
Solid-Phase Synthesis of Selectively Mono-Fluorobenz(o)ylated Polyamines as a Basis for the Development of 18F-Labeled Radiotracers



Abstract
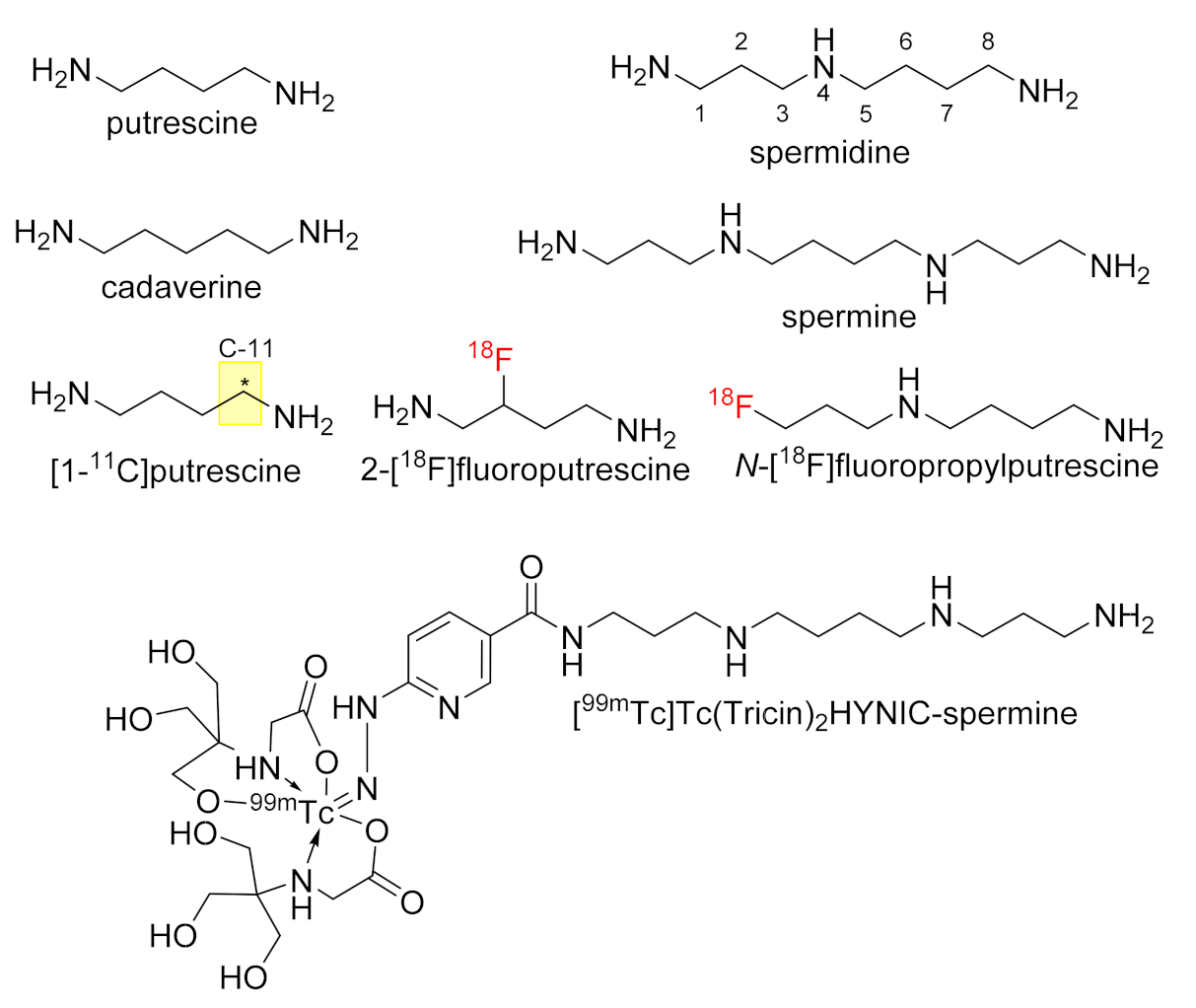
:1. Introduction
2. Results and Discussion
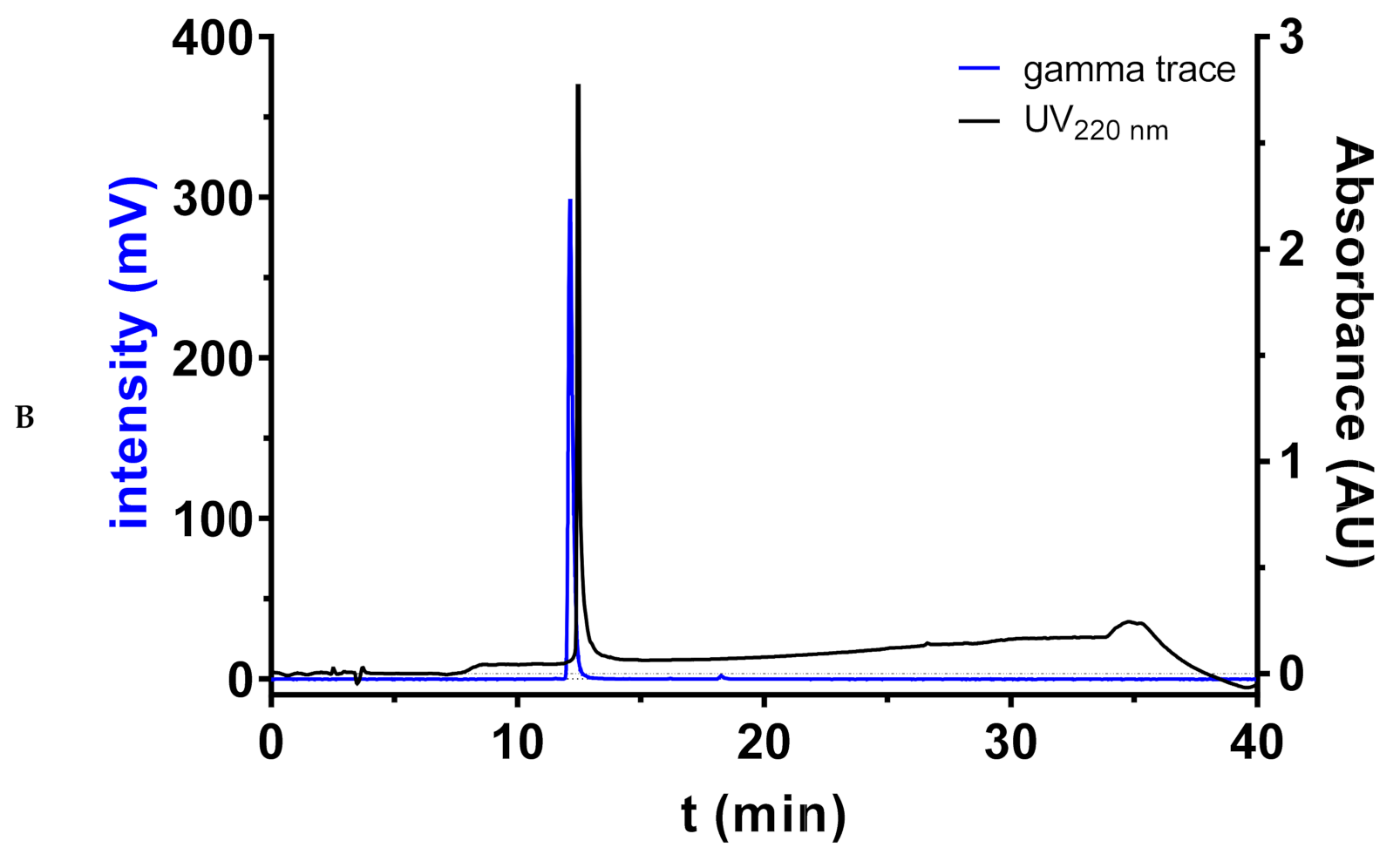
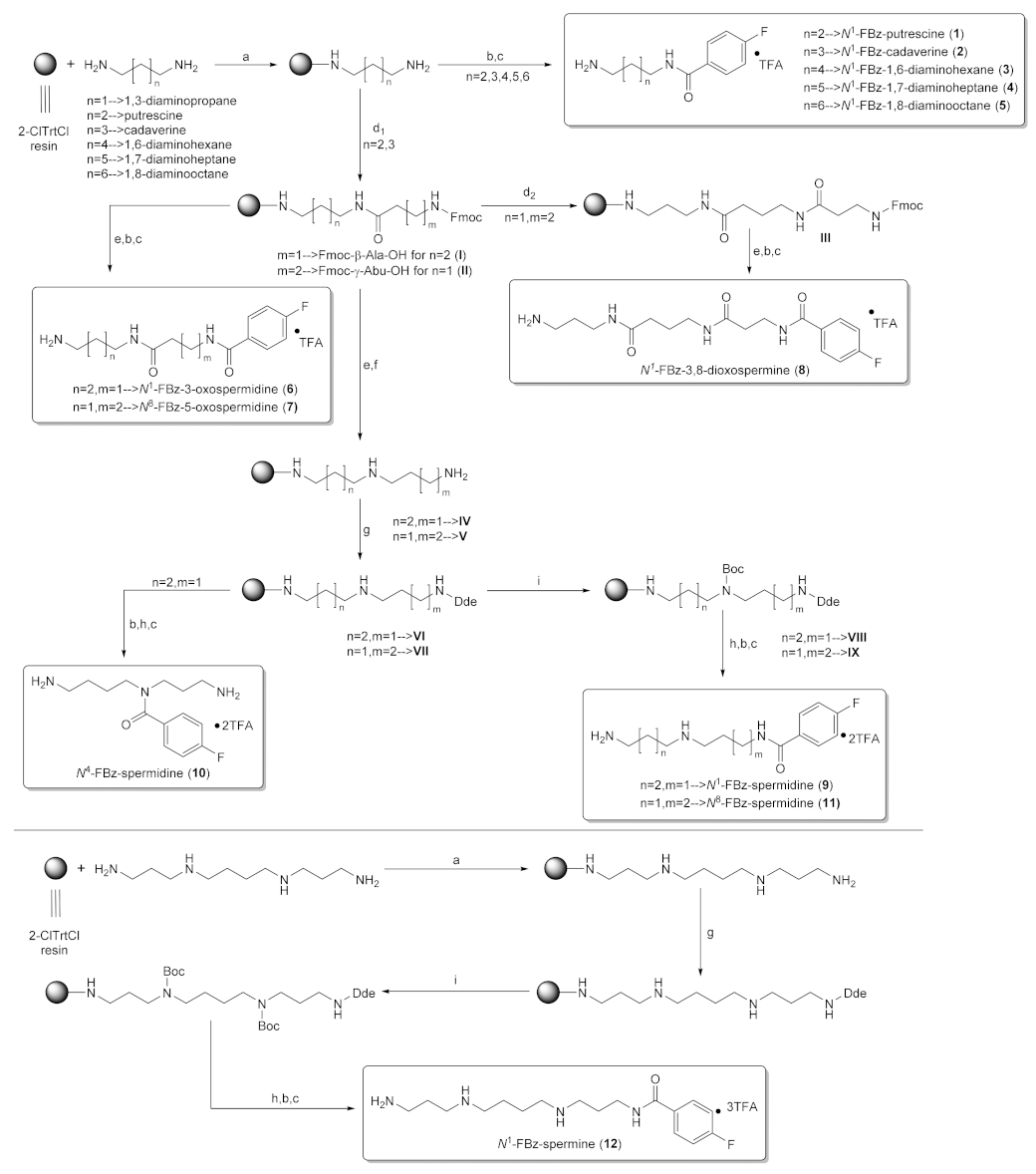
2.1. Solid-Phase Synthesis of Fluorobenzoylated Polyamines
