
Effect of Cosolvent on the Vesicle Formation Pathways under Solvent Exchange Process: A Dissipative Particle Dynamics Simulation


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Direct Self-Assembly of Diblock Copolymer in Water
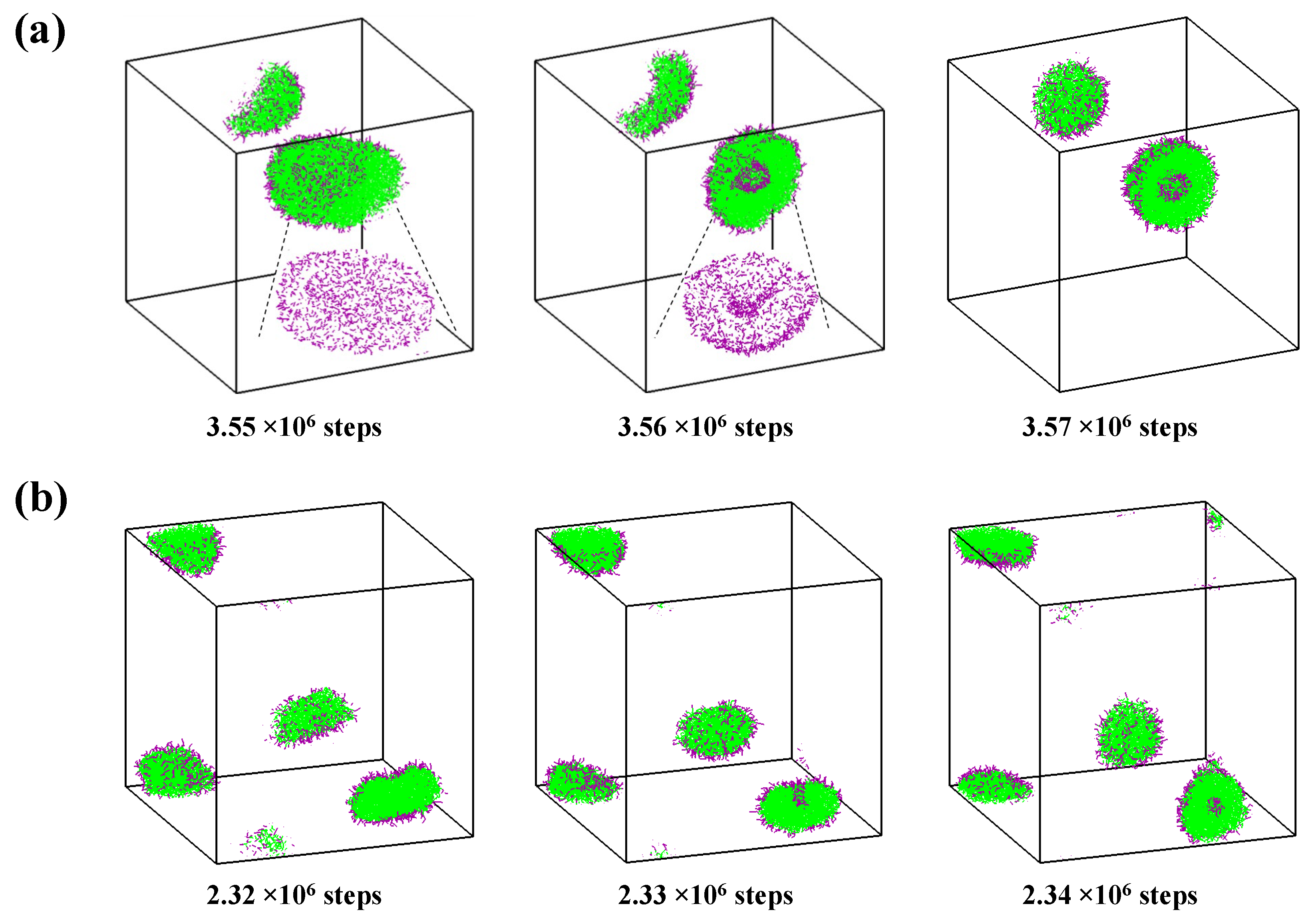
2.2. Self-Assembly in Liquid Mixture Where Cosolvent and Water Is Attractive
2.3. Self-Assembly in Liquid Mixture Where Cosolvent and Water Is Repulsive
2.4. The Physiochemical Principles behind the Different Self-Assembly Behaviors
3. Methods and Parameters
3.1. DPD Simulations
3.2. Solvent Exchange Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mai, Y.Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef]
- Deng, Z.Y.; Liu, S.Y. Emerging trends in solution self-assembly of block copolymers. Polymer 2020, 207, 122914. [Google Scholar] [CrossRef]
- Meng, F.; Zhong, Z.; Feijen, J. Stimuli-responsive polymersomes for programmed drug delivery. Biomacromolecules 2009, 10, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Gaurav, I.; Thakur, A.; Iyaswamy, A.; Wang, X.H.; Chen, X.Y.; Yang, Z.J. Factors affecting extracellular vesicles based drug delivery systems. Molecules 2021, 26, 1544. [Google Scholar] [CrossRef] [PubMed]
- Han, E.; Kim, D.; Cho, Y.; Lee, S.; Kim, J.; Kim, H. Development of polymersomes co-delivering doxorubicin and melittin to overcome multidrug resistance. Molecules 2023, 28, 1087. [Google Scholar] [CrossRef] [PubMed]
- Larrañaga, A.; Lomora, M.; Sarasua, J.R.; Palivan, C.G.; Pandit, A. Polymer capsules as micro-/nanoreactors for therapeutic applications: Current strategies to control membrane permeability. Prog. Mater. Sci. 2017, 90, 325–357. [Google Scholar] [CrossRef]
- Tu, Y.F.; Peng, F.; Adawy, A.; Men, Y.J.; Abdelmohsen, L.K.E.A.; Wilson, D.A. Mimicking the cell: Bio-inspired functions of supramolecular assemblies. Chem. Rev. 2016, 116, 2023–2078. [Google Scholar] [CrossRef]
- Tanner, P.; Baumann, P.; Enea, R.; Onaca, O.; Palivan, C.; Meier, W. Polymeric vesicles: From drug carriers to nanoreactors and artificial organelles. Acc. Chem. Res. 2011, 44, 1039–1049. [Google Scholar] [CrossRef]
- Du, J.Z.; O’Reilly, R.K. Advances and challenges in smart and functional polymer vesicles. Soft Matter 2009, 5, 3544–3561. [Google Scholar] [CrossRef]
- Noguchi, H.; Takasu, M. Self-assembly of amphiphiles into vesicles: A Brownian dynamics simulation. Phys. Rev. E 2001, 64, 041913. [Google Scholar] [CrossRef]
