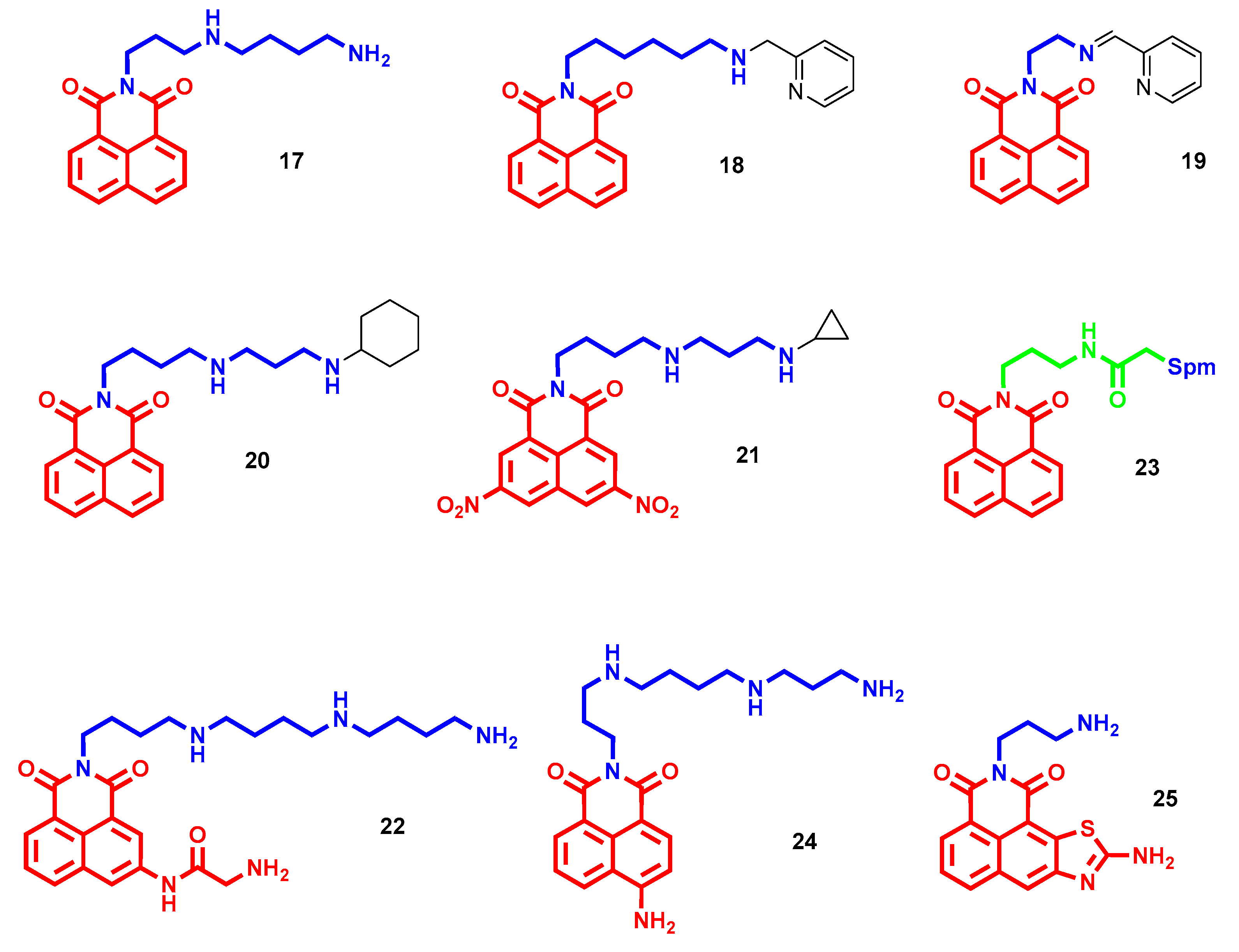
- Tian, Z.; Li, J.; Li, Q.; Zang, F.; Zhao, Z.; Wang, C. Study on the Synthesis, Biological Activity and Spectroscopy of Naphthalimide-Diamine Conjugates. Molecules 2014, 19, 7646–7668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Z.; Huang, Y.; Zhang, Y.; Song, L.; Qiao, Y.; Xu, X.; Wang, C. Spectroscopic and molecular modeling methods to study the interaction between naphthalimide-polyamine conjugates and DNA. J. Photochem. Photobiol. B-Biol. 2016, 158, 1–15. [Google Scholar] [CrossRef]
- Seliga, R.; Pilatova, M.; Sarissky, M.; Viglasky, V.; Walko, M.; Mojzis, J. Novel naphthalimide polyamine derivatives as potential antitumor agents. Mol. Biol. Rep. 2013, 40, 4129–4137. [Google Scholar] [CrossRef]
- Dai, F.; He, H.; Xu, X.; Chen, S.; Wang, C.; Feng, C.; Tian, Z.; Dong, H.; Xie, S. Synthesis and biological evaluation of naphthalimide-polyamine conjugates modified by alkylation as anticancer agents through p53 pathway. Bioorg. Chem. 2018, 77, 16–24. [Google Scholar] [CrossRef]
- Ma, J.; Li, Y.; Li, L.; Yue, K.; Liu, H.; Wang, J.; Xi, Z.; Shi, M.; Zhao, S.; Ma, Q.; et al. A Polyamine-Based Dinitro-Naphthalimide Conjugate as Substrates for Polyamine Transporters Preferentially Accumulates in Cancer Cells and Minimizes Side Effects. Front. Chem. 2020, 8, 166. [Google Scholar] [CrossRef]
- Ma, J.; Li, L.; Yue, K.; Zhang, Z.; Su, S.; Chen, Y.; Yu, L.; Zhang, P.; Ma, R.; Li, Y.; et al. A naphthalimide-polyamine conjugate preferentially accumulates in hepatic carcinoma metastases as a lysosome-targeted antimetastatic agent. Eur. J. Med. Chem. 2021, 221, 113469. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.; Zhang, J.; Xie, S.; Wang, C.; Wu, Y. Synthesis and Biological Evaluation of Novel Aromatic Imide-Polyamine Conjugates. Molecules 2016, 21, 1637. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Li, Q.; Zhang, Y.H.; Tian, Z.Y.; Ma, H.X.; Zhao, J.; Xie, S.Q.; Wang, C.J. Antitumor effects and preliminary systemic toxicity of ANISpm in vivo and in vitro. Anticancer Drugs 2013, 24, 32–42. [Google Scholar] [CrossRef]
- Ge, C.; Chang, L.; Zhao, Y.; Chang, C.; Xu, X.; He, H.; Wang, Y.; Dai, F.; Xie, S.; Wang, C. Design, Synthesis and Evaluation of Naphthalimide Derivatives as Potential Anticancer Agents for Hepatocellular Carcinoma. Molecules 2017, 22, 342. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Wang, B.; Jia, D.; Yang, X.; Huang, Y. Synthesis, electrochemistry, DNA binding and in vitro cytotoxic activity of tripodal ferrocenyl bis-naphthalimide derivatives. J. Inorg. Biochem. 2021, 219, 111425. [Google Scholar] [CrossRef] [PubMed]
- Rong, R.; Sun, Q.; Ma, C.; Chen, B.; Wang, W.; Wang, Z.; Wang, K.; Cao, Z.; Li, X. Development of novel bis-naphthalimide derivatives and their anticancer properties. MedChemComm 2016, 7, 679–685. [Google Scholar] [CrossRef]