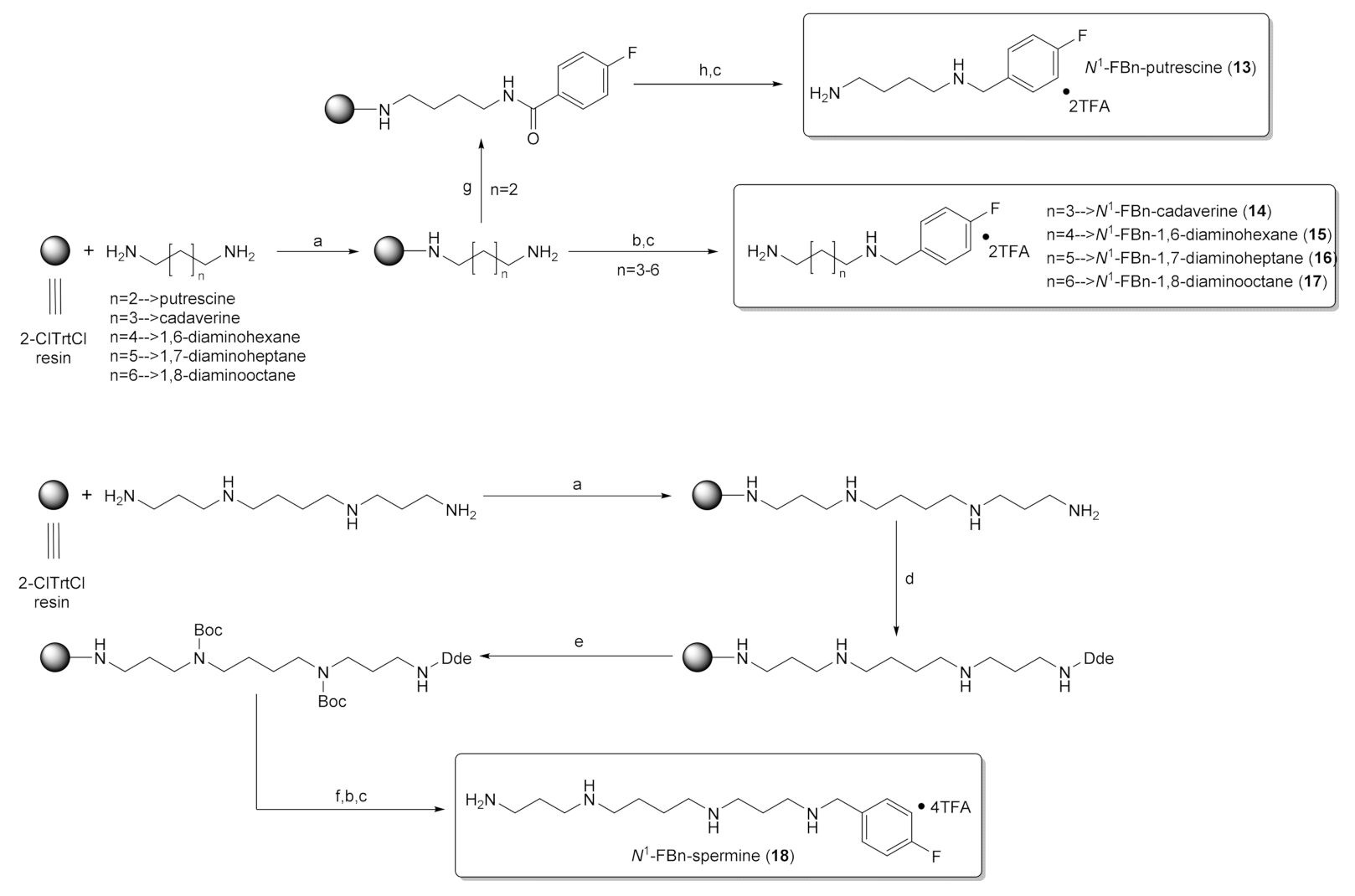
2.2. Solid-Phase Synthesis of Fluorobenzylated Polyamines (Diamines)
3. Materials and Methods
3.1. General
3.2. Synthetic Methods
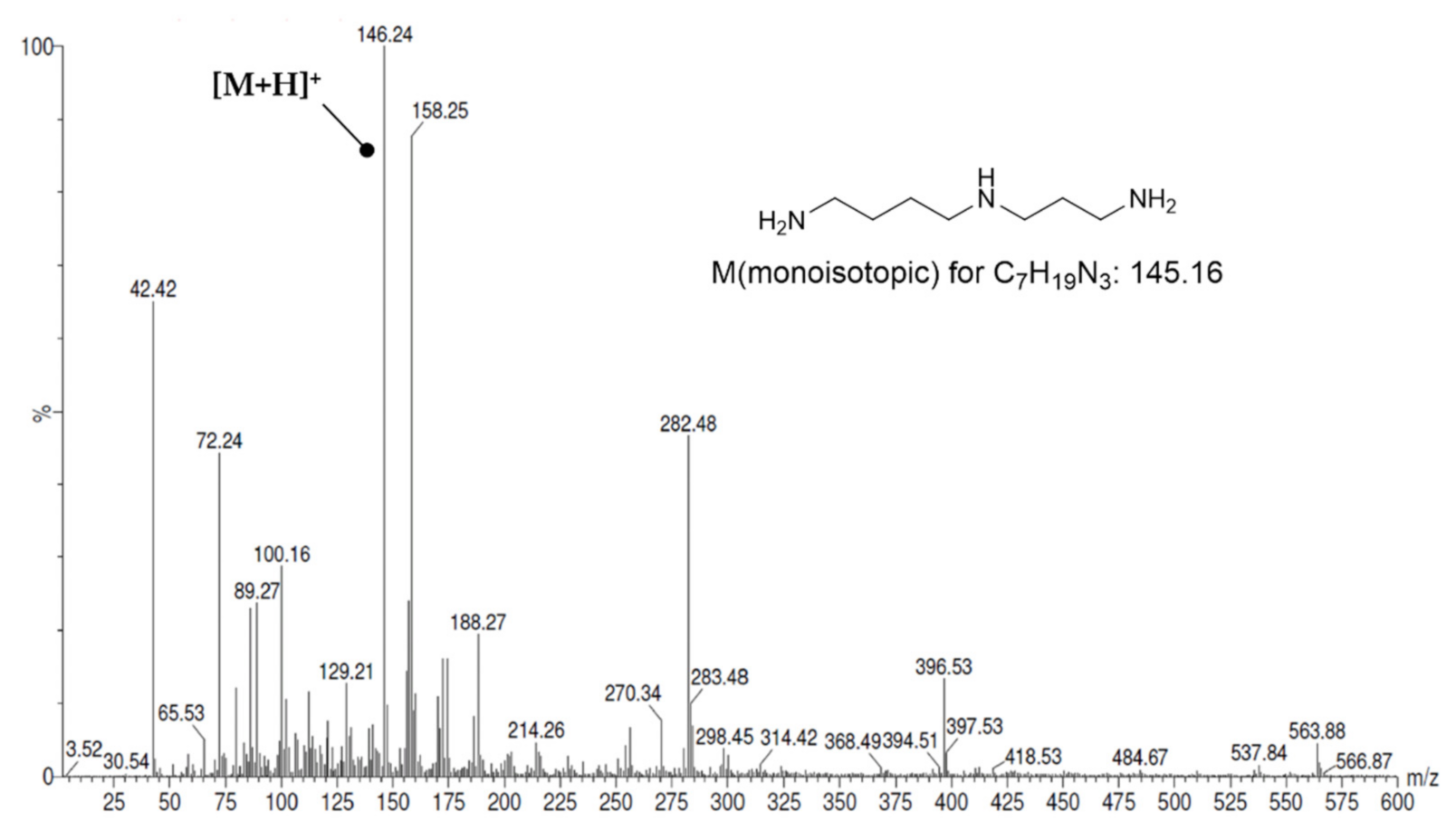
3.2.1. Loading of Symmetric Polyamines to the 2-ClTrtCl Resin
3.2.2. Assembly of Fmoc-Protected Oxopolyamines
3.2.3. Determination of Resin Loading Levels
3.2.4. Removal of the Fmoc Group