- He, X.H.; Schmid, F. Spontaneous formation of complex micelles from a homogeneous solution. Phys. Rev. Lett. 2008, 100, 137802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.H.; Schmid, F. Dynamics of spontaneous vesicle formation in dilute solutions of amphiphilic diblock copolymers. Macromolecules 2006, 39, 2654–2662. [Google Scholar] [CrossRef]
- Ji, S.C.; Ding, J.D. Spontaneous formation of vesicles from mixed amphiphiles with dispersed molecular weight: Monte Carlo simulation. Langmuir 2006, 22, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.M.; Narayanan, T.; Wolf, C.; Gradzielski, M.; Panine, P.; Finet, S.; Helsby, W.I. Dynamics of the self-assembly of unilamellar vesicles. Phys. Rev. Lett. 2005, 94, 038303. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, S. Spontaneous formation and fusion of raspberry vesicle self-assembled from star block terpolymers in aqueous solution. Materials 2021, 14, 7690. [Google Scholar] [CrossRef]
- Korchagina, E.V.; Qiu, X.-P.; Winnik, F.M. Effect of heating rate on the pathway for vesicle formation in salt-free aqueous solutions of thermosensitive cationic diblock copolymers. Macromolecules 2013, 46, 2341–2351. [Google Scholar] [CrossRef]
- Xiao, M.Y.; Xia, G.J.; Wang, R.; Xie, D.Q. Controlling the self-assembly pathways of amphiphilic block copolymers into vesicles. Soft Matter 2012, 8, 7865–7874. [Google Scholar] [CrossRef]
- Ianiro, A.; Wu, H.L.; van Rijt, M.M.J.; Vena, M.P.; Keizer, A.D.A.; Esteves, A.C.C.; Tuinier, R.; Friedrich, H.; Sommerdijk, N.A.J.M.; Patterson, J.P. Liquid-liquid phase separation during amphiphilic self-assembly. Nat. Chem. 2019, 11, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Takahashi, R. Competition between the micellization and the liquid-liquid phase separation in amphiphilic block copolymer solutions. Polym. J. 2017, 49, 273–277. [Google Scholar] [CrossRef]
- Rizvi, A.; Patel, U.; Ianiro, A.; Hurst, P.J.; Merham, J.G.; Patterson, J.P. Nonionic block copolymer coacervates. Macromolecules 2020, 53, 6078–6086. [Google Scholar] [CrossRef]
- Campos-Villalobos, G.; Siperstein, F.R.; Charles, A.; Patti, A. Solvent-induced morphological transitions in methacrylate-based block-copolymer aggregates. J. Colloid. Interface Sci. 2020, 572, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.-L.; Liu, D.; Wang, R. Self-assembly of amphiphilic diblock copolymers induced by liquid-liquid phase separation. Chin. J. Polym. Sci. 2021, 39, 1217–1224. [Google Scholar] [CrossRef]
