

Apitherapy in Post-Ischemic Brain Neurodegeneration of Alzheimer’s Disease Proteinopathy: Focus on Honey and Its Flavonoids and Phenolic Acids



Abstract
:1. Introduction
2. Search and Data Collection Criteria
3. Requirements for Natural Agents in the Treatment of Post-Ischemic Neurodegeneration
4. Apitherapy
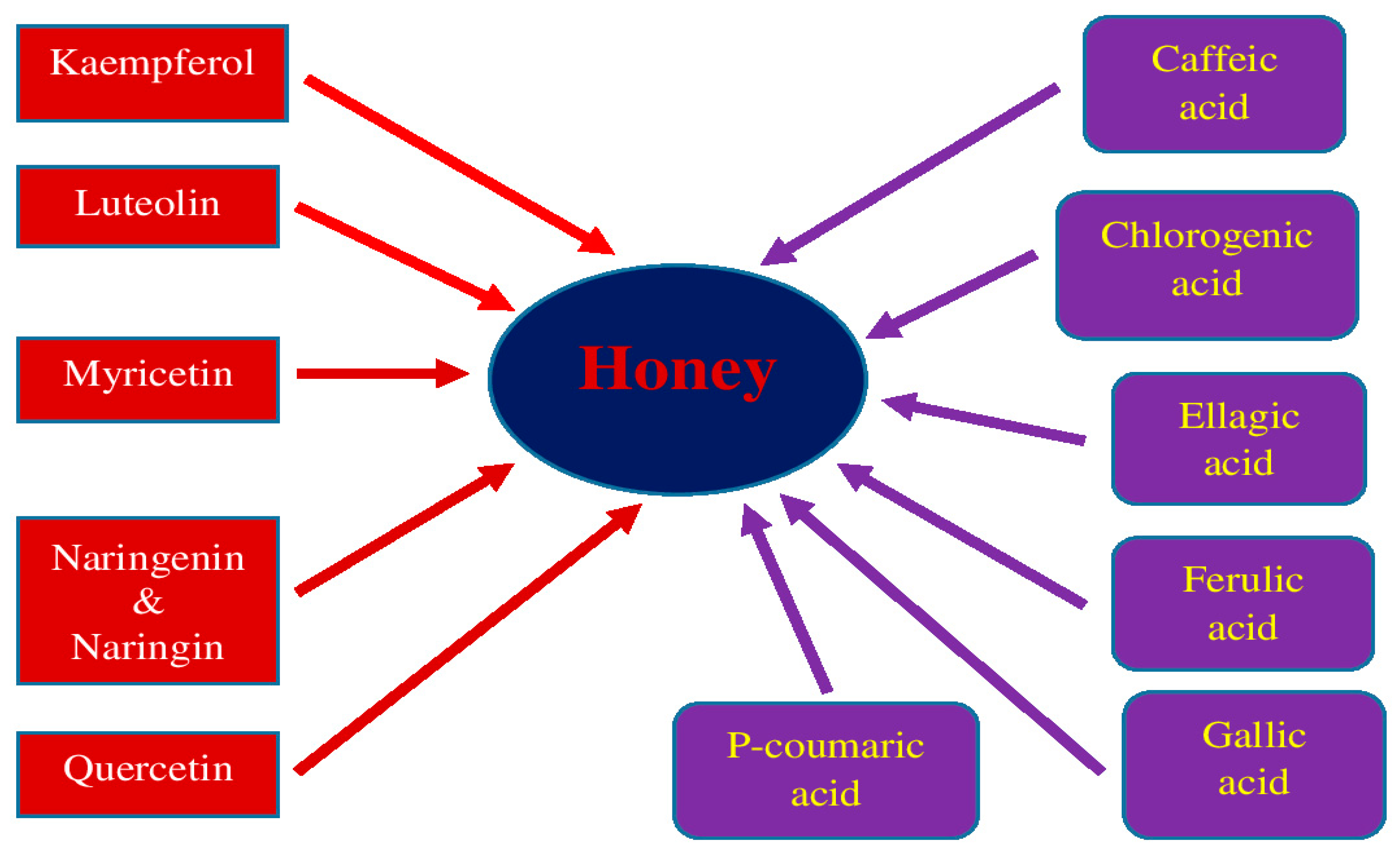
5. Honey and Its Medical Properties
6. Post-Ischemic Neurodegenerative Cascade in the Brain
6.1. Excitotoxicity
6.2. Neurotransmission
6.3. Blood–Brain Barrier
6.4. Neuroinflammation
6.5. Free Radicals
6.6. Amyloid and Tau Proteins
6.7. Vasospasm
6.8. Cerebral Amyloid Angiopathy
6.9. Neuronal Death
6.10. Dementia
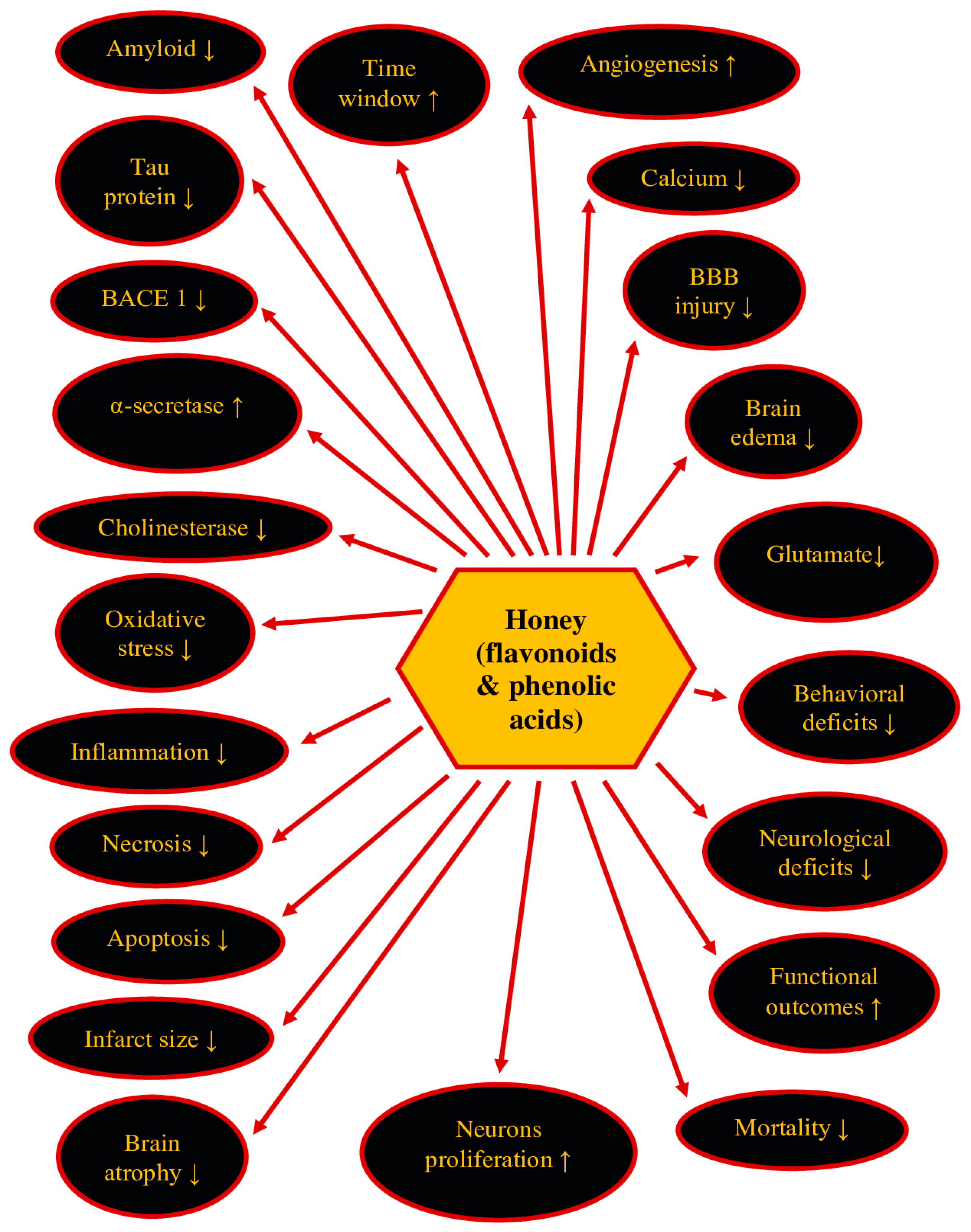
7. Therapeutic Potential of Honey and Its Ingredients in Post-Ischemic Neurodegeneration
8. Bioavailability, Safety and Side Effects of Honey
9. Conclusions
10. Clinical Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mandzia, J.; Cipriano, L.E.; Kapral, M.K.; Fang, J.; Hachinski, V.; Sposato, L.A. Intravenous thrombolysis after first-ever ischemic stroke and reduced incident dementia rate. Stroke 2022, 53, 1170–1177. [Google Scholar]
- Bejot, Y.; Daubail, B.; Giroud, M. Epidemiology of stroke and transient ischemic attacks: Current knowledge and perspectives. Rev. Neurol. 2016, 172, 59–68. [Google Scholar] [CrossRef]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the immune response in ischemic stroke. Front. Immunol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abedi, V.; et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Patabendige, A.; Singh, A.; Jenkins, S.; Sen, J.; Chen, R. Astrocyte activation in neurovascular damage and repair following ischaemic stroke. Int. J. Mol. Sci. 2021, 22, 4280. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Neuroinflammation in post-ischemic neurodegeneration of the brain: Friend, foe, or both? Int. J. Mol. Sci. 2021, 22, 4405. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics—2021 update: A report from the American heart association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Dang, H.; Mao, W.; Wang, S.; Sha, J.; Lu, M.; Cong, L.; Meng, X.; Li, H. Systemic inflammation response index as a prognostic predictor in patients with acute ischemic stroke: A propensity score matching analysis. Front. Neurol. 2023, 13, 1049241. [Google Scholar] [CrossRef]
- Hernández, I.H.; Villa-González, M.; Martín, G.; Soto, M.; Pérez-Álvarez, M.J. Glial Cells as therapeutic approaches in brain ischemia-reperfusion injury. Cells 2021, 10, 1639. [Google Scholar] [CrossRef]
- Kamarova, M.; Baig, S.; Patel, H.; Monks, K.; Wasay, M.; Ali, A.; Redgrave, J.; Majid, A.; Bell, S.M. Antiplatelet use in ischemic stroke. Ann. Pharmacother. 2022, 56, 1159–1173. [Google Scholar] [CrossRef]
- Wang, Y.; Leak, R.K.; Cao, G. Microglia-mediated neuroinflammation and neuroplasticity after stroke. Front. Cell Neurosci. 2022, 16, 980722. [Google Scholar] [CrossRef] [PubMed]
- Venketasubramanian, N.; Yoon, B.W.; Pandian, J.; Navarro, J.C. Stroke epidemiology in south, east, and south-east Asia: A review. J. Stroke 2017, 19, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.; Onuma, O.; Owolabi, M.; Sachdev, S. Stroke: A global response is needed. Bull. World Health Organ. 2016, 94, 634. [Google Scholar] [CrossRef]
- Owolabi, M.O.; Akarolo-Anthony, S.; Akinyemi, R.; Arnett, D.; Gebregziabher, M.; Jenkins, C.; Tiwari, H.; Arulogun, O.; Akpalu, A.; Sarfo, F.S.; et al. Members of the H3 Africa Consortium. Members of the H3 Africa Consortium. The burden of stroke in Africa: A glance at the present and a glimpse into the future. Cardiovasc. J. Afr. 2015, 26 (Suppl. 1), S27–S38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, M.; Vieira, L.E.; Buttari, B.; Profumo, E.; Saso, L. The Nrf2 pathway in ischemic stroke: A review. Molecules 2021, 26, 5001. [Google Scholar] [CrossRef] [PubMed]
- Bulygin, K.V.; Beeraka, N.M.; Saitgareeva, A.R.; Nikolenko, V.N.; Gareev, I.; Beylerli, O.; Akhmadeeva, L.R.; Mikhaleva, L.M.; Torres Solis, L.F.; Solís Herrera, A.; et al. Can miRNAs be considered as diagnostic and therapeutic molecules in ischemic stroke pathogenesis?—Current Status. Int. J. Mol. Sci. 2020, 21, 6728. [Google Scholar] [CrossRef] [PubMed]
- Howard, G.; Goff, D.C. Population shifts and the future of stroke: Forecasts of the future burden of stroke. Ann. N. Y. Acad. Sci. 2012, 1268, 14–20. [Google Scholar] [CrossRef]
- Simats, A.; Liesz, A. Systemic inflammation after stroke: Implications for post-stroke comorbidities. EMBO Mol. Med. 2022, 14, e16269. [Google Scholar] [CrossRef]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.Y.; Kim, B.; Lee, S.R.; Cha, S.H.; Lee, D.S.; Lee, H.J. Prospects of therapeutic target and directions for ischemic stroke. Pharmaceuticals 2021, 14, 321. [Google Scholar] [CrossRef]
- Goulay, R.; Mena Romo, L.; Hol, E.M.; Dijkhuizen, R.M. From stroke to dementia: A Comprehensive review exposing tight interactions between stroke and amyloid-β formation. Transl. Stroke Res. 2020, 11, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Rost, N.S.; Brodtmann, A.; Pase, M.P.; van Veluw, S.J.; Biffi, A.; Duering, M.; Hinman, J.D.; Dichgans, M. Post-stroke cognitive impairment and dementia. Circ. Res. 2022, 130, 1252–1271. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, M. Risk factors and their correlation with severity of cerebral microbleed in acute large artery atherosclerotic cerebral infarction patients. Clin. Neurol. Neurosurg. 2022, 221, 107380. [Google Scholar] [CrossRef]
- Snowdon, D.A.; Greiner, L.H.; Mortimer, J.A.; Riley, K.P.; Greiner, P.A.; Markesbery, W.R. Brain infarction and the clinical expression of Alzheimer disease: The Nun Study. JAMA 1997, 277, 813–817. [Google Scholar] [CrossRef]
- Van Groen, T.; Puurunen, K.; Mäki, H.M.; Sivenius, J.; Jolkkonen, J. Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 2005, 36, 1551–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Wu, H.; Yang, Y.; Wand, D.; Chen, Y.; Gu, Y.; Liu, T. Cerebral ischemia and Alzheimer’s disease: The expression of amyloid-β and apolipoprotein E in human hippocampus. J. Alzheimers Dis. 2007, 12, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek, M.; Jabłoński, M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat. Rec. 2009, 292, 1863–1881. [Google Scholar] [CrossRef]
- Sekeljic, V.; Bataveljic, D.; Stamenkovic, S.; Ułamek, M.; Jabłoński, M.; Radenovic, L.; Pluta, R.; Andjus, P.R. Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct. Funct. 2012, 217, 411–420. [Google Scholar] [CrossRef]
- Hatsuta, H.; Takao, M.; Nogami, A.; Uchino, A.; Sumikura, H.; Takata, T.; Morimoto, S.; Kanemaru, K.; Adachi, T.; Arai, T.; et al. Tau and TDP-43 accumulation of the basal nucleus of Meynert in individuals with cerebral lobar infarcts or hemorrhage. Acta Neuropathol. Commun. 2019, 7, 49. [Google Scholar] [CrossRef]
- Radenovic, L.; Nenadic, M.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J.; Andjus, P.R.; Pluta, R. Heterogeneity in brain distribution of activated microglia and astrocytes in a rat ischemic model of Alzheimer’s disease after 2 years of survival. Aging 2020, 12, 12251–12267. [Google Scholar] [CrossRef]
- Ihle-Hansen, H.; Thommessen, B.; Wyller, T.B.; Engedal, K.; Oksengard, A.R.; Stenset, V.; Loken, K.; Aaberg, M.; Fure, B. Incidence and subtypes of MCI and dementia 1 year after first-ever stroke in patients without pre-existing cognitive impairment. Dement. Geriatr. Cogn. Disord. 2011, 32, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Douiri, A.; Rudd, A.G.; Wolfe, C.D. Prevalence of poststroke cognitive impairment: South London Stroke Register 1995–2010. Stroke 2013, 44, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquin, A.; Binquet, C.; Rouaud, O.; Graule-Petot, A.; Daubail, B.; Osseby, G.V.; Bonithon-Kopp, C.; Giroud, M.; Bejot, Y. Post-stroke cognitive impairment: High prevalence and determining factors in a cohort of mild stroke. J. Alzheimers Dis. 2014, 40, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.W.; Crawford, J.D.; Desmond, D.W.; Godefroy, O.; Jokinen, H.; Mahinrad, S.; Bae, H.J.; Lim, J.S.; Kohler, S.; Douven, E.; et al. Stroke and Cognition (STROKOG) Collaboration. Profile of and risk factors for poststroke cognitive impairment in diverse ethnoregional groups. Neurology 2019, 93, e2257–e2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashim, S.; Ahmad, S.; Al Hatamleh, M.A.I.; Mustafa, M.Z.; Mohamed, M.; Mohamud, R.; Kadir, R.; Kub, T.N.T. Trigona honey as a potential supplementary therapy to halt the progression of post-stroke vascular cognitive impairment. Int. Med. J. 2021, 28, 335–338. [Google Scholar]