3.2.5. Amide Bond Reduction
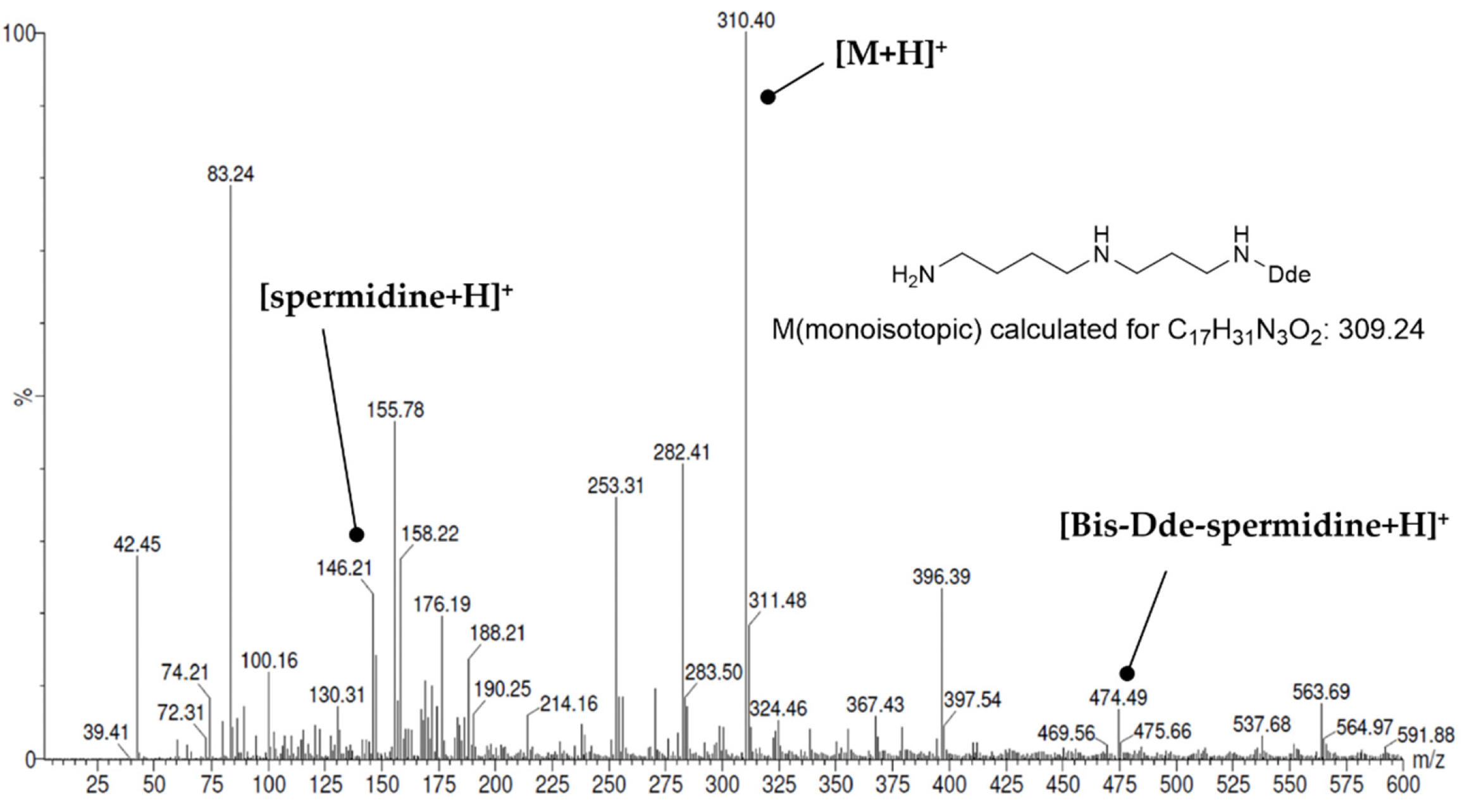
3.2.6. Dde Protection
3.2.7. Boc Protection
3.2.8. Removal of the Dde Group
3.2.9. Fluorobenzoylation of Amino Groups
3.2.10. Fluorobenzylation of Amino Groups
3.2.11. Cleavage of Polyamines/Oxopolyamines from the Resin
3.3. Yields and Analytical Data for Compounds 1–18
3.3.1. N1-(4-Fluorobenzoyl)-putrescine × TFA (1)

