




Computational Modeling of DNA 3D Structures: From Dynamics and Mechanics to Folding


Abstract
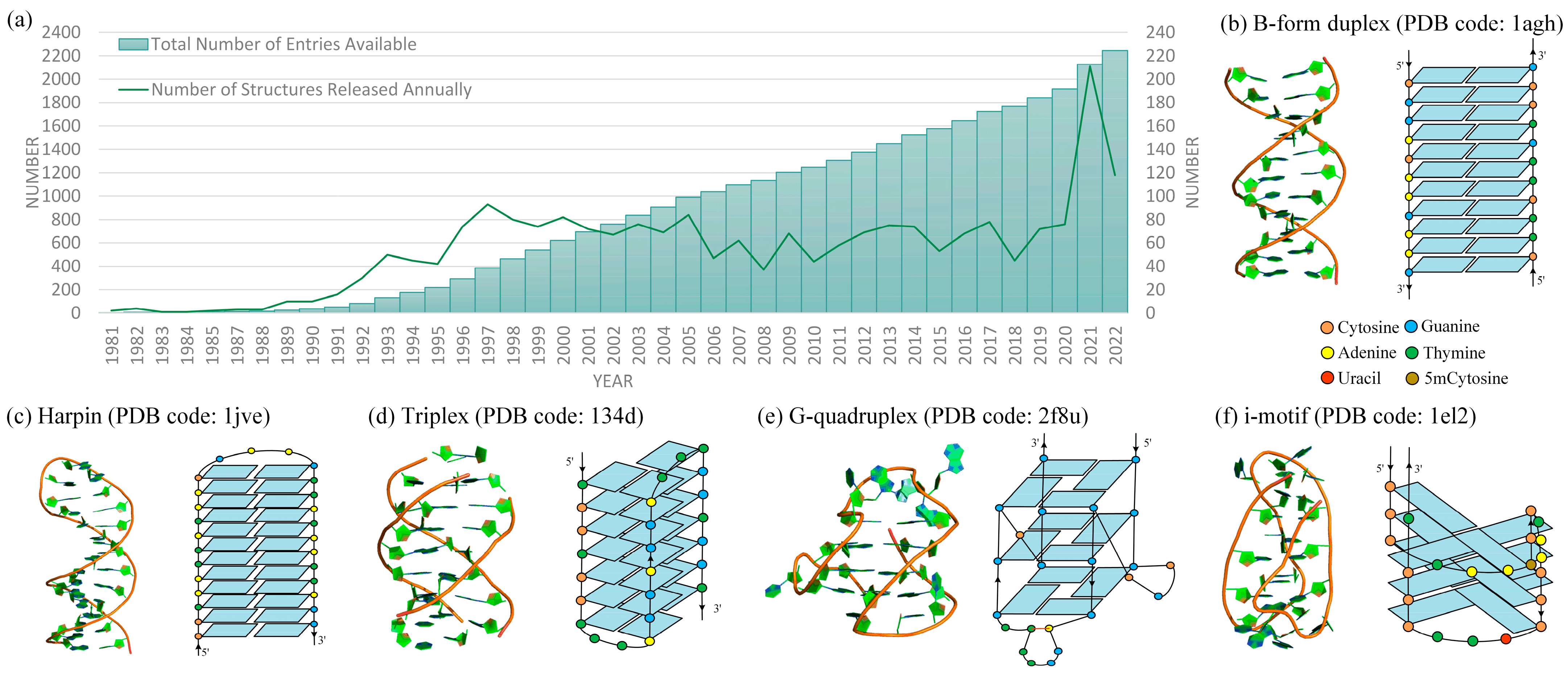
:1. Introduction
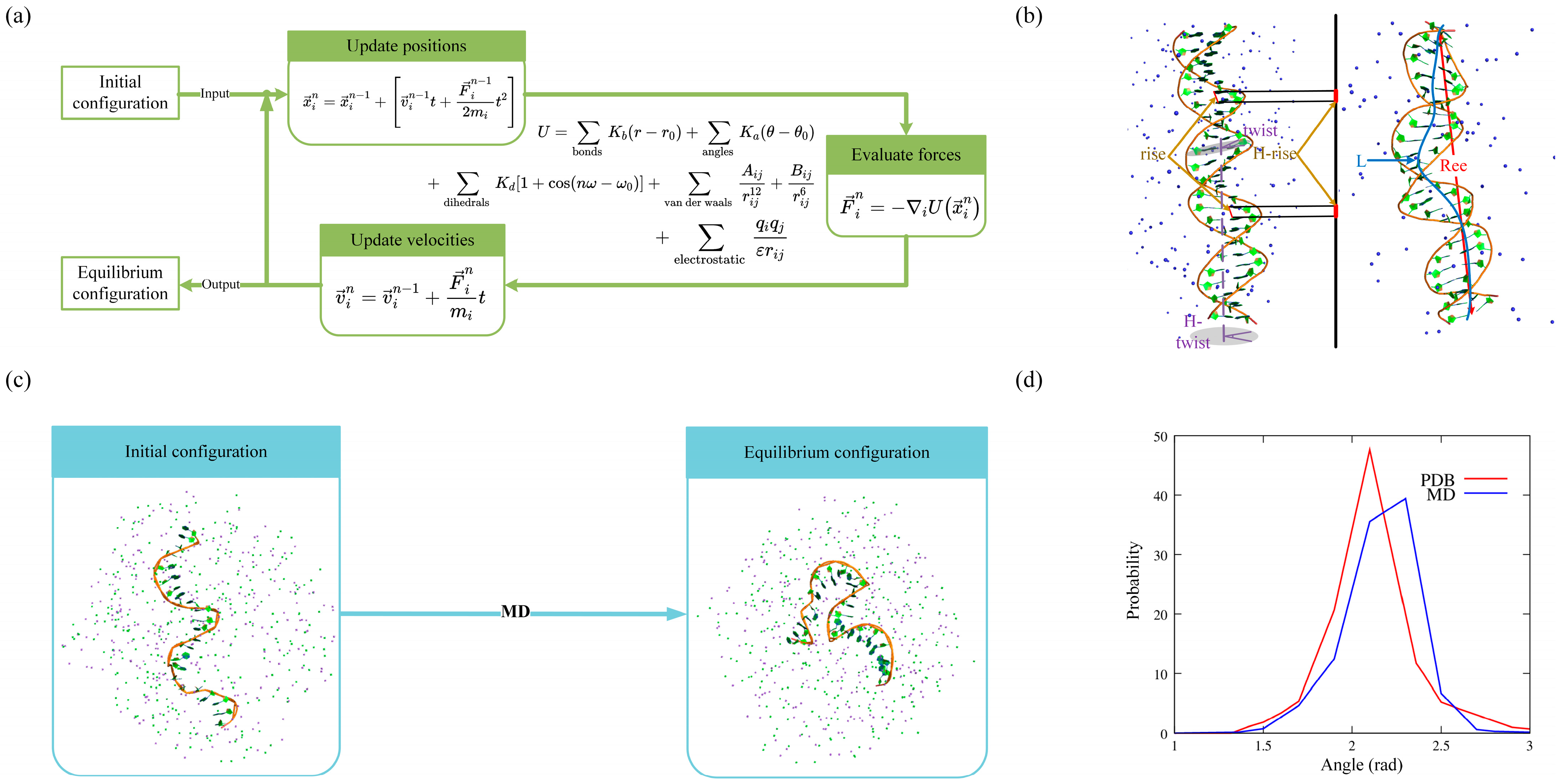
2. Molecular Dynamics Simulations for DNAs
2.1. Structural Dynamics
2.2. Structural Flexibility
2.3. DNA–Ion Interaction
2.4. Limitations
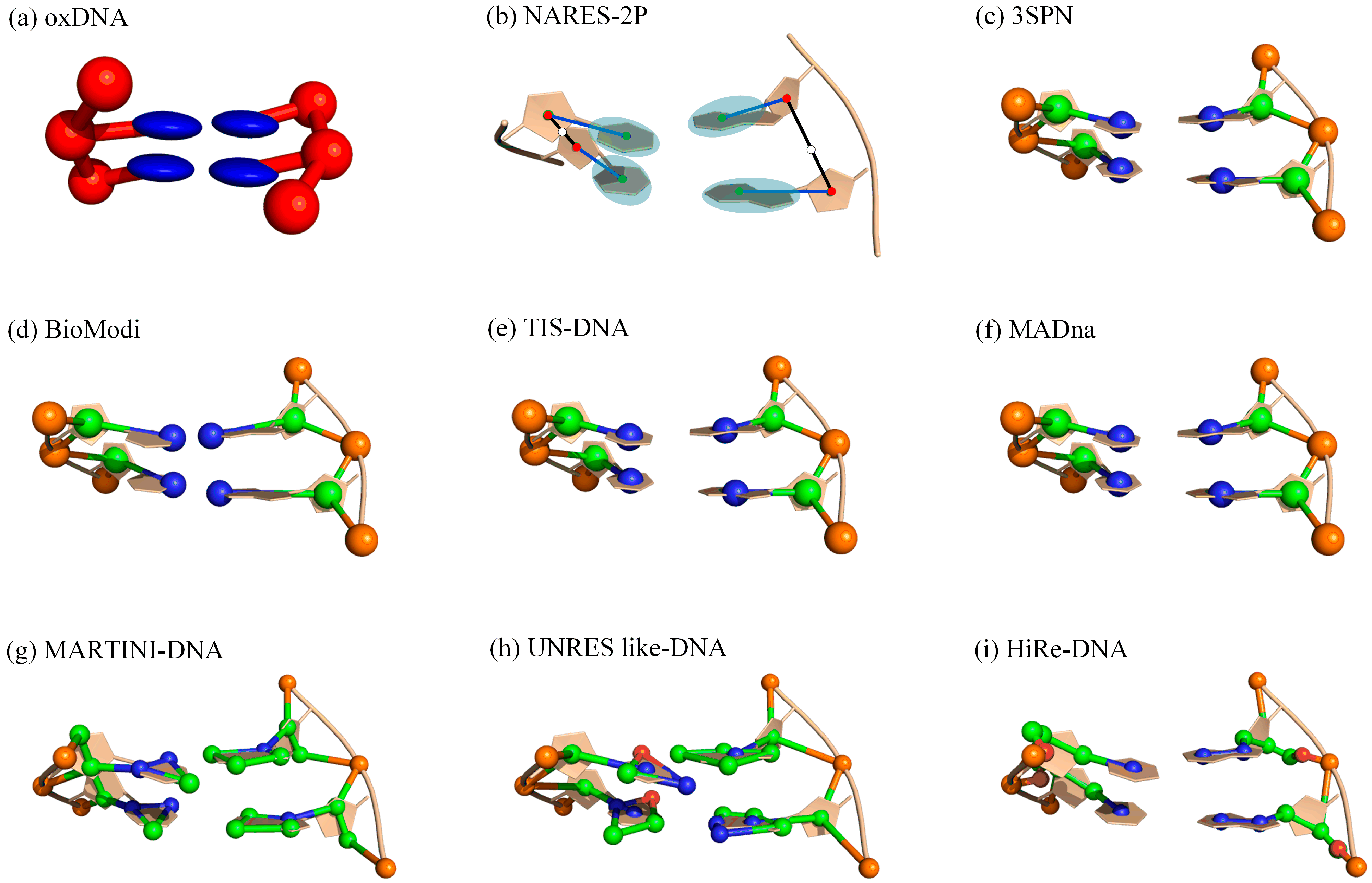
3. Coarse-Grained (CG) Modeling for DNAs
3.1. CG Models for DNA Structure Dynamics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
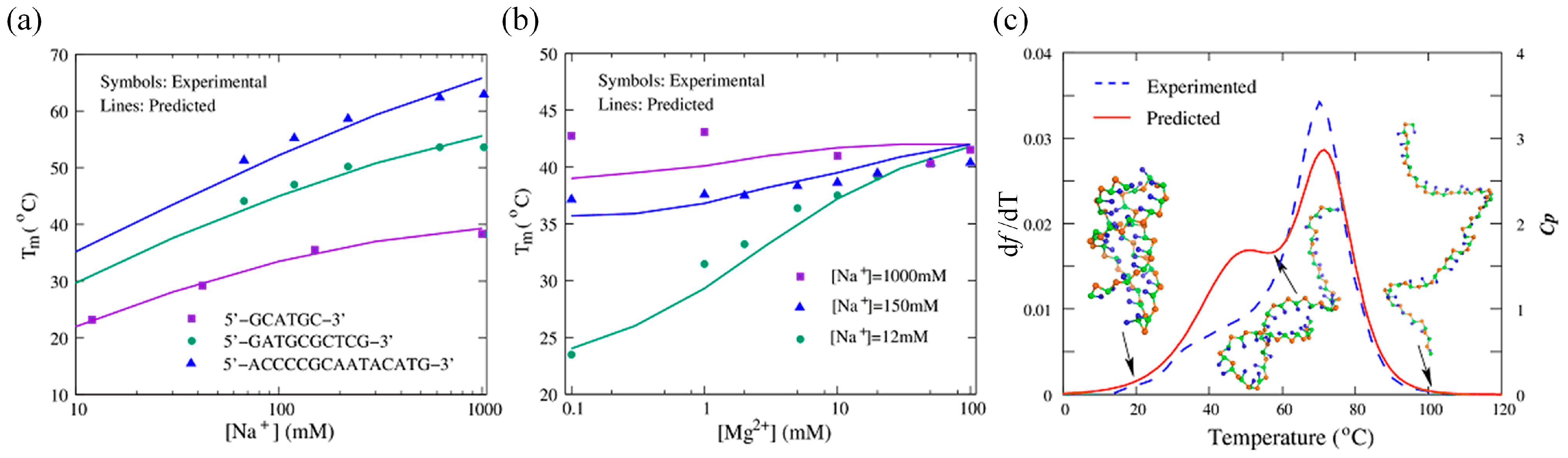{kind=link}
{kind=link}
| Models | Representation | Type a | Application b | Available c |
|---|---|---|---|---|
| Hall et al. [119] | 1 bead | Gō-like | Duplex/Triplex Tm | / |
| Aksimentiev et al. [77] | 2 beads | ab initio | Rg, Lp, force-extension | / |
| oxDNA [104,105,106,107] | 2 beads | Gō-like | Tm, Lp, force-extension, hybridization, dynamics, DNA–ion interaction, and nanotechnology | https://oxdna.org (accessed on 1 October 2022) |
| NARES-2P [108,109] | 2 beads | ab initio | 3D structure prediction, Tm, dynamics | / |
| Mittal et al. [120] | 2 beads | Gō-like | Tm, Particle interactions | / |
| MaDNA [116] | 3 beads | MD | dsDNA structure/elastic properties, Lp | https://github.com/saassenza/MADna (accessed on 1 October 2022) |
| 3SPN [110,111,112,113] | 3 beads | Gō-like | Tm, Lp, structure properties, dynamics, hybridization, DNA–ion interaction, nanotechnology | https://github.com/depablogroup (accessed on 1 October 2022) |
| TIS [115] | 3 beads | Gō-like | Rg, Lp, Tm, force extension, | / |
| Plotkin et al. [121] | 3 beads | ab initio | Lp, DNA twist, and stacking | / |
| Shi et al. [122] | 3 beads | ab initio | 3D structure prediction, salt effect, Tm, Lp | https://github.com/RNA-folding-lab/DNAfold (accessed on 1 October 2022) |
| BioModi [114] | 3 beads | Gō-like | Hybridization and self-assembly kinetics, salt-dependent Lp | / |
| Dorfman et al. [123,124,125] | 3 beads | ab initio | Tm, dynamics, structure properties, triplex forming | / |
| Nordenskiöld et al. [126] | 5 beads | MD | dsDNA Lp, LT | / |
| SIRAH [127] | 6 beads | MD | dsDNA Tm, transitions, and dynamics | / |
| “sugar” CG [128] | 6 beads | MD | dsDNA transitions, DNA–ion interaction | / |
| MARTINI [102] | 6/7 beads | MD | Rg, Lp, 3D structure, DNA–ion interaction, DNA–protein complexes | http://cgmartini.nl/ (accessed on 1 October 2022) |
| HiRe-DNA [118] | 6/7 beads | ab initio | dsDNA 3D structure, Tm | / |
| UNRES like-DNA [117] | 6/7/8 beads | ab initio | dsDNA 3D structure, structure properties, and hybridization | / |
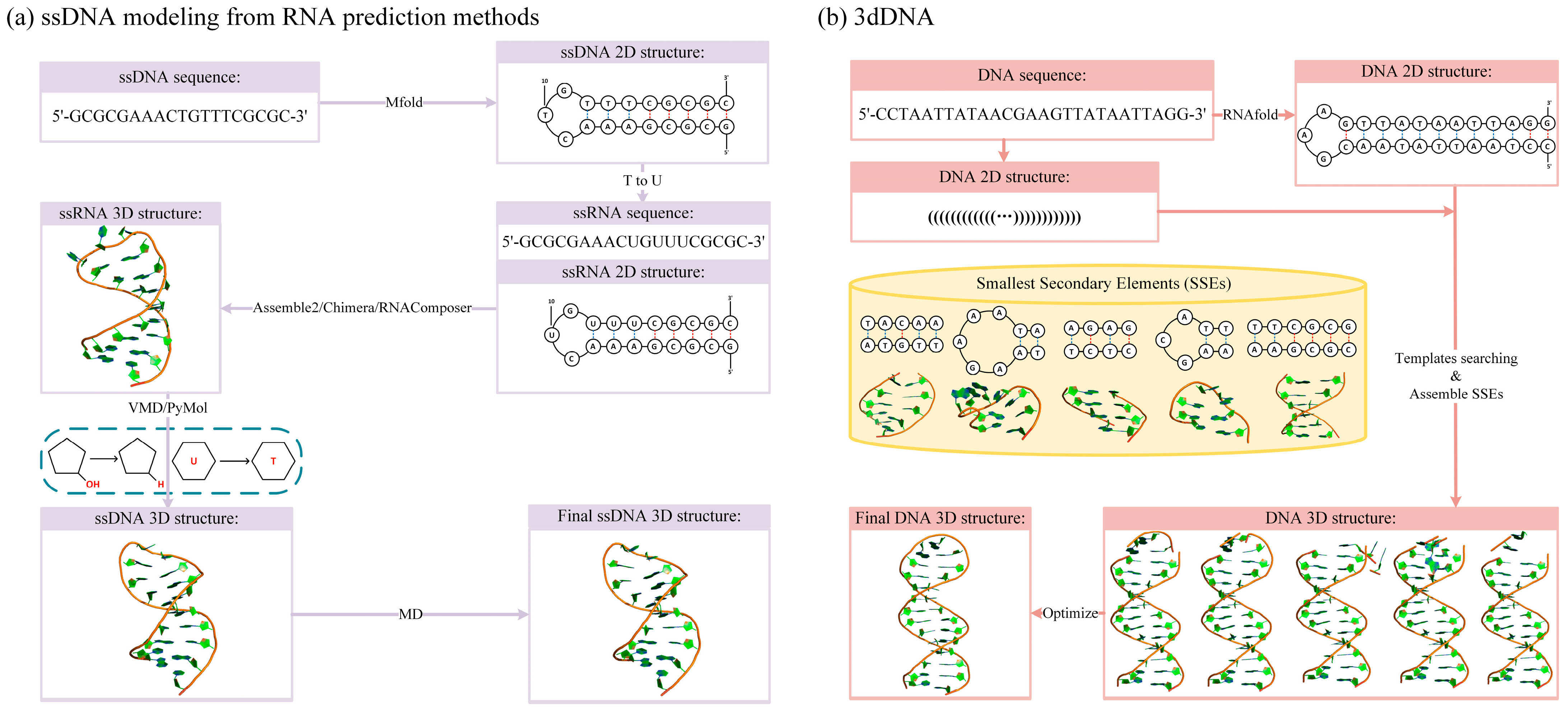
| 3dDNA [44] | all-atom | structure assembly | 3D structure prediction for DNAs with single, double, and multi-chains | http://biophy.hust.edu.cn/new/3dRNA (accessed on 1 October 2022) |
| Saiz et al. [129] | all-atom | structure assembly | ssDNA 3D structure prediction | / |
| Rahim et al. [130] | all-atom | structure assembly | ssDNA 3D structure prediction | / |
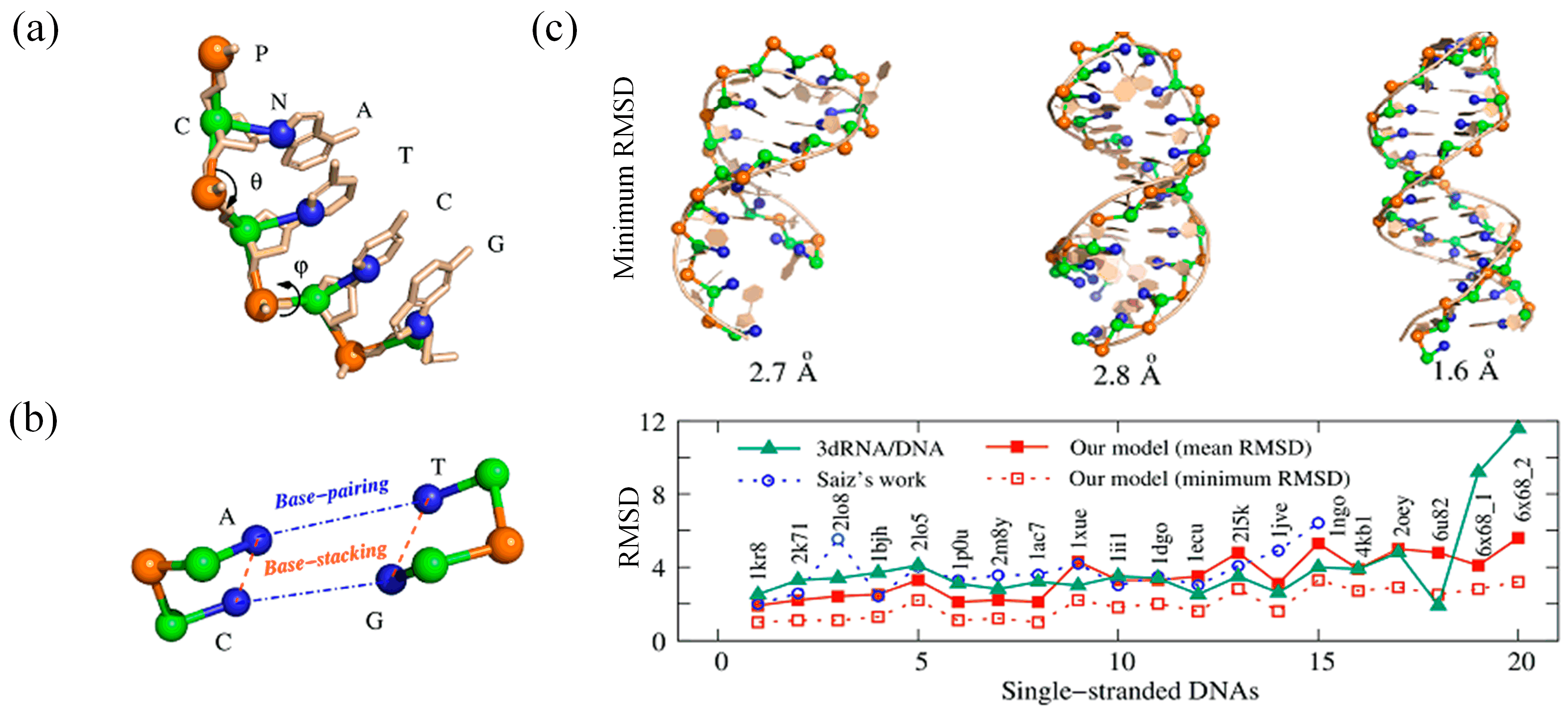
3.2. CG Models for DNA Structure Folding
3.3. Ab Initio CG Models
3.4. Discussion and Comparison of These CG Models
4. DNA Structure Assembly Method for 3D Structure Construction
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ferry, G. The structure of DNA. Nature 2019, 575, 35–36. [Google Scholar] [CrossRef] [Green Version]
- Neidle, S. Beyond the double helix: DNA structural diversity and the PDB. J. Biol. Chem. 2021, 296, 100553. [Google Scholar] [CrossRef] [PubMed]
- Nieuwland, C.; Hamlin, T.A.; Fonseca Guerra, C.; Barone, G.; Bickelhaupt, F.M. B-DNA structure and stability: The role of nucleotide composition and order. ChemistryOpen 2022, 11, e202100231. [Google Scholar]
- Guiblet, W.M.; Cremona, M.A.; Harris, R.S.; Chen, D.; Eckert, K.A.; Chiaromonte, F.; Huang, Y.-F.; Makova, K.D. Non-B DNA: A major contributor to small- and large-scale variation in nucleotide substitution frequencies across the genome. Nucleic Acids Res. 2021, 49, 1497–1516. [Google Scholar] [CrossRef]
- Bansal, A.; Kaushik, S.; Kukreti, S. Non-canonical DNA structures: Diversity and disease association. Front. Genet. 2022, 13, 959258. [Google Scholar] [CrossRef] [PubMed]
- Tateishi-Karimata, H.; Sugimoto, N. Roles of non-canonical structures of nucleic acids in cancer and neurodegenerative diseas-es. Nucleic Acids Res. 2021, 49, 7839–7855. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; De Silva, E.; Tornaletti, S.; Wang, G.; Vasquez, K.M.; Hanawalt, P.C. A Triplex-forming Sequence from the Human c-MYC Promoter Interferes with DNA Transcription. J. Biol. Chem. 2007, 282, 32433–32441. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.; Raguseo, F.; Nuccio, S.P.; Liano, D.; Di Antonio, M. DNA G-quadruplex structures: More than simple roadblocks to transcription? Nucleic Acids Res. 2021, 49, 8419–8431. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef]