- El Husseini, N.; Katzan, I.L.; Rost, N.S.; Blake, M.L.; Byun, E.; Pendlebury, S.T.; Aparicio, H.J.; Marquine, M.J.; Gottesman, R.F.; Smith, E.E.; et al. cognitive impairment after ischemic and hemorrhagic stroke: A scientific statement from the american heart association/american stroke association. Stroke 2023, 54, e272–e291. [Google Scholar] [CrossRef]
- Rasquin, S.M.; Lodder, J.; Verhey, F.R. Predictors of reversible mild cognitive impairment after stroke: A 2-year follow-up study. J. Neurol. Sci. 2005, 229–230, 21–25. [Google Scholar] [CrossRef]
- Liu, G.; Xie, W.; He, A.D.; Da, X.W.; Liang, M.L.; Yao, G.Q.; Xiang, J.Z.; Gao, C.J.; Ming, Z.Y. Antiplatelet activity of chrysin via inhibiting platelet αIIbβ3-mediated signaling pathway. Mol. Nutr. Food Res. 2016, 60, 1984–1993. [Google Scholar] [CrossRef]
- Dichgans, M.; Leys, D. Vascular cognitive impairment. Circ. Res. 2017, 120, 573–591. [Google Scholar] [CrossRef]
- Pendlebury, S.T.; Wadling, S.; Silver, L.E.; Mehta, Z.; Rothwell, P.M. Transient cognitive impairment in TIA and minor stroke. Stroke 2011, 42, 3116–3121. [Google Scholar] [CrossRef] [Green Version]
- Elman-Shina, K.; Efrati, S. Ischemia as a common trigger for Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1012779. [Google Scholar] [CrossRef]
- Neurology, T.L. A unified European action plan on stroke. Lancet Neurol. 2020, 19, 963. [Google Scholar] [CrossRef]
- Tao, T.; Liu, M.; Chen, M.; Luo, Y.; Wang, C.; Xu, T.; Jiang, Y.; Guo, Y.; Zha, J.H. Natural medicine in neuroprotection for ischemic stroke: Challenges and prospective. Pharmacol. Ther. 2020, 216, 107695. [Google Scholar] [CrossRef]
- Kremers, F.; Venema, E.; Duvekot, M.; Yo, L.; Bokkers, R.; Lycklama, À.; Nijeholt, G.; van Es, A.; van der Lugt, A.; Majoie, C.; et al. Outcome prediction models for endovascular treatment of ischemic stroke: Systematic review and external validation. Stroke 2022, 53, 825–836. [Google Scholar] [CrossRef]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Herson, P.S.; Traystman, R.J. Animal models of stroke: Translational potential at present and in 2050. Future Neurol. 2014, 9, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Sairazi, N.S.M.; Sirajudeen, K.N.S.; Asari, M.A.; Mummedy, S.; Muzaimi, M.; Sulaiman, S.A. Effect of Tualang Honey against KA-Induced Oxidative Stress and Neurodegeneration in the Cortex of Rats. BMC Complement. Altern. Med. 2017, 17, 31. [Google Scholar]
- Sairazi, N.S.M.; Sirajudeen, K.N.S.; Muzaimi, M.; Mummedy, S.; Asari, M.A.; Sulaiman, S.A. Tualang honey reduced neuroinflammation and caspase-3 activity in rat brain after kainic acid-induced status epilepticus. Evid. Based Complement. Altern. Med. 2018, 2018, e7287820. [Google Scholar]
- Saxena, A.K.; Phyu, H.P.; Al-Ani, I.M.; Talib, N.A. Potential protective effect of honey against chronic cerebral hypoperfusion-induced neurodegeneration in rats. J. Anat. Soc. India 2014, 63, 151–155. [Google Scholar] [CrossRef]
- Arshad, N.A.; Lin, T.S.; Yahaya, M.F. Stingless bee honey reduces anxiety and improves memory of the metabolic disease-induced rats. CNS Neurol. Disord.-Drug Targets 2020, 19, 115–126. [Google Scholar] [CrossRef]
- Zaidi, H.; Ouchemoukh, S.; Amessis-Ouchemoukh, N.; Debbache, N.; Pacheco, R.; Serralheiro, M.L.; Araujo, M.E. Biological properties of phenolic compound extracts in selected algerian honeys—The inhibition of acetylcholinesterase and α-glucosidase activities. Eur. J. Integr. Med. 2019, 25, 77–84. [Google Scholar] [CrossRef]
- Al-Himyari, F.A. P1-241: The Use of Honey as a natural preventive therapy of cognitive decline and dementia in the middle east. Alzheimers Dement. 2009, 5, P247. [Google Scholar] [CrossRef]
- Shaikh, A.; Ahmad, F.; Teoh, S.L.; Kumar, J.; Yahaya, M.F. Honey and Alzheimer’s Disease-current understanding and future prospects. Antioxidants 2023, 12, 427. [Google Scholar] [CrossRef] [PubMed]
- Lazarewicz, J.W.; Pluta, R.; Salinska, E.; Puka, M. Beneficial effect of nimodipine on metabolic and functional disturbances in rabbit hippocampus following complete cerebral ischemia. Stroke 1989, 20, 70–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarewicz, J.W.; Pluta, R.; Puka, M.; Salinska, E. Diverse mechanisms of neuronal protection by nimodipine in experimental rabbit brain ischemia. Stroke 1990, 21 (Suppl. 12), IV108–IV110. [Google Scholar]
- Lazarewicz, J.W.; Pluta, R.; Puka, M.; Salińska, E. Local nimodipine application improves early functional recovery in the rabbit hippocampus after 15-min global cerebral ischemia. Acta Neurobiol. Exp. 1993, 53, 499–510. [Google Scholar]
- Weis, W.A.; Ripari, N.; Conte, F.L.; da Silva Honorio, M.; Sartori, A.A.; Matucci, R.H.; Sforcin, J.M. An overview about apitherapy and its clinical applications. Phytomed. Plus 2022, 2, 100239. [Google Scholar] [CrossRef]
- Cooper, R. Honey in wound care: Antibacterial properties. GMS Krankenhhyg Interdiszi 2007, 2, Doc51. [Google Scholar]
- Putteeraj, M.; Lim, W.L.; Teoh, S.L.; Yahaya, M.F. Flavonoids and Its Neuroprotective Effects on Brain Ischemia and Neurodegenerative Diseases. Curr. Drug Targets 2018, 19, 1710–1720. [Google Scholar] [CrossRef]
- Ranneh, Y.; Ali, F.; Zarei, M.; Akim, A.M.; Hamid, H.A.; Khazaai, H. Malaysian stingless bee and tualang honeys: A comparative characterization of total antioxidant capacity and phenolic profile using liquid chromatography-mass spectrometry. LWT 2018, 89, 1–9. [Google Scholar] [CrossRef]
- Cheung, Y.; Meenu, M.; Yu, X.; Xu, B. Phenolic acids and flavonoids profiles of commercial honey from different floral sources and geographic sources. Int. J. Food Prop. 2019, 22, 290–308. [Google Scholar] [CrossRef] [Green Version]
- Olas, B. Honey and Its Phenolic Compounds as an Effective Natural Medicine for Cardiovascular Diseases in Humans? Nutrients 2020, 12, 283. [Google Scholar] [CrossRef] [Green Version]
- Parrella, E.; Gussago, C.; Porrini, V.; Benarese, M.; Pizzi, M. From preclinical stroke models to humans: Polyphenols in the prevention and treatment of stroke. Nutrients 2020, 29, 85. [Google Scholar] [CrossRef] [PubMed]
- Gośliński, M.; Nowak, D.; Szwengiel, A. Multidimensional comparative analysis of bioactive phenolic compounds of honeys of various origin. Antioxidants 2021, 10, 530. [Google Scholar] [CrossRef] [PubMed]
- Pashte, V.V.; Pashte, S.V.; Said, P.P. Nutraceutical properties of natural honey to fight health issues: A comprehensive review. J. Pharmacogn. Phytochem. 2020, 9, 234–242. [Google Scholar] [CrossRef]
- Combarros-Fuertes, P.; Estevinho, L.M.; Dias, L.G.; Castro, J.M.; Tomás-Barberán, F.A.; Tornadijo, M.E.; Fresno-Baro, J.M. Bioactive components and antioxidant and antibacterial activities of different varieties of honey: A screening prior to clinical application. J. Agric. Food Chem. 2019, 67, 688–698. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Salinska, E.; Puka, M.; Stafiej, A.; Lazarewicz, J.W. Early changes in extracellular amino acids and calcium concentrations in rabbit hippocampus following complete 15-min cerebral ischemia. Resuscitation 1988, 16, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Salinska, E.; Pluta, R.; Lazarewicz, J.W. Participation of NMDA-receptors in ischemic changes of calcium homeostasis in rabbit brain. Biomed. Biochim. Acta 1989, 48, S170–S173. [Google Scholar]
- Pluta, R.; Salińska, E.; Lazarewicz, J.W. Prostacyclin reduces early ischemic changes in central nervous system. Acta Neurobiol. Exp. 1990, 50, 295–302. [Google Scholar]
- Pluta, R.; Salińska, E.; Lazarewicz, J.W. Prostacyclin attenuates in the rabbit hippocampus early consequences of transient complete cerebral ischemia. Acta Neurol. Scand. 1991, 83, 370–377. [Google Scholar] [CrossRef]