| Yield: | 28 mg (63%), white solid |
| MS (ESI+): | m/z = 211.3 ([M + H]+), m/z = 432.6 ([2M + H]+) M (monoisotopic) calculated for C11H15FN2O: 210.12 g/mol |
3.3.2. N1-(4-Fluorobenzoyl)-cadaverine × TFA (2)

| Yield: | 53 mg (65%), white waxy solid |
| MS (ESI+): | m/z = 225.4 ([M + H]+) M (monoisotopic) calculated for C12H17FN2O: 224.13 g/mol |
3.3.3. N1-(4-Fluorobenzoyl)-1,6-diaminohexane × TFA (3)

| Yield: | 44 mg (84%), yellow waxy solid |
| MS (ESI+): | m/z = 238.92 ([M + H]+) M (monoisotopic) calculated for C13H19FN2O: 238.15 g/mol |
3.3.4. N1-(4-Fluorobenzoyl)-1,7-diaminoheptane × TFA (4)

| Yield: | 37 mg (64%), white solid |
| MS (ESI+): | m/z = 252.82 ([M + H]+) M (monoisotopic) calculated for C14H21FN2O: 252.16 g/mol |
3.3.5. N1-(4-Fluorobenzoyl)-1,8-diaminooctane × TFA (5)

| Yield: | 35 mg (64%), white solid |
| MS (ESI+): | m/z = 266.83 ([M + H]+) M (monoisotopic) calculated for C15H23FN2O: 266.16 g/mol |
3.3.6. N1-(4-Fluorobenzoyl)-3-oxospermidine × TFA (6)

| Yield: | 26 mg (88%), white waxy solid |
| MS (ESI+): | m/z = 282.5 ([M + H]+), m/z = 304.5 ([M + Na]+), m/z = 563.7 ([2M + H]+)M (monoisotopic) calculated for C14H20FN3O2: 281.15 g/mol |
3.3.7. N8-(4-Fluorobenzoyl)-5-oxospermidine × TFA (7)

| Yield: | 39 mg (quantitative containing residual TFA), colorless viscous oil |
| MS (ESI+): | m/z = 282.4 ([M + H]+), m/z = 304.4 ([M + Na]+), m/z = 563.7 ([2M + H]+) M (monoisotopic) calculated for C14H20FN3O2: 281.15 g/mol |
3.3.8. N1-(4-Fluorobenzoyl)-3,8-dioxospermine × TFA (8)

| Yield: | 69 mg (89%), colorless viscous oil |
| MS (ESI+): | m/z = 353.5 ([M + H]+) M (monoisotopic) calculated for C17H25FN4O3: 352.19 g/mol |
3.3.9. N1-(4-Fluorobenzoyl)-spermidine × 2TFA (9)

| Yield: | 28 mg (38%), pale yellow viscous oil |
| MS (ESI+): | m/z = 123.3 ([FBz]+), m/z = 146.4 ([M – FBz + 2H]+), m/z =268.5 ([M + H]+) M (monoisotopic) calculated for C14H22FN3O: 267.17 g/mol |
3.3.10. N4-(4-Fluorobenzoyl)-spermidine × 2TFA (10)

| Yield: | 21 mg (35%), colorless viscous oil |
| MS (ESI+): | m/z = 146.7 ([M – FBz + 2H]+), m/z = 268.4 ([M + H]+), m/z = 535.7 ([2M + H]+) M (monoisotopic) calculated for C14H22FN3O: 267.17 g/mol |
3.3.11. N8-(4-Fluorobenzoyl)-spermidine × 2TFA (11)

| Yield: | 14 mg (29%), pale yellow waxy solid |
| MS (ESI+): | m/z = 123.3 ([FBz]+), m/z = 146.7 ([M – FBz + 2H]+), m/z = 268.5 ([M + H]+) M (monoisotopic) calculated for C14H22FN3O: 267.17 g/mol |
3.3.12. N1-(4-Fluorobenzoyl)-spermine × 3TFA (12)

| Yield: | 16 mg (10%), white solid |
| MS (ESI+): | m/z = 102.3 ([M – FBz + 3H]2+), m/z = 123.1 ([FBz]+), m/z = 163.4 ([M + 2H]2+), m/z = 203.4 ([M – FBz + 2H]+), m/z = 325.5 ([M + H]+) M (monoisotopic) calculated for C17H29FN4O: 324.23 g/mol |
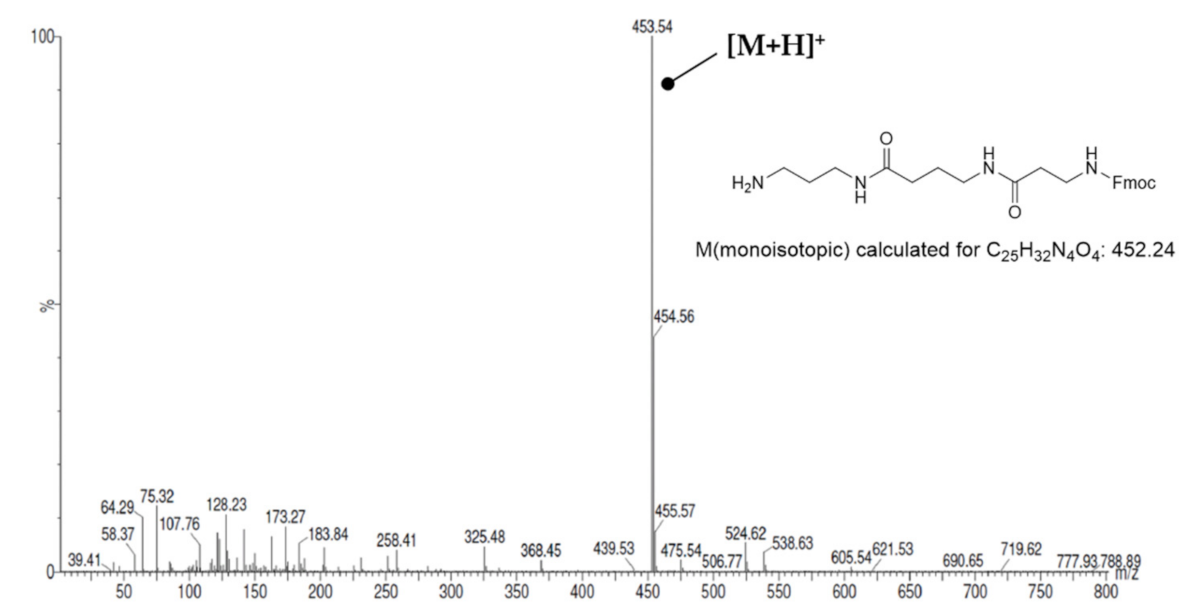
3.3.13. N1-(4-Fluorobenzyl)-putrescine × 2TFA (13)

| Yield: | 55 mg (40%), white solid |
| MS (ESI+): | m/z = 196.70 ([M + H]+) M (monoisotopic) calculated for C11H17FN2: 196.14 g/mol |
3.3.14. N1-(4-Fluorobenzyl)-cadaverine × 2TFA (14)