- Zok, T.; Kraszewska, N.; Miskiewicz, J.; Pielacinska, P.; Zurkowski, M.; Szachniuk, M. ONQUADRO: A database of experimentally determined quadruplex structures. Nucleic Acids Res. 2022, 50, D253–D258. [Google Scholar] [CrossRef]
- Seeman, N.; Sleiman, H. DNA nanotechnology. Nat. Rev. Mater. 2017, 3, 17068. [Google Scholar] [CrossRef]
- Hu, Q.; Li, H.; Wang, L.; Gu, H.; Fan, C. DNA Nanotechnology-Enabled Drug Delivery Systems. Chem. Rev. 2019, 119, 6459–6506. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhan, Y.; Mao, C.; Xie, X.; Lin, Y. The biological applications of DNA nanomaterials: Current challenges and future directions. Signal Transduct. Target. Ther. 2021, 6, 351. [Google Scholar] [CrossRef]
- Butovskaya, E.; Heddi, B.; Bakalar, B.; Richter, S.N.; Phan, A.T. Major G-Quadruplex Form of HIV-1 LTR Reveals a (3 + 1) Folding Topology Containing a Stem-Loop. J. Am. Chem. Soc. 2018, 140, 13654–13662. [Google Scholar] [CrossRef] [Green Version]
- Bryant, Z.; Oberstrass, F.C.; Basu, A. Recent developments in single-molecule DNA mechanics. Curr. Opin. Struct. Biol. 2012, 22, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Kriegel, F.; Ermann, N.; Lipfert, J. Probing the mechanical properties, conformational changes, and interactions of nucleic acids with magnetic tweezers. J. Struct. Biol. 2017, 197, 26–36. [Google Scholar] [CrossRef]
- Haynes, P.J.; Main, K.H.S.; Akpinar, B.; Pyne, A.L.B. Atomic Force Microscopy of DNA and DNA-Protein Interactions. Methods Mol. 2022, 2476, 43–62. [Google Scholar] [CrossRef]
- Di, W.; Gao, X.; Huang, W.; Sun, Y.; Lei, H.; Liu, Y.; Li, W.; Li, Y.; Wang, X.; Qin, M.; et al. Direct Measurement of Length Scale Dependence of the Hydrophobic Free Energy of a Single Collapsed Polymer Nanosphere. Phys. Rev. Lett. 2019, 122, 047801. [Google Scholar] [CrossRef] [PubMed]
- Minhas, V.; Sun, T.; Mirzoev, A.; Korolev, N.; Lyubartsev, A.P.; Nordenskiöld, L. Modeling DNA Flexibility: Comparison of Force Fields from Atomistic to Multiscale Levels. J. Phys. Chem. B 2020, 124, 38–49. [Google Scholar] [CrossRef]
- Jones, M.S.; Ashwood, B.; Tokmakoff, A.; Ferguson, A.L. Determining Sequence-Dependent DNA Oligonucleotide Hybridization and Dehybridization Mechanisms Using Coarse-Grained Molecular Simulation, Markov State Models, and Infrared Spectroscopy. J. Am. Chem. Soc. 2021, 143, 17395–17411. [Google Scholar] [CrossRef]
- He, J.; Wang, J.; Tao, H.; Xiao, Y.; Huang, S.-Y. HNADOCK: A nucleic acid docking server for modeling RNA/DNA–RNA/DNA 3D complex structures. Nucleic Acids Res. 2019, 47, W35–W42. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, H.; Ou-Yang, Z.-C. Stretching Single-Stranded DNA: Interplay of Electrostatic, Base-Pairing, and Base-Pair Stacking Interactions. Biophys. J. 2001, 81, 1133–1143. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2005, 26, 114. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; McCready, M.J.; Maginn, E.J. Evaluation and refinement of the general AMBER force field for nineteen pure organic electrolyte solvents. J. Chem. Eng. 2018, 3488–3502. [Google Scholar] [CrossRef]
- Hart, K.; Foloppe, N.; Baker, C.M.; Denning, E.J.; Nilsson, L.; MacKerell, A.D., Jr. Optimization of the CHARMM additive force field for DNA: Improved treatment of the BI/BII conformational equilibrium. J. Chem. Theory Comput. 2012, 8, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.; Allen, J.E.; Yang, Y.; Bennett, W.F.D.; Gokhale, M.; Moshiri, N.; Rosing, T.S. Accelerators for Classical Molecular Dynamics Simulations of Biomolecules. J. Chem. Theory Comput. 2022, 18, 4047–4069. [Google Scholar] [CrossRef]
- Nomidis, S.K.; Kriegel, F.; Vanderlinden, W.; Lipfert, J.; Carlon, E. Twist-Bend Coupling and the Torsional Response of Double-Stranded DNA. Phys. Rev. Lett. 2017, 118, 217801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marko, J.F.; Siggia, E.D. Fluctuations and Supercoiling of DNA. Science 1994, 265, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Toan, N.M.; Thirumalai, D. On the origin of the unusual behavior in the stretching of single-stranded DNA. J. Chem. Phys. 2012, 136, 235103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SantaLucia, J., Jr.; Hicks, D. The Thermodynamics of DNA Structural Motifs. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 415–440. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406. [Google Scholar] [CrossRef]