- Rong, R.; Wang, S.; Liu, X.; Li, R.; Wang, K.; Cao, Z.; Li, X. Lysosomes-targeting imaging and anticancer properties of novel bis-naphthalimide derivatives. Bioorganic Med. Chem. Lett. 2018, 28, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, J.; Liu, S.; Song, L.; Wang, H.; Wang, Q.; Liang, S.; Lu, L.; Wei, J.; Huang, R.; et al. Design, synthesis, and antitumor evaluation of morpholine substituted bisnaphthalimides as DNA targeting agents. Bioorganic Med. Chem. Lett. 2023, 85, 129218. [Google Scholar] [CrossRef]
- Tumiatti, V.; Milelli, A.; Minarini, A.; Micco, M.; Gasperi Campani, A.; Roncuzzi, L.; Baiocchi, D.; Marinello, J.; Capranico, G.; Zini, M.; et al. Design, synthesis, and biological evaluation of substituted naphthalene imides and diimides as anticancer agent. J. Med. Chem. 2009, 52, 7873–7877. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Zhao, J.; Xie, S.; Wang, C. Nonhematotoxic naphthalene diimide modified by polyamine: Synthesis and biological evaluation. J. Med. Chem. 2012, 55, 3502–3512. [Google Scholar] [CrossRef]
- Milelli, A.; Tumiatti, V.; Micco, M.; Rosini, M.; Zuccari, G.; Raffaghello, L.; Bianchi, G.; Pistoia, V.; Fernando Díaz, J.; Pera, B.; et al. Structure-activity relationships of novel substituted naphthalene diimides as anticancer agents. Eur. J Med. Chem. 2012, 57, 417–428. [Google Scholar] [CrossRef]
- Parise, A.; Milelli, A.; Tumiatti, V.; Minarini, A.; Neviani, P.; Zuccari, G. Preparation, characterization and in vitro evaluation of sterically stabilized liposome containing a naphthalenediimide derivative as anticancer agent. Drug Deliv. 2015, 22, 590–597. [Google Scholar] [CrossRef] [Green Version]
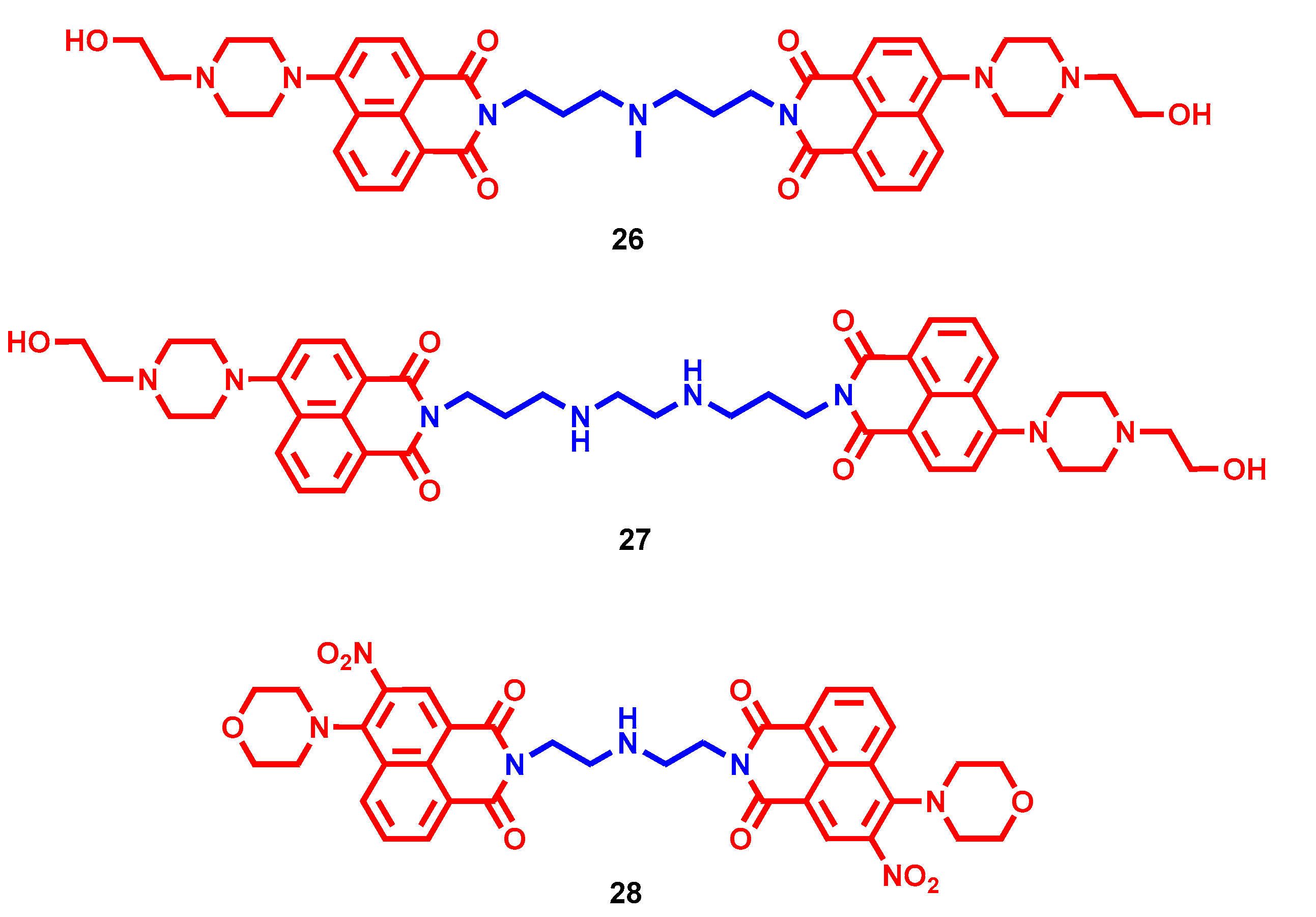
- Milelli, A.; Marchetti, C.; Greco, M.L.; Moraca, F.; Costa, G.; Turrini, E.; Catanzaro, E.; Betari, N.; Calcabrini, C.; Sissi, C.; et al. Naphthalene diimide-polyamine hybrids as antiproliferative agents: Focus on the architecture of the polyamine chains. Eur. J. Med. Chem. 2017, 128, 107–122. [Google Scholar] [CrossRef]
- Pasini, A.; Marchetti, C.; Sissi, C.; Cortesi, M.; Giordano, E.; Minarini, A.; Milelli, A. Novel Polyamine-Naphthalene Diimide Conjugates Targeting Histone Deacetylases and DNA for Cancer Phenotype Reprogramming. ACS Med. Chem. Lett. 2017, 8, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Recagni, M.; Greco, M.; Milelli, A.; Minarini, A.; Zaffaroni, N.; Folini, M.; Sissi, C. Distinct biological responses of metastatic castration resistant prostate cancer cells upon exposure to G-quadruplex interacting naphthalenediimide derivatives. Eur. J. Med. Chem. 2019, 177, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Minarini, A.; Tumiatti, V.; Moraca, F.; Parrotta, L.; Alcaro, S.; Rigo, R.; Sissi, C.; Gunaratnam, M.; Ohnmacht, S.A.; et al. Macrocyclic naphthalene diimides as G-quadruplex binders. Bioorg. Med. Chem. 2015, 23, 3819–3830. [Google Scholar] [CrossRef]