- Salińska, E.; Pluta, R.; Puka, M.; Lazarewicz, J.W. Blockade of N-methyl-D-aspartate-sensitive excitatory amino acid receptors with 2-amino-5-phosphonovalerate reduces ischemia-evoked calcium redistribution in rabbit hippocampus. Exp. Neurol. 1991, 112, 89–94. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Participation of Amyloid and Tau Protein in Neuronal Death and neurodegeneration after brain ischemia. Int. J. Mol. Sci. 2020, 21, 4599. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Kocki, J.; Bogucki, J.; Januszewski, S.; Bogucka-Kocka, A.; Czuczwar, S.J. Expression of the tau protein and amyloid protein precursor processing genes in the ca3 area of the hippocampus in the ischemic model of Alzheimer’s Disease in the rat. Mol. Neurobiol. 2020, 57, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Ouyang, L.; Januszewski, S.; Li, Y.; Czuczwar, S.J. Participation of amyloid and tau protein in post-ischemic neurodegeneration of the hippocampus of a nature identical to Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 2460. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. neuroinflammation, stroke, blood-brain barrier dysfunction, and imaging modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Januszewski, S.; Jabłoński, M. Acetylated tau protein: A new piece in the puzzle between brain ischemia and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 9174. [Google Scholar] [CrossRef]
- Pluta, R.; Lossinsky, A.S.; Wisniewski, H.M.; Mossakowski, M.J. Early blood-brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res. 1994, 633, 41–52. [Google Scholar] [CrossRef]
- Pluta, R.; Lossinsky, A.S.; Walski, M.; Wisniewski, H.M.; Mossakowskim, M.J. Platelet occlusion phenomenon after short- and long-term survival following complete cerebral ischemia in rats produced by cardiac arrest. J. Brain Res. 1994, 35, 463–471. [Google Scholar]
- Wardlow, J.M.; Sandercock, P.A.; Dennis, M.S.; Starr, J. Is breakdown of the blood-brain barrier responsible for lacunar stroke, leukoaraiosis, and dementia? Stroke 2003, 34, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Song, Y.; Rocha, M.; Shi, Y. Ischemic brain edema: Emerging cellular mechanisms and therapeutic approaches. Neurobiol. Dis. 2023, 178, 106029. [Google Scholar] [CrossRef]
- Pluta, R. The role of apolipoprotein E in the deposition of β-amyloid peptide during ischemia-reperfusion brain injury. A model of early Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 903, 324–334. [Google Scholar] [CrossRef]
- Jabłoński, M.; Maciejewski, R.; Januszewski, S.; Ułamek, M.; Pluta, R. One year follow up in ischemic brain injury and the role of Alzheimer factors. Physiol. Res. 2011, 60 (Suppl. 1), S113–S119. [Google Scholar] [CrossRef] [PubMed]
- Gemmell, E.; Bosomworth, H.; Allan, L.; Hall, R.; Khundakar, A.; Oakley, A.E.; Deramecourt, V.; Polvikoski, T.M.; O’Brien, J.T.; Kalaria, R.N. Hippocampal neuronal atrophy and cognitive function in delayed poststroke and aging-related dementias. Stroke 2012, 43, 808–814. [Google Scholar] [CrossRef] [Green Version]
- Kiryk, A.; Pluta, R.; Figiel, I.; Mikosz, M.; Ułamek, M.; Niewiadomska, G.; Jabłoński, M.; Kaczmarek, L. Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav. Brain Res. 2011, 219, 1–7. [Google Scholar] [CrossRef]
- Koton, S.; Pike, J.R.; Johansen, M.; Knopman, D.S.; Lakshminarayan, K.; Mosley, T.; Patole, S.; Rosamond, W.D.; Schneider, A.L.C.; Sharrett, A.R.; et al. Association of ischemic stroke incidence, severity, and recurrence with dementia in the atherosclerosis risk in communities cohort study. JAMA Neurol. 2022, 79, 271–280. [Google Scholar] [CrossRef]
- Gao, Z.; Li, J.; Wang, L.; Li, Y. A systematic review of auricular therapy for poststroke cognitive impairment and dementia: A protocol for systematic review and meta-analysis. Medicine 2023, 102, e32933. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Kiś, J.; Januszewski, S.; Jabłoński, M.; Czuczwar, S.J. Cross-talk between amyloid, tau protein and free radicals in post-ischemic brain neurodegeneration in the form of Alzheimer’s Disease proteinopathy. Antioxidants 2022, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shan, X.; Men, W.; Zhai, H.; Qiao, X.; Geng, L.; Li, C. The effect of crocin on memory, hippocampal acetylcholine level, and apoptosis in a rat model of cerebral ischemia. Biomed. Pharmacother. 2020, 130, 110543. [Google Scholar] [CrossRef]
- Shadfar, S.; Hwang, C.J.; Lim, M.S.; Choi, D.Y.; Hong, J.T. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharmacal Res. 2015, 38, 2106–2119. [Google Scholar] [CrossRef]
- Nakai, M.; Akino, H.; Kaneda, T.; Matsuta, Y.; Shiyama, R.; Tanase, K.; Ito, H.; Aoki, Y.; Oyama, N.; Miwa, Y.; et al. Acetylcholinesterase inhibitor acting on the brain improves detrusor overactivity caused by cerebral infarction in rats. Neuroscience 2006, 142, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, M.; Hyakawa, N.; Kato, H.; Araki, T. Time dependent changes of striatal interneurons after focal cerebral ischemia in rats. J. Neural Transm. 2008, 115, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Ben Assayag, E.; Shenhar-Tsarfaty, S.; Ofek, K.; Soreq, L.; Bova, I.; Shopin, L.; Berg, R.M.; Berliner, S.; Shapira, I.; Bornstein, N.M.; et al. Serum cholinesterase activities distinguish between stroke patients and controls and predict 12-month mortality. Mol. Med. 2010, 16, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Fu, Q.; Liu, X.; Zhang, H.; Dong, M. Increased acetylcholinesterase and capase-3 expression in the brain and peripheral immune system of focal cerebral ischemic rats. J. Neuroimmunol. 2009, 211, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Boisgard, R.; Thézé, B.; Van Camp, N.; Kuhnast, B.; Damont, A.; Kassiou, M.; Dollé, F.; Tavitian, B. Evaluation of the PBR/TSPO radioligand [18F]DPA-714 in a rat model of focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 230–241. [Google Scholar] [CrossRef] [Green Version]
- Tracey, K.J. Physiology and immunology of the cholinergic antiinflammatory pathway. J. Clin. Investig. 2007, 117, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.S.; Lee, S.H.; Jung, J.H.; Song, I.H.; Park, H.S.; Moon, B.S.; Kim, S.E.; Lee, B.C. TSPO Expression modulatory effect of acetylcholinesterase inhibitor in the ischemic stroke rat model. Cells 2021, 10, 1350. [Google Scholar] [CrossRef]
- Muhammad, A.; Odunola, O.A.; Gbadegesin, M.A.; Sallau, A.B.; Ndidi, U.S.; Ibrahim, M.A. Inhibitory effects of sodium arsenite and acacia honey on acetylcholinesterase in rats. Int. J. Alzheimers Dis. 2015, 2015, 903603. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, X.; He, J.; Xie, Z.; Xia, S.; Lu, G. The roles of GABA in ischemia-reperfusion injury in the central nervous system and peripheral organs. Oxidative Med. Cell. Longev. 2019, 2019, 4028394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gürler, G.; Soylu, K.O.; Yemisci, M. Importance of pericytes in the pathophysiology of cerebral ischemia. Arch. Neuropsychiatry 2022, 59 (Suppl. 1), S29–S35. [Google Scholar]
- Mossakowski, M.J.; Lossinsky, A.S.; Pluta, R.; Wisniewski, H.M. Changes in cerebral microcirculation system following experimentally induced cardiac arrest: A SEM and TEM study. In Microcirculatory Stasis in the Brain; Tomita, M., Ed.; Elsevier Science Publishers B.V.: Amsterdam, The Netherlands, 1993; pp. 99–106. [Google Scholar]
- Mossakowski, M.J.; Lossinsky, A.S.; Pluta, R.; Wisniewski, H.M. Abnormalities of the blood-brain barrier in global cerebral ischemia in rats due to experimental cardiac arrest. Acta Neurochir. Suppl. 1994, 60, 274–276. [Google Scholar] [PubMed]
- Wisniewski, H.M.; Pluta, R.; Lossinsky, A.S.; Mossakowski, M.J. Ultrastructural studies of cerebral vascular spasm after cardiac arrest-related global cerebral ischemia in rats. Acta Neuropathol. 1995, 90, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R. Pathological opening of the blood-brain barrier to horseradish peroxidase and amyloid precursor protein following ischemia-reperfusion brain injury. Chemotherapy 2005, 51, 223–226. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek, M.; Januszewski, S. Micro-blood-brain barrier openings and cytotoxic fragments of amyloid precursor protein accumulation in white matter after ischemic brain injury in long-lived rats. Acta Neurochir. 2006, 96, 267–271. [Google Scholar]
- Pluta, R.; Miziak, B.; Czuczwar, S.J. post-ischemic permeability of the blood–brain barrier to amyloid and platelets as a factor in the maturation of Alzheimer’s Disease-type brain neurodegeneration. Int. J. Mol. Sci. 2023, 24, 10739. [Google Scholar] [CrossRef]