- Ingólfsson, H.I.; Lopez, C.A.; Uusitalo, J.J.; de Jong, D.H.; Gopal, S.M.; Periole, X.; Marrink, S.J. The power of coarse graining in biomolecular simulations. WIREs Comput. Mol. Sci. 2014, 4, 225–248. [Google Scholar] [CrossRef]
- Dans, P.D.; Walther, J.; Gómez, H.; Orozco, M. Multiscale simulation of DNA. Curr. Opin. Struct. Biol. 2016, 37, 29–45. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Minhas, V.; Korolev, N.; Mirzoev, A.; Lyubartsev, A.P.; Nordenskiöld, L. Bottom-Up Coarse-Grained Modeling of DNA. Front. Mol. Biosci. 2021, 8, 645527. [Google Scholar] [CrossRef]
- Reshetnikov, R.; Stolyarova, A.; Zalevsky, A.; Panteleev, D.Y.; Pavlova, G.V.; Klinov, D.V.; Golovin, A.V.; Protopopova, A.D. A coarse-grained model for DNA origami. Nucleic Acids Res. 2018, 46, 1102–1112. [Google Scholar] [CrossRef] [Green Version]
- Walther, J.; Dans, P.D.; Balaceanu, A.; Hospital, A.; Bayarri, G.; Orozco, M. A multi-modal coarse grained model of DNA flexibility mappable to the atomistic level. Nucleic Acids Res. 2020, 48, e29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krokhotin, A.; Houlihan, K.; Dokholyan, N.V. iFoldRNA v2: Folding RNA with constraints. Bioinformatics 2015, 31, 2891–2893. [Google Scholar] [CrossRef] [Green Version]
- Jossinet, F.; Ludwig, T.E.; Westhof, E. Assemble: An interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics 2010, 26, 2057–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Zhang, D.; Chen, S.-J. Vfold-Pipeline: A web server for RNA 3D structure prediction from sequences. Bioinformatics 2022, 38, 4042–4043. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, Y.; Gong, Z.; Wang, Y.; Man, J.; Xiao, Y. Automated and fast building of three-dimensional RNA structures. Sci. Rep. 2012, 2, 734. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Mao, K.; Zhao, Y.; Zeng, C.; Xiang, J.; Zhang, Y.; Xiao, Y. Optimization of RNA 3D structure prediction using evolutionary restraints of nucleotide–nucleotide interactions from direct coupling analysis. Nucleic Acids Res. 2017, 45, 6299–6309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, J.; Huang, Y.; Xiao, Y. 3dRNA v2.0: An Updated Web Server for RNA 3D Structure Prediction. Int. J. Mol. Sci. 2019, 20, 4116. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiong, Y.; Xiao, Y. 3dDNA: A Computational Method of Building DNA 3D Structures. Molecules 2022, 27, 5936. [Google Scholar] [CrossRef]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Kruger, P.; Van Gunsteren, W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem. 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [Green Version]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.A.; García, A.E. High-resolution reversible folding of hyperstable RNA tetraloops using molecular dynamics simula-tions. Proc. Natl. Acad. Sci. USA 2013, 110, 16820–16825. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, A.; Bogart, B.M.; Dutagaci, B. Protein–Nucleic Acid Interactions for RNA Polymerase II Elongation Factors by Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 3079–3089. [Google Scholar] [CrossRef] [PubMed]
- Salsbury, A.M.; A Lemkul, J. Recent developments in empirical atomistic force fields for nucleic acids and applications to studies of folding and dynamics. Curr. Opin. Struct. Biol. 2021, 67, 9–17. [Google Scholar] [CrossRef]
- Kameda, T.; Awazu, A.; Togashi, Y. Molecular dynamics analysis of biomolecular systems including nucleic acids. Biophys. Physicobiology 2022, 19, e190027. [Google Scholar] [CrossRef]
- Ghoshdastidar, D.; Bansal, M. Dynamics of physiologically relevant noncanonical DNA structures: An overview from experi-mental and theoretical studies. Brief. Funct. Genom. 2018, 18, 192–204. [Google Scholar] [CrossRef]
- Frank-Kamenetskii, M.D.; Prakash, S. Fluctuations in the DNA double helix: A critical review. Phys. Life Rev. 2014, 11, 153–170. [Google Scholar] [CrossRef]
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, T.E., III; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar–Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736. [Google Scholar] [CrossRef] [PubMed]
- Strelnikov, I.A.; Kovaleva, N.A.; Klinov, A.P.; Zubova, E.A. C-B-A test of DNA force fields. ACS Omega 2023, 8, 10253–10265. [Google Scholar] [CrossRef] [PubMed]