- Collie, G.; Promontorio, R.; Hampel, S.; Micco, M.; Neidle, S.; Parkinson, G. Structural Basis for Telomeric G-Quadruplex Targeting by Naphthalene Diimide Ligands. J. Am. Chem. Soc. 2012, 134, 2723–2731. [Google Scholar] [CrossRef]
- Ohnmacht, S.; Marchetti, C.; Gunaratnam, M.; Besser, R.; Haider, S.; Di Vita, G.; Lowe, H.; Mellinas-Gomez, M.; Diocou, S.; Robson, M.; et al. A G-quadruplex-binding compound showing anti-tumour activity in an in vivo model for pancreatic cancer. Sci. Rep. 2015, 5, 11385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, C.; Zyner, K.; Ohnmacht, S.; Robson, M.; Haider, S.; Morton, J.; Marsico, G.; Vo, T.; Laughlin-Toth, S.; Ahmed, A.; et al. Targeting Multiple Effector Pathways in Pancreatic Ductal Adenocarcinoma with a G-Quadruplex-Binding Small Molecule. J. Med. Chem. 2018, 61, 2500–2517. [Google Scholar] [CrossRef]
- Ahmed, A.; Angell, R.; Oxenford, S.; Worthington, J.; Williams, N.; Barton, N.; Fowler, T.; O’Flynn, D.; Sunose, M.; McConville, M.; et al. Asymmetrically Substituted Quadruplex-Binding Naphthalene Diimide Showing Potent Activity in Pancreatic Cancer Models. ACS Med. Chem. Lett. 2020, 11, 1634–1644. [Google Scholar] [CrossRef]
- Vo, T.; Oxenford, S.; Angell, R.; Marchetti, C.; Ohnmacht, S.; Wilson, W.; Neidle, S. Substituted Naphthalenedimide Compounds Bind Selectively to Two Human Quadruplex Structures with Parallel Topology. ACS Med. Chem. Lett. 2020, 11, 991–999. [Google Scholar] [CrossRef]
- Micco, M.; Collie, G.; Dale, A.; Ohnmacht, S.; Pazitna, I.; Gunaratnam, M.; Reszka, A.; Neidle, S. Structure-Based Design and Evaluation of Naphthalene Diimide G-Quadruplex Ligands as Telomere Targeting Agents in Pancreatic Cancer Cells. J. Med. Chem. 2013, 56, 2959–2974. [Google Scholar] [CrossRef]
- Ahmed, A.; Marchetti, C.; Ohnmacht, S.; Neidle, S. A G-quadruplex-binding compound shows potent activity in human gemcitabine-resistant pancreatic cancer cells. Sci. Rep. 2020, 10, 12192. [Google Scholar] [CrossRef]
- Li, J.; Tian, R.; Ge, C.; Chen, Y.; Liu, X.; Wang, Y.; Yang, Y.; Luo, W.; Dai, F.; Wang, S.; et al. Discovery of the Polyamine Conjugate with Benzo[cd]indol-2(1H)-one as a Lysosome-Targeted Antimetastatic Agent. J. Med. Chem. 2018, 61, 6814–6829. [Google Scholar] [CrossRef]