| Yield: | 27 mg (36%), colorless solid |
| MS (ESI+): | m/z = 210.81 ([M + H]+) M (monoisotopic) calculated for C12H19FN2: 210.15 g/mol |
3.3.15. N1-(4-Fluorobenzyl)-1,6-diaminohexane × 2TFA (15)

| Yield: | 15 mg (20%), white, waxy solid |
| MS (ESI+): | m/z = 224.91 ([M + H]+) M (monoisotopic) calculated for C13H21FN2: 224.17 g/mol |
3.3.16. N1-(4-Fluorobenzyl)-1,7-diaminoheptane × 2TFA (16)

| Yield: | 11 mg (14%), colorless oil |
| MS (ESI+): | m/z = 238.92 ([M + H]+) M (monoisotopic) calculated for C14H23FN2: 238.18 g/mol |
3.3.17. N1-(4-Fluorobenzyl)-1,7-diaminooctane × 2TFA (17)

| Yield: | 22 mg (28%), white, waxy solid |
| MS (ESI+): | m/z = 252.92 ([M + H]+) M (monoisotopic) calculated for C15H25FN2: 252.20 g/mol |
3.3.18. N1-(4-Fluorobenzyl)-spermine × 4TFA (18)

| Yield: | 11 mg (14%), white solid |
| MS (ESI+): | m/z = 311.06 ([M + H]+) M (monoisotopic) calculated for C17H31FN4: 310.25 g/mol |
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Agostinelli, E.; Marques, M.P.; Calheiros, R.; Gil, F.P.; Tempera, G.; Viceconte, N.; Battaglia, V.; Grancara, S.; Toninello, A. Polyamines: Fundamental characters in chemistry and biology. Amino Acids 2010, 38, 393–403. [Google Scholar] [CrossRef]
- Jancewicz, A.L.; Gibbs, N.M.; Masson, P.H. Cadaverine’s Functional Role in Plant Development and Environmental Response. Front. Plant. Sci. 2016, 7, 870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienz, S.; Detterbeck, R.; Ensch, C.; Guggisberg, A.; Hausermann, U.; Meisterhans, C.; Wendt, B.; Werner, C.; Hesse, M. Putrescine, spermidine, spermine, and related polyamine alkaloids. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: Cambridge, MA, USA, 2002; Volume 58, pp. 83–338. [Google Scholar]
- Bienz, S.; Bisegger, P.; Guggisberg, A.; Hesse, M. Polyamine alkaloids. Nat. Prod. Rep. 2005, 22, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Benz, H.; Fiedler, W.; Guggisberg, A.; Bienz, S.; Hesse, M. Polyamine Toxins from Spiders and Wasps. In The Alkaloids: Chemistry and Pharmacology; Cordell, G.A., Brossi, A., Eds.; Academic Press: Cambridge, MA, USA, 1994; Volume 45, pp. 1–125. [Google Scholar]
- Michael, A.J. Biosynthesis of polyamines and polyamine-containing molecules. Biochem. J. 2016, 473, 2315–2329. [Google Scholar] [CrossRef] [PubMed]
- Murray-Stewart, T.R.; Woster, P.M.; Casero, R.A., Jr. Targeting polyamine metabolism for cancer therapy and prevention. Biochem. J. 2016, 473, 2937–2953. [Google Scholar] [CrossRef] [Green Version]
- D′Agostino, L.; di Pietro, M.; Di Luccia, A. Nuclear aggregates of polyamines. IUBMB Life 2006, 58, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Lightfoot, H.L.; Hall, J. Endogenous polyamine function-The RNA perspective. Nucleic Acids Res. 2014, 42, 11275–11290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, E.C.; Park, M.H.; Folk, J.E. Cleavage of spermidine as the first step in deoxyhypusine synthesis. The role of NAD. J. Biol. Chem. 1990, 265, 4793–4799. [Google Scholar] [CrossRef]
- Wolff, E.C.; Park, M.H. Role of the Polyamine Spermidine as a Precursor for Hypusine Modification in eIF5A. In Polyamines; Kusano, T., Suzuki, H., Eds.; Springer: Tokyo, Japan, 2015; Chapter 10; pp. 121–129. [Google Scholar]
- Igarashi, K.; Kashiwagi, K. The functional role of polyamines in eukaryotic cells. Int. J. Biochem. Cell Biol. 2019, 107, 104–115. [Google Scholar] [CrossRef]
- Kim, J.H.; Marton, J.; Ametamey, S.M.; Cumming, P. A Review of Molecular Imaging of Glutamate Receptors. Molecules 2020, 25, 4749. [Google Scholar] [CrossRef]
- Kurata, H.T. Polyamine Block of Inwardly Rectifying Potassium (Kir) Channels. In Polyamines; Kusano, T., Suzuki, H., Eds.; Springer: Tokyo, Japan, 2015; Chapter 18; pp. 217–228. [Google Scholar]
- Munoz-Esparza, N.C.; Latorre-Moratalla, M.L.; Comas-Baste, O.; Toro-Funes, N.; Veciana-Nogues, M.T.; Vidal-Carou, M.C. Polyamines in Food. Front. Nutr. 2019, 6, 108. [Google Scholar] [CrossRef]
- Ha, H.C.; Sirisoma, N.S.; Kuppusamy, P.; Zweier, J.L.; Woster, P.M.; Casero, R.A., Jr. The natural polyamine spermine functions directly as a free radical scavenger. Proc. Natl. Acad. Sci. USA 1998, 95, 11140–11145. [Google Scholar] [CrossRef] [Green Version]
- Seiler, N. Functions of polyamine acetylation. Can. J. Physiol. Pharmacol. 1987, 65, 2024–2035. [Google Scholar] [CrossRef] [PubMed]
- Lentini, A.; Abbruzzese, A.; Caraglia, M.; Marra, M.; Beninati, S. Protein-polyamine conjugation by transglutaminase in cancer cell differentiation: Review article. Amino Acids 2004, 26, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Beninati, S.; Folk, J.E. Covalent polyamine-protein conjugates: Analysis and distribution. Adv. Exp. Med. Biol. 1988, 250, 411–422. [Google Scholar] [PubMed]
- Beninati, S.; Piacentini, M.; Cocuzzi, E.T.; Autuori, F.; Folk, J.E. Covalent incorporation of polyamines as γ-glutamyl derivatives into CHO cell protein. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1988, 952, 325–333. [Google Scholar] [CrossRef]
- Song, Y.; Kirkpatrick, L.L.; Schilling, A.B.; Helseth, D.L.; Chabot, N.; Keillor, J.W.; Johnson, G.V.; Brady, S.T. Transglutaminase and polyamination of tubulin: Posttranslational modification for stabilizing axonal microtubules. Neuron 2013, 78, 109–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phanstiel, O. An overview of polyamine metabolism in pancreatic ductal adenocarcinoma. Int. J. Cancer 2018, 142, 1968–1976. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.-J.; Volkow, N.D.; Wolf, A.P.; Madajewicz, S.; Fowler, J.S.; Schlyer, D.J.; MacGregor, R.R. Positron emission tomography study of human prostatic adenocarcinoma using carbon-11 putrescine. Nucl. Med. Biol. 1994, 21, 77–82. [Google Scholar] [CrossRef]
- Hwang, D.-R.; Mathias, C.J.; Welch, M.J.; McGuire, A.H.; Kadmon, D. Imaging prostate derived tumors with PET and N-(3-[18F]fluoropropyl)putrescine. Int. J. Rad. Appl. Instr. B 1990, 17, 525–532. [Google Scholar] [CrossRef]
- Hwang, D.R.; Jerabek, P.A.; Kadmon, D.; Kilbourn, M.R.; Patrick, T.B.; Welch, M.J. 2-[18F]fluoroputrescine: Preparation, biodistribution, and mechanism of defluorination. Int. J. Rad. Appl. Instr. A 1986, 37, 607–612. [Google Scholar] [CrossRef]