- Panczyk, T.; Wojton, P.; Wolski, P. Mechanism of unfolding and relative stabilities of G-quadruplex and I-motif noncanonical DNA structures analyzed in biased molecular dynamics simulations. Biophys. Chem. 2019, 250, 106173. [Google Scholar] [CrossRef] [PubMed]
- Panczyk, T.; Wojton, P.; Wolski, P. Molecular Dynamics Study of the Interaction of Carbon Nanotubes with Telomeric DNA Fragment Containing Noncanonical G-Quadruplex and i-Motif Forms. Int. J. Mol. Sci. 2020, 21, 1925. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Yu, T.; Zhang, S.; Wang, Y.; Zhang, W. Thermodynamic and kinetic properties of a single base pair in A-DNA and B-DNA. Phys. Rev. E 2021, 103, 042409. [Google Scholar] [CrossRef]
- Xu, S.; Zhan, J.; Man, B.; Jiang, S.; Yue, W.; Gao, S.; Guo, C.; Liu, H.; Li, Z.; Wang, J.; et al. Real-time reliable determination of binding kinetics of DNA hybridization using a multi-channel graphene biosensor. Nat. Commun. 2017, 8, 14902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galindo-Murillo, R.; Roe, D.R. Cheatham TE 3rd. On the absence of intrahelical DNA dynamics on the μs to ms timescale. Nat. Commun. 2014, 5, 5152. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Murillo, R.; Roe, D.R.; Cheatham, T.E. Convergence and reproducibility in molecular dynamics simulations of the DNA duplex d(GCACGAACGAACGAACGC). Biochim. Biophys. Acta 2015, 1850, 1041–1058. [Google Scholar] [CrossRef] [Green Version]
- Nikolova, E.N.; Kim, E.; Wise, A.A.; O’brien, P.J.; Andricioaei, I.; Al-Hashimi, H.M. Transient Hoogsteen base pairs in canonical duplex DNA. Nature 2011, 470, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Kim, E.; Pak, Y. Free energy landscape and transition pathways from Watson–Crick to Hoogsteen base pairing in free duplex DNA. Nucleic Acids Res. 2015, 43, 7769–7778. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Wales, D.J. Probing helical transitions in a DNA duplex. Phys. Chem. Chem. Phys. 2017, 19, 878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Gonzalez, A.; Vilhena, J.G.; Perez, R.; Moreno-Herrero, F. A molecular view of DNA flexibility. Q. Rev. Biophys. 2021, 54, e8. [Google Scholar] [CrossRef]
- Ghoshdastidar, D.; Bansal, M. Flexibility of flanking DNA is a key determinant of transcription factor affinity for the core motif. Biophys. J. 2022, 121, 3987–4000. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.-W.; Dong, H.-L.; Xiong, K.-X.; Zhang, W.; Tan, Z.-J. Understanding sequence effect in DNA bending elasticity by molecular dynamic simulations. Commun. Theor. Phys. 2021, 73, 075601. [Google Scholar] [CrossRef]
- Liebl, K.; Dršata, T.; Lankas, F.; Lipfert, J.; Zacharias, M. Explaining the striking difference in twist-stretch coupling between DNA and RNA: A comparative molecular dynamics analysis. Nucleic Acids Res. 2015, 43, 10143–10156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Pettitt, B.M. The Effects of Flexibility on dsDNA-dsDNA Interactions. Life 2022, 12, 699. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Bao, L.; Zhang, X.; Tan, Z.-J. Flexibility of short DNA helices with finite-length effect: From base pairs to tens of base pairs. J. Chem. Phys. 2015, 142, 125103. [Google Scholar] [CrossRef] [Green Version]
- Marin-Gonzalez, A.; Vilhena, J.G.; Moreno-Herrero, F.; Perez, R. DNA Crookedness Regulates DNA Mechanical Properties at Short Length Scales. Phys. Rev. Lett. 2019, 122, 048102. [Google Scholar] [CrossRef] [Green Version]
- Liebl, K.; Zacharias, M. Accurate modeling of DNA conformational flexibility by a multivariate Ising model. Proc. Natl. Acad. Sci. USA 2021, 118, e2021263118. [Google Scholar] [CrossRef] [PubMed]
- Marin-Gonzalez, A.; Vilhena, J.G.; Perez, R.; Moreno-Herrero, F. Understanding the mechanical response of double-stranded DNA and RNA under constant stretching forces using all-atom molecular dynamics. Proc. Natl. Acad. Sci. USA 2017, 114, 7049–7054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Zhang, X.; Shi, Y.-Z.; Wu, Y.-Y.; Tan, Z.-J. Understanding the Relative Flexibility of RNA and DNA Duplexes: Stretching and Twist-Stretch Coupling. Biophys. J. 2017, 112, 1094–1104. [Google Scholar] [CrossRef] [Green Version]
- Bouchal, T.; Durník, I.; Kulhánek, P. Bending of Canonical and G/T Mismatched DNAs. J. Chem. Inf. Model. 2021, 61, 6000–6011. [Google Scholar] [CrossRef]
- Sharma, M.; Predeus, A.V.; Mukherjee, S.; Feig, M. DNA Bending Propensity in the Presence of Base Mismatches: Implications for DNA Repair. J. Phys. Chem. B 2013, 117, 6194–6205. [Google Scholar] [CrossRef] [Green Version]
- Maffeo, C.; Ngo, T.T.M.; Ha, T.; Aksimentiev, A. A Coarse-Grained Model of Unstructured Single-Stranded DNA Derived from Atomistic Simulation and Single-Molecule Experiment. J. Chem. Theory Comput. 2014, 10, 2891–2896. [Google Scholar] [CrossRef] [Green Version]