- Hallenbeck, J.M.; Dutka, A.J.; Tanishima, T.; Kochanek, P.M.; Kumaroo, K.K.; Thompson, C.B.; Obrenovitch, T.P.; Contreras, T.J. Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke 1986, 17, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Barcikowska, M.; Januszewski, S.; Misicka, A.; Lipkowski, A.W. Evidence of blood-brain barrier permeability/leakage for circulating human Alzheimer’s β-amyloid-(1-42)-peptide. Neuroreport 1996, 7, 1261–1265. [Google Scholar] [CrossRef]
- Pluta, R.; Misicka, A.; Januszewski, J.; Barcikowska, M.; Lipkowski, A.W. Transport of human β-amyloid peptide through the rat blood-brain barrier after global cerebral ischemia. Acta Neurochir. 1997, 70, 247–249. [Google Scholar]
- Pluta, R.; Barcikowska, M.; Misicka, A.; Lipkowski, A.W.; Spisacka, S.; Januszewski, S. Ischemic rats as a model in the study of the neurobiological role of human β-amyloid peptide. Time-dependent disappearing diffuse amyloid plaques in brain. Neuroreport 1999, 10, 3615–3619. [Google Scholar] [CrossRef]
- Lee, P.H.; Bang, O.Y.; Hwang, E.M.; Lee, J.S.; Joo, U.S.; Mook-Jung, I.; Huh, K. Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J. Neural Transm. 2005, 112, 1371–1379. [Google Scholar] [CrossRef]
- Liu, Y.H.; Cao, H.Y.; Wang, Y.R.; Jiao, S.S.; Bu, X.L.; Zeng, F.; Wang, Q.H.; Li, J.; Deng, J.; Zhou, H.D.; et al. Serum Aβ is predictive for short-term neurological deficits after acute ischemic stroke. Neurotox. Res. 2015, 27, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Eisenmenger, L.B.; Peret, A.; Famakin, B.M.; Spahic, A.; Roberts, G.S.; Bockholt, J.H.; Johnson, K.M.; Paulsen, J.S. Vascular contributions to Alzheimer’s disease. Transl. Res. 2023, 254, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Ugidos, I.F.; González-Rodríguez, P.; Santos-Galdiano, M.; Font-Belmonte, E.; Anuncibay-Soto, B.; Pérez-Rodríguez, D.; Gonzalo-Orden, J.M.; Fernández-López, A. Neuroprotective effects of meloxicam on transient brain ischemia in rats: The two faces of anti-inflammatory treatments. Neural Regen. Res. 2023, 18, 1961–1967. [Google Scholar]
- Kaur, N.; Chugh, H.; Sakharkar, M.K.; Dhawan, U.; Chidambaram, S.B.; Chandra, R. Neuroinflammation mechanisms and phytotherapeutic intervention: A systematic review. ACS Chem. Neurosci. 2020, 11, 3707–3731. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, Z.; Liao, Y.; Sun, S.; Dai, Y.; Tang, Y. Ischemic stroke: From pathological mechanisms to neuroprotective strategies. Front. Neurol. 2022, 13, 1013083. [Google Scholar] [CrossRef]
- Otani, K.; Shichita, T. Cerebral sterile inflammation in neurodegenerative diseases. Inflamm. Regen. 2020, 40, 28. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M.; Simons, M. CNS remyelination and inflammation: From basic mechanisms to therapeutic opportunities. Neuron 2022, 110, 3549–3565. [Google Scholar] [CrossRef]
- Passarelli, J.P.; Nimjee, S.M.; Townsend, K.L. Stroke and neurogenesis: Bridging clinical observations to new mechanistic insights from animal models. Transl. Stroke Res. 2022, 1–6. [Google Scholar] [CrossRef]
- Zhang, W.F.; Jin, Y.C.; Li, X.M.; Yang, Z.; Wang, D.; Cui, J.J. Protective effects of leptin against cerebral ischemia/reperfusion injury. Exp. Ther. Med. 2019, 17, 3282–3290. [Google Scholar] [CrossRef] [Green Version]
- Lorek, A.; Takei, Y.; Cady, E.B.; Wyatt, J.S.; Penrice, J.; Edwards, A.D.; Peebles, D.; Wylezinska, M.; Owen-Reece, H.; Kirkbride, V.; et al. Delayed (“secondary”) cerebral energy failure after acute hypoxia-ischemia in the newborn piglet: Continuous 48-hour studies by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1994, 36, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendharkar, A.V.; Levy, S.L.; Ho, A.L.; Sussman, E.S.; Cheng, M.Y.; Steinberg, G.K. Optogenetic modulation in stroke recovery. Neurosurg. Focus 2016, 40, E6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleuskens, D.G.; Gonçalves Costa, F.; Annink, K.V.; van den Hoogen, A.; Alderliesten, T.; Groenendaal, F.; Benders, M.J.N.; Dudink, J. Pathophysiology of cerebral hyperperfusion in term neonates with hypoxic-ischemic encephalopathy: A systematic review for future research. Front. Pediatr. 2021, 9, 631258. [Google Scholar] [CrossRef]
- Chen, L.; Ma, S.; Shi, M.; Wang, Q.; Miao, Y. A new nitronyl nitroxide radical with salicylic acid framework attenuates blood-brain barrier disruption and oxidative stress in a rat model of middle cerebral artery occlusion. Neuroreport 2022, 33, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Kida, E.; Lossinsky, A.S.; Golabek, A.A.; Mossakowski, M.J.; Wisniewski, H.M. Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s β-amyloid protein precursor in the brain. Brain Res. 1994, 649, 323–328. [Google Scholar] [CrossRef]
- Kato, T.; Hirano, A.; Katagiri, T.; Sasaki, H.; Yamada, S. Neurofibrillary tangle formation in the nucleus basalis of meynert ipsilateral to a massive cerebral infarct. Ann. Neurol. 1988, 23, 620–623. [Google Scholar] [CrossRef]
- Wen, Y.; Yang, S.-H.; Liu, R.; Perez, E.J.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2007, 1772, 473–483. [Google Scholar] [CrossRef]
- Khan, S.; Yuldasheva, N.Y.; Batten, T.F.C.; Pickles, A.R.; Kellett, K.A.B.; Saha, S. Tau pathology and neurochemical changes associated with memory dysfunction in an optimized murine model of global cerebral ischaemia—A potential model for vascular dementia? Neurochem. Int. 2018, 118, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Kocki, J.; Ułamek-Kozioł, M.; Bogucka-Kocka, A.; Januszewski, S.; Jabłonski, M.; Gil-Kulik, P.; Brzozowska, J.; Petniak, A.; Furmaga-Jabłonska, W.; Bogucki, J.; et al. Dysregulation of amyloid precursor protein, b-secretase, presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J. Alzheimers Dis. 2015, 47, 1047–1056. [Google Scholar] [CrossRef]
- Pluta, R.; Kocki, J.; Ułamek-Kozioł, M.; Petniak, A.; Gil-Kulik, P.; Januszewski, S.; Bogucki, J.; Jabłonski, M.; Brzozowska, J.; Furmaga-Jabłonska, W.; et al. Discrepancy in expression of β-secretase and amyloid-β protein precursor in Alzheimer-related genes in the rat medial temporal lobe cortex following transient global brain ischemia. J. Alzheimers Dis. 2016, 51, 1023–1031. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Kocki, J.; Ułamek-Kozioł, M.; Bogucka-Kocka, A.; Gil-Kulik, P.; Januszewski, S.; Jabłonski, M.; Petniak, A.; Brzozowska, J.; Bogucki, J.; et al. Alzheimer-associated presenilin 2 gene is dysregulated in rat medial temporal lobe cortex after complete brain ischemia due to cardiac arrest. Pharmacol. Rep. 2016, 68, 155–161. [Google Scholar] [CrossRef]
- Zetterberg, H.; Mortberg, E.; Song, L.; Chang, L.; Provuncher, G.K.; Patel, P.P.; Ferrell, E.; Fournier, D.R.; Kan, C.W.; Campbell, T.G.; et al. Hypoxia due to cardiac arrest induces a time dependent increase in serum amyloid levels in humans. PLoS ONE 2011, 6, e28263. [Google Scholar] [CrossRef] [Green Version]
- Schiefecker, A.J.; Putzer, G.; Braun, P.; Martini, J.; Strapazzon, G.; Antunes, A.P.; Mulino, M.; Pinggera, D.; Glodny, B.; Brugger, H.; et al. Total tau protein as investigated by cerebral microdialysis increases in hypothermic cardiac arrest: A Pig Study. Ther. Hypothermia Temp. Manag. 2021, 11, 28–34. [Google Scholar] [CrossRef]
- Bitsch, A.; Horn, C.; Kemmling, Y.; Seipelt, M.; Hellenbrand, U.; Stiefel, M.; Ciesielczyk, B.; Cepek, L.; Bahn, E.; Ratzka, P.; et al. Serum tau protein level as a marker of axonal damage in acute ischemic stroke. Eur. Neurol. 2002, 47, 45–51. [Google Scholar] [CrossRef]
- Kurzepa, J.; Bielewicz, J.; Grabarska, A.; Stelmasiak, Z.; Stryjecka-Zimmer, M.; Bartosik-Psujek, H. Matrix metalloproteinase-9 contributes to the increase of tau protein in serum during acute ischemic stroke. J. Clin. Neurosci. 2010, 17, 997–999. [Google Scholar] [CrossRef]
- Bielewicz, J.; Kurzepa, J.; Czekajska-Chehab, E.; Stelmasiak, Z.; Bartosik-Psujek, H. Does serum tau protein predict the outcome of patients with ischemic stroke? J. Mol. Neurosci. 2011, 43, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Mörtberg, E.; Zetterberg, H.; Nordmark, J.; Blennow, K.; Catry, C.; Decraemer, H.; Vanmechelen, E.; Rubertsson, S. Plasma tau protein in comatose patients after cardiac arrest treated with therapeutic hypothermia. Acta Anaesthesiol. Scand. 2011, 55, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Randall, J.; Mörtberg, E.; Provuncher, G.K.; Fournier, D.R.; Duffy, D.C.; Rubertsson, S.; Blennow, K.; Zetterberg, H.; Wilson, D.H. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: Results of a pilot study. Resuscitation 2012, 84, 351–356. [Google Scholar] [CrossRef]