- Szumilak, M.; Galdyszynska, M.; Dominska, K.; Bak-Sypien, I.I.; Merecz-Sadowska, A.; Stanczak, A.; Karwowski, B.T.; Piastowska-Ciesielska, A.W. Synthesis, Biological Activity and Preliminary in Silico ADMET Screening of Polyamine Conjugates with Bicyclic Systems. Molecules 2017, 22, 794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szulawska-Mroczek, A.; Szumilak, M.; Szczesio, M.; Olczak, A.; Nazarski, R.B.; Lewgowd, W.; Czyz, M.; Stanczak, A. Synthesis and biological evaluation of new bischromone derivatives with antiproliferative activity. Arch. Pharm. 2013, 346, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Szumilak, M.; Merecz, A.; Strek, M.; Stanczak, A.; Inglot, T.W.; Karwowski, B.T. DNA Interaction Studies of Selected Polyamine Conjugates. Int. J. Mol. Sci. 2016, 17, 1560. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ma, J.; Li, Y.; Yue, K.; Li, L.; Xi, Z.; Zhang, X.; Liu, J.; Feng, K.; Ma, Q.; et al. Polyamine-Based Pt(IV) Prodrugs as Substrates for Polyamine Transporters Preferentially Accumulate in Cancer Metastases as DNA and Polyamine Metabolism Dual-Targeted Antimetastatic Agents. J. Med. Chem. 2019, 62, 11324–11334. [Google Scholar] [CrossRef]
- Galiana-Rosello, C.; Aceves-Luquero, C.; Gonzalez, J.; Martinez-Camarena, A.; Villalonga, R.; de Mattos, S.; Soriano, C.; Llinares, J.; Garcia-Espana, E.; Villalonga, P.; et al. Toward a Rational Design of Polyamine-Based Zinc-Chelating Agents for Cancer Therapies. J. Med. Chem. 2020, 63, 1199–1215. [Google Scholar] [CrossRef]
- Kang, C.S.; Ren, S.; Sun, X.; Chong, H.S. Theranostic Polyaminocarboxylate-Cyanine-Transferrin Conjugate for Anticancer Therapy and Near-Infrared Optical Imaging. ChemMedChem 2016, 11, 2188–2193. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Sun, X.; Wang, H.; Nguyen, T.H.; Sadeghipour, N.; Xu, X.; Kang, C.S.; Liu, Y.; Xu, H.; Wu, N.; et al. Design, Synthesis, and Biological Evaluation of Polyaminocarboxylate Ligand-Based Theranostic Conjugates for Antibody-Targeted Cancer Therapy and Near-Infrared Optical Imaging. ChemMedChem 2018, 13, 2606–2617. [Google Scholar] [CrossRef]
- Corcé, V.; Renaud, S.; Cannie, I.; Julienne, K.; Gouin, S.G.; Loréal, O.; Gaboriau, F.; Deniaud, D. Synthesis and biological properties of Quilamines II, new iron chelators with antiproliferative activities. Bioconjug. Chem. 2014, 25, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.L.; Campilongo, R.; Casalino, M.; Micheli, G.; Colonna, B.; Prosseda, G. Polyamines: Emerging players in bacteria-host interactions. Int. J. Med. Microbiol. 2013, 303, 484–491. [Google Scholar] [CrossRef]