- Rossetti, G.; Dans, P.D.; Gomez-Pinto, I.; Ivani, I.; Gonzalez, C.; Orozco, M. The structural impact of DNA mismatches. Nucleic Acids Res. 2015, 43, 4309–4321. [Google Scholar] [CrossRef] [Green Version]
- Bouchal, T.; Durník, I.; Illík, V.; Réblová, K.; Kulhánek, P. Importance of base-pair opening for mismatch recognition. Nucleic Acids Res. 2020, 48, 11322–11334. [Google Scholar] [CrossRef]
- Lavery, R.; Maddocks, J.H.; Pasi, M.; Zakrzewska, K. Analyzing ion distributions around DNA. Nucleic Acids Res. 2014, 42, 8138–8149. [Google Scholar] [CrossRef] [PubMed]
- Tolokh, I.S.; Thomas, D.G.; Onufriev, A.V. Explicit ions/implicit water generalized Born model for nucleic acids. J. Chem. Phys. 2018, 148, 195101. [Google Scholar] [CrossRef]
- Sun, L.Z.; Qian, J.L.; Cai, P.; Xu, X. Mutual effects between single-stranded DNA conformation and Na+-Mg2+ ion competition in mixed salt solutions. Phys. Chem. Chem. Phys. 2022, 24, 20867–20881. [Google Scholar] [CrossRef]
- Xue, J.; Wang, P.; Li, X.; Tan, R.; Zong, W. Transformation characteristics of A-DNA in salt solution revealed through molecular dynamics simulations. Biophys. Chem. 2022, 288, 106845. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Singh, P.C. The combined action of cations and anions of ionic liquids modulates the formation and stability of G-quadruplex DNA. Phys. Chem. Chem. Phys. 2021, 23, 24497–24504. [Google Scholar] [CrossRef] [PubMed]
- Pasi, M.; Maddocks, J.H.; Lavery, R. Analyzing ion distributions around DNA: Sequence-dependence of potassium ion distributions from microsecond molecular dynamics. Nucleic Acids Res. 2015, 43, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Zhang, C.; Qiang, X.-W.; Yang, Y.-J.; Dai, L.; Tan, Z.-J.; Zhang, X.-H. Opposite Effects of High-Valent Cations on the Elasticities of DNA and RNA Duplexes Revealed by Magnetic Tweezers. Phys. Rev. Lett. 2020, 124, 058101. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Leon, S.; Vanderlinden, W.; Muller, P.; Forster, T.; Staudt, G.; Lin, Y.Y.; Lipfert, J.; Schwierz, N. Twisting DNA by salt. Nucleic Acids Res. 2022, 50, 5726–5738. [Google Scholar] [CrossRef]
- Long, M.P.; Alland, S.; Martin, M.E.; Isborn, C.M. Molecular dynamics simulations of alkaline earth metal ions binding to DNA reveal ion size and hydration effects. Phys. Chem. Chem. Phys. 2020, 22, 5584–5596. [Google Scholar] [CrossRef]
- Xi, K.; Wang, F.-H.; Xiong, G.; Zhang, Z.-L.; Tan, Z.-J. Competitive Binding of Mg2+ and Na+ Ions to Nucleic Acids: From Helices to Tertiary Structures. Biophys. J. 2018, 114, 1776–1790. [Google Scholar] [CrossRef] [Green Version]
- Krüger, A.; Zimbres, F.M.; Kronenberger, T.; Wrenger, C. Molecular Modeling Applied to Nucleic Acid-Based Molecule Development. Biomolecules 2018, 8, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oweida, T.J.; Kim, H.S.; Donald, J.M.; Singh, A.; Yingling, Y.G. Faculty Opinions recommendation of Assessment of AMBER Force Fields for Simulations of ssDNA. J. Chem. Theory Comput. 2021, 17, 1208–1217. [Google Scholar] [CrossRef]
- Dans, P.D.; Ivani, I.; Hospital, A.; Portella, G.; González, C.; Orozco, M. How accurate are accurate force-fields for B-DNA? Nucleic Acids Res. 2017, 45, 4217–4230. [Google Scholar] [CrossRef] [Green Version]
- Cruz-León, S.; Grotz, K.K.; Schwierz, N. Extended magnesium and calcium force field parameters for accurate ion–nucleic acid interactions in biomolecular simulations. J. Chem. Phys. 2021, 154, 171102. [Google Scholar] [CrossRef]
- Castelli, M.; Doria, F.; Freccero, M.; Colombo, G.; Moroni, E. Studying the Dynamics of a Complex G-Quadruplex System: Insights into the Comparison of MD and NMR Data. J. Chem. Theory Comput. 2022, 18, 4515–4528. [Google Scholar] [CrossRef]
- Havrila, M.; Stadlbauer, P.; Islam, B.; Otyepka, M.; Šponer, J. Effect of Monovalent Ion Parameters on Molecular Dynamics Simula-tions of G-Quadruplexes. J. Chem. Theory Comput. 2017, 13, 3911–3926. [Google Scholar] [CrossRef]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef] [PubMed]
- van Gunsteren, W.F.; Daura, X.; Hansen, N.; Mark, A.E.; Oostenbrink, C.; Riniker, S.; Smith, L.J. Validation of Molecular Simulation: An Overview of Issues. Angew. Chem. Int. Ed. 2018, 57, 884–902. [Google Scholar] [CrossRef] [PubMed]
- Betz, R.M.; Dror, R.O. How Effectively Can Adaptive Sampling Methods Capture Spontaneous Ligand Binding? J. Chem. Theory Comput. 2019, 15, 2053–2063. [Google Scholar] [CrossRef]