- Sato, T. Kinetics of micellization and liquid-liquid phase separation in dilute block copolymer solutions. Polymers 2023, 15, 708. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.Y.; Yu, H.Z.; Du, H.B.; Jiang, W. Effect of selective solvent addition rate on the pathways for spontaneous vesicle formation of ABA amphiphilic triblock copolymers. J. Am. Chem. Soc. 2010, 132, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Keßler, S.; Drese, K.; Schmid, F. Simulating copolymeric nanoparticle assembly in the co-solvent method: How mixing rates control final particle sizes and morphologies. Polymer 2017, 126, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Wang, Y.Y.; Han, Y.Y.; Cui, J.; Jiang, W. A facile method for preparation of uniform polymeric vesicles with tunable size. Nanoscale 2018, 10, 14860–14867. [Google Scholar] [CrossRef]
- Bovone, G.; Cousin, L.; Steiner, F.; Tibbitt, M.W. Solvent controls nanoparticle size during nanoprecipitation by limiting block copolymer assembly. Macromolecules 2022, 55, 8040–8048. [Google Scholar] [CrossRef]
- Choucair, A.; Lavigueur, C.; Eisenberg, A. Polystyrene-b-poly(acrylic acid) vesicle size control using solution properties and hydrophilic block length. Langmuir 2004, 20, 3894–3900. [Google Scholar] [CrossRef]
- Yu, Y.S.; Zhang, L.F.; Eisenberg, A. Morphogenic effect of solvent on crew-cut aggregates of apmphiphilic diblock copolymers. Macromolecules 1998, 31, 1144–1154. [Google Scholar] [CrossRef]
- Simonutti, R.; Bertani, D.; Marotta, R.; Ferrario, S.; Manzone, D.; Mauri, M.; Gregori, M.; Orlando, A.; Masserini, M. Morphogenic effect of common solvent in the self-assembly behavior of amphiphilic PEO-b-PLA. Polymer 2021, 218, 123511. [Google Scholar] [CrossRef]
- Borsdorf, L.; Herkert, L.; Baeumer, N.; Rubert, L.; Soberats, B.; Korevaar, P.A.; Bourque, C.; Gatsogiannis, C.; Fernandez, G. Pathway-controlled aqueous supramolecular polymerization via solvent-dependent chain conformation effects. J. Am. Chem. Soc. 2023, 145, 8882–8895. [Google Scholar] [CrossRef]
- Pijpers, I.A.B.; Meng, F.H.; van Hest, J.C.M.; Abdelmohsen, L.K.E.A. Investigating the self-assembly and shape transformation of poly(ethylene glycol)-b-poly(d,l-lactide) (PEG-PDLLA) polymersomes by tailoring solvent-polymer interactions. Polym. Chem. 2020, 11, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Mukherji, D.; Marques, C.M.; Kremer, K. Polymer collapse in miscible good solvents is a generic phenomenon driven by preferential adsorption. Nat. Commun. 2014, 5, 4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.-Y.; Li, Y.-C.; Lu, Z.-Y. The important role of cosolvent in the amphiphilic diblock copolymer self-assembly process. Polymer 2019, 171, 1–7. [Google Scholar] [CrossRef]
- Buglakov, A.I.; Vasilevskaya, V.V. Fibrillar gel self-assembly via cononsolvency of amphiphilic polymer. J. Colloid. Interface Sci. 2022, 614, 181–193. [Google Scholar] [CrossRef]