- Lasek-Bal, A.; Jedrzejowska-Szypulka, H.; Rozycka, J.; Bal, W.; Kowalczyk, A.; Holecki, M.; Dulawa, J.; Lewin-Kowalik, J. The presence of Tau protein in blood as a potential prognostic factor in stroke patients. J. Physiol. Pharmacol. 2016, 67, 691–696. [Google Scholar] [PubMed]
- De Vos, A.; Bjerke, M.; Brouns, R.; De Roeck, N.; Jacobs, D.; Van den Abbeele, L.; Guldolf, K.; Zetterberg, H.; Blennow, K.; Engelborghs, S.; et al. Neurogranin and tau in cerebrospinal fluid and plasma of patients with acute ischemic stroke. BMC Neurol. 2017, 17, 170. [Google Scholar] [CrossRef] [Green Version]
- Sayas, C.L.; Ávila, J. GSK-3 and tau: A key duet in Alzheimer’s disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- You, G.; Yao, J.; Liu, Q.; Li, N. The Strategies for Treating “Alzheimer’s Disease”: Insulin Signaling May Be a Feasible Target. Curr. Issues Mol. Biol. 2022, 44, 6172–6188. [Google Scholar] [CrossRef]
- Alonso, A.C.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef]
- Oakley, S.S.; Maina, M.B.; Marshall, K.E.; Al-Hilaly, Y.K.; Harrington, C.R.; Wischik, C.M.; Serpell, L.C. Tau filament self-assembly and structure: Tau as a therapeutic target. Front. Neurol. 2020, 11, 590754. [Google Scholar] [CrossRef]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau post-translational modifications: Dynamic transformers of tau function, degradation, and aggregation. Front. Neurol. 2021, 11, 595532. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Clavaguera, F.; Potier, M.C. The prion-like propagation hypothesis in Alzheimer’s and Parkinson’s disease. Curr. Opin. Neurol. 2019, 32, 266–271. [Google Scholar] [CrossRef]
- Brunello, C.A.; Merezhko, M.; Uronen, R.L.; Huttunen, H.J. Mechanisms of secretion and spreading of pathological tau protein. Cell. Mol. Life Sci. 2020, 77, 1721–1744. [Google Scholar] [CrossRef] [Green Version]
- Polanco, J.C.; Götz, J. Exosomal and vesicle-free tau seeds-propagation and convergence in endolysosomal permeabilization. FEBS J. 2022, 289, 6891–6907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cao, Y.; Ma, L.; Wei, Y.; Li, H. Possible mechanisms of tau spread and toxicity in Alzheimer’s disease. Front. Cell Dev. Biol. 2021, 9, 707268. [Google Scholar] [CrossRef] [PubMed]
- Hirunpattarasilp, C.; Barkaway, A.; Davis, H.; Pfeiffer, T.; Sethi, H.; Attwell, D. Hyperoxia evokes pericyte-mediated capillary constriction. J. Cereb. Blood Flow Metab. 2022, 42, 2032–2047. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef]
- Pluta, R.; Lossinsky, A.S.; Mossakowski, M.J.; Faso, L.; Wisniewski, H.M. Reassessment of a new model of complete cerebral ischemia in rats. Method of induction of clinical death, pathophysiology and cerebrovascular pathology. Acta Neuropathol. 1991, 83, 1–11. [Google Scholar] [CrossRef]
- Wang, W.Z.; Guo, S.Z.; Tsai, T.M.; Anderson, G.L.; Miller, F.N. Platelet-activating factor contributes to postischemic vasospasm. J. Surg. Res. 2000, 89, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R. Blood-brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia-reperfusion brain injury with 1-year survival. Acta Neurochir. 2003, 86, 117–122. [Google Scholar]
- Guo, J.; Tuo, Q.Z.; Lei, P. Iron, ferroptosis, and ischemic stroke. J. Neurochem. 2023, 165, 487–520. [Google Scholar] [CrossRef] [PubMed]
- Lasoń, W.; Jantas, D.; Leśkiewicz, M.; Regulska, M.; Basta-Kaim, A. Vitamin D3 and Ischemic Stroke: A Narrative Review. Antioxidants 2022, 11, 2120. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Zhang, Y.; Wu, R.; Dou, L.; Cao, F.; Yan, Y.; Tang, Y.; Huang, C.; Zhao, Y.; Zhang, J. Network pharmacology approach to investigate the multitarget mechanisms of Zhishi Rhubarb Soup on acute cerebral infarction. Pharm. Biol. 2022, 60, 1394–1406. [Google Scholar] [CrossRef]
- Saxena, A.K.; Phyu, H.; Al-Ani, I.; Oothuman, P. Improved spatial learning and memory performance following tualang honey treatment during cerebral hypoperfusion-induced neurodegeneration. J. Transl. Sci. 2016, 2, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Rivera, F.; Urbanavicius, J.; Gervaz, E.; Morquio, A.; Dajas, F. Some aspects of the in vivo neuroprotective capacity of flavonoids: Bioavailability and structure-activity relationship. Neurotox. Res. 2004, 6, 543–553. [Google Scholar] [CrossRef]
- Cho, J.Y.; Kim, I.S.; Jang, Y.H.; Kim, A.R.; Lee, S.R. Protective effect of quercetin, a natural flavonoid against neuronal damage after transient global cerebral ischemia. Neurosci. Lett. 2006, 404, 330–335. [Google Scholar] [CrossRef]
- Lee, J.K.; Kwak, H.J.; Piao, M.S.; Jang, J.W.; Kim, S.H.; Kim, H.S. Quercetin reduces the elevated matrix metalloproteinases-9 level and improves functional outcome after cerebral focal ischemia in rats. Acta Neurochir. 2011, 153, 1321–1329. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, H.; Zhang, J.; Yan, M. Isoquercetin attenuates oxidative stress and neuronal apoptosis after ischemia/reperfusion injury via Nrf2-mediated inhibition of the NOX4/ROS/NF-κB pathway. Chem. Biol. Interact. 2018, 284, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Park, D.J.; Kang, J.B.; Shah, M.A.; Koh, P.O. Quercetin alleviates the injury-induced decrease of protein phosphatase 2A subunit B in cerebral ischemic animal model and glutamate-exposed HT22 cells. J. Vet. Med. Sci. 2019, 81, 1047–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.J.; Kang, J.B.; Shah, F.A.; Jin, Y.B.; Koh, P.O. Quercetin attenuates decrease of thioredoxin expression following focal cerebral ischemia and glutamate-induced neuronal cell Damage. Neuroscience 2020, 428, 38–49. [Google Scholar] [CrossRef]
- Park, D.J.; Jeonm, S.J.; Kang, J.B.; Koh, P.O. Quercetin Reduces Ischemic Brain Injury by Preventing Ischemia-induced decreases in the neuronal calcium sensor protein hippocalcin. Neuroscience 2020, 430, 47–62. [Google Scholar] [CrossRef]
- Park, D.J.; Kang, J.B.; Shah, F.A.; Koh, P.O. Quercetin attenuates the reduction of parvalbumin in middle cerebral artery occlusion animal model. Lab. Anim. Res. 2021, 37, 9. [Google Scholar] [CrossRef]
- Yang, R.; Shen, Y.-J.; Chen, M.; Zhao, J.-Y.; Chen, S.-H.; Zhang, W.; Song, J.-K.; Li, L.; Du, G.-H. Quercetin attenuates ischemia reperfusion injury by protecting the blood-brain barrier through Sirt1 in MCAO rats. J. Asian Nat. Prod. Res. 2021, 24, 278–289. [Google Scholar] [CrossRef]
- Wu, S.; Yue, Y.; Peng, A.; Zhang, L.; Xiang, J.; Cao, X.; Ding, H.; Yin, S. Myricetin ameliorates brain injury and neurological deficits via Nrf2 activation after experimental stroke in middle-aged rats. Food Funct. 2016, 7, 2624–2634. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Myricetin as a Promising Molecule for the Treatment of Post-Ischemic Brain Neurodegeneration. Nutrients 2021, 13, 342. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, K.; Wang, C.; Ma, C.; Gong, L.; Zhou, H.; Xue, X.; Peng, C.; Li, Y. Protective effects of dietary quercetin on cerebral ischemic injury: Pharmacology, pharmacokinetics and bioavailability-enhancing nanoformulations. Food Funct. 2023, 14, 4470–4489. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xu, P.; Fu, T.; Huang, X.; Song, J.; Chen, M.; Tian, X.; Yin, H.; Han, J. Myricetin against ischemic cerebral injury in rat middle cerebral artery occlusion model. Mol. Med. Rep. 2018, 17, 3274–3280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Sánchez, C.; Martín-Romero, F.J.; Sun, F.; Luis, L.; Samhan-Arias, A.K.; García-Martínez, V.; Gutiérrez-Merino, C. Blood micromolar concentrations of kaempferol afford protection against ischemia/reperfusion-induced damage in rat brain. Brain Res. 2007, 1182, 123–137. [Google Scholar] [CrossRef]
- Yu, L.; Chen, C.; Wang, L.F.; Kuang, X.; Liu, K.; Zhang, H.; Du, J.R. Neuroprotective effect of kaempferol glycosides against brain injury and neuroinflammation by inhibiting the activation of NF-κB and STAT3 in transient focal stroke. PLoS ONE 2013, 8, e55839. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Cheng, X.; Yang, Y.L.; Liu, M.; Zhang, S.S.; Wang, Y.H.; Du, G.H. Kaempferol attenuates neuroinflammation and blood brain barrier dysfunction to improve neurological deficits in cerebral ischemia/reperfusion rats. Brain Res. 2019, 1722, 146361. [Google Scholar] [CrossRef]