- Markthaler, D.; Fleck, M.; Stankiewicz, B.; Hansen, N. Exploring the Effect of Enhanced Sampling on Protein Stability Prediction. J. Chem. Theory Comput. 2022, 18, 2569–2583. [Google Scholar] [CrossRef] [PubMed]
- Kasavajhala, K.; Lam, K.; Simmerling, C. Exploring Protocols to Build Reservoirs to Accelerate Temperature Replica Exchange MD Simulations. J. Chem. Theory Comput. 2020, 16, 7776–7799. [Google Scholar] [CrossRef]
- de Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2012, 9, 687–697. [Google Scholar] [CrossRef]
- Uusitalo, J.J.; Ingólfsson, H.I.; Akhshi, P.; Tieleman, D.P.; Marrink, S.J. Martini Coarse-Grained Force Field: Extension to DNA. J. Chem. Theory Comput. 2015, 11, 3932–3945. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Ouldridge, T.E.; Louis, A.A.; Doye, J.P.K. Structural, mechanical, and thermodynamic properties of a coarse-grained DNA model. J. Chem. Phys. 2011, 134, 085101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouldridge, T.; Louis, A.; Doye, J. DNA Nanotweezers Studied with a Coarse-Grained Model of DNA. Phys. Rev. Lett. 2010, 104, 178101. [Google Scholar] [CrossRef] [PubMed]
- Šulc, P.; Romano, F.; Ouldridge, T.E.; Rovigatti, L.; Doye, J.P.; Louis, A.A. Sequence-dependent thermodynamics of a coarse-grained DNA model. J. Chem. Phys. 2012, 137, 135101. [Google Scholar] [CrossRef] [Green Version]
- Snodin, B.E.K.; Randisi, F.; Mosayebi, M.; Šulc, P.; Schreck, J.S.; Romano, F.; Ouldridge, T.E.; Tsukanov, R.; Nir, E.; Louis, A.A.; et al. Introducing improved structural properties and salt dependence into a coarse-grained model of DNA. J. Chem. Phys. 2015, 142, 234901. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Maciejczyk, M.; Ołdziej, S.; Scheraga, H.A.; Liwo, A. Mean-Field Interactions between Nucleic-Acid-Base Dipoles can Drive the Formation of a Double Helix. Phys. Rev. Lett. 2013, 110, 098101. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Liwo, A.; Scheraga, H.A. Optimization of a Nucleic Acids united-RESidue 2-Point model (NARES-2P) with a maximum-likelihood approach. J. Chem. Phys. 2015, 143, 243111. [Google Scholar] [CrossRef] [Green Version]
- Knotts, T.A.; Rathore, N.; Schwartz, D.C.; De Pablo, J.J. A coarse grain model for DNA. J. Chem. Phys. 2007, 126, 084901. [Google Scholar] [CrossRef] [Green Version]
- Sambriski, E.; Schwartz, D.; de Pablo, J. A Mesoscale Model of DNA and Its Renaturation. Biophys. J. 2009, 96, 1675–1690. [Google Scholar] [CrossRef] [Green Version]
- Hinckley, D.M.; Freeman, G.S.; Whitmer, J.K.; De Pablo, J.J. An experimentally-informed coarse-grained 3-site-per-nucleotide model of DNA: Structure, thermodynamics, and dynamics of hybridization. J. Chem. Phys. 2013, 139, 144903. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.S.; Hinckley, D.M.; Lequieu, J.P.; Whitmer, J.K.; de Pablo, J.J. Coarse-grained modeling of DNA curvature. J. Chem. Phys. 2014, 141, 165103. [Google Scholar] [CrossRef] [Green Version]
- Markegard, C.B.; Fu, I.W.; Reddy, K.A.; Nguyen, H.D. Coarse-Grained Simulation Study of Sequence Effects on DNA Hybridization in a Concentrated Environment. J. Phys. Chem. B 2015, 119, 1823–1834. [Google Scholar] [CrossRef]
- Chakraborty, D.; Hori, N.; Thirumalai, D. Sequence-Dependent Three Interaction Site Model for Single- and Double-Stranded DNA. J. Chem. Theory Comput. 2018, 14, 3763–3779. [Google Scholar] [CrossRef]
- Assenza, S.; Perez, R. Accurate sequence-dependent coarse-grained model for conformational and elastic properties of dou-ble-stranded DNA. J. Chem. Theory Comput. 2022, 18, 3239–3256. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Spasic, A.; Liwo, A.; Scheraga, H.A. DNA Duplex Formation with a Coarse-Grained Model. J. Chem. Theory Comput. 2014, 10, 5020–5035. [Google Scholar] [CrossRef] [Green Version]
- Cragnolini, T.; Derreumaux, P.; Pasquali, S. Coarse-Grained Simulations of RNA and DNA Duplexes. J. Phys. Chem. B 2013, 117, 8047–8060. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.W.; Barker, K.; Benner, S.; Betancourt, T.; Hall, C.K. Development of a simple coarse-grained DNA model for analysis of oligonucleotide complex formation. Mol. Simul. 2018, 44, 1004–1015. [Google Scholar] [CrossRef]
- Ding, Y.; Mittal, J. Insights into DNA-mediated interparticle interactions from a coarse-grained model. J. Chem. Phys. 2014, 141, 184901. [Google Scholar] [CrossRef] [Green Version]
- Morriss-Andrews, A.; Rottler, J.; Plotkin, S.S. A systematically coarse-grained model for DNA and its predictions for persistence length, stacking, twist, and chirality. J. Chem. Phys. 2010, 132, 035105. [Google Scholar] [CrossRef] [Green Version]