- Bharadwaj, S.; van der Vegt, N.F.A. Does preferential adsorption drive cononsolvency? Macromolecules 2019, 52, 4131–4138. [Google Scholar] [CrossRef]
- Zuo, T.S.; Ma, C.L.; Jiao, G.S.; Han, Z.H.; Xiao, S.Y.; Liang, H.J.; Hong, L.; Bowron, D.; Soper, A.; Han, C.C.; et al. Water/cosolvent attraction induced phase separation: A molecular picture of cononsolvency. Macromolecules 2019, 52, 457–464. [Google Scholar] [CrossRef]
- Dudowicz, J.; Freed, K.F.; Douglas, J.F. Communication: Cosolvency and cononsolvency explained in terms of a Flory-Huggins type theory. J. Chem. Phys. 2015, 143, 131101. [Google Scholar] [CrossRef] [Green Version]
- Hoogerbrugge, P.J.; Koelman, J.M.V.A. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys. Lett. 1992, 19, 155–160. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef] [Green Version]
- Clever, H.L.; Pigott, S.P. Enthalpies of mixing of dimethylsulfoxide with water and with several ketones at 298.15 K. J. Chem. Thermodyn. 1971, 3, 221–225. [Google Scholar] [CrossRef]
- Yang, C.; Li, W.; Wu, C. Laser light-scattering study of solution dynamics of water/cycloether mixtures. J. Phys. Chem. B 2004, 108, 11866–11870. [Google Scholar] [CrossRef]
- Seaton, M.A.; Anderson, R.L.; Metz, S.; Smith, W. DL_MESO: Highly scalable mesoscale simulations. Mol. Simul. 2013, 39, 796–821. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| W | G | A | B | |
|---|---|---|---|---|
| W | 25 | |||
| G | 15, 30 | 25 | ||
| A | 25 | 25 | 25 | |
| B | 50 | 25 | 50 | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Z.; Shu, Z.; Jiang, Y.; Wang, B. Effect of Cosolvent on the Vesicle Formation Pathways under Solvent Exchange Process: A Dissipative Particle Dynamics Simulation. Molecules 2023, 28, 5113. https://doi.org/10.3390/molecules28135113
Luo Z, Shu Z, Jiang Y, Wang B. Effect of Cosolvent on the Vesicle Formation Pathways under Solvent Exchange Process: A Dissipative Particle Dynamics Simulation. Molecules. 2023; 28(13):5113. https://doi.org/10.3390/molecules28135113
Chicago/Turabian StyleLuo, Zhonglin, Zhou Shu, Yi Jiang, and Biaobing Wang. 2023. "Effect of Cosolvent on the Vesicle Formation Pathways under Solvent Exchange Process: A Dissipative Particle Dynamics Simulation" Molecules 28, no. 13: 5113. https://doi.org/10.3390/molecules28135113
APA StyleLuo, Z., Shu, Z., Jiang, Y., & Wang, B. (2023). Effect of Cosolvent on the Vesicle Formation Pathways under Solvent Exchange Process: A Dissipative Particle Dynamics Simulation. Molecules, 28(13), 5113. https://doi.org/10.3390/molecules28135113