- Hwang, D.R.; Lang, L.; Mathias, C.J.; Kadmon, D.; Welch, M. N-3-[18F]Fluoropropylputrescine as Potential PET Imaging Agent for Prostate and Prostate Derived Tumors. J. Nucl. Med. 1989, 30, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Pesnel, S.; Guminski, Y.; Pillon, A.; Lerondel, S.; Imbert, T.; Guilbaud, N.; Kruczynski, A.; Bailly, C.; Le Pape, A. 99mTc-HYNIC-spermine for imaging polyamine transport system-positive tumours: Preclinical evaluation. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, R.; Kahler, J.P.; Martin, S.; van Veen, S.; Verhelst, S.H.L. Clickable Polyamine Derivatives as Chemical Probes for the Polyamine Transport System. ChemBioChem 2018, 19, 907–911. [Google Scholar] [CrossRef]
- Palmer, A.J.; Ghani, R.A.; Kaur, N.; Phanstiel, O.; Wallace, H.M. A putrescine-anthracene conjugate: A paradigm for selective drug delivery. Biochem. J. 2009, 424, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traquete, R.; Ghani, R.A.; Phanstiel, O.; Wallace, H.M. Ant 4,4, a polyamine-anthracene conjugate, induces cell death and recovery in human promyelogenous leukemia cells (HL-60). Amino Acids 2013, 44, 1193–1203. [Google Scholar] [CrossRef]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in health and disease. Science 2018, 359, 2788. [Google Scholar] [CrossRef] [Green Version]
- Viltard, M.; Durand, S.; Perez-Lanzon, M.; Aprahamian, F.; Lefevre, D.; Leroy, C.; Madeo, F.; Kroemer, G.; Friedlander, G. The metabolomic signature of extreme longevity: Naked mole rats versus mice. Aging 2019, 11, 4783–4800. [Google Scholar] [CrossRef]
- Nichugovskiy, A.; Tron, G.C.; Maslov, M. Recent Advances in the Synthesis of Polyamine Derivatives and Their Applications. Molecules 2021, 26, 6579. [Google Scholar] [CrossRef]
- Fiedler, W.J.; Hesse, M. Synthese von selektiv N- funktionalisierten Polyamin-Derivaten. Helv. Chim. Acta 1993, 76, 1511–1519. [Google Scholar] [CrossRef]
- Li, Y.; Popaj, K.; Lochner, M.; Geneste, H.; Budriesi, R.; Chiarini, A.; Melchiorre, C.; Hesse, M. Analogues of polyamine alkaloids and their synthetic advantages. Il Farmaco 2001, 56, 127–131. [Google Scholar] [CrossRef]
- Kariya, Y.; Asanuma, Y.; Inai, M.; Asakawa, T.; Ohashi-Ito, K.; Fukuda, H.; Egi, M.; Kan, T. Practical Synthesis of Spermine, Thermospermine and Norspermine. Chem. Pharm. Bull. 2016, 64, 1403–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, C.A.; Kristensen, A.S.; Strømgaard, K. Small Molecules from Spiders Used as Chemical Probes. Angew. Chem. Int. Ed. Engl. 2011, 50, 11296–11311. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.; Schepers, U. Solid Phase Chemistry for the Directed Synthesis of Biologically Active Polyamine Analogs, Derivatives, and Conjugates. Top. Curr. Chem. 2007, 278, 135–208. [Google Scholar]
- Olsen, C.A.; Franzyk, H.; Jaroszewski, J.W. N-Alkylation Reactions and Indirect Formation of Amino Functionalities in Solid-Phase Synthesis. Synthesis 2005, 16, 2631–2653. [Google Scholar] [CrossRef]
- Karigiannis, G.; Papaioannou, D. Structure, Biological Activity and Synthesis of Polyamine Analogues and Conjugates. Eur. J. Org. Chem. 2000, 1841–1863. [Google Scholar] [CrossRef]
- Manov, N.; Bienz, S. A new approach in the solid-phase synthesis of polyamine derivatives: Construction of polyamine backbones from the center. Tetrahedron 2001, 57, 7893–7898. [Google Scholar] [CrossRef]
- Chhabra, S.R.; Khan, A.N.; Bycroft, B.W. Solid-phase synthesis of polyamines using a Dde-linker: Philanthotoxin-4.3.3 via an on-resin Mitsunobu reaction. Tetrahedron Lett. 2000, 41, 1099–1102. [Google Scholar] [CrossRef]
- Olsen, C.A.; Witt, M.; Jaroszewski, J.W.; Franzyk, H. Solid-phase synthesis of rigid acylpolyamines using temporary N-4,4’-dimethoxytrityl protection in the presence of trtyl linkers. J. Org. Chem. 2004, 69, 6149–6152. [Google Scholar] [CrossRef]
- Chhabra, S.R.; Khan, A.N.; Bycroft, B.W. Solid-phase synthesis of symmetrical and unsymmetrical polyamine analogues of philanthotoxins using a Dde-linker. Tetrahedron Lett. 2000, 41, 1095–1098. [Google Scholar] [CrossRef]
- Wang, F.; Manku, S.; Hall, D.G. Solid phase syntheses of polyamine toxins HO-416b and PhTX-433. Use of an efficient polyamide reduction strategy that facilitates access to branched analogues. Org. Lett. 2000, 2, 1581–1583. [Google Scholar] [CrossRef] [PubMed]
- Manku, S.; Laplante, C.; Kopac, D.; Chan, T.; Hall, D.G. A mild and general solid-phase method for the synthesis of chiral polyamines. Solution studies on the cleavage of borane-amine intermediates from the reduction of secondary amides. J. Org. Chem. 2001, 66, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.G.; Laplante, C.; Manku, S.; Nagendran, J. Mild oxidative cleavage of borane−amine adducts from amide reductions: Efficient solution- and solid-phase synthesis of N-alkylamino acids and chiral oligoamines. J. Org. Chem. 1999, 64, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Nash, I.A.; Bycroft, B.W.; Chan, W.C. Dde—a selective primary amine protecting group: A facile solid phase synthetic approach to polyamine conjugates. Tetrahedron Lett. 1996, 37, 2625–2628. [Google Scholar] [CrossRef]
- Mäding, P.; Füchtner, F.; Wüst, F. Module-assisted synthesis of the bifunctional labelling agent N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB). Appl. Radiat. Isot. 2005, 63, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Nymann Petersen, I.; Madsen, J.; Bernard Matthijs Poulie, C.; Kjaer, A.; Herth, M.M. One-Step Synthesis of N-Succinimidyl-4-[18F]Fluorobenzoate ([18F]SFB). Molecules 2019, 24, 3436. [Google Scholar] [CrossRef] [Green Version]
- Vaidyanathan, G.; Zalutsky, M.R. Synthesis of N-succinimidyl 4-[18F]fluorobenzoate, an agent for labeling proteins and peptides with 18F. Nat. Protoc. 2006, 1, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Kuchar, M.; Pretze, M.; Kniess, T.; Steinbach, J.; Pietzsch, J.; Löser, R. Site-selective radiolabeling of peptides by (18)F-fluorobenzoylation with [18F]SFB in solution and on solid phase: A comparative study. Amino Acids 2012, 43, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Kuchar, M.; Neuber, C.; Belter, B.; Bergmann, R.; Lenk, J.; Wodtke, R.; Kniess, T.; Steinbach, J.; Pietzsch, J.; Löser, R. Evaluation of Fluorine-18-Labeled α1(I)-N-Telopeptide Analogs as Substrate-Based Radiotracers for PET Imaging of Melanoma-Associated Lysyl Oxidase. Front. Chem. 2018, 6, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steglich, W.; Steffan, B.; Stroech, K.; Wolf, M. Pistillarin, ein charakteristischer Inhaltsstoff der Herkuleskeule (Clavariadelphus pistillaris) und einiger Ramaria-Arten (Basidiomycetes)/Pistillarin, a Characteristic Metabolite of Clavariadelphus pistillaris and Several Ramaria Species (Basidiomycetes). Z. Naturforsch. C 1984, 39, 10–12. [Google Scholar]