- Mu, Z.-C.; Tan, Y.-L.; Zhang, B.-G.; Liu, J.; Shi, Y.-Z. Ab initio predictions for 3D structure and stability of single- and double-stranded DNAs in ion solutions. PLoS Comput. Biol. 2022, 18, e1010501. [Google Scholar] [CrossRef]
- Kenward, M.; Dorfman, K.D. Brownian dynamics simulations of single-stranded DNA hairpins. J. Chem. Phys. 2009, 130, 095101. [Google Scholar] [CrossRef]
- Linak, M.C.; Dorfman, K.D. Analysis of a DNA simulation model through hairpin melting experiments. J. Chem. Phys. 2010, 133, 125101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linak, M.C.; Tourdot, R.; Dorfman, K.D. Moving beyond Watson–Crick models of coarse grained DNA dynamics. J. Chem. Phys. 2011, 135, 205102. [Google Scholar] [CrossRef] [Green Version]
- Korolev, N.; Luo, D.; Lyubartsev, A.P.; Nordenskiöld, L. A Coarse-Grained DNA Model Parameterized from Atomistic Simulations by Inverse Monte Carlo. Polymers 2014, 6, 1655–1675. [Google Scholar] [CrossRef] [Green Version]
- Dans, P.D.; Zeida, A.; Machado, M.R.; Pantano, S. A coarse grained model for atomic-detailed DNA simulations with explicit elec-trostatics. J. Chem. Theory Comput. 2010, 6, 1711–1725. [Google Scholar] [CrossRef]
- Kovaleva, N.A.; Koroleva Kikot, I.P.; Mazo, M.A.; Zubova, E.A. The “sugar” coarse-grained DNA model. J. Mol. Model. 2017, 23, 66. [Google Scholar] [CrossRef] [Green Version]
- Jeddi, I.; Saiz, L. Three-dimensional modeling of single stranded DNA hairpins for aptamer-based biosensors. Sci. Rep. 2017, 7, 1178. [Google Scholar] [CrossRef] [Green Version]
- Sabri, M.Z.; Hamid, A.A.A.; Hitam, S.M.S.; Rahim, M.Z.A. The assessment of three dimensional modelling design for single strand DNA aptamers for computational chemistry application. Biophys. Chem. 2020, 267, 106492. [Google Scholar] [CrossRef]
- Prieto, L.; de Sancho, D.; Rey, A. Thermodynamics of Go-type models for protein folding. J. Chem. Phys. 2005, 123, 154903. [Google Scholar] [CrossRef]
- Šulc, P.; Romano, F.; Ouldridge, T.E.; Doye, J.P.; Louis, A.A. A nucleotide-level coarse-grained model of RNA. J. Chem. Phys. 2014, 140, 235102. [Google Scholar] [CrossRef] [Green Version]
- Hyeon, C.; Thirumalai, D. Mechanical unfolding of RNA hairpins. Proc. Natl. Acad. Sci. USA 2005, 102, 6789–6794. [Google Scholar] [CrossRef] [Green Version]
- Hyeon, C.; Thirumalai, D. Capturing the essence of folding and functions of biomolecules using coarse-grained models. Nat. Commun. 2011, 2, 487. [Google Scholar] [CrossRef] [Green Version]
- Denesyuk, N.A.; Thirumalai, D. Coarse-Grained Model for Predicting RNA Folding Thermodynamics. J. Phys. Chem. B 2013, 117, 4901–4911. [Google Scholar] [CrossRef] [Green Version]
- Pasquali, S.; Derreumaux, P. HiRE-RNA: A high resolution coarse-grained energy model for RNA. J. Phys. Chem. B. 2010, 114, 11957–11966. [Google Scholar] [CrossRef]
- Shi, Y.-Z.; Wang, F.-H.; Wu, Y.-Y.; Tan, Z.-J. A coarse-grained model with implicit salt for RNAs: Predicting 3D structure, stability and salt effect. J. Chem. Phys. 2014, 141, 105102. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.-Z.; Jin, L.; Wang, F.-H.; Zhu, X.-L.; Tan, Z.-J. Predicting 3D Structure, Flexibility, and Stability of RNA Hairpins in Monovalent and Divalent Ion Solutions. Biophys. J. 2015, 109, 2654–2665. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.-Z.; Jin, L.; Feng, C.-J.; Tan, Y.-L.; Tan, Z.-J. Predicting 3D structure and stability of RNA pseudoknots in monovalent and divalent ion solutions. PLoS Comput. Biol. 2018, 14, e1006222. [Google Scholar] [CrossRef]
- Jin, L.; Shi, Y.-Z.; Feng, C.-J.; Tan, Y.-L.; Tan, Z.-J. Modeling Structure, Stability, and Flexibility of Double-Stranded RNAs in Salt Solutions. Biophys. J. 2018, 115, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Tan, Y.-L.; Wu, Y.; Wang, X.; Shi, Y.-Z.; Tan, Z.-J. Structure folding of RNA kissing complexes in salt solutions: Predicting 3D structure, stability, and folding pathway. RNA 2019, 25, 1532–1548. [Google Scholar] [CrossRef]
- SantaLucia, J., Jr.; Allawi, H.T.; Seneviratne, P.A. Improved Nearest-Neighbor Parameters for Predicting DNA Duplex Stability. Biochemistry 1996, 35, 3555–3562. [Google Scholar] [CrossRef]
- Tan, Z.J.; Chen, S.J. Electrostatic correlations and fluctuations for ion binding to a finite length polyelectrolyte. J. Chem. Phys. 2005, 122, 44903. [Google Scholar] [CrossRef]
- Tan, Z.; Zhang, W.; Shi, Y.; Wang, F. RNA folding: Structure prediction, folding kinetics and ion electrostatics. Adv. Exp. Med. Biol. 2015, 827, 143–183. [Google Scholar]