- Goytia, M.; Shafer, W.M. Polyamines can increase resistance of Neisseria gonorrhoeae to mediators of the innate human host defense. Infect Immun. 2010, 78, 3187–3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dela Vega, A.L.; Delcour, A.H. Polyamines decrease Escherichia coli outer membrane permeability. J. Bacteriol. 1996, 178, 3715–3721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, D.H.; Lu, C.D. Polyamine effects on antibiotic susceptibility in bacteria. Antimicrob. Agents Chemother. 2007, 51, 2070–2077. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.N.; Shinnar, A.E.; Noecker, L.A.; Chao, T.L.; Feibush, B.; Snyder, B.; Sharkansky, I.; Sarkahian, A.; Zhang, X.; Jones, S.R.; et al. Aminosterols from the dogfish shark Squalus acanthias. J. Nat. Prod. 2000, 63, 631–635. [Google Scholar] [CrossRef]
- Douafer, H.; Andrieu, V.; Phanstiel, O.; Brunel, J.M. Antibiotic Adjuvants: Make Antibiotics Great Again! J. Med. Chem. 2019, 62, 8665–8681. [Google Scholar] [CrossRef]
- Blanchet, M.; Borselli, D.; Brunel, J.M. Polyamine derivatives: A revival of an old neglected scaffold to fight resistant Gram-negative bacteria? Future Med. Chem. 2016, 8, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.B.; Li, C.; Taotafa, U.; Ding, B.; Guan, Q. Antibacterial properties of cationic steroid antibiotics. FEMS Microbiol. Lett. 2002, 217, 1–7. [Google Scholar] [CrossRef]
- Chen, W.H.; Wennersten, C.; Moellering, R.C.; Regen, S.L. Towards squalamine mimics: Synthesis and antibacterial activities of head-to-tail dimeric sterol-polyamine conjugates. Chem. Biodivers. 2013, 10, 385–393. [Google Scholar] [CrossRef]
- Lu, Y.M.; Deng, L.Q.; Huang, X.; Chen, J.X.; Wang, B.; Zhou, Z.Z.; Hu, G.S.; Chen, W.H. Synthesis and anionophoric activities of dimeric polyamine-sterol conjugates: The impact of rigid vs. flexible linkers. Org. Biomol. Chem. 2013, 11, 8221–8227. [Google Scholar] [CrossRef]
- Blanchet, M.; Borselli, D.; Rodallec, A.; Peiretti, F.; Vidal, N.; Bolla, J.M.; Digiorgio, C.; Morrison, K.R.; Wuest, W.M.; Brunel, J.M. Claramines: A New Class Of Broad-Spectrum Antimicrobial Agents with Bimodal Activity. ChemMedChem 2018, 13, 1018–1027. [Google Scholar] [CrossRef]
- Sue, K.; Cadelis, M.; Troia, T.; Rouvier, F.; Bourguet-Kondracki, M.; Brunel, J.; Copp, B. Investigation of alpha,omega-Disubstituted Polyamine-Cholic Acid Conjugates Identifies Hyodeoxycholic and Chenodeoxycholic Scaffolds as Non-Toxic, Potent Antimicrobials. Antibiotics 2023, 12, 404. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Khalitova, R.R.; Nedopekina, D.A.; Gubaidullin, R.R. Antimicrobial properties of amine- and guanidine-functionalized derivatives of betulinic, ursolic and oleanolic acids: Synthesis and structure/activity evaluation. Steroids 2020, 154, 108530. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, E.F.; Sinou, V.; Babkov, D.A.; Kazakova, O.; Brunel, J.M. Development of New Antimicrobial Oleanonic Acid Polyamine Conjugates. Antibiotics 2022, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, I.; Petrova, A.; Giniyatullina, G.; Smirnova, A.; Volobueva, A.; Pavlyukova, J.; Zarubaev, V.; Loc, T.V.; Tran Thi Phoung, T.; Hau, V.T.B.; et al. Synthesis, Anti-Influenza H1N1 and Anti-Dengue Activity of A-Ring Modified Oleanonic Acid Polyamine Derivatives. Molecules 2022, 27, 8499. [Google Scholar] [CrossRef]