- Tait, G.H. The identification and biosynthesis of siderochromes formed by Micrococcus denitrificans. Biochem. J. 1975, 146, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Alemayehu, G.; Abegaz, B.; Snatzke, G.; Duddeck, H. Bianthraquinones and a spermidine alkaloid from Cassia floribunda. Phytochemistry 1988, 27, 3255–3258. [Google Scholar] [CrossRef]
- Doll, M.K.H.; Guggisberg, A.; Hesse, M. N4-Benzoylspermidine from Oncinotis tenuiloba: Analytical differentiation of the three isomeric N-benzoylspermidines. Helv. Chim. Acta 1994, 77, 1229–1235. [Google Scholar] [CrossRef]
- Bergeron, R.J.; Burton, P.S.; McGovern, K.A.; St Onge, E.J.; Streiff, R.R. Synthesis, absorption, and toxicity of N1,N8-bis(2,3-dihydroxybenzoyl)spermidine, a potent iron chelator. J. Med. Chem. 1980, 23, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Choomuenwai, V.; Schwartz, B.D.; Beattie, K.D.; Andrews, K.T.; Khokhar, S.; Davis, R.A. The discovery, synthesis and antimalarial evaluation of natural product-based polyamine alkaloids. Tetrahedron Lett. 2013, 54, 5188–5191. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Posseme, F.; Le Barbier, C.; Carreaux, F.; Carboni, B.; Seiler, N.; Moulinoux, J.P.; Delcros, J.G. N-benzylpolyamines as vectors of boron and fluorine for cancer therapy and imaging: Synthesis and biological evaluation. J. Med. Chem. 2001, 44, 3653–3664. [Google Scholar] [CrossRef] [PubMed]
- Egner, B.J.; Cardno, M.; Bradley, M. Linkers for combinatorial chemistry and reaction analysis using solid phase in situ mass spectrometry. J. Chem. Soc. Chem. Commun. 1995, 2163–2164. [Google Scholar] [CrossRef]
- Gude, M.; Ryf, J.; White, P.D. An accurate method for the quantitation of Fmoc-derivatized solid phase supports. Lett. Peptide Sci. 2002, 9, 203–206. [Google Scholar] [CrossRef]
- Brown, H.C.; Heim, P. Selective reductions. XVIII. The fast reaction of primary, secondary and tertiary amides with diborane. A simple, convenient procedure for the conversion of amides to the corresponding amines. J. Org. Chem. 1973, 38, 912–916. [Google Scholar] [CrossRef]
- Brown, H.C.; Heim, P. Diborane as a mild reducing agent for the conversion of primary, secondary and tertiary amides into the corresponding amines. J. Am. Chem. Soc. 1964, 86, 3566–3567. [Google Scholar] [CrossRef]
- de Carvalho Alcântara, A.F.; dos Santos Barroso, H.; Piló-Veloso, D. Redução de amidas por boranos. Quím. Nova 2002, 25, 300–311. [Google Scholar] [CrossRef]
- Lane, C.F. The borane-amine complexes. Aldrichim. Acta 1973, 6, 51–58. [Google Scholar]
- Schwartz, M.A.; Rose, B.F.; Vishnuvajjala, B. Intramolecular oxidative phenol coupling. III. Two-electron oxidation with thallium(II) trifluoroacetate. J. Am. Chem. Soc. 1973, 95, 612–613. [Google Scholar] [CrossRef]
- Choi, S.; Bruce, I.; Fairbanks, A.J.; Fleet, G.W.J.; Jones, A.H.; Nash, R.J.; Fellows, L.E. Alexines from heptonolactones. Tetrahedron Lett. 1991, 32, 5517–5520. [Google Scholar] [CrossRef]
- Curran, W.V.; Angier, R.B. Selective borane reduction of a trifluoroacetamide substituent in the presence of a carbamate. J. Org. Chem. 1966, 31, 3867–3868. [Google Scholar] [CrossRef]
- Wen, J.J.; Crews, C.M. Synthesis of 9-fluorenylmethoxycarbonyl-protected amino aldehydes. Tetrahedron Asymmetry 1998, 9, 1855–1858. [Google Scholar] [CrossRef]
- Bollhagen, R.; Schmiedberger, M.; Barlos, K.; Grell, E. A new reagent for the cleavage of fully protected peptides synthesised on 2-chlorotrityl chloride resin. J. Chem. Soc. Chem. Comm. 1994, 2559–2560. [Google Scholar] [CrossRef]
- Caravan, P.; Amedio, J.C., Jr.; Dunham, S.U.; Greenfield, M.T.; Cloutier, N.J.; McDermid, S.A.; Spiller, M.; Zech, S.G.; Looby, R.J.; Raitsimring, A.M.; et al. When are two waters worse than one? Doubling the hydration number of a Gd-DTPA derivative decreases relaxivity. Chem. Eur. J. 2005, 11, 5866–5874. [Google Scholar] [CrossRef]
- Amedio, J.C.; Bernard, P.J.; Fountain, M.; Wagenen, G.V. A practical manufacturing synthesis of 1-(R)-hydroxymethyl-Dtpa: An important intermediate in the synthesis of MRI contrast agents. Synth. Commun. 1999, 29, 2377–2391. [Google Scholar] [CrossRef]
- Bycroft, B.W.; Chan, W.C.; Chhabra, S.R.; Hone, N.D. A novel lysine-protecting procedure for continuous flow solid phase synthesis of branched peptides. J. Chem. Soc. Chem. Commun. 1993, 778–779. [Google Scholar] [CrossRef]
- Bycroft, B.W.; Chan, W.C.; Chhabra, S.R.; Teesdale-Spittle, P.H.; Hardy, P.M. A novel amino protection-deprotection procedure and its application in solid phase peptide synthesis. J. Chem. Soc. Chem. Commun. 1993, 776–777. [Google Scholar] [CrossRef]
- Bycroft, B.W.; Chan, W.C.; Hone, N.D.; Millington, S.; Nash, I.A. Synthesis of the spider toxins nephilatoxin-9 and -11 by a novel solid-phase strategy. J. Am. Chem. Soc. 1994, 116, 7415–7416. [Google Scholar] [CrossRef]
- Echalier, C.; Al-Halifa, S.; Kreiter, A.; Enjalbal, C.; Sanchez, P.; Ronga, L.; Puget, K.; Verdie, P.; Amblard, M.; Martinez, J.; et al. Heating and microwave assisted SPPS of C-terminal acid peptides on trityl resin: The truth behind the yield. Amino Acids 2013, 45, 1395–1403. [Google Scholar] [CrossRef]
- Wallace, H.M.; Mackarel, A.J. Regulation of polyamine acetylation and efflux in human cancer cells. Biochem. Soc. Trans. 1998, 26, 571–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinsky, S.A.; Christianson, D.W. Polyamine Deacetylase Structure and Catalysis: Prokaryotic Acetylpolyamine Amidohydrolase and Eukaryotic HDAC10. Biochemistry 2018, 57, 3105–3114. [Google Scholar] [CrossRef] [PubMed]