- Raza, S.S.; Khan, M.M.; Ahmad, A.; Ashafaq, M.; Islam, F.; Wagner, A.P.; Safhi, M.M.; Islam, F. Neuroprotective effect of naringenin is mediated through suppression of NF-κB signaling pathway in experimental stroke. Neuroscience 2013, 230, 157–171. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, X.; Chen, L.; Zhang, J.; Zhang, L.; Zhao, X.; Zhao, T.; Zhao, Y. Protective effect of naringenin in experimental ischemic stroke: Down-regulated NOD2. RIP2. NF-κB. MMP-9 and up-regulated claudin-5 expression. Neurochem. Res. 2014, 39, 1405–1415. [Google Scholar] [CrossRef]
- Wang, K.; Chen, Z.; Huang, J.; Huang, L.; Luo, N.; Liang, X.; Liang, M.; Xie, W. Naringenin prevents ischaemic stroke damage via anti-apoptotic and anti-oxidant effects. Clin. Exp. Pharmacol. Physiol. 2017, 44, 862–871. [Google Scholar] [CrossRef]
- Li, Q.; Tian, Z.; Wang, M.; Kou, J.; Wang, C.; Rong, X.; Li, J.; Xie, X.; Pang, X. Luteoloside attenuates neuroinflammation in focal cerebral ischemia in rats via regulation of the PPAR/Nrf2/NF-κB signaling pathway. Int. Immunopharmacol. 2019, 66, 309–316. [Google Scholar] [CrossRef]
- Luo, S.; Li, H.; Mo, Z.; Lei, J.; Zhu, L.; Huang, Y.; Fu, R.; Li, C.; Huang, Y.; Liu, K.; et al. Connectivity map identifies luteolin as a treatment option of ischemic stroke by inhibiting MMP9 and activation of the PI3K/Akt signaling pathway. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Qiao, H.; Dong, L.; Zhang, X.; Zhu, C.; Wang, L.; Liu, Z.; Chen, L.; Xing, Y.; Wang, C.; Li, Y. Protective effect of luteolin in experimental ischemic stroke: Upregulated SOD1. CAT. Bcl-2 and claudin-5. down-regulated MDA and Bax expression. Neurochem. Res. 2012, 37, 2014–2024. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Zhang, X.; Zhu, C.; Dong, L.; Wang, L.; Xing, Y.; Wang, C.; Ji, Y.; Cao, X. Luteolin downregulates TLR4. TLR5. NF-kappaB and p-p38MAPK expression. upregulates the p-ERK expression. and protects rat brains against focal ischemia. Brain Res. 2012, 1448, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.K.; Lin, M.J.; Liao, P.H.; Yang, C.Y.; Lin, S.M.; Liu, S.M.; Lin, R.H.; Chih, C.L.; Huang, S.S. Caffeic acid phenethyl ester ameliorates cerebral infarction in rats subjected to focal cerebral ischemia. Life Sci. 2006, 78, 2758–2762. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fang, S.H.; Ye, Y.L.; Chu, L.S.; Zhang, W.P.; Wang, M.L.; Wei, E.Q. Caffeic acid ameliorates early and delayed brain injuries after focal cerebral ischemia in rats. Acta Pharmacol. Sin. 2006, 27, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altug, M.E.; Serarslan, Y.; Bal, R.; Kontaş, T.; Ekici, F.; Melek, I.M.; Aslan, H.; Duman, T. Caffeic acid phenethyl ester protects rabbit brains against permanent focal ischemia by antioxidant action: A biochemical and planimetric study. Brain Res. 2008, 1201, 135–142. [Google Scholar] [CrossRef]
- Lee, K.; Lee, B.J.; Bu, Y. Protective Effects of Dihydrocaffeic Acid, a Coffee Component Metabolite, on a Focal Cerebral Ischemia Rat Model. Molecules 2015, 20, 11930–11940. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Shi, B.; Luo, W.; Yang, J. The protective effect of caffeic acid on global cerebral ischemia-reperfusion injury in rats. Behav. Brain Funct. 2015, 11, 18. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.Y.; Ho, T.Y.; Lee, E.J.; Su, S.Y.; Tang, N.Y.; Hsieh, C.L. Ferulic acid reduces cerebral infarct through its antioxidative and anti-inflammatory effects following transient focal cerebral ischemia in rats. Am. J. Chin. Med. 2008, 36, 1105–1119. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Su, S.Y.; Tang, N.Y.; Ho, T.Y.; Chiang, S.Y.; Hsieh, C.L. Ferulic acid provides neuroprotection against oxidative stress-related apoptosis after cerebral ischemia/reperfusion injury by inhibiting ICAM-1 mRNA expression in rats. Brain Res. 2008, 1209, 136–150. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Su, S.Y.; Tang, N.Y.; Ho, T.Y.; Lo, W.Y.; Hsieh, C.L. Ferulic acid inhibits nitric oxide-induced apoptosis by enhancing GABA(B1) receptor expression in transient focal cerebral ischemia in rats. Acta Pharmacol. Sin. 2010, 31, 889–899. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, H.; Wang, T.; Jiang, N.; Yu, P.; Chong, Y.; Fu, F. Ferulic acid ameliorates nerve injury induced by cerebral ischemia in rats. Exp. Ther. Med. 2015, 9, 972–976. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.Y.; Tang, N.Y.; Kao, S.T.; Hsieh, C.L. Ferulic Acid Administered at Various Time Points Protects against Cerebral Infarction by Activating p38 MAPK/p90RSK/CREB/Bcl-2 Anti-Apoptotic Signaling in the Subacute Phase of Cerebral Ischemia-Reperfusion Injury in Rats. PLoS ONE 2016, 11, e0155748. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Zhang, R.; Li, Y.; Li, Y.; Yang, Z.; Yang, H. Ferulic acid exerts neuroprotective effects against cerebral ischemia/reperfusioninduced injury via antioxidant and anti-apoptotic mechanisms in vitro and in vivo. Int. J. Mol. Med. 2017, 40, 1444–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guven, M.; Aras, A.B.; Akman, T.; Sen, H.M.; Ozkan, A.; Salis, O.; Sehitoglu, I.; Kalkan, Y.; Silan, C.; Deniz, M.; et al. Neuroprotective effect of p-coumaric acid in rat model of embolic cerebral ischemia. Iran. J. Basic Med. Sci. 2015, 18, 356–363. [Google Scholar] [PubMed]
- Sakamula, R.; Thong-Asa, W. Neuroprotective effect of p-coumaric acid in mice with cerebral ischemia reperfusion injuries. Metab. Brain Dis. 2018, 33, 765–773. [Google Scholar] [CrossRef]
- Lee, K.; Lee, J.S.; Jang, H.J.; Kim, S.M.; Chang, M.S.; Park, S.H.; Kim, K.S.; Bae, J.; Park, J.W.; Lee, B.; et al. Chlorogenic acid ameliorates brain damage and edema by inhibiting matrix metalloproteinase-2 and 9 in a rat model of focal cerebral ischemia. Eur. J. Pharmacol. 2012, 689, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Miao, M.; Cao, L.; Li, R.; Fang, X.; Miao, Y. Protective effect of chlorogenic acid on the focal cerebral ischemia reperfusion rat models. Saudi Pharm. J. 2017, 25, 556–563. [Google Scholar] [CrossRef]
- Liu, D.; Wang, H.; Zhang, Y.; Zhang, Z. Protective Effects of Chlorogenic Acid on Cerebral Ischemia/Reperfusion Injury Rats by Regulating Oxidative Stress-Related Nrf2 Pathway. Drug Des. Dev. Ther. 2020, 14, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.A.; Kang, J.B.; Park, D.J.; Kim, M.O.; Koh, P.O. Chlorogenic acid alleviates neurobehavioral disorders and brain damage in focal ischemia animal models. Neurosci. Lett. 2021, 760, 136085. [Google Scholar] [CrossRef]
- Fan, Y.; Li, Y.; Yang, Y.; Lin, K.; Lin, Q.; Luo, S.; Zhou, X.; Lin, Q.; Zhang, F. Chlorogenic acid promotes angiogenesis and attenuates apoptosis following cerebral ischaemia-reperfusion injury by regulating the PI3K-Akt signalling. Pharm. Biol. 2022, 60, 1646–1655. [Google Scholar] [CrossRef]
- Shah, M.A.; Kang, J.B.; Kim, M.O.; Koh, P.O. Chlorogenic acid alleviates the reduction of Akt and Bad phosphorylation and of phospho-Bad and 14-3-3 binding in an animal model of stroke. J. Vet. Sci. 2022, 23, e84. [Google Scholar] [CrossRef]
- Shah, M.A.; Kang, J.B.; Park, D.J.; Kim, M.O.; Koh, P.O. Chlorogenic acid alleviates cerebral ischemia-induced neuroinflammation via attenuating nuclear factor kappa B activation. Neurosci. Lett. 2022, 773, 136495. [Google Scholar] [CrossRef] [PubMed]
- Lapchak, P.A. The phenylpropanoid micronutrient chlorogenic acid improves clinical rating scores in rabbits following multiple infarct ischemic strokes: Synergism with tissue plasminogen activator. Exp. Neurol. 2007, 205, 407–413. [Google Scholar] [CrossRef]
- Liu, Q.S.; Deng, R.; Li, S.; Li, X.; Li, K.; Kebaituli, G.; Li, X.; Liu, R. Ellagic acid protects against neuron damage in ischemic stroke through regulating the ratio of Bcl-2/Bax expression. Appl. Physiol. Nutr. Metab. 2017, 42, 855–860. [Google Scholar] [CrossRef]