- Djouhri-Bouktab, L.; Vidal, N.; Rolain, J.M.; Brunel, J.M. Synthesis of new 3,20-bispolyaminosteroid squalamine analogues and evaluation of their antimicrobial activities. J. Med. Chem. 2011, 54, 7417–7421. [Google Scholar] [CrossRef]
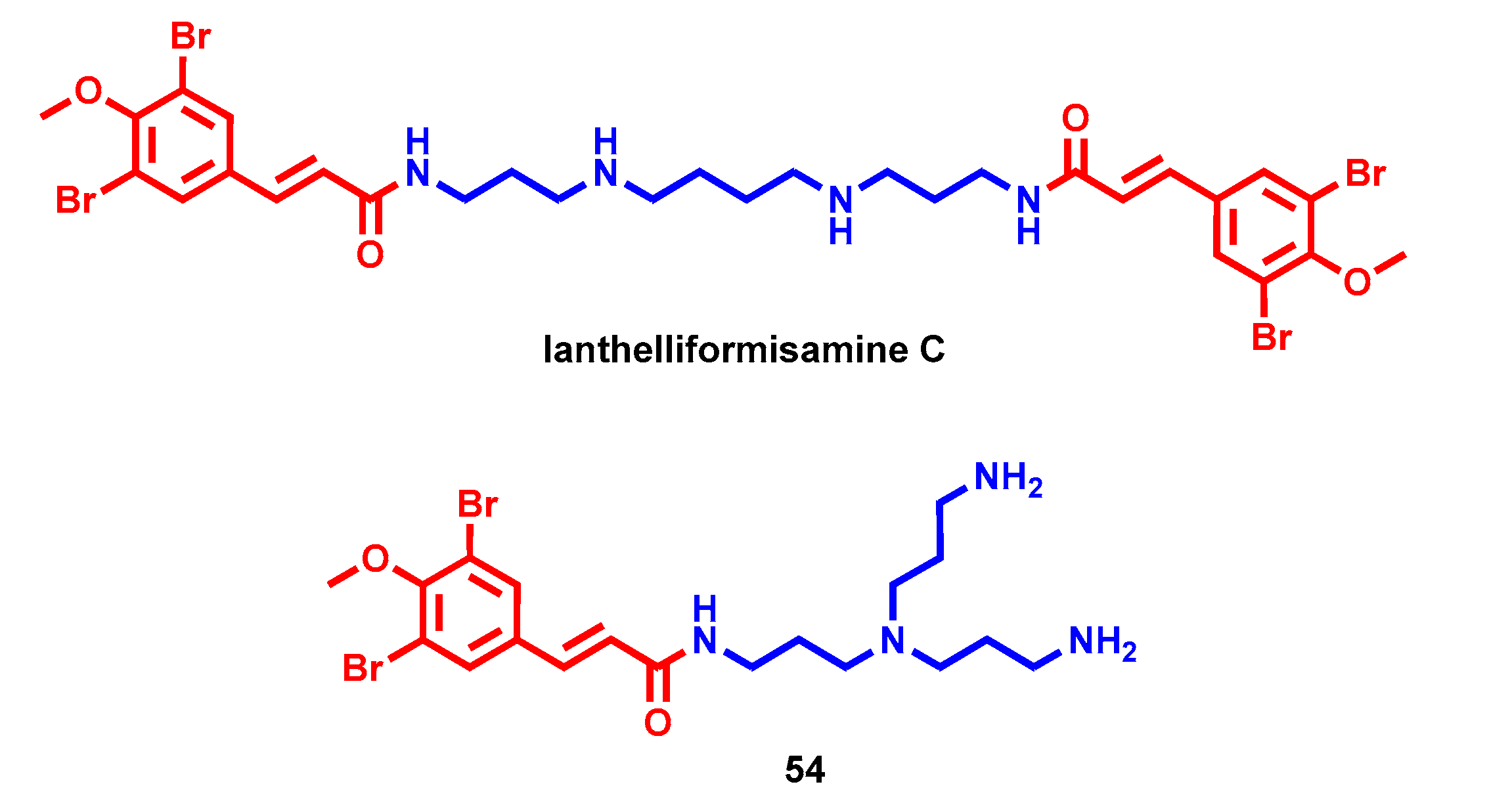
- Xu, M.; Davis, R.A.; Feng, Y.; Sykes, M.L.; Shelper, T.; Avery, V.M.; Camp, D.; Quinn, R.J. Ianthelliformisamines A-C, antibacterial bromotyrosine-derived metabolites from the marine sponge Suberea ianthelliformis. J. Nat. Prod. 2012, 75, 1001–1005. [Google Scholar] [CrossRef]
- Pieri, C.; Borselli, D.; Di Giorgio, C.; De Méo, M.; Bolla, J.M.; Vidal, N.; Combes, S.; Brunel, J.M. New Ianthelliformisamine derivatives as antibiotic enhancers against resistant Gram-negative bacteria. J. Med. Chem. 2014, 57, 4263–4272. [Google Scholar] [CrossRef]
- Kostopoulou, O.N.; Kouvela, E.C.; Magoulas, G.E.; Garnelis, T.; Panagoulias, I.; Rodi, M.; Papadopoulos, G.; Mouzaki, A.; Dinos, G.P.; Papaioannou, D.; et al. Conjugation with polyamines enhances the antibacterial and anticancer activity of chloramphenicol. Nucleic Acids Res. 2014, 42, 8621–8634. [Google Scholar] [CrossRef] [PubMed]
- Pearce, A.N.; Chen, D.; Edmeades, L.R.; Cadelis, M.M.; Troudi, A.; Brunel, J.M.; Bourguet-Kondracki, M.L.; Copp, B.R. Repurposing primaquine as a polyamine conjugate to become an antibiotic adjuvant. Bioorg. Med. Chem. 2021, 38, 116110. [Google Scholar] [CrossRef]
- Birkholtz, L.M.; Williams, M.; Niemand, J.; Louw, A.I.; Persson, L.; Heby, O. Polyamine homoeostasis as a drug target in pathogenic protozoa: Peculiarities and possibilities. Biochem. J. 2011, 438, 229–244. [Google Scholar] [CrossRef] [Green Version]
- Liew, L.P.; Kaiser, M.; Copp, B.R. Discovery and preliminary structure-activity relationship analysis of 1,14-sperminediphenylacetamides as potent and selective antimalarial lead compounds. Bioorg. Med. Chem. Lett. 2013, 23, 452–454. [Google Scholar] [CrossRef]
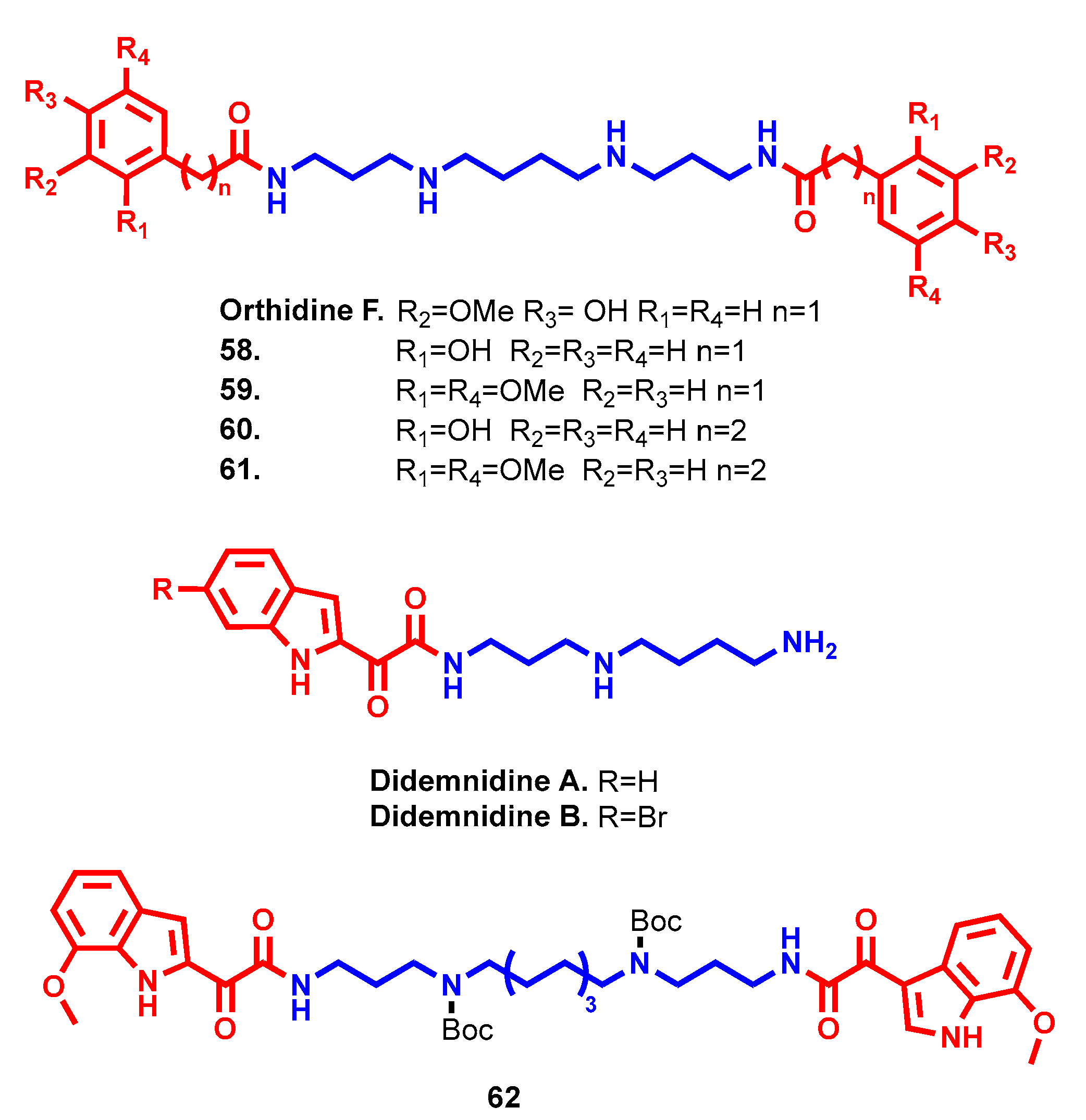
- Finlayson, R.; Pearce, A.N.; Page, M.J.; Kaiser, M.; Bourguet-Kondracki, M.L.; Harper, J.L.; Webb, V.L.; Copp, B.R. Didemnidines A and B, indole spermidine alkaloids from the New Zealand ascidian Didemnum sp. J. Nat. Prod. 2011, 74, 888–892. [Google Scholar] [CrossRef]
- Liew, L.P.; Pearce, A.N.; Kaiser, M.; Copp, B.R. Synthesis and in vitro and in vivo evaluation of antimalarial polyamines. Eur. J. Med. Chem. 2013, 69, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kaiser, M.; Copp, B.R. Investigation of indolglyoxamide and indolacetamide analogues of polyamines as antimalarial and antitrypanosomal agents. Mar. Drugs 2014, 12, 3138–3160. [Google Scholar] [CrossRef] [Green Version]