- Herbst-Gervasoni, C.J.; Christianson, D.W. Binding of N(8)-Acetylspermidine Analogues to Histone Deacetylase 10 Reveals Molecular Strategies for Blocking Polyamine Deacetylation. Biochemistry 2019, 58, 4957–4969. [Google Scholar] [CrossRef]
- Herbst-Gervasoni, C.J.; Christianson, D.W. X-ray Crystallographic Snapshots of Substrate Binding in the Active Site of Histone Deacetylase 10. Biochemistry 2021, 60, 303–313. [Google Scholar] [CrossRef]
- Geraldy, M.; Morgen, M.; Sehr, P.; Steimbach, R.R.; Moi, D.; Ridinger, J.; Oehme, I.; Witt, O.; Malz, M.; Nogueira, M.S.; et al. Selective Inhibition of Histone Deacetylase 10: Hydrogen Bonding to the Gatekeeper Residue is Implicated. J. Med. Chem. 2019, 62, 4426–4443. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, R.; Popov, V.; Laws, M.T.; Gelovani, D.; Majhi, A.; Shavrin, A.; Lu, X.; Muzik, O.; Turkman, N.; Liu, R.; et al. Molecular Imaging of Sirtuin1 Expression-Activity in Rat Brain Using Positron-Emission Tomography-Magnetic-Resonance Imaging with [18F]-2-Fluorobenzoylaminohexanoicanilide. J. Med. Chem. 2018, 61, 7116–7130. [Google Scholar] [CrossRef]
- Cullis, P.M.; Green, R.E.; Merson-Davies, L.; Travis, N.G. Chemical highlights of polyamine transport. Biochem. Soc. Trans. 1998, 26, 595–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mischiati, C.; Feriotto, G.; Tabolacci, C.; Domenici, F.; Melino, S.; Borromeo, I.; Forni, C.; De Martino, A.; Beninati, S. Polyamine Oxidase Is Involved in Spermidine Reduction of Transglutaminase Type 2-Catalyzed betaH-Crystallins Polymerization in Calcium-Induced Experimental Cataract. Int. J. Mol. Sci. 2020, 21, 5427. [Google Scholar] [CrossRef] [PubMed]
- Lentini, A.; Mattioli, P.; Provenzano, B.; Abbruzzese, A.; Caraglia, M.; Beninati, S. Role of the FAD-dependent polyamine oxidase in the selective formation of N1,N8-bis(γ-glutamyl)spermidine protein cross-links. Biochem. Soc. Trans. 2007, 35, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Hultsch, C.; Rode, K.; Kniess, T.; Wüst, F.; Bergmann, R.; Pietzsch, J. Synthesis of fluorinated N-benzoylpolyamines as substrates of tissue transglutaminase. In Annual Report 2003, Institute of Bioinorganic and Radiopharmaceutical Chemistry; Spies, H., Ed.; Research Center Rossendorf: Dresden, Germany, 2004; p. 36. [Google Scholar]
- Mäding, P.; Füchtner, F.; Johannsen, B.; Steinbach, J.; Hilger, C.S.; Friebe, M.; Halks-Miller, M.; Horuk, R.; Mohan, R. 18F-labelling of a potent nonpeptide CCR1 antagonist: Synthesis of 1-(5-chloro-2-{2-[(2R)-4-(4-[18F]fluorobenzyl)-2-methylpiperazin-1-yl]-2-oxoethoxy}phenyl)urea in an automated module. J. Label. Compd. Radiopharm. 2006, 49, 253–262. [Google Scholar] [CrossRef]
- Van der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuest, F. Fluorine-18 labeling of small molecules: The use of 18F-labeled aryl fluorides derived from no-carrier-added [18F]fluoride as labeling precursors. In PET Chemistry; Schubiger, P.A., Lehmann, L., Friebe, M., Eds.; Springer: Berlin, Germany, 2007; pp. 51–78. [Google Scholar]
- Kügler, F.; Ermert, J.; Coenen, H.H. Labeling of benzodioxin piperazines with fluorine-18 as prospective radioligands for selective imaging of dopamine D4 receptors. J. Labelled Comp. Radiopharm. 2013, 56, 609–618. [Google Scholar] [CrossRef]
- Horne, W.S.; Wiethoff, C.M.; Cui, C.L.; Wilcoxen, K.M.; Amorin, M.; Ghadiri, M.R.; Nemerow, G.R. Antiviral cyclic D,L-α-peptides: Targeting a general biochemical pathway in virus infections. Bioorg. Med. Chem. 2005, 13, 5145–5153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méret, M.; Bienz, S. Efficient and Flexible Solid-Phase Synthesis of N-Hydroxypolyamine Derivatives. Eur. J. Org. Chem. 2008, 5518–5525. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Name | Mass (mg) | Yield (%) 1 |
|---|---|---|---|
| 4-Fluorobenzoylated (FBz) Diamines and Polyamines | |||
| 1 | N1-FBz-putrescine × TFA | 28 | 63 |
| 2 | N1-FBz-cadaverine × TFA | 53 | 65 |
| 3 | N1-FBz-1,6-diaminohexane × TFA | 44 | 84 |
| 4 | N1-FBz-1,7-diaminoheptane × TFA | 37 | 64 |
| 5 | N1-FBz-1,8-diaminooctane × TFA | 35 | 64 |
| 6 | N1-FBz-3-oxospermidine × TFA | 26 | 88 |
| 7 | N8-FBz-5-oxospermidine × TFA | 39 | >100 2 |
| 8 | N1-FBz-3,8-dioxospermine × TFA | 69 | 89 |
| 9 | N1-FBz-spermidine × 2TFA | 28 | 38 |
| 10 | N4-FBz-spermidine × 2TFA | 21 | 35 |
| 11 | N8-FBz-spermidine × 2TFA | 14 | 29 |
| 12 | N1-FBz-spermine × 3TFA | 16 | 10 |
| 4-Fluorobenzylated (FBn) Diamines and Polyamines | |||
| 13 | N1-FBn-putrescine × 2TFA | 55 | 40 |
| 14 | N1-FBn-cadaverine × 2TFA | 27 | 36 |
| 15 | N1-FBn-1,6-diaminohexane × 2TFA | 15 | 20 |
| 16 | N1-FBn-1,7-diaminoheptane × 2TFA | 11 | 14 |
| 17 | N1-FBn-1,8-diaminooctane × 2TFA | 22 | 28 |
| 18 | N1-FBn-spermine × 4TFA | 11 | 14 |
| Pump (2x): | Varian, PrepStar 218 Solvent Delivery Module |
| UV/VIS detector: | Varian, ProStar 325 |
| wave length: | 254 nm |
| Software: | AlphaCrom, Star |
| stationary phase: | Varian, Dynamax 250 × 21.4 mm, Microsorb 60-8 C18 |
| eluent: | A: 0.1% TFA in Wasser B: 0.1% TFA in CH3CN |
| elution mode: | Gradient (Table 3) |
| flow rate: | 10 mL/min |
| t (min) | A (%) | B (%) |
|---|---|---|
| 0–7 | 90 | 10 |
| 7–22 | 20 | 90 |
| 22–30 | 20 | 90 |
| 30–31 | 90 | 10 |
| 31–35 | 90 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wodtke, R.; Pietzsch, J.; Löser, R. Solid-Phase Synthesis of Selectively Mono-Fluorobenz(o)ylated Polyamines as a Basis for the Development of 18F-Labeled Radiotracers. Molecules 2021, 26, 7012. https://doi.org/10.3390/molecules26227012
Wodtke R, Pietzsch J, Löser R. Solid-Phase Synthesis of Selectively Mono-Fluorobenz(o)ylated Polyamines as a Basis for the Development of 18F-Labeled Radiotracers. Molecules. 2021; 26(22):7012. https://doi.org/10.3390/molecules26227012
Chicago/Turabian StyleWodtke, Robert, Jens Pietzsch, and Reik Löser. 2021. "Solid-Phase Synthesis of Selectively Mono-Fluorobenz(o)ylated Polyamines as a Basis for the Development of 18F-Labeled Radiotracers" Molecules 26, no. 22: 7012. https://doi.org/10.3390/molecules26227012
APA StyleWodtke, R., Pietzsch, J., & Löser, R. (2021). Solid-Phase Synthesis of Selectively Mono-Fluorobenz(o)ylated Polyamines as a Basis for the Development of 18F-Labeled Radiotracers. Molecules, 26(22), 7012. https://doi.org/10.3390/molecules26227012