- Sun, J.; Li, Y.Z.; Ding, Y.H.; Wang, J.; Geng, J.; Yang, H.; Ren, J.; Tang, J.Y.; Gao, J. Neuroprotective effects of gallic acid against hypoxia/reoxygenation-induced mitochondrial dysfunctions in vitro and cerebral ischemia/reperfusion injury in vivo. Brain Res. 2014, 1589, 126–139. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Habtemariam, S.; Di Lorenzo, A.; Sureda, A.; Khanjani, S.; Nabavi, S.M.; Daglia, M. Post-Stroke depression modulation and in vivo antioxidant activity of gallic acid and its synthetic derivatives in a murine model system. Nutrients 2016, 8, 248. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Li, D.; Zhu, Z.; Sun, Y. Improved Neuroprotective effects of gallic acid-loaded chitosan nanoparticles against ischemic stroke. Rejuvenation Res. 2019, 23, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Mirshekari Jahangiri, H.; Sarkaki, A.; Farbood, Y.; Dianat, M.; Goudarzi, G. Gallic acid affects blood-brain barrier permeability, behaviors, hippocampus local EEG, and brain oxidative stress in ischemic rats exposed to dusty particulate matter. Environ. Sci. Pollut. Res. 2020, 27, 5281–5292. [Google Scholar] [CrossRef]
- Abdelsalam, S.A.; Renu, K.; Zahra, H.A.; Abdallah, B.M.; Ali, E.M.; Veeraraghavan, V.P.; Sivalingam, K.; Ronsard, L.; Ammar, R.B.; Vidya, D.S.; et al. Polyphenols Mediate Neuroprotection in Cerebral Ischemic Stroke—An Update. Nutrients 2023, 15, 1107. [Google Scholar] [CrossRef] [PubMed]
- Rezai-Zadeh, K.; Shytle, R.D.; Bai, Y.; Tian, J.; Hou, H.; Mori, T.; Zeng, J.; Obregon, D.; Town, T.; Tan, J. Flavonoid-mediated presenilin-1 phosphorylation reduces Alzheimer’s disease β-amyloid production. J. Cell. Mol. Med. 2009, 13, 574–588. [Google Scholar] [CrossRef]
- Mori, T.; Koyama, N.; Guillot-Sestier, M.-V.; Tan, J.; Town, T. Ferulic Acid Is a Nutraceutical β-Secretase Modulator That Improves Behavioral Impairment and Alzheimer-like Pathology in Transgenic Mice. PLoS ONE 2013, 8, e55774. [Google Scholar] [CrossRef] [Green Version]
- Ogunsuyi, O.B.; Oboh, G.; Oluokun, O.O.; Ademiluyi, A.O.; Ogunruku, O.O. Gallic Acid Protects against Neurochemical Alterations in Transgenic Drosophila Model of Alzheimer’s Disease. Adv. Tradit. Med. 2020, 20, 89–98. [Google Scholar] [CrossRef]
- Zheng, Q.; Kebede, M.T.; Kemeh, M.M.; Islam, S.; Lee, B.; Bleck, S.D.; Wurfl, L.A.; Lazo, N.D. Inhibition of the Self-Assembly of Aβand of Tau by Polyphenols: Mechanistic Studies. Molecules 2019, 24, 2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harakeh, S.; Qari, M.H.; Ramadan, W.S.; Al Jaouni, S.K.; Almuhayawi, M.S.; Al Amri, T.; Ashraf, G.M.; Bharali, D.J.; Mousa, S.A. A Novel Nanoformulation of Ellagic Acid Is Promising in Restoring Oxidative Homeostasis in Rat Brains with Alzheimer’s Disease. Curr. Drug Metab. 2021, 22, 299–307. [Google Scholar]
- Rashno, M.; Gholipour, P.; Salehi, I.; Komaki, A.; Rashidi, K.; Khoshnam, S.E.; Ghaderi, S. P-Coumaric acid mitigates passive avoidance memory and hippocampal synaptic plasticity impairments in aluminum chloride-induced Alzheimer’s Disease rat model. J. Funct. Foods 2022, 94, 105117. [Google Scholar] [CrossRef]
- Hussain, H.; Ahmad, S.; Shah, S.W.A.; Ghias, M.; Ullah, A.; Rahman, S.U.; Kamal, Z.; Khan, F.A.; Khan, N.M.; Muhammad, J.; et al. Neuroprotective Potential of Synthetic Mono-Carbonyl Curcumin Analogs Assessed by Molecular Docking Studies. Molecules 2021, 26, 7168. [Google Scholar] [CrossRef]
- Hussain, H.; Ahmad, S.; Shah, S.W.A.; Ullah, A.; Ali, N.; Almehmadi, M.; Ahmad, M.; Khalil, A.A.K.; Jamal, S.B.; Ahmad, H.; et al. Attenuation of Scopolamine-Induced Amnesia via Cholinergic Modulation in Mice by Synthetic Curcumin Analogs. Molecules 2022, 27, 2468. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, R.; Saitou, K.; Suzukamo, C.; Osaki, N.; Asada, T. Effect of Chlorogenic Acids on Cognitive Function in Mild Cognitive Impairment: A Randomized Controlled Crossover Trial. J. Alzheimers Dis. 2019, 72, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Riche, K.; Lenard, N.R. Quercetin’s effects on glutamate cytotoxicity. Molecules 2022, 27, 7620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, J.; Yang, F.; Li, S.; Ma, W.; Chang, X.; Yang, L. Neuroprotective Effects of Quercetin on Ischemic Stroke: A Literature Review. Front. Pharmacol. 2022, 13, 854249. [Google Scholar] [CrossRef]
- Bauer, L.; Kohlich, A.; Hirschwehr, R.; Siemann, U.; Ebner, H.; Scheiner, O.; Kraft, D.; Ebner, C. Food Allergy to Honey: Pollen or Bee Products?: Characterization of Allergenic Proteins in Honey by Means of Immunoblotting. J. Allergy Clin. Immunol. 1996, 97, 65–73. [Google Scholar] [CrossRef]
- Aguiar, R.; Duarte, F.C.; Mendes, A.; Bartolomé, B.; Barbosa, M.P. Anaphylaxis Caused by Honey: A Case Report. Asia Pac. Allergy 2017, 7, 48–50. [Google Scholar] [CrossRef] [Green Version]
- Di Costanzo, M.; De Paulis, N.; Peveri, S.; Montagni, M.; Canani, R.B.; Biasucci, G. Anaphylaxis Caused by Artisanal Honey in a Child: A Case Report. J. Med. Case Rep. 2021, 15, 235. [Google Scholar] [CrossRef]
- Al-Waili, N.; SAlom, K.; Al-Ghamdi, A.; Ansari, M.J. Antibiotic, pesticide, and microbial contaminants of honey: Human health hazards. Sci. World J. 2012, 2012, 930849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaylaci, S.; Kocayigit, I.; Aydin, E.; Osken, A.; Genc, A.B.; Cakar, M.A.; Tamer, A. Clinical and laboratory findings in mad honey poisoning: A single center experience. Niger. J. Clin. Pract. 2014, 17, 589–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dur, A.; Sonmez, E.; Civelek, C.; Tutkdogan, K.A.; Vatankulu, M.A.; Sogut, O. Mad honey intoxication mimicking acute coronary syndrome. J. Pak. Med. Assoc. 2014, 9, 1078–1080. [Google Scholar]
- Karabag, T.; Sayin, R.; Yavuz, N.; Aktop, Z. Type 2 myocardial infarction after ingestion of md honey in a patient with normal coronary arteries. Korean J. Intern. Med. 2015, 30, 540–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenler, A.K. Cardiac effects of mad honey poisoning and its management in emergency department: A review from Turkey. Cardiovasc. Toxicol. 2016, 16, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Morand, C.; Manach, C.; Remesy, C. Absorption and metabolism of polyphenols in the gut and impact on health. BioMed Pharmacother. 2002, 56, 276–282. [Google Scholar] [CrossRef]
- Renouf, M.; Marmet, C.; Giuffrida, F.; Lepage, M.; Barron, D.; Beaumont, M.; Williamson, G.; Dionisi, F. Dose-response plasma appearance of coffee chlorogenic and phenolic acids in adults. Mol. Nutr. Food Res. 2014, 58, 301–309. [Google Scholar] [CrossRef]
- Thilakarathna, S.H.; Rupasinghe, H.P. Flavonoid bioavailability and attempts for bioavailability enhancement. Nutrients 2013, 5, 3367–3387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.K.; Barreto, T.A.; Martinez, F.J.; Comstock, A.T.; Sajjan, U.S. Randomised clinical trial to determine the safety of quercetin supplementation in patients with chronic obstructive pulmonary disease. BMJ Open Respir. Res 2020, 7, e000392. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Substance | Model | Treatment | Effects | References |
|---|---|---|---|---|
| Honey | ||||
| Malaysian Tualang honey | p2VO | Pre: 1.2 g/kg for 10 days with Post: 10 weeks | ↓ Hippocampal CA1 region damage ↑ Spatial learning, memory performance | [50,161] |
| Flavonoids | ||||
| Quercetin | tMCAO | Post: 20 mg/kg/d for 3 days | ↓ Oxidative stress, necrosis, apoptosis, brain edema, brain injury, neurological deficits | [165] |
| Quercetin | pMCAO | Post: 30 mg/kg single dose | ↓ Brain injury | [162] |
| Quercetin | Photothrombotic model | Post: 25 µmol/kg every 12 h for 3 days | ↓ BBB injury, brain edema, neurological deficits ↑ Functional outcomes | [164] |
| Quercetin | 2VO | Pre: 50 mg/kg 30 min before and immediately post-ischemia, then daily for 2 days | ↓ BBB injury, delayed neuronal damage in CA1, CA2, brain injury | [163] |
| Quercetin | tMCAO | Pre: 10 mg/kg 30 min before | ↓ Neurological deficits, behavioral changes ↑ Parvalbumin expression | [169] |
| Quercetin | tMCAO | Pre: 10 mg/kg 1 h before | ↓ Brain edema, damage in brain cortex, neurological deficits ↑ Thioredoxin, interaction of apoptosis signal-regulating kinase 1 and thioredoxin | [167] |
| Quercetin | tMCAO | Pre: 10 mg/kg 30 min before | ↓ Infarct volume, neurological deficit ↑ Protein phosphatase 2A | [166] |
| Quercetin | tMCAO | Post: 10, 30, 50 mg/kg at the onset of reperfusion | ↓ BBB injury, ROS, infarct volume, neurological deficit | [170] |
| Quercetin | pMCAO | Pre: 10 mg/kg 1 h before | ↓ Intracellular calcium overload, glutamate excitotoxicity, caspase-3. | [168] |