- Niemand, J.; Burger, P.; Verlinden, B.K.; Reader, J.; Joubert, A.M.; Kaiser, A.; Louw, A.I.; Kirk, K.; Phanstiel, O.; Birkholtz, L.M. Anthracene-polyamine conjugates inhibit in vitro proliferation of intraerythrocytic Plasmodium falciparum parasites. Antimicrob. Agents Chemother. 2013, 57, 2874–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vennerstrom, J.L.; Arbe-Barnes, S.; Brun, R.; Charman, S.A.; Chiu, F.C.; Chollet, J.; Dong, Y.; Dorn, A.; Hunziker, D.; Matile, H.; et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 2004, 430, 900–904. [Google Scholar] [CrossRef]
- Pearce, A.N.; Kaiser, M.; Copp, B.R. Synthesis and antimalarial evaluation of artesunate-polyamine and trioxolane-polyamine conjugates. Eur. J. Med. Chem. 2017, 140, 595–603. [Google Scholar] [CrossRef]
- Reigada, C.; Phanstiel, O.; Miranda, M.R.; Pereira, C.A. Targeting polyamine transport in Trypanosoma cruzi. Eur. J. Med. Chem. 2018, 147, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Fiorillo, A.; Genovese, I.; Colotti, G. Polyamine-trypanothione pathway: An update. Future Med. Chem. 2017, 9, 61–77. [Google Scholar] [CrossRef]
- Jagu, E.; Pomel, S.; Pethe, S.; Loiseau, P.M.; Labruère, R. Polyamine-based analogs and conjugates as antikinetoplastid agents. Eur. J. Med. Chem. 2017, 139, 982–1015. [Google Scholar] [CrossRef]
- Martín-Escolano, R.; Molina-Carreño, D.; Delgado-Pinar, E.; Martin-Montes, Á.; Clares, M.P.; Medina-Carmona, E.; Pitarch-Jarque, J.; Martín-Escolano, J.; Rosales, M.J.; García-España, E.; et al. New polyamine drugs as more effective antichagas agents than benznidazole in both the acute and chronic phases. Eur. J. Med. Chem. 2019, 164, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Martìn-Escolano, R.; Molina-Carreño, D.; Martìn-Escolano, J.; Paz Clares, M.; Galiana-Roselló, C.; González-García, J.; Cirauqui, N.; LLinares, J.M.; José Rosales, M.A.; García-España, E.; et al. Identification of Aryl Polyamines Derivatives as Anti-Trypanosoma cruzi Agents Targeting Iron Superoxide Dismutase. Pharmaceutics 2023, 15, 140. [Google Scholar] [CrossRef]
- Maiocchi, S.; Ku, J.; Hawtrey, T.; De Silvestro, I.; Malle, E.; Rees, M.; Thomas, S.; Morris, J. Polyamine-Conjugated Nitroxides Are Efficacious Inhibitors of Oxidative Reactions Catalyzed by Endothelial-Localized Myeloperoxidase. Chem. Res. Toxicol. 2021, 34, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
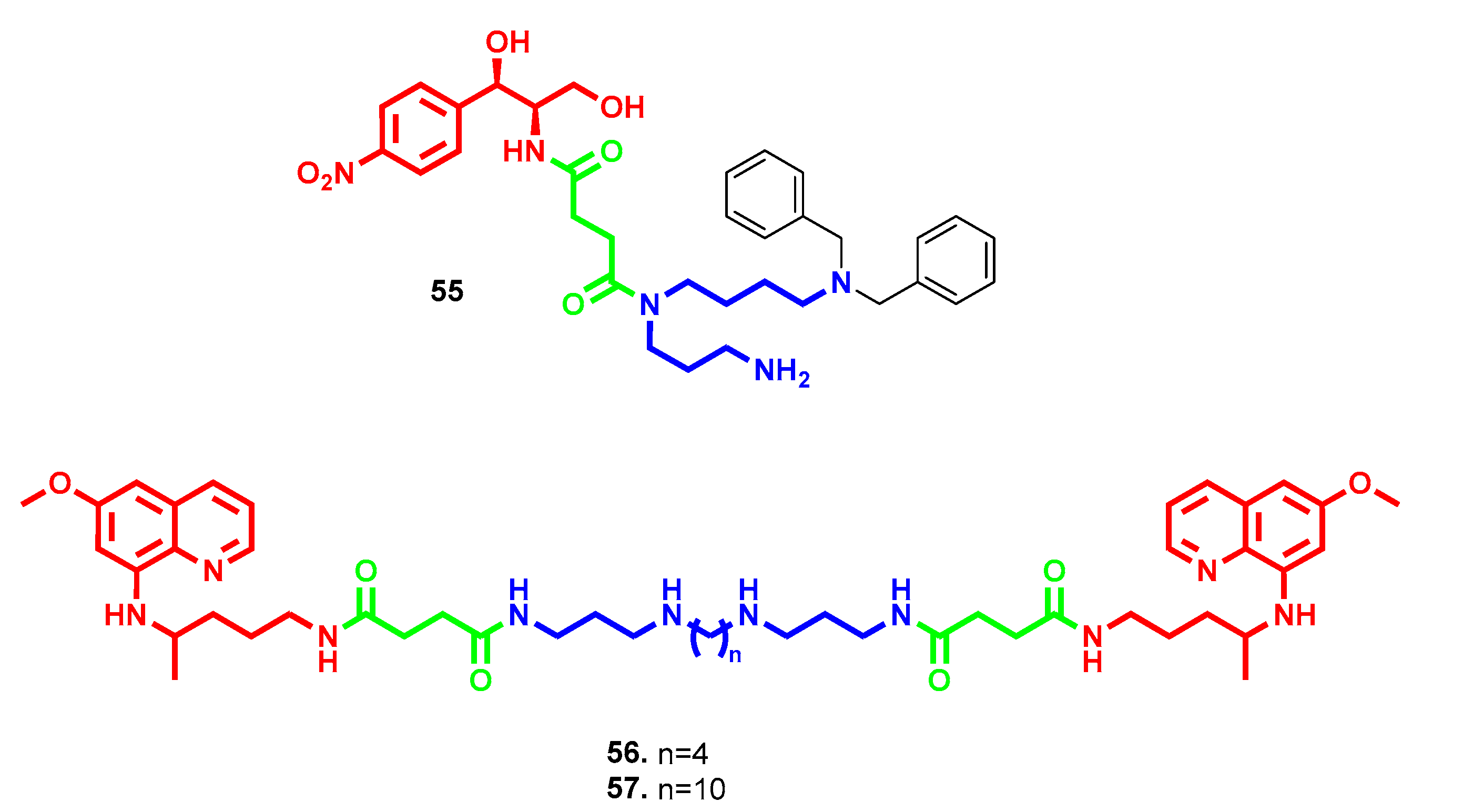
- Hadjipavlou-Litina, D.; Magoulas, G.E.; Bariamis, S.E.; Tsimali, Z.; Avgoustakis, K.; Kontogiorgis, C.A.; Athanassopoulos, C.M.; Papaioannou, D. Synthesis and evaluation of the antioxidative potential of minoxidil-polyamine conjugates. Biochimie 2013, 95, 1437–1449. [Google Scholar] [CrossRef]