- Tan, Y.-L.; Wang, X.; Shi, Y.-Z.; Zhang, W.; Tan, Z.-J. rsRNASP: A residue-separation-based statistical potential for RNA 3D structure evaluation. Biophys. J. 2022, 121, 142–156. [Google Scholar] [CrossRef]
- Tan, Y.-L.; Wang, X.; Yu, S.; Zhang, B.; Tan, Z.-J. cgRNASP: Coarse-grained statistical potentials with residue separation for RNA structure evaluation. NAR Genom. Bioinform. 2023, 5, lqad016. [Google Scholar] [CrossRef]
- Li, Z.; Yang, Y.; Zhan, J.; Dai, L.; Zhou, Y. Energy Functions in De Novo Protein Design: Current Challenges and Future Prospects. Annu. Rev. Biophys. 2013, 42, 315–335. [Google Scholar] [CrossRef] [Green Version]
- Xiong, P.; Wu, R.; Zhan, J.; Zhou, Y. Pairing a high-resolution statistical potential with a nucleobase-centric sampling algorithm for improving RNA model refinement. Nat. Commun. 2021, 12, 2777. [Google Scholar] [CrossRef]
- Maffeo, C.; Aksimentiev, A. MrDNA: A multi-resolution model for predicting the structure and dynamics of DNA systems. Nucleic Acids Res. 2020, 48, 5135–5146. [Google Scholar] [CrossRef] [Green Version]
- Veneziano, R.; Ratanalert, S.; Zhang, K.; Zhang, F.; Yan, H.; Chiu, W.; Bathe, M. Designer nanoscale DNA assemblies programmed from the top down. Science 2016, 352, 1534. [Google Scholar] [CrossRef] [Green Version]
- de Llano, E.; Miao, H.; Ahmadi, Y.; Wilson, A.J.; Beeby, M.; Viola, I.; Barisic, I. Adenita: Interactive 3D modelling and visualization of DNA nanostructures. Nucleic Acids Res. 2020, 48, 8269–8275. [Google Scholar] [CrossRef]
- Zeng, C.; Jian, Y.; Vosoughi, S.; Zeng, C.; Zhao, Y. Evaluating native-like structures of RNA-protein complexes through the deep learning method. Nat. Commun. 2023, 14, 1060. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Yan, C. Improved protein contact prediction using dimensional hybrid residual networks and singularity enhanced loss function. Briefings Bioinform. 2021, 22, bbab341. [Google Scholar] [CrossRef]
- Si, Y.; Yan, C. Improved inter-protein contact prediction using dimensional hybrid residual networks and protein language models. Briefings Bioinform. 2023, 24, bbad039. [Google Scholar] [CrossRef]
- Huang, B.; Du, Y.; Zhang, S.; Li, W.; Wang, J.; Zhang, J. Computational prediction of RNA tertiary structures using machine learning methods. Chin. Phys. B 2020, 29, 08704. [Google Scholar] [CrossRef]
- Li, J.; Zhu, W.; Wang, J.; Li, W.; Gong, S.; Zhang, J.; Wang, W. RNA3DCNN: Local and global quality assessments of RNA 3D structures using 3D deep convolutional neural networks. PLoS Comput. Biol. 2018, 14, e1006514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Schlick, T.; Portillo-Ledesma, S.; Myers, C.G.; Beljak, L.; Chen, J.; Dakhel, S.; Darling, D.; Ghosh, S.; Hall, J.; Jan, M.; et al. Biomolecular modeling and simulation: A prospering multi-disciplinary field. Annu. Rev. Biophys. 2021, 50, 267–301. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Hu, G.; Tao, P. Allosteric Regulation at the Crossroads of New Technologies: Multiscale Modeling, Networks, and Machine Learning. Front. Mol. Biosci. 2020, 7, 136. [Google Scholar] [CrossRef]






| Potential | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model | |||||||||||||||
| oxDNA | √ | √ | √ | √ | √ | √ | √ | ||||||||
| 3SPN | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| TIS | √ | √ | √ | √ | √ | √ | |||||||||
| Plotkin et al. | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
| UNRES-like DNA | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| HiRE-DNA | √ | √ | √ | √ | √ | ||||||||||
| NARES-2P | √ | √ | √ | √ | |||||||||||
| Shi et al. | √ | √ | √ | √ | √ | √ | √ | √ | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, Z.-C.; Tan, Y.-L.; Liu, J.; Zhang, B.-G.; Shi, Y.-Z. Computational Modeling of DNA 3D Structures: From Dynamics and Mechanics to Folding. Molecules 2023, 28, 4833. https://doi.org/10.3390/molecules28124833
Mu Z-C, Tan Y-L, Liu J, Zhang B-G, Shi Y-Z. Computational Modeling of DNA 3D Structures: From Dynamics and Mechanics to Folding. Molecules. 2023; 28(12):4833. https://doi.org/10.3390/molecules28124833
Chicago/Turabian StyleMu, Zi-Chun, Ya-Lan Tan, Jie Liu, Ben-Gong Zhang, and Ya-Zhou Shi. 2023. "Computational Modeling of DNA 3D Structures: From Dynamics and Mechanics to Folding" Molecules 28, no. 12: 4833. https://doi.org/10.3390/molecules28124833
APA StyleMu, Z. -C., Tan, Y. -L., Liu, J., Zhang, B. -G., & Shi, Y. -Z. (2023). Computational Modeling of DNA 3D Structures: From Dynamics and Mechanics to Folding. Molecules, 28(12), 4833. https://doi.org/10.3390/molecules28124833