| Myricetin | tMCAO | Pre: 20 mg/kg 2 h before and daily for 2 days after ischemia Pre: 25 mg/kg daily for 7 days | ↓ Oxidative stress, apoptosis, neuronal loss, inflammation, infarct volume, ROS, neurological deficits ↑ Antioxidant enzymes, mitochondrial function, Nrf2 nuclear translocation, HO-1 expression | [171] |
| Myricetin | pMCAO | Pre: 1 mg/kg, 5 mg/kg, 25 mg/kg for 7 days | ↓ IL-1β, IL-6, TNF-α, MDA, p38 MAPK, NF-κB/p65, apoptosis, infarct area, neurological deficit ↑ GSH/GSSG ratio, SOD, phosphorylated AKT | [174] |
| Myricetin | tMCAO | Pre: 25 mg/kg for 7 days | ↓ Excitotoxicity, oxidative stress, inflammation, apoptosis | [173] |
| Kaempferol | tMCAO | Pre: 10,15 μmol/l 30 min before and immediately after ischemia Post: 7.5, 10 mg/kg single dose Post: 25, 50, 100 mg/kg daily for 7 days | ↓ Metalloproteinase, anti-laminin staining, nitrosative-oxidative stress, caspase-9, apoptosis, poly-(ADP-ribose) polymerase, amyloid protein precursor, glial fibrillary acidic protein, phosphorylated STAT3, NF-κB p65, nuclear content of NF-κB p65, tumor necrosis factor α, interleukin 1β, intercellular adhesion molecule 1, matrix metallopeptidase 9, inducible nitric oxide synthase, myeloperoxidase, neuroinflammation, BBB injury, microglia activity, brain injury, neurological deficits | [175,176,181] |
| Naringenin | pMACO | Pre: 100 mg/kg daily for 4 days | ↓ Neuroinflammation, edema, NOD2, RIP2, NF-κB, MMP-9, BBB injury, infarct volume, neurological deficits ↑ Claudin-5 | [179] |
| Naringenin | tMCAO | Pre: 50 mg/kg daily for 21 days Post: 80 µM single dose | ↓ Apoptosis, oxidative stress, edema, NF-κB, myeloperoxidase, nitric oxide, cytokines, neuroinflammation, glial activation, injury volume, neurological deficits ↑ Cortical neurons proliferation | [178,180] |
| Luteolin | tMCAO | Post: 20, 40, 80 mg/kg 0 and 12 h after ischemia | ↓ Injury volume, edema, IL-1β, TNF-α, iNOS, COX-2, NF-κB, inflammation, neurological deficits ↑ Nrf2, PPARγ. | [181] |
| Luteolin | tMCAO | Post: 5, 10, 25 mg/kg single dose | ↓ Oxidative stress, apoptosis, mRNA and protein of MMP9, infarct volume, neurological deficits ↑ PI3K/Akt | [182] |
| Luteolin | pMCAO | Post: 10, 25 mg/kg single dose post-ischemia | ↓ MDA, Bax, oxidative stress, apoptosis, edema, infarct volume, neurological deficits ↑ SOD1, CAT, Bcl-2, claudin-5 | [183] |
| Luteolin | pMCAO | Post: 5, 10 mg/kg 0 h and daily for 3 days survival | ↓ Brain edema, TLR4, TLR5, p-p38, NF-κB infarct size, neurological deficit ↑Phospho-ERK | [184] |
| Phenolic acids | ||||
| Caffeic acid | tMCAO | Pre: 10, 50 mg/kg 30 min before, 0, 1, 2 h, and every 12 h for 4 days after ischemia Pre: 0.1, 1, 10 µg/kg 15 min before, single dose | ↓ Neuroinflammation, leukotrienes, neuron loss, 5-lipoxygenase, astrocyte proliferation, infarct volume, brain atrophy, neurological dysfunction ↑ NO | [185,186] |
| Caffeic acid | pMCAO | Post: 10 µmol/kg daily for 7 days | ↓ MDA, CAT, XO, oxidative stress, lipid peroxidation, infarct size, neurological deficits ↑ GSH, NO | [187] |
| Caffeic acid | Global ischemia | Post: 10, 30, 50 mg/kg single dose | ↓ Hippocampus injury, NF-κBp65, MDA, 5-LO, oxidative stress, memory deficits ↑ SOD | [189] |
| Caffeic acid | tMCAO | Post: 3, 10, 30 mg/kg 0, 2 h after ischemia | ↓ MMP-2, MMP-9, edema, damage in penumbra, infarct volume, sensory-motor deficits, behavioral deficits | [188] |
| Ferulic acid | Global ischemia | Post: 28, 56, 112 mg/kg daily for 5 days | ↓ Oxidative stress, mRNA caspase 3, mRNA Bax, hippocampus apoptosis, memory impairment ↑ mRNA Bcl-2, SOD | [195] |
| Ferulic acid | tMCAO | Post: 50, 100, 200 mg/kg daily for 7 days | ↓ Hippocampus injury, neurological deficits ↑ In hippocampus, EPO and granulocyte colony-stimulating factor | [193] |
| Ferulic acid | tMCAO | Post: 100 mg/kg 0 h post-ischemia Post: 100 mg/kg 2 h post-ischemia Post: 100 mg/kg 24 h Pre: 100 mg/kg 24 h before ischemia Pre: 100 mg/kg 2 h before ischemia | Pretreatment 2 h before ischemia and posttreatment 2 h after ischemia ↓ Bax, astrocytosis, infarction volume | [194] |
| Ferulic acid | tMCAO | Pre: 80, 100 mg/kg Post: 100 mg/kg 30 min after ischemia | ↓ Superoxide radicals, ICAM-1, NF-κB, infarct size, neurological deficits | [190] |
| Ferulic acid | tMCAO | Post; 100 mg/kg 0 h after ischemia | ↓ ICAM-1 mRNA, Mac-1 mRNA, Mac-1, 4-HNE, 8-OHdG positive cells, TUNEL positive cells, caspase 3, microglia activity, apoptosis, macrophages, oxidative stress, inflammation | [191] |
| Ferulic acid | tMCAO | Post: 100 mg/kg 0 h, or 30 min or 2 h after ischemia | ↓ PSD-95, nNOS, iNOS, nitrotyrosine, caspase-3, apoptosis, Bax, cytochrome c, MAP kinase ↑ Gamma-aminobutyric acid type B receptor, therapeutic window | [192] |
| P-coumaric acid | pMCAO | Post: 100 mg/kg single dose | ↓ Oxidative damage, MDA, apoptosis, caspase-3, caspase-9, edema, infarct volume, neurological deficits ↑ SOD, NRF-1 | [196] |
| P-coumaric acid | Global ischemia | Pre: 100 mg/kg for 2 weeks before ischemia | ↓ MDA, oxidative stress, hippocampal neuronal death, infarct volume, brain damage ↑ Catalase, superoxide dismutase | [197] |
| Chlorogenic acid | tMCAO | Post: 3, 10, 30 mg/kg 0, 2 h after ischemia Pre: 15, 30, 60 mg/kg for 1 week | ↓ BBB, oxidative stress, MMP-2, MMP-9, edema, infarct volume, sensory-motor deficits, behavioral deficits | [198,199] |
| Chlorogenic acid | tMCAO | Post: 30 mg/kg 2 h after ischemia | ↓ Cytochrome c, caspase-3, cleaved caspase-3, neurological deficits ↑ Phospho-PDK1, phospho-Akt, phospho-Bad | [202] |
| Chlorogenic acid | tMCAO | Pre: 15, 30, 60 mg/kg once a day for 1 week | ↓ Mortality, infarction area, injury of hippocampus, cortex lesions, neurological deficit ↑ EPO, HIF-1α, NGF | [199] |
| Chlorogenic acid | Repeated global ischemia | Post: 20, 100, 500 mg/kg single dose | ↓ Oxidative stress, apoptosis, MMPs, infarct volume, memory deficits ↑ SOD, GSH | [200] |
| Chlorogenic acid | Embolic strokes with rtPA | Post: 50 mg/kg 5 min, 1, 3 h after ischemia | ↓ Behavioral deficits ↑ Therapeutic window | [205] |
| Chlorogenic acid | tMCAO | Post: 30 mg/kg 2 h after ischemia | ↓ TUNEL-positive cells, caspase-3 and -7, oxidative stress, edema, infarct size, neurological damage | [201] |
| Chlorogenic acid | tMCAO | Post: 30 mg/kg 2 h post-ischemia | ↓ Reactive oxygen species, oxidative stress, NF-κB, IL-1β, TNF-α, microglia, astrocyte activation, inflammation, cortex pathology | [204] |
| Chlorogenic acid | tMCAO | Post: 30 mg/kg/d 3 days after ischemia | ↓ Cerebral cortex apoptosis, infarct volume ↑ Angiogenesis, VEGFA, PI3K/Akt signaling | [202] |
| Gallic acid | tMCAO | Pre: 50 mg/kg daily for 7 days Pre: 50 mg/kg single dose | ↓ Oxidative stress, apoptosis, neuroinflammation, mitochondrial dysfunction, injury size, neurological deficits | [207,209] |
| Gallic acid | Global ischemia | Pre: 100 mg/kg/d for 10 days | ↓ BBB injury, MDA, oxidative stress, hippocampus EEG changes, anxiety, behavioral deficits | [210] |
| Gallic acid | Global ischemia | Post: 25, 50 mg/kg/d for 1 week | ↓ Oxidative stress, depressive symptoms | [208] |
| Ellagic acid | Photothrombotic model | Pre: 10, 30 mg/kg 24 h before and 0 h post-ischemia | ↓ Apoptotic cells, infarct size, neurological deficits | [206] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pluta, R.; Miziak, B.; Czuczwar, S.J. Apitherapy in Post-Ischemic Brain Neurodegeneration of Alzheimer’s Disease Proteinopathy: Focus on Honey and Its Flavonoids and Phenolic Acids. Molecules 2023, 28, 5624. https://doi.org/10.3390/molecules28155624
Pluta R, Miziak B, Czuczwar SJ. Apitherapy in Post-Ischemic Brain Neurodegeneration of Alzheimer’s Disease Proteinopathy: Focus on Honey and Its Flavonoids and Phenolic Acids. Molecules. 2023; 28(15):5624. https://doi.org/10.3390/molecules28155624
Chicago/Turabian StylePluta, Ryszard, Barbara Miziak, and Stanisław J. Czuczwar. 2023. "Apitherapy in Post-Ischemic Brain Neurodegeneration of Alzheimer’s Disease Proteinopathy: Focus on Honey and Its Flavonoids and Phenolic Acids" Molecules 28, no. 15: 5624. https://doi.org/10.3390/molecules28155624
APA StylePluta, R., Miziak, B., & Czuczwar, S. J. (2023). Apitherapy in Post-Ischemic Brain Neurodegeneration of Alzheimer’s Disease Proteinopathy: Focus on Honey and Its Flavonoids and Phenolic Acids. Molecules, 28(15), 5624. https://doi.org/10.3390/molecules28155624