- Hong, C.; Luo, W.; Yao, D.; Su, Y.B.; Zhang, X.; Tian, R.G.; Wang, C.J. Novel aromatic-polyamine conjugates as cholinesterase inhibitors with notable selectivity toward butyrylcholinesterase. Bioorg. Med. Chem. 2014, 22, 3213–3219. [Google Scholar] [CrossRef]
- Kumamoto, T.; Nakajima, M.; Uga, R.; Ihayazaka, N.; Kashihara, H.; Katakawa, K.; Ishikawa, T.; Saiki, R.; Nishimura, K.; Igarashi, K. Design, synthesis, and evaluation of polyamine-memantine hybrids as NMDA channel blockers. Bioorg. Med. Chem. 2018, 26, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Simoni, E.; Bergamini, C.; Fato, R.; Tarozzi, A.; Bains, S.; Motterlini, R.; Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Hrelia, P.; et al. Polyamine conjugation of curcumin analogues toward the discovery of mitochondria-directed neuroprotective agents. J. Med. Chem. 2010, 53, 7264–7268. [Google Scholar] [CrossRef]
- Simoni, E.; Caporaso, R.; Bergamini, C.; Fiori, J.; Fato, R.; Miszta, P.; Filipek, S.; Caraci, F.; Giuffrida, M.L.; Andrisano, V.; et al. Polyamine Conjugation as a Promising Strategy To Target Amyloid Aggregation in the Framework of Alzheimer’s Disease. ACS Med. Chem. Lett. 2016, 7, 1145–1150. [Google Scholar] [CrossRef] [Green Version]
- Li, L.-B.; Fan, Y.-G.; Wu, W.-X.; Bai, C.-Y.; Jia, M.-Y.; Hu, J.-P.; Gao, H.-L.; Wang, T.; Zhong, M.-L.; Huang, X.-S.; et al. Novel melatonin-trientine conjugate as potential therapeutic agents for Alzheimer’s disease. Bioorg. Chem. 2022, 128, 106100. [Google Scholar] [CrossRef]
- Azfar, M.; van Veen, S.; Houdou, M.; Hamouda, N.; Eggermont, J.; Vangheluwe, P. P5B-ATPases in the mammalian polyamine transport system and their role in disease. Biochim. Biophys. Acta-Mol. Cell Res. 2022, 1869, 119354. [Google Scholar] [CrossRef]





















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basagni, F.; Marotta, G.; Rosini, M.; Minarini, A. Polyamine–Drug Conjugates: Do They Boost Drug Activity? Molecules 2023, 28, 4518. https://doi.org/10.3390/molecules28114518
Basagni F, Marotta G, Rosini M, Minarini A. Polyamine–Drug Conjugates: Do They Boost Drug Activity? Molecules. 2023; 28(11):4518. https://doi.org/10.3390/molecules28114518
Chicago/Turabian StyleBasagni, Filippo, Giambattista Marotta, Michela Rosini, and Anna Minarini. 2023. "Polyamine–Drug Conjugates: Do They Boost Drug Activity?" Molecules 28, no. 11: 4518. https://doi.org/10.3390/molecules28114518
APA StyleBasagni, F., Marotta, G., Rosini, M., & Minarini, A. (2023). Polyamine–Drug Conjugates: Do They Boost Drug Activity? Molecules, 28(11), 4518. https://doi.org/10.3390/molecules28114518